

Abstract
Postischemic myocardial fibrosis is a factor for the development of cardiac dysfunction and malignant cardiac arrhythmias, and no effective therapy is currently available. Circular RNAs are emerging as important epigenetic players in various biological functions; however, their roles in cardiac fibrosis are unknown. With the use of a rat model of postischemic myocardial fibrosis, we identified an increase in circHNRNPH1 in the ischemic myocardium after myocardial infarction, particularly in cardiac fibroblasts. In cardiac fibroblasts, circHNRNPH1 was responsive to transforming growth factor β1 (TGF-β1), the principal profibrotic factor. The downregulation of circHNRNPH1, in contrast to its overexpression, promoted myofibroblast migration and α-smooth muscle actin and collagen I expression and inhibited myofibroblast apoptosis. The recombinant adeno-associated virus 9 (rAAV9)-mediated, cardiac-specific knockdown of circHNNRPH1 accordingly facilitated cardiac fibrosis and aggravated cardiac dysfunction. Mechanistically, circHNRNPH1 colocalized with and sponged microRNA (miR)-216-5p in the cytoplasm of cardiac fibroblasts to induce SMAD7 (protein family of signal transduction component of the canonical transforming growth factor-β signaling pathway) expression, accelerating the degradation of TGF-β receptor I. Thus, our results indicated that circHNRNPH1 negatively regulates the fibrogenesis of cardiac fibroblasts and may provide a new therapeutic strategy for postischemic myocardial fibrosis.
Keywords: cardiac fibrosis, circHNRNPH1, apoptosis, miR216-5p
Graphical Abstract
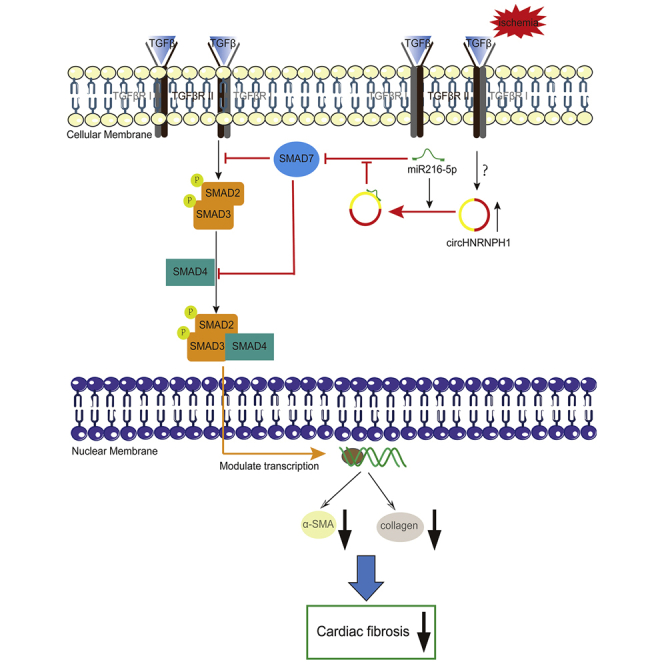
So far, there are no effective therapies for cardiac fibrosis, a severe, adverse cardiac ischemia. circHNRNPH1 was found first in the study and increased obviously in postischemia cardiac myocardium, which promoted SMAD7 expression as an miR216-5p sponge to accelerate TβRI degradation and decrease SMAD2/3 phosphorylation and then inhibited cardiac fibrosis by reducing the expression of α-SMA and collagen I. circHNRNPH1 was a potential therapeutic target for cardiac fibrosis.
Introduction
Myocardial fibrosis is widespread in ischemic heart disease and is characterized by excessive deposition of myofibroblast-secreted collagen in the cardiac extracellular matrix (ECM). Following scar formation after myocardial infarction (MI), myocardial fibrosis results in the loss of ventricular compliance and thereafter, cardiac dysfunction. Therefore, fibrosis and scarring synergistically contribute to the initiation and maintenance of cardiac arrhythmias and even to sudden cardiac death.1, 2, 3
Fibrosis originates from the activation of cardiac fibroblasts (CFs), which are highly enriched in the heart.4 Once activated, quiescent fibroblasts transdifferentiate into myofibroblasts, which express the contractile protein α-smooth muscle actin (α-SMA) and secrete ECM proteins such as periostin, collagen I and III, fibronectin, and fibronectin isoforms.5,6 The expression of these ECM proteins is mainly regulated by transforming growth factor β1 (TGF-β1), the principal profibrotic factor secreted by macrophages, monocytes, and resident fibroblasts. In contrast to the positive effects of certain members of the SMAD family, such as SMAD2/3, on TGF-β1 signaling, SMAD7 negatively regulates SMAD2/3 phosphorylation by blocking access to TGF-β receptor 1 (TβRI) or inducing the degradation of TβRI via ubiquitination by recruiting smurf2.7, 8, 9
The key roles of epigenetic events in controlling profibrotic gene expression have attracted increased attention,10 but the underlying epigenetic regulators and their therapeutic potential for treating fibrotic cardiac diseases remain largely unknown. Circular RNAs (circRNAs) are emerging as a novel class of epigenetic regulators and are characterized by stability in blood, cells, and organisms. This closed-loop, noncoding RNA is formed by backsplicing and ligating the upstream 5′ donor site to a downstream 3′ acceptor site through a phosphodiester bond, resulting in no 5′ or 3′ terminus.11 These molecules are evolutionarily conserved across eukaryotic cells and have various biological functions. In the cardiovascular system, circRNAs, such as circHRCR, circCdr1as, circFoxo3, and circANRIL, have been documented to participate in the development of pathological cardiac remodeling.12, 13, 14, 15, 16 The roles of circRNAs in cardiac fibrosis await examination.
In this study, we identified an increase in circHNRNPH1 expression in postischemic myocardial tissues, predominantly in CFs. Enforced expression of circHNRNPH1 depressed TGF-β1 signaling to restrict the differentiation of CFs into myofibroblasts through the microRNA (miR)-216-5p-SMAD7-mediated degradation of TβRI, whereas a reduction in circHNRNPH1 worsened cardiac fibrosis and dysfunction.
Results
circHNRNPH1 Is Increased in the Postischemic Heart and Predominantly Expressed in CFs
A set of cardiac-enriched circRNAs have been described previously,17 but few have been studied in relation to cardiac fibrosis. With the use of a well-established rat model of MI to predict circRNA expression by high-throughput sequencing, we found that circHNRNPH1, a novel circRNA, was upregulated in fibrotic tissue. Importantly, this molecule was resistant to RNase R, validating the circular characteristics of circHNRNPH1 (Figure 1A). Next, we integrated an in silico prediction with sequencing to define the form of circHNRNPH1. RNA Sanger sequencing and BLAST analysis on the NCBI website revealed the backspliced junction of circHNRNPH1 and indicated that circHNRNPH1 was formed by exon 6 and exon 7 of the HNRNPH1 gene (Figures 1B and 1C). As shown in Figure 1D, the expression of circHNRNPH1 was significantly increased in the ischemic ventricular tissues compared with the sham controls. There was no change in HNRNPH1 mRNA (Figure 1E). Additionally, circHNRNPH1 was mainly expressed in CFs compared with vessel endothelial cells and cardiomyocytes (CMs) (Figure 1F).
Figure 1.

circHNRNPH1 Was Increased in the Postischemic Heart
(A) Validation of circHNRNPH1 by RNase R. (B) Schematic illustration of circHNRNPH1 formation. (C) Validation of the junction site of circHNRNPH1 by Sanger sequencing. (D and E) Cardiac circHNRNPH1 (D) and linear HNRNPH1 (E) expression assays in the rat ischemic heart at the 5th week after myocardial infarction (MI). Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (F) circHNRNPH1 expression in primary cardiomyocytes, cardiac fibroblasts, and vascular endothelial cells (VECs). Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05).
circHNRNPH1 Regulates the Fibrogenic Activity of CFs
To examine the function of circHNRNPH1, we transfected circHNRNPH1 or small interfering (si)-circHNRNPH1 into isolated primary CFs in vitro. The overexpression of circHNRNPH1 downregulated the RNA and protein levels of α-SMA and collagen I, which were upregulated by knockdown of the circRNA (Figures 2A–2D). Additionally, wound healing and Transwell assays indicated that circHNRNPH1 overexpression decreased the migratory capacity of CFs, in contrast to the knockdown of circHNRNPH1 (Figures 2E–2I). In summary, circHNRNPH1 regulates the fibrogenic activity of CFs.
Figure 2.

circHNRNPH1 Inhibited Cardiac Fibroblast Fibrogenic Activity In Vitro
(A and B) Quantitative PCR examination of α-SMA (A) and collagen I (B) mRNA expression with circHNRNPH1overexpression or knockdown. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (C and D) Western blot (C) and densitometric (D) assays of α-SMA and collagen I expression with circHNRNPH1 overexpression and knockdown. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (E–G) Migration assay (E), distance between edges (F) and migration velocity analysis (G) of cardiac fibroblasts at 12 h and 24 h after circHNRNPH1 overexpression or knockdown. The images are representative of three independent experiments. Magnification 40×. Scale bars, 100 μm. si-NC, si-circHNRNPH1-NC; si-circ, si-circHNRNPH1; circ-NC, circHNRNPH1-NC; circ, circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (H and I) Transwell analysis (H) and cell number evaluation (I) of cardiac fibroblasts at 24 h after circHNRNPH1 overexpression or knockdown. The images are representative of three independent experiments. Magnification 100×. Scale bars, 100 μm. The number of migrated cells was plotted as the average number of cells per field of view from three different experiments. si-NC, si-circHNRNPH1-NC; si-circ, si-circHNRNPH1; circ-NC, circHNRNPH1-NC; circ, circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05).
Downregulation of Cardiac circHNRNPH1 Aggravates Cardiac Fibrosis and Dysfunction
To determine the in vivo effects of circHNRNPH1 on myocardial fibrogenesis, we directly injected recombinant adeno-associated virus 9 (rAAV9)-short hairpin (sh)-circHNRNPH1 into the peri-infarcted area immediately after surgery to induce MI in rats. At the 5th week post-MI, we found that this approach could downregulate circHNRNPH1 in the myocardium but had little effect on mRNA-HNRNPH1 (Figures 3A and 3B). Moreover, echocardiography was applied to assess cardiac function and indicated the deterioration of cardiac function in rats with circHNRNPH1 knockdown (Figure 3C), showing a decreased cardiac ejection fraction (EF) and fractional shortening (FS) and increased left-ventricular end-diastolic diameter (LVEDD), left-ventricular end-systolic diameter (LVESD), left-ventricular end-diastolic volume (LVEDV), and left-ventricular end-systolic volume (LVESV) (Figures 3D–3I). Additionally, cardiac circHNRNPH1 knockdown increased the mRNA levels of α-SMA and collagen I and the protein levels of TβRI, α-SMA, and collagen I but decreased SMAD7 protein expression in the ischemic myocardium (Figures 3J–3M). Myocardial structural analysis via Masson’s staining revealed aggravated fibrosis in the circHNRNPH1-knockdown myocardium (Figure 3N). Immunohistochemistry further demonstrated that circHNRNPH1 knockdown promoted the protein expression of α-SMA and collagen I (Figures 3O–3R). Thus, depression of circHNRNPH1 in the ischemic myocardium is detrimental in postischemic fibrosis.
Figure 3.

circHNRNPH1 Regulates Cardiac Fibrogenesis and Function Postinfarction
(A and B) The expression of circHNRNPH1 (A) and mRNA-HNRNPH1 (B) after rAAV9-sh-circHNRNPH1 injection into the peri-infarcted area. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (C) The cardiac function was assayed by transthoracic echocardiography at the 5th week post-MI. (D–I) Echocardiography assay. (D) EF, ejection fraction; (E) FS, fractional shortening; (F) LVEDD, left-ventricular end-diastolic diameter; (G) LVESD, left-ventricular end-systolic diameter; (H) LVEDV, left-ventricular end-diastolic volume; and (I) LVESV, left-ventricular end-systolic volume. Data are presented as the mean ± SD (n = 6 for the sham and rAAV9-vector + MI groups; n = 4 for the MI and rAAV9-sh-circ + MI groups, ∗p < 0.05). (J and K) The α-SMA (J) and collagen I (K) RNA expression assay by quantitative PCR. Data are presented as the mean ± SD (n = 6 for the sham and rAAV9-vector + MI groups; n = 4 for the MI and rAAV9-sh-circ + MI groups, ∗p < 0.05). (L and M) Western blot (L) and densitometric assays (M) of TGF-β receptor I (TβRI), α-SMA, collagen I, and SMAD7. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (N) The samples and Masson’s trichrome staining of the heart in the sham, MI, rAAV9-vector + MI, and rAAV9-sh-circ + MI groups. Magnification 100×. Scale bar, 1 mm. (O–R) Immunohistochemistry of α-SMA (O) and collagen I (Q) and evaluation of the expression of α-SMA (P) and collagen I (R). Magnification 200×. Scale bars, 100 μm.
circHNRNPH1 Is a Downstream Effector of Fibrotic TGF-β1 Signaling
TGF-β1 is the principal profibrotic factor that induces the differentiation of CFs to myofibroblasts and therefore collagen production and α-SMA expression.18, 19, 20, 21, 22, 23, 24, 25 Thus, we analyzed the expression of circHNRNPH1 and linear HNRNPH1 mRNA in isolated CFs exposed to TGF-β1 at different concentrations and observed upregulated expression of circHNRNPH1 and slight downregulation of linear HNRNPH1 mRNA (Figure 4A).
Figure 4.

circHNRNPH1 Was Regulated by TGF-β1 Signaling
(A) circHNRNPH1 and mRNA-HNRNPH1 expression assays of cardiac fibroblasts treated with TGF-β1 at different concentrations. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (B and C) Western blot (B) and densitometric (C) analyses of ADAR1 expression affected by TGF-β1 at different concentrations. ∗p < 0.05. (D) Assays of ADAR1 RNA expression affected by TGF-β1 at different concentrations. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (E and F) Quantitative PCR examination showing the effects of ADAR1 on HNRNPH1 mRNA (E) and circHNRNPH1 (F) expression. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (G) The expression of mRNA-HNRNPH1 and circHNRNPH1 with Staufen1 knockdown. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (H and I) The expression analysis of mRNA-HNRNPH1 (H) and circHNRNPH1 (I) regulated by ADAR1 with/without Staufen1 knockdown via quantitative PCR. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (J) Assay of Staufen1 RNA expression affected by different concentrations of TGF-β1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (K) Staufen1 protein expression was affected by TGF-β1 at different concentrations by western blot analyses. (L) Assay of TGF-β-induced circHNRNPH1 expression with either ADAR1 or Staufen1 overexpression. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05).
Next, we investigated the potential mechanism underlying the TGF-β1-mediated regulation of circHNRNPH1. We analyzed the flanking introns of exon 6 and exon 7 and found that they had a complementary sequence with enrichment of adenine-uracil (A-U) (Figure S1), which may be key to circHNRNPH1 formation and the docking site of adenosine deaminase acting on RNA 1 (ADAR1). ADAR1 regulates the transcription and translation of target genes by RNA editing via the conversion of adenosine to inosine.26 This enzyme affects circRNA formation, not only by impairing intron-pairing catalysis but also by changing the rate of transcription by inhibiting Staufen1-mediated mRNA decay.27,28
We conjectured that ADAR1 may be a potential regulator of TGF-β1-dependent circHNRNPH1 expression. We found that TGF-β1 inhibited the mRNA and protein expression of this molecule (Figures 4B–4D). ADAR1 overexpression could upregulate HNRNPH1 mRNA but downregulate circHNRNPH1, and ADAR1 knockdown induced the opposite effects on linear HNRNPH1 mRNA and circHNRNPH1 (Figures 4E and 4F). Furthermore, to test whether Staufen1 participated in ADAR1-mediated circHNRNPH1 biogenesis, we downregulated Staufen1 by transfecting siRNA into primary CFs. The results showed that Staufen1 knockdown could decrease HNRNPH1 mRNA expression and increase circHNRNPH1 expression (Figure 4G). Furthermore, when the ADAR1 overexpression plasmid or si-ADAR1 was cotransfected into primary CFs with si-Staufen1, Staufen1 knockdown weakened the effect of ADAR1 and enhanced the effect of si-ADAR1 on circHNRNPH1 and HNRNPH1 mRNA (Figures 4H and 4I). To verify whether Staufen1 directly mediates the regulation of circHNRNPH1 by TGF-β1, we also tested the expression of Staufen1 in primary CFs treated with TGF-β1. TGF-β1 substantially reduced the mRNA and protein expression of Staufen1 (Figures 4J and 4K). Either ADAR1 or Staufen1 inhibited TGF-β1-induced circHNRNPH1 formation (Figure 4L). We also studied whether circHNRNPH1 interacted with its parent RNA and found that HNRNPH1 mRNA knockdown decreased the expression of circHNRNPH1, and circHNRNPH1 positively regulated the expression of mRNA-HNRNPH1 (Figure S2). Thus, circHNRNPH1 is a downstream target of the profibrotic TGF-β1 signaling pathway, but the regulation of its biogenesis is complicated.
circHNRNPH1 Sponges miR216-5p to Promote SMAD7 Expression and Fibroblast Apoptosis
To assess further whether and how circHNRNPH1 affects profibrotic TGF-β1 signals, we adopted in silico prediction and experimental validation to identify its targets. The miRNA sponging effects underlie the circRNA-based regulation of the targeted transcript’s translation.13,14,29
With the use of RegRNA 2.0, we predicted miR216-5p as a potential target of circHNRNPH1, with 3 binding sites and a low minimum free energy (http://regrna2.mbc.nctu.edu.tw/detection_output.php). Then, we analyzed the stable secondary structure of circHNRNPH1 by using mfold software (http://unafold.rna.albany.edu/?q=mfold) and the site of circHNRNPH1 that interacts with miR216-5p (Figure S3). Fluorescence in situ hybridization (FISH) analysis demonstrated the physical colocalization of circHNRNPH1 with miR216-5p in the cytoplasm of primary CFs (Figure 5A). To verify the direct interaction between circHNRNPH1 and miR-216-5p, we constructed circHNRNPH1 fragments or circHNRNPH1 mutants (Mut) and inserted them downstream of the luciferase reporter gene (LUC + circHNRNPH1) (Figure 5B). miR216-5p mimics and the reporters were cotransfected in HEK293T cells. Compared with the control, miR216-5p decreased the luciferase activity by at least 20% (Figure 5C). In addition, to test whether miR216-5p could bind circHNRNPH1, we used biotin-coupled miR216-5p to test whether miR216-5p could pull down circHNRNPH1 (Figure 5D). The results showed that circHNRNPH1 was enriched more in the miR216-5p-captured fraction than with the introduction of a mutation that disrupted the base pairing between circHNRNPH1 and miR216-5p (Figure 5E). Given that Argonaute 2 (AGO2) buttresses circRNA and miRNA interactions to form RNA-induced silencing complexes,30 we also conducted AGO2-based RNA-binding protein immunoprecipitation (RIP) assays using an AGO2 antibody to identify the effects of enrichment between circHNRNPH1 and miR-216-5p. As shown in Figures 5F and 5G, circHNRNPH1 overexpression resulted in increased enrichment of miR216-5p in the AGO2-immunoprecipitated transcripts, as well as miR216-5p mimics. Together, these findings indicated that miR216-5p is a target of circHNRNPH1.
Figure 5.

circHNRNPH1 Mediated Primary Cardiac Fibroblast Apoptosis by Sponging miR216-5p
(A) FISH experiment showing that circHNRNPH1 colocalized with miR216-5p in the cytoplasm of primary cardiac fibroblasts. Red, circHNRNPH1; green, miR216-5p; blue, 4′,6-diamidino-2-phenylindole (DAPI). The images are representative of three independent experiments. Magnification 400×. Scale bars, 50 μm. (B) Schematic graphics of the pmirGLO plasmids with circHNRNPH1-WT and circHNRNPH1-Mut for the dual-luciferase reporter assay. (C) Dual-luciferase reporter assay. The luciferase activity was analyzed by quantitative PCR. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (D) Diagram of the circHNRNPH1 sponge with biotin-coupled miR-216-5p WT and its Mut (top). (Bottom) The WT (bio-216-wt) and Mut (bio-216-mut) biotin-coupled miR-216-5p sequences are shown. (E) The WT (bio-216-wt) or Mut (bio-216-mut) biotin-coupled miR-216-5p was transfected into primary cardiac fibroblasts. After streptavidin capture, circHNRNPH1 and GAPDH mRNA were quantified by quantitative PCR, and the relative pellet/input ratio was plotted. Input: 10% samples were loaded; pellet: 100% samples were loaded. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (F and G) RIP assay. AGO2 RNA immunoprecipitation (IP) in primary cardiac fibroblasts transfected with circHNRNPH1 or its Mut (F) or with miR216-5p or its Mut (G). The level of circHNRNPH1 or miR216-5p was quantified by quantitative PCR. RIP represents an RNA-binding protein IP assay; input: 10% samples were loaded. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (H) Quantitative PCR examination of the expression of SMAD7 regulated by circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (I and J) Western blot (I) and densitometric analysis (J) of SMAD7 expression regulated by circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (K and L) Western blot (K) and densitometric analysis (L) of SMAD7 expression regulated by circHNRNPH1 via miR216-5p. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (M and N) Western blot (M) and densitometric analysis (N) of apoptotic proteins in primary cardiac fibroblasts with circHNRNPH1 overexpression or knockdown. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (O and P) TUNEL assay (O) and quantitative analysis (P) of primary cardiac fibroblasts with circHNRNPH1 overexpression or knockdown. si-NC, si-circHNRNPH1-NC; si-circ, si-circHNRNPH1; circ-NC, circHNRNPH1-NC; circ, circHNRNPH1; DAPI, 4′,6-diamidino-2-phenylindole. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). The images are representative of three independent experiments. Green, TUNEL-positive cells; blue, DAPI. Magnification 200×. Scale bars, 50 μm. (Q) Primary cardiac fibroblast apoptosis assay by flow cytometry with overexpression or knockdown of circHNRNPH1. si-NC, si-circHNRNPH1-NC; si-circ, si-circHNRNPH1; circ-NC, circHNRNPH1-NC; circ, circHNRNPH1.
SMAD7, the negative effector of TGF-β signaling, is the target of miR216-5p.31,32 We thus examined whether circHNRNPH1 regulated SMAD7 expression by inhibiting miR216-5p. circHNRNPH1 overexpression enhanced the mRNA and protein expression of SMAD7, whereas circHNRNPH1 knockdown slightly reduced its expression (Figures 5H–5J). When circHNRNPH1 and miR216-5p were coexpressed in cells, there was no significant change in SMAD7 expression compared with that in the circHNRNPH1 control group or miR216-5p control group (Figures 5K and 5L), suggesting that circHNRNPH1 promotes SMAD7 expression by sponging miR216-5p.
SMAD7 has been reported to mediate cellular apoptosis.33, 34, 35, 36 Thus, we conjectured that circHNRNPH1 could induce CF apoptosis. Therefore, primary CFs were transfected with circHNRNPH1 or si-circHNRNPH1, and after 48 h, cellular apoptosis was assayed. We found that circHNRNPH1 knockdown downregulated the expression of cleaved-PARP1), cleaved caspase-3, and BAX, which was opposite to the effects of circHNRNPH1 overexpression. Accordingly, BCL2 protein, an anti-apoptosis marker, was upregulated in the circHNRNPH1 knockdown group and downregulated in the circHNRNPH1 overexpression group (Figures 5M and 5N). Terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) assays confirmed that circHNRNPH1 promoted CF apoptosis (Figures 5O and 5P). Flow cytometric assays validated that circHNRNPH1 not only led to CF apoptosis but also induced cell death (Figure 5Q). Therefore, circHNRNPH1 could promote SMAD7 expression by inhibiting miR216-5p function and expediting CF apoptosis and death.
circHNRNPH1 Regulates the Membrane Stability of TβRI via SMAD7
TGF-β1 signaling is mainly mediated by TβRI.37 We found that the expression levels of TβRI protein and mRNA were decreased by circHNRNPH1 overexpression compared with its knockdown (Figures 6A–6C). Given the circHNRNPH1-dependent expression of SMAD7 (Figures 5E and 5F) and the SMAD7/smurf2-mediated expression of TβRI,7,38, 39, 40 we also assessed the influence of circHNRNPH1 on the stability of the TβRI protein. With the use of a cycloheximide chase analysis, which mainly inhibits protein synthesis in eukaryotes to facilitate the detection of protein degradation, we showed that circHNRNPH1 overexpression promoted TβRI degradation, which was especially obvious at 6 h after cycloheximide treatment (Figures 6D and 6E). Moreover, when si-SMAD7 and circHNRNPH1 were cotransfected in CFs, the degradation of the TβRI protein was significantly inhibited (Figures 6F and 6G). Thus, circHNRNPH1 upregulation promoted the degradation of the TβRI protein, and SMAD7 served as an important mediator.
Figure 6.

circHNRNPH1 Regulated TβRI Stability Mainly via SMAD7
(A) Quantitative PCR examination of TβRI expression affected by circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (B and C) Western blot (B) and densitometric analysis (C) of TβRI expression affected by circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (D and E) Western blot (D) and densitometric analysis (E) showing the degradation of the TβRI protein affected by circHNRNPH1 after 6 h and 12 h with cycloheximide treatment. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05). (F and G) Western blot (F) and densitometric analysis (G) of TβRI protein expression regulated by circHNRNPH1 via SMAD7. si-NC, si-circHNRNPH1-NC; si-circ, si-circHNRNPH1; circ-NC, circHNRNPH1-NC; circ, circHNRNPH1. Data are presented as the mean ± SD (n = 3 per group, ∗p < 0.05).
Discussion
Fibrosis and the resultant organ failure account for at least one-third of deaths worldwide. In the heart, myocardial fibrosis is associated with a fifty percent increase in the risk of adverse cardiac events.41 This scenario is particularly prominent in postischemic myocardium fibrogenesis. Although the complex profibrotic signaling pathways have been clarified, effective treatment regimens are lacking.42 In this study, the negative-feedback regulation of circHNRNPH1, a downstream effector of profibrotic signals on TβR1, suggested potential new therapeutics for postischemic cardiac fibrosis.
circRNAs are mostly derived from precursor mRNAs by backsplicing-based exon circularization. Herein, we found that circHNRNPH1, composed of exon 6 and exon 7 (628 bp) of HNRNPH1, was localized in the cytoplasm of CFs, in contrast to nuclear HNRNPH1, which encodes an RNA-binding protein mediating post-transcriptional regulation.43 The short flanking introns in circHNRNPH1 are enriched with A-U base pairs, which form the double-stranded RNA (dsRNA) structure to maintain circHNPNPH1 and provide the regulatory site for dsRNA-binding protein (dsRBP). These features of circHNRNPH1 define its functional modality. However, the formation and regulation of circHNNRPH1 biogenesis need to be fully elucidated and require further research.
In this study, we showed that circHNRNPH1 was highly responsive to TGF-β1. The upregulation of circHNRNPH1 may be mediated by TGF-β1 by downregulating ADAR1 and Stafufen1 expression. More interestingly, we also identified the reciprocal effect between circHNRNPH1 and HNRNPH1 mRNA (Figure S2). Thus, the regulatory mechanism of circHNRNPH1 remains complex.
miRNA sponging is the principal mechanism by which circRNA regulates its targets. Our results showed that circHNRNPH1 inhibited the gene expression responsible for the activation and migration of CFs. The regulation of profibrotic gene expression by circHNRNPH1 was achieved by direct interaction with miR216-5p. Importantly, the circHNRNPH1-induced upregulation of SMAD7 promoted TβRI degradation, terminating profibrotic TGF-β signaling. Thus, our findings revealed the biological function of circHNRNPH1 for the first time. Importantly, the regulatory cascade of circHNRNPH1-miR216-5p-SMAD7-TβRI could be used to design new regimens for fibrotic diseases, in addition to postischemic myocardial fibrosis. However, we realized that circHNRNPH1 may exert many other biological functions by sponging different miRNAs. To ensure the safety and effectiveness of therapeutic applications, researchers must not only investigate the function and mechanism of circHNRNPH1 but also engineer and deliver circular RNA in a targeted manner.
In summary, we identified the in vitro and in vivo biological functions of circHNRNPH1 in postischemic myocardial fibrosis and the underlying mechanism. circHNRNPH1 acts as a negative regulator of cardiac fibrosis in ischemic heart disease and may be a potential new treatment.
Materials and Methods
Animal Model Construction
All animal experiments were performed according to the relevant guidelines and regulations of the Animal Care Committee (publication number 85-23, revised 1996) and approved by the Shanghai General Hospital Institutional Animal Care and Use Committee, and all animal procedures conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes or the NIH guidelines.
The MI model was constructed by ligating the left anterior descending (LAD) coronary artery, and the control group was treated with a sham operation as described previously.44 The specific operation procedure was as follows: the rat was anesthetized using 3% sodium pentobarbital (30 mg/kg) by intravenous injection and then connected to a small animal ventilator, and the skin was cut through the fourth intercostal space on the left side. Then, we isolated the muscle tissue, opened the chest cavity, cut the pericardium, extruded the heart, and ligated the LAD coronary artery (approximately 1 to 2 mm at the starting point of the branch). An electrocardiogram was recorded, and the ST-segment elevation of the electrocardiogram showed successful MI. Then, the heart was quickly returned into the chest, and the chest and skin were sutured. In the sham operation group (negative control [NC] group), the coronary artery was not ligated, the other operations were the same as those of the experimental animals, and gentamicin was used locally. 5 weeks later, the rats were euthanized using sodium pentobarbital at three times the anesthetic dose with intravenous injection. The hearts were removed for further studies.
Isolation and Culture of Primary CFs and CMs
The isolation of primary CFs and CMs was performed according to a previous protocol described with little modification.45 Primary CFs and CMs were obtained from 1- to 3-day-old Norway rats (Shanghai Jie Si Jie Laboratory Animal, Shanghai, China) via enzymatic dispersion. In brief, the hearts were isolated under sterile conditions and rinsed with PBS (HyClone, Logan, UT, USA) and then minced into 1 mm3 pieces. The pieces were digested with 0.125% trypsin solution (HyClone, Logan, UT, USA) and 0.1% collagenase II solution (Sigma, Darmstadt, Germany) (1:1 volume) at 37°C for 7 min, 5 times. The cells were resuspended in DMEM (HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Waltham, MA, USA) and seeded onto sterile culture dishes (Corning, Corning, NY, USA) for 90 min in a humidified atmosphere of 5% CO2 at 37°C. The detached cells were purified from CFs and cultured in DMEM. The cell suspension with CMs was collected and cultured in DMEM/F12 culture medium with bromodeoxyuridine (BrdU; 0.1 mM) for 24 h, supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and then used in the study. The CFs were passaged every 3 days, and the first and second generations were used in the study. Rat aortic endothelial cells (RAOECs) were purchased from BioVector NTCC (Beijing, China).
RNAi Studies
The knockdown of circHNRNPH1, HNRNPH1, SMAD7, ADAR1, and Staufen1 was achieved by siRNAs, designed and synthesized by Guangzhou RiboBio (Guangzhou, China), and validated by quantitative real-time polymerase chain reaction for RNA expression analysis and western blots for protein expression analysis. The target sequence of each siRNA was blasted on the NCBI website to ensure the specificity of each siRNA. The siRNA sequences are listed in Table 1, and the effectiveness of the siRNAs is shown in Figure S4.
Table 1.
Sequences of siRNAs Used in the Study
| Name | Target Sequence (5′–3′) |
|---|---|
| si-Staufen1 | GGAGATGGAAGGAAAGTAA |
| si-HNRNPH1 | GCACAGGTATATTGAAATA |
| si-cirHNRNPH1 | ATGCACCTCAATTATTGTT |
| si-ADAR1 | CCATGAACCTCGATTTAAA |
| si-SMAD7 | GGCTAAGTTCAGAAGTGA |
RNA Extraction, RNase R Treatment, and Quantitative Real-Time PCR
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from CFs according to the manufacturer’s instructions. Total RNA (5 μg) was incubated with 8 U of RNase R at 37°C for 10 min in 1× buffer (Epicenter, Madison, WI, USA). Then, the RNA was reverse transcribed to a cDNA library using the Superscript II Reverse Transcriptase Kit (TaKaRa, Otsu, Shiga, Japan). The cDNA was amplified for 35 cycles using Taq MasterMix (TaKaRa, Otsu, Shiga, Japan), and quantitative real-time PCR was performed in triplicate using a SYBR Fast Kit (TaKaRa, Otsu, Shiga, Japan). The expression levels were normalized to the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) by the 2−ΔΔCt method. The primers used in the study are listed in Table 2.
Table 2.
Primers Used in the Study
| Name | Sequence (5′–3′) |
|---|---|
| circHNRNPH1 | forward: GGTGAAGCAGATGTTGAGTTTG, reverse: TTACTCTGCCATCAGGTCCAAT |
| HNRNPH1 | forward: GCTGGCTTTGAGAGGATGAG, reverse: ATCCTGAAGAAATGGTAGCACA |
| hnRNA-HNRNPH1 | forward: ATCCTGAAGAAATGGTAGCACA, reverse: AGACATCCCTCCCATTCCAT |
| SMAD7 | forward: AGGCTGTGTTGCTGTGAATCT, reverse: GGTTTCATCGGAGGAAGGTA |
| ADAR1 | forward: GGTTTCATCGGAGGAAGGTA, reverse: TGCTCTGGATTCTGACTGATGC |
| Staufen1 | forward: AAGACTTCCCCAAGAACAACA, reverse: GGCATCTCTGTGCTCTGTTG |
| α-SMA | forward: ACTGGTATTGTGCTGGACTCTG, reverse: TCAGCAGTAGTCACGAAGGAAT |
| Collagen I | forward: CGCAGAAGTCATAGGAGTCG, reverse: TAGGACATCTGGGAAGCAAA |
| GAPDH | forward: CCAAGGTCATCCATGACAAC, reverse: GCTTCACCACCTTCTTGATG |
| TβR I | forward: TCACCGAGACCACAGACAAAG, reverse: CAGCAATACGTAACTGCCCCT |
hnRNA, heterogeneous nuclear RNA.
FISH
The specific Cy5-labeled probe (Servicebio, Wuhan, China) targeting the backsplice of circHNRNPH1 and a carboxyfluorescein (FAM)-labeled probe (Servicebio, Wuhan, China) targeting miR216-5p were used for FISH. In brief, for localization of circHNRNPH1, CFs were grown to the exponential phase and 80%–90% confluence and then hybridized with the Cy5-labeled probe in hybridized buffer after prehybridization at 60°C overnight. For the colocalization of circHNRNPH1 and miR216-5p, Cy5-labeled and FAM-labeled probes were used for double FISH assays, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime Biotechnology, Shanghai, China). All images were acquired on a microscope (Leica Microsystems, Mannheim, Germany).
RIP Assay
RIP assays were performed using the Magana RIP Kit (Millipore, MA, USA), according to the manufacturer’s instructions. circHNRNPH1 overexpression plasmid or miR216-5p mimics were transfected into HEK293T cells and incubated for 48 h. Then, the cells were lysed in 100 μL of RIP lysis buffer containing proteinase inhibitor cocktail and RNase inhibitor (Promega, Madison, WI, USA). Next, the lysate was treated with DNase I at 37°C for 10 min and centrifuged at 12,000 × g for 30 min. The lysate was diluted with 900 μL of RIP buffer and incubated overnight at 4°C with anti-AGO2 (1:100; Abcam, Cambridge, UK) or anti-immunoglobulin G (IgG; 1:100; Abcam, Cambridge, UK) bound on magnetic beads (Millipore, MA, USA). A 10-μL aliquot of the RIP mixture was saved as the input. The beads were washed five times with RIP wash buffer. The immunoprecipitate was treated with proteinase K at 37°C for 30 min. Total RNA was extracted by TRIzol reagent, and quantitative real-time PCR was used to assay the expression of selected RNAs.
Dual-Luciferase Reporter Assay
The wild-type (WT) and Mut circHNTNPH1 fragments were amplified by RT-PCR and inserted into the pmirGLO dual-luciferase vector (Promega, Madison, WI, USA) to construct pmirGLO-circHNTNPH1-WT and pmirGLO-circHNTNPH1-Mut. For the luciferase assay, luciferase reporter plasmids were cotransfected with miR216-5p mimics into HEK293T cells using Lipofectamine 3000 (Thermo Scientific, MA, USA). The firefly and Renilla luciferase activities were detected using a Thermo Scientific Varioskan LUX Multimode Microplate Reader (Thermo Scientific, MA, USA). Renilla luciferase was used as an internal control.
Pulldown Assay
The biotin-miR216-5p-WT or biotin-miR216-5p-Mut was transfected into HEK293T cells and incubated for 48 h. Then, the total RNA was extracted, and 100 μg of total RNA was incubated with 500 μg of streptavidin magnetic beads (Millipore, MA, USA). The bound RNA was eluted, and the relative levels were detected by quantitative PCR.
TUNEL Assay
TUNEL assays were used to analyze apoptosis in the CFs treated with circHNRNPH1 or si-circHNRNPH1 for 18 h using the In Situ Cell Death Detection Kit (TUNEL fluorescence fluorescein isothiocyanate [FITC] kit; Roche, Basel, Switzerland). In brief, cryosections were stained for CFs and then immersed in DAPI for staining of the nuclei. The images were detected via microscope (Leica Microsystems, Mannheim, Germany).
Protein Degradation Assay
When the CFs reached 70%–80% confluence, they were seeded on 6 cm culture dishes and transfected with circHNRNPH1 or circHNRNPH1-NC. After 24 h, the cells were treated with cycloheximide (APExBIO, Houston, TX, USA) at 100 μg/mL for 0 h, 6 h, and 12 h. For the analysis of degradation, the cells were cultured in 6 cm dishes, as described above, transfected with circHNRNPH1 or si-SMAD7, or cotransfected with circHNRNPH1 and si-SMAD7 and then treated with cycloheximide at 100 μg/mL for 12 h. Western blots were used to analyze the relative expression of proteins.
Flow Cytometry
Cell apoptosis was assessed by flow cytometry. In brief, CFs transfected with cirHNRNPH1 or si-circHNRNPH1 or cotransfected with cirHNRNPH1 and si-SMAD7 for 24 h were detached with trypsin without EDTA and washed with PBS 3 times, stained with 5 μL of FITC and 5 μL of propidium iodide (PI) for 20 min at room temperature (RT), and measured using flow cytometry (BD Accuri C6; BD Biosciences, NJ, USA).
Transwell Migration and Wound-Healing Assays
Transwell migration assays were performed using a 24-well Transwell chamber (8 μm pores; Corning, Corning, NY, USA). CFs transfected with cirHNRNPH1 or si-circHNRNPH1 and coated with 30 μL of Matrigel (BD Biosciences, NJ, USA) were seeded onto the upper chamber with 600 μL of complete medium in the lower chamber. The migrated cells were stained with 0.25% crystal violet after incubation at 37°C for 24 h, and the nonmigrated cells were removed by a cotton swab.
CFs were cultured in 6-well chambers and transfected with cirHNRNPH1 or si-circHNRNPH1. When the cells reached 90% confluence, a 200-μL pipette tip was applied to scratch the cell layer. Then, the cells were washed with warm PBS and incubated in medium without FBS at 37°C for 0 h, 12 h, and 24 h. The edges of the scratch were photographed with a digital camera system (Olympus, Tokyo, Japan). All experiments were performed three times independently.
Western Blot Analysis
The protein was extracted with RIP assay (RIPA) lysis buffer and quantified using the Bicinchoninic Acid (BCA) Protein Assay Kit (Beyotime Biotechnology, Shanghai, China). A total of 20 μg of protein from each sample was resolved on a 12.5% gel for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, MA, USA). Nonspecific binding was blocked by incubation in 5% nonfat milk in PBS-Tween 20 (PBST) for 2 h at RT, and the membrane was then washed with Tris-buffered saline-Tween 20 (TBST). The membrane was incubated overnight at 4°C with appropriately diluted primary antibodies, such as anti-bax (1:1,000; Beyotime Biotechnology, Shanghai, China), anti-BCL2 (1:1,000; Beyotime Biotechnology, Shanghai, China), anti-α-SMA (1:1,000; Abcam, Cambridge, UK), anti-collagen I (1:1,000; Abcam, Cambridge, UK), anti-cleaved PARP1 (1:1,000; Abcam, Cambridge, UK), anti-cleaved caspase-3 (1:1,000; Abcam, Cambridge, UK), anti-SMAD7 (1:1,000; Proteintech, Rosemont, IL, USA), anti-TβRI (1:1,000; Proteintech, Rosemont, IL, USA), and anti-GAPDH (1:5,000; Cell Signaling Technology [CST], USA). After washes in TBST, the membrane was incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at RT. The signals were detected with the Enhanced Chemiluminescence (ECL) Reagent Kit (Millipore, MA, USA) and recorded by Gel Doc XR+ and ChemiDoc XRS+ Imaging Systems (Bio-Rad, CA, USA).
rAAV9-Mediated Gene Delivery in the Heart and Cardiac Function Assessment
Twenty-eight male Norway rats (6 weeks) weighing 200–250 g were randomly divided into the MI group (n = 22) and sham group (n = 6). The rat models were constructed as described above. 1 week after the LAD surgical procedure, the remaining surviving rats were further divided into MI group A (n = 6), MI-rAAV9-vector group B (n = 6), and MI-rAAV9-sh-circHNRNPH1 group C (n = 6). Groups B and C were intramyocardially injected with 5 × 1011 vg/mL rAAV9-sh-NC or rAAV9-sh-circHNRNPH1 virus (Vigene Biosciences, Shandong, China), respectively, and diluted to 20 μL using normal saline. The rats in the sham group or A group were injected with 20 μL of normal saline only at the same sites. At 5 weeks post-MI, the cardiac function indices of the remaining rats of each group were measured using echocardiography with a 14-Hz ultrasound probe (Hewlett-Packard Sonos 5500; USA), and the EF, FS, LVEDD, LVESD, LVEDV, and LVESV were used to evaluate cardiac function. All parameters were measured over 6 consecutive cardiac cycles.
Histology
Cardiac fibrosis was measured by Masson’s staining. The rats were euthanized by injection with an overdose of sodium pentobarbital, after which, the hearts were removed quickly and fixed with 4% paraformaldehyde, embedded in optimal cutting temperature (O.C.T.) compound, frozen at −80°C, and then cryosectioned (6 μm thickness) at 3 mm intervals. Three sections of each group were stained with Masson’s trichrome. The areas of infarcted tissue and left ventricle were determined by a stereoscope and camera (Leica Microsystems, Mannheim, Germany).
Immunohistochemistry
Paraffin-embedded sections of cardiac tissue were used to detect protein expression. According to the manufacturer, the sections were processed by antigen retrieval, blocked with Immunol Staining Blocking Buffer (Beyotime Biotechnology, Shanghai, China) for 1 h, and incubated at 4°C overnight with rabbit anti-mouse primary antibodies, anti-α-SMA (1:1,000; Abcam, Cambridge, UK), and anti-collagen I (1:1,000; Abcam, Cambridge, UK). Then, the sections were washed 3 times with PBS and labeled with biotinylated secondary antibody. The sections were washed with PBS and incubated in 3,3-diaminobenzidine (DAB) chromogen, and the reactions were stopped with tap water. Then, the tissue sections were counterstained with hematoxylin and coverslipped. The photomicrographs were taken by a senior pathologist, and the proteins were semiquantified with ImageJ analysis software (National Institutes of Health, USA).
Statistical Analyses
All quantitative data were obtained from three independent biologically replicated experiments and are presented as the mean ± SD. Comparison of two groups was performed with Student’s t test with a two-tailed p value. Multiple groups were compared with one-way ANOVA. p values <0.05 were considered statistically significant.
Author Contributions
F.W. and W.L. designed the study. F.W. and J.L. supervised the project. W.L., Y.W., and Y.D. performed all experiments. W.L., Y.W., Y.D., H.N., G.S., and X.L. analyzed the data. W.L. and Y.W. wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to thank the Shanghai Science and Technology Development Foundation for funding assistance. This work was supported by the Shanghai Science and Technology Development Foundation (0601N1691).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.08.008.
Contributor Information
Jun Li, Email: jun.li01@hotmail.com.
Fang Wang, Email: onlyfang1@163.com.
Supplemental Information
References
- 1.Bruder O., Wagner A., Jensen C.J., Schneider S., Ong P., Kispert E.M., Nassenstein K., Schlosser T., Sabin G.V., Sechtem U., Mahrholdt H. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010;56:875–887. doi: 10.1016/j.jacc.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 2.O’Hanlon R., Grasso A., Roughton M., Moon J.C., Clark S., Wage R., Webb J., Kulkarni M., Dawson D., Sulaibeekh L. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010;56:867–874. doi: 10.1016/j.jacc.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Harris K.M., Spirito P., Maron M.S., Zenovich A.G., Formisano F., Lesser J.R., Mackey-Bojack S., Manning W.J., Udelson J.E., Maron B.J. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114:216–225. doi: 10.1161/CIRCULATIONAHA.105.583500. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee I., Yekkala K., Borg T.K., Baudino T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N Y Acad. Sci. 2006;1080:76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 5.Serini G., Bochaton-Piallat M.L., Ropraz P., Geinoz A., Borsi L., Zardi L., Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomasek J.J., Gabbiani G., Hinz B., Chaponnier C., Brown R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 7.Kavsak P., Rasmussen R.K., Causing C.G., Bonni S., Zhu H., Thomsen G.H., Wrana J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 8.Verrecchia F., Chu M.L., Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001;276:17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 9.Bujak M., Frangogiannis N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao H., Shi K.H., Yang J.J., Huang C., Liu L.P., Li J. Epigenetic regulation of cardiac fibrosis. Cell. Signal. 2013;25:1932–1938. doi: 10.1016/j.cellsig.2013.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Hsiao K.Y., Sun H.S., Tsai S.J. Circular RNA - New member of noncoding RNA with novel functions. Exp. Biol. Med. (Maywood) 2017;242:1136–1141. doi: 10.1177/1535370217708978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K., Long B., Liu F., Wang J.-X., Liu C.-Y., Zhao B., Zhou L.Y., Sun T., Wang M., Yu T. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 2016;37:2602–2611. doi: 10.1093/eurheartj/ehv713. [DOI] [PubMed] [Google Scholar]
- 13.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 14.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 15.Du W.W., Yang W., Liu E., Yang Z., Dhaliwal P., Yang B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858. doi: 10.1093/nar/gkw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burd C.E., Jeck W.R., Liu Y., Sanoff H.K., Wang Z., Sharpless N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan W.L., Lim B.T., Anene-Nzelu C.G., Ackers-Johnson M., Dashi A., See K., Tiang Z., Lee D.P., Chua W.W., Luu T.D. A landscape of circular RNA expression in the human heart. Cardiovasc. Res. 2017;113:298–309. doi: 10.1093/cvr/cvw250. [DOI] [PubMed] [Google Scholar]
- 18.Weber K.T. Fibrosis and hypertensive heart disease. Curr. Opin. Cardiol. 2000;15:264–272. doi: 10.1097/00001573-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y., Weber K.T. Cells expressing angiotensin II receptors in fibrous tissue of rat heart. Cardiovasc. Res. 1996;31:518–525. [PubMed] [Google Scholar]
- 20.Campbell S.E., Janicki J.S., Weber K.T. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. J. Mol. Cell. Cardiol. 1995;27:1545–1560. doi: 10.1016/s0022-2828(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 21.Sun Y., Zhang J.Q., Zhang J., Lamparter S. Cardiac remodeling by fibrous tissue after infarction in rats. J. Lab. Clin. Med. 2000;135:316–323. doi: 10.1067/mlc.2000.105971. [DOI] [PubMed] [Google Scholar]
- 22.Butt R.P., Laurent G.J., Bishop J.E. Collagen production and replication by cardiac fibroblasts is enhanced in response to diverse classes of growth factors. Eur. J. Cell Biol. 1995;68:330–335. [PubMed] [Google Scholar]
- 23.Eghbali M., Tomek R., Sukhatme V.P., Woods C., Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ. Res. 1991;69:483–490. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 24.Petrov V.V., Fagard R.H., Lijnen P.J. Transforming growth factor-beta(1) induces angiotensin-converting enzyme synthesis in rat cardiac fibroblasts during their differentiation to myofibroblasts. J. Renin Angiotensin Aldosterone Syst. 2000;1:342–352. doi: 10.3317/jraas.2000.064. [DOI] [PubMed] [Google Scholar]
- 25.Eghbali M., Tomek R., Woods C., Bhambi B. Cardiac fibroblasts are predisposed to convert into myocyte phenotype: specific effect of transforming growth factor beta. Proc. Natl. Acad. Sci. USA. 1991;88:795–799. doi: 10.1073/pnas.88.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanov A., Memczak S., Wyler E., Torti F., Porath H.T., Orejuela M.R., Piechotta M., Levanon E.Y., Landthaler M., Dieterich C., Rajewsky N. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–177. doi: 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Sakurai M., Shiromoto Y., Ota H., Song C., Kossenkov A.V., Wickramasinghe J., Showe L.C., Skordalakes E., Tang H.Y., Speicher D.W., Nishikura K. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2017;24:534–543. doi: 10.1038/nsmb.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebbesen K.K., Hansen T.B., Kjems J. Insights into circular RNA biology. RNA Biol. 2017;14:1035–1045. doi: 10.1080/15476286.2016.1271524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Q., Bao C., Guo W., Li S., Chen J., Chen B., Luo Y., Lyu D., Li Y., Shi G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasquinelli A.E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012;13:271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J., Ning X., Cui W., Bi M., Zhang D., Zhang J. Transforming growth factor (TGF)-β-induced microRNA-216a promotes acute pancreatitis via Akt and TGF-β pathway in mice. Dig. Dis. Sci. 2015;60:127–135. doi: 10.1007/s10620-014-3261-9. [DOI] [PubMed] [Google Scholar]
- 32.Xia H., Ooi L.L.P.J., Hui K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. doi: 10.1002/hep.26369. [DOI] [PubMed] [Google Scholar]
- 33.Schiffer M., Bitzer M., Roberts I.S.D., Kopp J.B., ten Dijke P., Mundel P., Böttinger E.P. Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okado T., Terada Y., Tanaka H., Inoshita S., Nakao A., Sasaki S. Smad7 mediates transforming growth factor-beta-induced apoptosis in mesangial cells. Kidney Int. 2002;62:1178–1186. doi: 10.1111/j.1523-1755.2002.kid583.x. [DOI] [PubMed] [Google Scholar]
- 35.Landström M., Heldin N.E., Bu S., Hermansson A., Itoh S., ten Dijke P., Heldin C.H. Smad7 mediates apoptosis induced by transforming growth factor beta in prostatic carcinoma cells. Curr. Biol. 2000;10:535–538. doi: 10.1016/s0960-9822(00)00470-x. [DOI] [PubMed] [Google Scholar]
- 36.Lallemand F., Mazars A., Prunier C., Bertrand F., Kornprost M., Gallea S., Roman-Roman S., Cherqui G., Atfi A. Smad7 inhibits the survival nuclear factor kappaB and potentiates apoptosis in epithelial cells. Oncogene. 2001;20:879–884. doi: 10.1038/sj.onc.1204167. [DOI] [PubMed] [Google Scholar]
- 37.Vander Ark A., Cao J., Li X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018;52:112–120. doi: 10.1016/j.cellsig.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Hanyu A., Ishidou Y., Ebisawa T., Shimanuki T., Imamura T., Miyazono K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001;155:1017–1027. doi: 10.1083/jcb.200106023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakao A., Afrakhte M., Morén A., Nakayama T., Christian J.L., Heuchel R., Itoh S., Kawabata M., Heldin N.E., Heldin C.H., ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 40.Nie J., Dou X., Hao W., Wang X., Peng W., Jia Z., Chen W., Li X., Luo N., Lan H.Y., Yu X.Q. Smad7 gene transfer inhibits peritoneal fibrosis. Kidney Int. 2007;72:1336–1344. doi: 10.1038/sj.ki.5002533. [DOI] [PubMed] [Google Scholar]
- 41.Dimmeler S., Zeiher A.M. Netting Insights into Fibrosis. N. Engl. J. Med. 2017;376:1475–1477. doi: 10.1056/NEJMcibr1616598. [DOI] [PubMed] [Google Scholar]
- 42.González A., Schelbert E.B., Díez J., Butler J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018;71:1696–1706. doi: 10.1016/j.jacc.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 43.Han S.P., Tang Y.H., Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem. J. 2010;430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D., Zhu L., Li C., Mu J., Fu Y., Zhu Q., Zhou Z., Liu P., Han C. Sialyltransferase7A, a Klf4-responsive gene, promotes cardiomyocyte apoptosis during myocardial infarction. Basic Res. Cardiol. 2015;110:28. doi: 10.1007/s00395-015-0484-7. [DOI] [PubMed] [Google Scholar]
- 45.Thum T., Gross C., Fiedler J., Fischer T., Kissler S., Bussen M., Galuppo P., Just S., Rottbauer W., Frantz S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.