

Abstract
The coronavirus disease 2019 (COVID‐19) pandemic has spread around the globe very rapidly. Previously, the evolution pattern and similarity among the COVID‐19 causative organism severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and causative organisms of other similar infections have been determined using a single type of genetic marker in different studies. Herein, the SARS‐CoV‐2 and related β coronaviruses Middle East respiratory syndrome coronavirus (MERS‐CoV), SARS‐CoV, bat coronavirus (BAT‐CoV) were comprehensively analyzed using a custom‐built pipeline that employed phylogenetic approaches based on multiple types of genetic markers including the whole genome sequences, mutations in nucleotide sequences, mutations in protein sequences, and microsatellites. The whole‐genome sequence‐based phylogeny revealed that the strains of SARS‐CoV‐2 are more similar to the BAT‐CoV strains. The mutational analysis showed that on average MERS‐CoV and BAT‐CoV genomes differed at 134.21 and 136.72 sites, respectively, whereas the SARS‐CoV genome differed at 26.64 sites from the reference genome of SARS‐CoV‐2. Furthermore, the microsatellite analysis highlighted a relatively higher number of average microsatellites for MERS‐CoV and SARS‐CoV‐2 (106.8 and 107, respectively), and a lower number for SARS‐CoV and BAT‐CoV (95.8 and 98.5, respectively). Collectively, the analysis of multiple genetic markers of selected β viral genomes revealed that the newly born SARS‐COV‐2 is closely related to BAT‐CoV, whereas, MERS‐CoV is more distinct from the SARS‐CoV‐2 than BAT‐CoV and SARS‐CoV.
Keywords: COVID‐19, MERS, pandemic, phylogenetic, SARS, SARS‐CoV‐2
1. INTRODUCTION
The Coronaviridae family comprises viruses of positive‐sense, single‐stranded RNA that has a size of 27–32 kb. It has α, β, δ, and γ categories. 1 , 2 As the name implies, the spherical external spike protein has a crown shape. 3 , 4 The virus has been found to infect a wide range of hosts including humans, other mammals, and birds.
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is a new type of coronavirus that causes severe respiratory disease with several other manifestations and having a fatality rate of ~2%–4%. 5 , 6 It belongs to the β‐coronavirus genus of the Coronaviridae family and is 96% identical gnomically with a previously detected SARS‐like bat coronavirus (BAT‐CoV). 7 , 8 It was first identified in the Wuhan province of China in December 2019.* * It progressed rapidly via human‐to‐human interaction and spread in all major countries of the world. WHO declared a health emergency on January 30, 2020 for international concerns. 9 The first case was confirmed on February 26, 2020 in Pakistan and the total number of 506,701 confirmed cases and 10,717 deaths in Pakistan had been registered till January 12, 2021. 10
During the course of transmission and replication within the host, the viruses acquire gene mutations in the genome. Rapid genomic sequencing enabled us to find and analyze the genetic mutations in thousands of viral genome sequences. For the identification of potential vaccine targets of the virus, it is necessary to identify the region in which the virus is highly mutated.
Several studies have conducted a phylogenetic tree‐based analysis to study the evolutionary relationship of SARS‐CoV‐2 with other beta coronaviruses using the genomic sequences (Table 1). By comparison of the genome sequence with phylogenetic tree and multisequence alignment, 88% identity was found for SARS‐CoV‐2 with BAT‐CoV. 11 Li et al. 12 used the genomic sequences of around 70 SARS‐CoV‐2 isolated from coronavirus disease 2019 (COVID‐19) patients and analyzed the spike glycoprotein gene that is most related to COVID‐19 mutations. 12 They also predicted that the BetaCoV‐bat‐Yunnan‐RaTG13‐2013 virus is almost identical to SARS‐CoV‐2. A unique peptide (PRPA) was also identified in the genomic sequence of SARS‐CoV‐2 patients.
Table 1.
Overview and comparison of previous studies based on the analysis type
| Reference | Phylogenetic analysis | MSA | Homology | Recombinant regions | Variant identification | Statistical analysis | Microsatellite analysis |
|---|---|---|---|---|---|---|---|
| Lu et al. 11 | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| Li et al. 12 | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| Petrosillo et al. 13 | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ |
| Rehman et al. 14 | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ |
| Our study | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The study performed by Petrosillo et al., 13 also used the DNA sequences of SARS‐CoV, Middle East respiratory syndrome coronavirus (MERS‐CoV), and SARS‐CoV‐2 but BAT‐CoV sequences were not included in the analysis. In this study, a review of the differences between beta coronaviruses was provided. Pathogenesis and epidemiology techniques were used to compare the genome samples. It compared SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 with clinical features and identified that SARS‐CoV is more related to SARS‐CoV‐2 than MERS‐CoV. In a study by Rehman et al., 14 six recombination regions and the homology between the genome sequence of SARS‐CoV, MERS‐CoV, BAT‐SARS‐CoV, and SARS‐CoV‐2 were found. Comparative analysis, comprising variant and statistical analysis of these four virus types, that is, SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2 has not yet been addressed so far. The microsatellite comparison of SARS‐CoV, MERS‐CoV, BAT‐SARS‐CoV, and SARS‐CoV‐2 is also important for the identification of structural composition analysis. 15
In this study, four types of coronaviruses, that is, SARS‐CoV, MERS‐CoV, BAT‐SARS‐CoV, and SARS‐CoV‐2 were analyzed using multiple genetic markers including the single nucleotide polymorphisms (SNPs), whole‐genome sequence phylogeny, mutations in proteins, and microsatellites in comparison with the SARS‐CoV‐2 reference genome of Wuhan strain (Wuhan‐Wu‐I). Statistical analysis was then performed on the predicted SNPs, and microsatellites for inferring more comprehensive phylogeny insights.
2. MATERIALS AND METHODS
2.1. Data acquisition
The whole‐genome sequences of SARS‐CoV, BAT‐CoV, and SARS‐CoV‐2 to date were extracted from the NCBI Genbank. For the MERS‐CoV genome sequences, the NCBI BLAST search was applied using the reference sequence of MERS‐CoV (Accession id: NC_019842) as a query. Similar sequences of MERS‐CoV were extracted by BLAST search and were added to the data set. The sequences were filtered based on the host and nucleotide completeness. Only the genome sequences with the label of “complete genome” were retrieved. For the MERS‐CoV, SARS‐CoV, and novel SARS‐CoV‐2 sequences, the genomes with the human host were retained. The genome sequences of BAT‐CoV were filtered with the BAT as a host. The whole‐genome sequence of the SARS‐CoV‐2 strain of Wuhan, China was also downloaded from Genbank in fasta and genbank format. The structural protein sequences of selected viruses were also extracted from the virus portal of NCBI in fasta format. The summarized description of the samples of our data set is given in Table 2.
Table 2.
Characteristics of the data set samples used in this study
| Genome | Query | Host | Sequences | Total unique |
|---|---|---|---|---|
| SARS | SARSr‐CoV | Not available | 76 | 83 |
| SARS‐CoV Exon1 | Not available | 33 | ||
| SARS‐CoV Wtic‐MB | Not available | 31 | ||
| SARS Urbani | Homo Sapiens | 8 | ||
| SARS Tor2 | Homo Sapiens | 7 | ||
| MERS | MERS‐CoV | Homo Sapiens | 100 | 100 |
| COVID | SARS‐CoV‐2 | Homo Sapiens | 5955 | 5955 |
| BAT‐CoV | BAT‐CoV | Any BAT | 14 | 40 |
| BAT‐SARS | BAT‐SARS | Not available | 13 | |
| BAT‐SARS‐like | BAT‐SARS‐like | Not available | 13 |
Abbreviations: BAT‐CoV, bat coronavirus; BAT‐SARS, bat severe acute respiratory syndrome; COVID, coronavirus disease; SARS, severe acute respiratory syndrome.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The workflow for the analysis is shown in Figure 1. The analyses were performed in three categories that is, whole‐genome sequence‐based analysis, variants‐based analysis, and microsatellite‐based analysis. The whole‐genome‐based sequence alignment and a phylogenetic tree were drawn after cleaning the sequences. Single nucleotide variants (SNVs) and insertions/deletions were determined after the alignment. The similarity of proteins was identified by similarity plots and multi‐sequence alignment. For the identification of simple sequence repeat structural differences, microsatellite analysis was performed. The detailed workflow is described in Figure 1.
Figure 1.
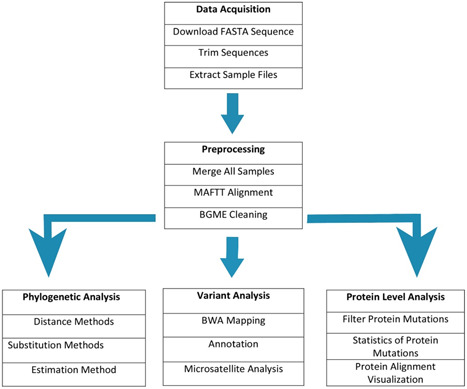
The data acquisition and analysis workflow used in this study for inferring evolutionary insights in four types of beta coronaviruses
2.2. Phylogenetic analysis
For the genomic phylogenetic analysis of different coronaviruses, 73 whole genome sequences (WGSs) of different viruses including 15 SARS‐CoV, 13 BAT SARS‐CoV, 20 MERS‐CoV, and 25 SARS‐CoV‐2 were selected from the data set (Table S1). The reference genome sequence of the SARS‐CoV‐2 of Wuhan (accession‐id: NC_045512) was included for phylogenetic analysis. All the genomes were aligned by the Multiple Alignments Fast Fourier transform algorithm and sequences were cleaned with Block Mapping and Gathering with Entropy 16 cleaning algorithm. The Mega X 17 desktop application was used to generate and visualize the phylogenetic tree. The maximum likelihood approach with a 1000 bootstrap value was used for the best interfacing of a tree. The Hasegawa‐Kishino‐Yano model was calculated as the best substitution model for the phylogenetic. Simplot software was used to visualize the similarity plot between four selected species.
2.3. Variant analysis
For the variant‐based analysis, the genome sequence of the SARS‐CoV‐2 of Wuhan (Accession id: NC_045512) was selected as the reference (termed Wuhan‐Wu‐I hereafter). All the genome sequences of SARS‐CoV (n = 83), MERS‐CoV (n = 100), BAT‐CoV (n = 40), and SARS‐CoV‐2 (n = 5955) in the data set were mapped to the reference genome by using the BWA‐MEM algorithm of Burrows–Wheeler Aligner (BWA). 18 The conversion of SAM files to BAM files and sorting of BAM files were performed with Samtools. 19 The genomic variations including SNVs and insertions/deletions (indels) were determined by using the bcfTools. 20 The gene regions of all the identified mutations were determined by the annotation with SnpEff tool. 21 The SnpEff predicted the impact of the mutations as well as the gene name, gene‐id and transcript features. The Wuhan‐Wu‐I reference genome was manually registered as a targeted database in SnpEff (version 4.3). The complete working of variant calling and variant annotation is described in Algorithm 1 and the Python script is publicly available at “https://github.com/Abdul194/Variant_Calling”.
Algorithm 1
Variant calling and Variant annotation algorithm
| Input: Reference Sequence, Genome Sequences of Selected CoV's, Reference Genbank Sequence |
| Output: Genome Mutations, Annotated Mutations |
|
|
|
|
|
| SAM‐File = BWA‐MEM map → Sequence to → Ref‐Sequence |
| BAM‐File = Samtools convert → SAM‐File to Bam File |
| Sorted‐BAM‐File = Samtools Sort → BAM‐File |
| VCF‐Variants = bcfTools call‐variants on → Sorted‐BAM‐File comparing → Ref‐sequence |
| Filtered‐Variants = vcfutils filer variants from → VCF‐Variants base quality |
|
|
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Then, the BrowseVcf 22 tool was used to filter the variants based on quality scores. All the variants with a low‐quality score were discarded. Furthermore, a custom Python script was written for the analysis of variants. The script identified the number of variants that mutated in 50% of the samples and the variants with the highest mutation frequency.
2.4. Gene product analysis
The genome sequence of SARS‐CoV‐2 encodes a total of 27 proteins. The 5ʹ‐terminal region encodes 15 nonstructural proteins, whereas, the 3ʹ‐terminal region encodes four structural proteins labeled as envelope protein (E), spike protein (S), nucleocapsid protein (N), and membrane protein (M) with eight accessory proteins. As the structural proteins play a key role in the viral assembly, particular emphasis was given to mutations in structural proteins. The Python script filtered the mutations in the structural proteins only. The homology between the structural proteins of all species was calculated by variants analysis. The multi‐sequence analysis was also performed on structural proteins using the Multialin 23 online tool.
2.5. Microsatellite analysis
Microsatellite analysis tracks the repetitive sequences in the genome that have a significant impact on diseases and evolution. For the comparison of SARS‐CoV, MERS‐CoV, BAT‐SARS, and SARS‐CoV‐2, the microsatellite analysis was performed using imperfect microsatellite extractor (IMEX) 24 and fast microsatellite discovery (FMSD) 15 online tools. From IMEX, 20 genome sequences of SARS‐CoV (n = 5), MERS‐CoV (n = 5), BAT‐CoV (n = 5), SARS‐CoV‐2 (n = 5), and the reference of Wuhan (accession‐id: NC_045512) were selected from the data set (Table S2) for microsatellite analysis. The microsatellites in selected genome sequences were discovered using the IMEX online tool. All repeat size microsatellites were discovered with the following parameters: type of repeat = imperfect, minimum repeat number = 6, 3, 3, 3, 3, 3 (as reported in Alam et al. 25 ), the distance allowed between two adjacent microsatellites = 10 bp, and the default value for the remaining parameters. The density and GC content of selected WGS were calculated with the Python script. From FMSD, the microsatellites with atomic core lengths ≤ 7 were discovered for the selected 20 genome sequences. Unlike the IMEX, FMSD allows a single repeat value for all core lengths of the microsatellite. Hence, the minimum repeat number was set to 3 for all core lengths of microsatellites.
3. RESULTS AND DISCUSSION
3.1. Phylogenetic analysis
The whole‐genome sequences (n = 73) of SARS‐CoV, BAT‐CoV, MERS‐CoV, and SARS‐CoV‐2 were selected from the data set (Table S1) and downloaded from NCBI Genbank. The samples of SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 were collected only for the host Homo sapiens, whereas the BAT‐CoV sequences were collected from eight different types of bats including Rhinolophus sinicus. To analyze the relationship between selected coronaviruses, a phylogenetic tree was constructed (Figure 2). The best substitution model was estimated and the HKY (Hasegawa, Kishino, and Yano) substitution model was selected with the maximum likelihood approach for interfacing. Maximum likelihood estimation gives a better performance than the maximum parsimony and other traditional methods. It also indicates the error of the estimated tree. 26
Figure 2.
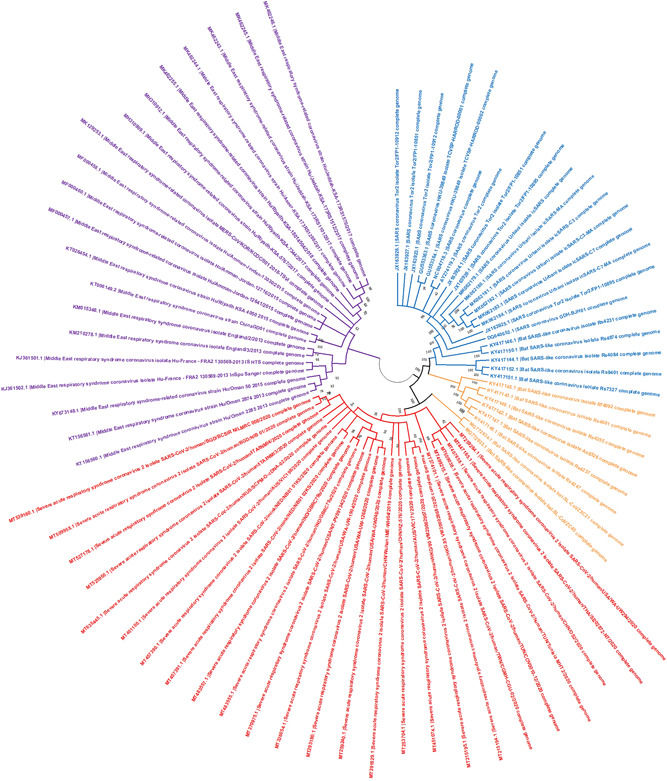
Phylogenetic tree of four types of beta coronaviruses genome sequences. The four genomes constituted four different clades. The MERS‐CoV was found as the outgroup clade, whereas the SARS‐CoV‐2, and SARS‐CoV and BAT‐CoV forming two descendant clades. Blue, SARS‐CoV; orange, BAT SARS‐CoV; red, SARS‐CoV‐2, and purple, MERS‐CoV. BAT‐CoV, bat coronavirus; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2
Our phylogenetic analysis showed different lineages for coronaviruses. The tree displays the branches of SARS‐CoV samples (blue), BAT SARS‐CoV (orange), MERS‐CoV (purple), and SARS‐CoV‐2 (red). The whole genome‐based phylogenetic analysis indicated MERS‐CoV as outgroup species and SARS‐CoV, SARS‐CoV‐2, and BAT‐CoV as ingroup species. Within the ingroup, two lineages were found comprising of SARS‐CoV‐2 in one lineage, and SARS‐CoV and BAT‐CoV in the other lineage. The branch length of the phylogeny indicated that the SARS‐CoV had diverged very early from the BAT‐CoV. It was noteworthy that two of the BAT genomes (accession‐id MG772933 and GM772934) were found as outgroups within the lineage of SARS‐CoV‐2 (Figure 2). The closely related outer neighbors of SARS‐CoV‐2 were BAT‐CoV's indicating an independent divergence of SARS‐CoV‐2 from the BAT‐CoV's. The phylogeny placed the MERS‐CoV far from the SARS‐CoV‐2 as compared to BAT‐CoV and SARS‐CoV.
The similarity between the four selected coronaviruses of the beta family was also calculated by constructing a similarity plot. The randomly selected WGS of SARS‐CoV (accession‐id: JX162923), MERS‐CoV (accession‐id: MT387202), BAT SARS‐CoV (accession‐id: GQ153539), and BAT SARS‐like (accession‐id: MG772933) were aligned with the Wuhan‐Wu‐I reference sequence. Aligned sequences were plotted for the estimation of similarity by using Simplot 27 software. The reference sequence of Wuhan SARS‐CoV‐2 was selected as a query and the rest of the sequences were compared with the query sequence. Simplot allows the analysis of up to ten genome sequences by automatically neglecting the sites that contain gaps. The similarity graph showed ~98% homology of BAT SARS‐like CoV with the reference sequence of SARS‐CoV‐2 of Wuhan (Figure 3). However, SARS‐CoV and MERS‐CoV showed ~92 and ~58 similarities, respectively, with NC_045512 (Figure 3).
Figure 3.
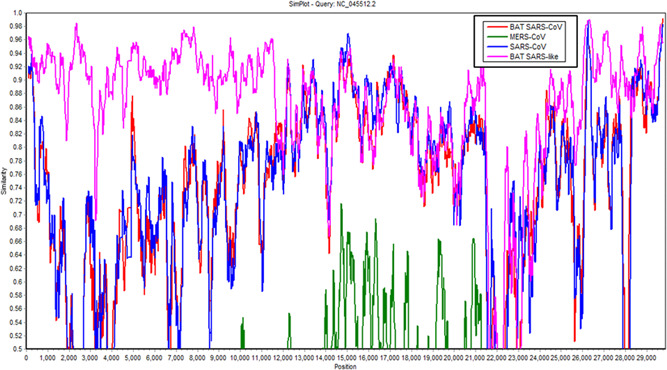
Similarity of BAT‐CoV, MERS‐CoV, and BAT‐SARS like genomes with Wuhan‐Wu‐I genome. The BAT‐SARS like showed highest similarity with SARS‐CoV‐2 throughout the genome (~98%) except at the positions 22K–24K. BAT‐CoV, bat coronavirus; BAT‐SARS, bat severe acute respiratory syndrome; MERS‐CoV, Middle East respiratory syndrome coronavirus
3.2. Analysis of genetic variants
For the variant‐based analysis, the genome sequences of four selected coronaviruses species were retrieved (see Section 2.1). The reference genome of the Wuhan SARS‐CoV‐2 strain was indexed and all genomes of the data set were mapped to the reference sequence. The total number of annotated variants was calculated for SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2 (Table 3). On average, the MERS‐CoV genome differed from the Wuhan‐Wu‐I reference strain at 134.21 sites, the BAT‐CoV genome differed at 136.72 sites, the SARS‐CoV genome differed at 26.64 sites, and the SARS‐CoV‐2 genome differed at 0.66 sites. It was noteworthy that despite a higher mutational rate in BAT‐CoV, it had the lowest nonsynonymous/synonymous ratio of 0.29, whereas this ratio was 0.31, 1.46, 1.57 for SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 genomes. This analysis represents the higher potential of MERS‐CoV and SARS‐CoV‐2 of acquiring mutations at missense sites. The lower number of missense variations in SARS‐CoV and BAT‐CoV may be due to the selection pressure on missense sites. Furthermore, we determined highly recurrent mutations, which were detected in ≥50% of the analyzed genomes in each of four CoV's. The filtration of variants, SARS‐CoV‐2 showed four major mutations that occurred in more than 50% of the samples at position 241(C/T, upstream), 3037(C/T), 14408(C/T), and 23403(A/G). The BAT‐CoV, SARS‐CoV, and MERS‐CoV showed 1690, 2178, and 4390 mutations, respectively, that occurred in more than 50% of samples.
Table 3.
Number of SNVs found in all four types of CoVs with respect to the Wuhan SARS‐CoV‐2 strain
| Genome | Number of samples | Reference sequence | Total SNV sites | Synonymous | Missense |
|---|---|---|---|---|---|
| SARS‐CoV | 83 | NC_045512.2 | 2211 | 1714 | 527 |
| MERS‐CoV | 100 | NC_045512.2 | 13421 | 5460 | 7961 |
| SARS‐CoV‐2 | 5955 | NC_045512.2 | 3935 | 1406 | 2209 |
| BAT‐CoV | 40 | NC_045512.2 | 5469 | 4224 | 1239 |
Abbreviations: BAT‐CoV, bat coronavirus; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV, severe acute respiratory syndrome coronavirus; SNV, single nucleotide variant.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Given that all the changes in nucleotides sequences do not affect the resultant protein (due to silent mutations); the number of mutations in S, E, M, and N structural proteins was calculated (Table 4). The SNPs in S, M, E, and N gene regions were filtered by our python script and then all the SNPs within at least 50% samples were retained (Table 5). The S, M, E, and N genes showed a different number of SNPs, where the BAT‐CoV showed the smallest number of SNPs as compared to SARS‐CoV in S, M, E, and N genes. The effect of SNPs was calculated by comparing the S, M, E, and N protein sequences of SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2. To compare the similarity between four selected coronaviruses, the S, M, E, and N proteins were aligned using the Multialin online tool. Multialin performed multiple sequence analysis (MSA) and produced a consensus sequence (Figures SF1–SF4). The multi‐sequence alignment of envelope protein has been displayed in Figure SF1 with different base colors. The high, low, and normal consensus have been displayed with red, blue, and black colors, respectively. The envelope protein of the MERS‐CoV strain (accession‐id: QJX19962.1) showed three variations at a high consensus position (Figure SF1).
Table 4.
Number of variants discovered in four structural proteins
| Genome | Number of samples | S gene SNPs | E gene SNPs | M gene SNPs | N gene SNPs |
|---|---|---|---|---|---|
| SARS‐CoV | 83 | 259 | 18 | 101 | 147 |
| MERS‐CoV | 100 | 92 | 0 | 0 | 0 |
| SARS‐CoV‐2 | 5955 | 449 | 26 | 79 | 260 |
| BAT‐CoV | 40 | 570 | 28 | 160 | 286 |
Abbreviations: BAT‐CoV, bat coronavirus; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV, severe acute respiratory syndrome coronavirus.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 5.
Number of variants discovered in four structural proteins in more than 50% of samples
| Genome | Number of samples | S gene SNPs | E gene SNPs | M gene SNPs | N gene SNPs |
|---|---|---|---|---|---|
| SARS‐CoV | 83 | 249 | 12 | 91 | 141 |
| MERS‐CoV | 100 | 66 | 0 | 0 | 0 |
| SARS‐CoV‐2 | 5955 | 1 | 0 | 0 | 0 |
| BAT‐CoV | 40 | 139 | 10 | 84 | 126 |
Abbreviations: BAT‐CoV, bat coronavirus; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV, severe acute respiratory syndrome coronavirus.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Microsatellites were determined for the selected genome sequences (detailed in Section 2). By using IMEX, the average 95.8, 106.8, 98.5, and 107 microsatellites for SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2 were discovered, respectively. The MERS‐CoV genome (Accession id: DQ022305) had the highest incidence of microsatellites and the BAT‐CoV (Accession id: KX574227) had the lowest number of microsatellites. The incidence of microsatellite in all genomes is presented in Figure S5A. The compound microsatellites were also discovered for the selected genomes with a 10 bp maximum distance between two adjacent microsatellites. The SARS‐CoV‐2 genome (Accession id: MT446350) had the largest incidence of compound microsatellites (cSSR = 7) (Figure S5B). The GC_Content of selected genomes showed a smaller variation between selected viruses with the average values of 40.82, 41.17, 41.10, and 37.97 for SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2, respectively. The BAT‐CoV genomes (Accession id: GQ153539) showed the highest microsatellite density (SSR = 3770.54) and the BAT genome (Accession id: KX574227) showed the lowest microsatellites density (SSR = 2732.61), whereas, the highest compound microsatellite was observed in the SARS‐CoV‐2 genome (Accession id: MT446350) and the lowest in the BAT‐CoV genome (Accession id: KY417142) (Table 6).
Table 6.
SSR, cSSR, GC_content, and density of the selected SARS genomes
| Genbank ID | SSRs | cSSRs | GC‐content | SSR density | cSSR density |
|---|---|---|---|---|---|
| SARS‐CoV genomes | |||||
| AY274119 | 97 | 2 | 40.76 | 3260.39 | 67.22 |
| FJ882926 | 96 | 2 | 40.83 | 3260.39 | 67.40 |
| JF292921 | 96 | 2 | 40.82 | 3238.21 | 67.46 |
| JX162087 | 94 | 1 | 40.89 | 3166.26 | 33.68 |
| KF514388 | 96 | 2 | 40.82 | 3233.74 | 67.37 |
| MERS‐CoV genomes | |||||
| KC164505 | 107 | 2 | 41.17 | 3553.52 | 66.42 |
| KM015348 | 105 | 2 | 41.20 | 3495.11 | 66.57 |
| KP209306 | 106 | 2 | 41.18 | 3518.91 | 66.39 |
| KT006149 | 110 | 3 | 41.12 | 3652.79 | 99.62 |
| KY581684 | 106 | 2 | 41.18 | 3518.91 | 66.39 |
| BAT‐CoV genomes | |||||
| DQ022305 | 114 | 2 | 41.11 | 3834.77 | 67.28 |
| GQ153539 | 112 | 2 | 41.16 | 3770.54 | 67.33 |
| KU182964 | 101 | 3 | 40.98 | 3511.70 | 104.31 |
| KX574227 | 81 | 1 | 41.16 | 2732.61 | 33.74 |
| KY417142 | 86 | 1 | 41.06 | 2893.19 | 33.64 |
| SARS‐CoV‐2 genomes | |||||
| MN908947 | 107 | 6 | 37.97 | 3578.24 | 200.65 |
| MT007544 | 107 | 6 | 37.96 | 3579.43 | 200.72 |
| MT418883 | 107 | 6 | 37.97 | 3582.43 | 200.88 |
| MT446350 | 107 | 7 | 37.98 | 3589.76 | 234.84 |
| MT449665 | 107 | 6 | 37.97 | 3579.55 | 200.72 |
Abbreviations: BAT‐CoV, bat coronavirus; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The FMSD tool discovered microsatellites for the 20 selected genome sequences of SARS‐CoV, MERS‐CoV, BAT‐CoV, and SARS‐CoV‐2. All the SSRs were discovered with a core length of 3 to 7. FMSD discovered 23.6%, 15%, 19.8%, and 23% microsatellites on average for SARS‐CoV, MERS‐CoV and BAT‐CoV, and SARS‐CoV‐2, respectively (Table 7). The average score of discovered SSRs for SARS‐CoV‐2 was closest to the average score of SARS‐CoV and then to the BAT‐CoV. Moreover, for the MERS‐CoV, the average score of the discovered SSRs was the largest as compared to SARS‐CoV‐2. The inconsistency in discovered SSR of IMEX and FMSD is due to mainly two reasons: firstly, the IMEX allows a minimum number of repeats for every core length separately while the FMSD allows a single number as a minimum repeat for all core length SSRs. Secondly, the IMEX is built for the discovery of microsatellites from all genomes while the FMSD is built for the discovery of structural microsatellites from SARS‐CoV and SARS‐CoV‐2.
Table 7.
Microsatellites of selected genomes with FMSD online tool
| Genbank ID | SSRs | Genbank ID | SSRs |
|---|---|---|---|
| SARS‐CoV genomes | |||
| AY274119 | 27 | JX162087 | 22 |
| FJ882926 | 22 | KF514388 | 23 |
| JF292921 | 24 | ||
| MERS‐CoV genomes | |||
| KC164505 | 15 | KT006149 | 14 |
| KM015348 | 16 | KY581684 | 15 |
| KP209306 | 15 | ||
| BAT‐CoV genomes | |||
| DQ022305 | 18 | KX574227 | 19 |
| GQ153539 | 17 | KY417142 | 26 |
| KU182964 | 19 | ||
| SARS‐CoV‐2 genomes | |||
| MN908947 | 23 | MT446350 | 21 |
| MT007544 | 23 | MT449665 | 25 |
| MT418883 | 23 | ||
Abbreviations: BAT‐CoV, bat coronavirus; FMSD, fast microsatellite discovery; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
4. CONCLUSION
In this study, the genome sequences of four β coronaviruses were analyzed using the phylogenetic technique. The Phylogenetic tree placed the BAT‐CoV strains as the closest neighbor of SARS‐CoV‐2 The SARS‐CoV strains were the second nearest neighbor of SARS‐CoV‐2 and the phylogenetic tree placed all MERS‐CoV strains at the most outer clade of SARS‐CoV‐2. The variant analysis identified more mutations for MERS‐CoV than SARS‐CoV and BAT‐CoV. MERS‐CoV also showed three variations at a high consensus position in comparison with the envelope protein (Figure S4). Microsatellite analysis using IMEX did not form significant results but microsatellites using FMSD showed more satellites for MERS‐CoV that differ from the reference as compared to SARS‐CoV and BAT‐CoV. By the phylogenetic, variant, multisequence, and microsatellite analysis, it is concluded that BAT is the native host of SARS‐CoV‐2 and BAT‐CoV is closely related to SARS‐CoV‐2. It may be possible there is an intermediate host for the transmission of COVID‐19 from BAT to humans that needs to conduct further studies. Although the FMSD tool discovered that the SARS‐CoV is closer to SARS‐CoV‐2 than BAT‐CoV, its results were inconsistent relative to the IMEX tool. Hence, From the four selected coronaviruses, BAT is the native host and closely related to SARS‐CoV‐2 and MERS‐CoV is less similar to SARS‐CoV‐2 than BAT‐CoV and SARS‐CoV.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Hafiz Abdul Rehman and Farheen Ramzan conceived the original idea. Hafiz Abdul Rehman developed the theory and performed the computations. Farheen Ramzan, Zarrin Basharat, and Muhammad Shakeel verified the analytical methods. Hafiz Abdul Rehman wrote the manuscript with support from Farheen Ramzan, Zarrin Basharat, and Muhammad Shakeel. Muhammad Usman Ghani Khan and Ishtiaq Ahmad Khan helped supervise the project. All authors discussed the results and contributed to the final manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jmv.26974
Supporting information
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
This study work is supported by National Center for Artificial Intelligence (NCAI), Al‐Khawarizmi Institute of Computer Science (KICS), University of Engineering and Technology (UET) Lahore, Pakistan.
Rehman HA, Ramzan F, Basharat Z, Shakeel M, Khan MUG, Khan IA. Comprehensive comparative genomic and microsatellite analysis of SARS, MERS, BAT‐SARS, and COVID‐19 coronaviruses. J Med Virol. 2021;93:4382‐4391. 10.1002/jmv.26974
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in NCBI Virus at https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide%26Completeness_s=complete with the accession IDs given in Table S1.
REFERENCES
- 1. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181‐192. 10.1038/s41579-018-0118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monchatre‐Leroy E, Boué F, Boucher JM, et al. Identification of alpha and beta coronavirus in wildlife species in France: bats, rodents, rabbits, and hedgehogs. Viruses. 2017;9(12):364. 10.3390/v9120364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cong Y, Ren X. Coronavirus entry and release in polarized epithelial cells: a review. Rev Med Virol. 2014;24(5):308‐315. 10.1002/rmv.1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tortorici MA, Veesler D. Structural insights into coronavirus entry. Advances in Virus Research. 105. Academic Press Inc.; 2019:93‐116. 10.1016/bs.aivir.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bassetti M, Vena A, Giacobbe DR. The novel Chinese coronavirus (2019‐nCoV) infections: challenges for fighting the storm. Eur J Clin Invest. 2020;50(3). 10.1111/eci.13209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID‐19) outbreak. J Autoimmun. 2020;109:102433. 10.1016/j.jaut.2020.102433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S. Full‐genome evolutionary analysis of the novel corona virus (2019‐nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol. 2020;79:79. 10.1016/j.meegid.2020.104212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. WHO. World Health Organization . https://www.who.int/. Accessed May 21, 2020.
- 10. Pakistan coronavirus: 506,701 cases and 10,717 deaths—worldometer . https://www.worldometers.info/coronavirus/country/pakistan/. Accessed January 12, 2021.
- 11. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2. J Med Virol. 2020;92(6):602‐611. 10.1002/jmv.25731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E. COVID‐19, SARS and MERS: are they closely related? Clin Microbiol Infect. 2020;26(6):729‐734. 10.1016/j.cmi.2020.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rehman Sur, Shafique L, Ihsan A, Liu Q. Evolutionary trajectory for the emergence of novel coronavirus SARS‐CoV‐2. Pathogens. 2020;9(3):240. 10.3390/pathogens9030240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naghibzadeh M, Savari H, Savadi A, Saadati N, Mehrazin E. Developing an ultra‐efficient microsatellite discoverer to find structural differences between SARS‐CoV‐1 and COVID‐19. Informatics Med Unlocked. 2020;19:100356. 10.1016/j.imu.2020.100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10(1):210. 10.1186/1471-2148-10-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms, Molecular Biology and Evolution. Oxford Academic . https://academic.oup.com/mbe/article/35/6/1547/4990887. Accessed August 5, 2020. [DOI] [PMC free article] [PubMed]
- 18. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bcftools by samtools . http://samtools.github.io/bcftools/. Accessed January 12, 2021.
- 21. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salatino S, Ramraj V. BrowseVCF: a web‐based application and workflow to quickly prioritize disease‐causative variants in VCF files. Brief Bioinform. 2017;18(5):774‐779. 10.1093/bib/bbw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MultAlin–multiple sequence alignment. Bioinformatics, Oxford Academic . https://academic.oup.com/bioinformatics/article-abstract/9/5/614/349386?redirectedFrom=fulltext. Accessed August 5, 2020.
- 24. Mudunuri SB, Nagarajaram HA. IMEx: imperfect microsatellite extractor. Bioinformatics. 2007;23(10):1181‐1187. 10.1093/bioinformatics/btm097 [DOI] [PubMed] [Google Scholar]
- 25. Alam CM, Iqbal A, Sharma A, Schulman AH, Ali S. Microsatellite diversity, complexity, and host range of mycobacteriophage genomes of the Siphoviridae family. Front Genet. 2019;10(MAR):207. 10.3389/fgene.2019.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17(6):368‐376. 10.1007/BF01734359 [DOI] [PubMed] [Google Scholar]
- 27. SimPlot v1.3 Documentation . https://sray.med.som.jhmi.edu/RaySoft/simplot_old/Version1/SimPlot_Doc_v13.html. Accessed August 5, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Data Availability Statement
The data that support the findings of this study are openly available in NCBI Virus at https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide%26Completeness_s=complete with the accession IDs given in Table S1.