

Abstract
Recently, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) variants (B.1.1.7 and B.1351) have emerged harbouring mutations that make them highly contagious. The N501Y mutation within the receptor‐binding domain (RBD) of the spike protein of these SARS‐CoV‐2 variants may enhance binding to the human angiotensin‐converting enzyme 2 (hACE2). However, no molecular explanation for such an enhanced affinity has so far been provided. Here, using all‐atom molecular dynamics simulations, we show that Y501 in the mutated RBD can be well‐coordinated by Y41 and K353 in hACE2 through hydrophobic interactions, which may increase the overall binding affinity of the RBD for hACE2 by approximately 0.81 kcal·mol−1. The binding dynamics revealed in our study may provide a working model to facilitate the design of more effective antibodies.
Keywords: ACE2, antibody, N501Y, SARS‐CoV‐2, spike protein
Abbreviations
FEP, free energy perturbation
hACE2, human angiotensin‐converting enzyme 2
MD, molecular dynamics
RBD, receptor‐binding domain
RMSF, root mean square fluctuation
SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
SASA, solvent accessible surface area
WHO, World Health Organization
The ongoing pandemic of COVID‐19 was first detected in China with a cluster of infections caused by a novel coronavirus subsequently identified as the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [1, 2]. The disease has spread rapidly across the world with a grave impact that has reverberated through every corner of the globe, affecting almost every aspect of human life. For example, since the World Health Organization (WHO) declared the COVID‐19 outbreak a global pandemic in March 2020, confirmed cases of COVID‐19 worldwide have exceeded 102 million, with the death toll surpassing 2 million as of 1 February 2021, according to the WHO COVID‐19 Dashboard (https://covid19.who.int). Furthermore, the global economic recession caused by the COVID‐19 pandemic is unusually severe, resulting in a dramatic impact on livelihoods and loss of income on a global scale. Under these unprecedented circumstances, numerous scientists and researchers are racing around the clock to find vaccines and therapeutics aimed at halting the coronavirus pandemic. As a result, a wealth of knowledge about SARS‐CoV‐2 has been accumulated in the past year.
However, there is growing concern about the impact of viral genome changes because the D614G mutation [3] alters both the fitness [4] and neutralization susceptibility [5] of SARS‐CoV‐2 and has quickly become the dominating variant (> 85%) since it was first identified in late January 2020. Recently, WHO identified the rapid and widespread emergent SARS‐CoV‐2 D614G variant with an additional mutation of N501Y [6], which has completely and independently emerged in lineage B.1.1.7 (also known as 20B/501Y.v1) in the UK and in lineage B.1.351 (also known as 20C/501Y.v2) in South Africa. So far, the molecular mechanism underlying the characteristics of the N501Y mutation is still enigmatic.
Despite a variety of proteins present in coronaviruses, one such major vaccine and antibody target is the spike glycoprotein (S‐protein) of the coronavirus, which plays a key role in the receptor recognition and cell membrane fusion process, facilitating viral entry into the host cell [7, 8, 9]. The S‐protein is present in the form of trimers on the virion surface forming the distinctive ‘corona’ and is composed of two subunits, S1 and S2. The S1 subunit contains a receptor‐binding domain (RBD), which is responsible for virus attachment at the cell surface, whereas the S2 subunit is responsible for viral cell membrane fusion by forming a six‐helical bundle via the two‐heptad repeat domain. Connecting the heads (S1 and S2) to the viral membrane, the stalk domain of S‐protein contains three hinges that provide the head substantial freedom to scan the host cell surface [10]. Previous studies on the pathophysiology of SARS‐CoV‐2 infection revealed that the human angiotensin‐converting enzyme 2 (hACE2), an integral membrane protein and a zinc metalloprotease of the ACE family, serves as a high‐affinity receptor where RBD of the SARS‐CoV‐2 S‐protein (hereafter referred to as sRBD) binds to promote the formation of endosomes to trigger viral fusion activity [8, 11, 12]. Importantly, the N501Y mutation occurs at the hACE2 binding site on sRBD. Previous experiments with adaptation of SARS‐CoV‐2 in the mouse [13] and high‐throughput screening of all possible mutations in sRBD [14] have predicted that the N501Y mutation can enhance the binding between sRBD and hACE2. As shown by previous experimental [8] and theoretical [15] results, the wild‐type SARS‐CoV‐2 sRBD can already bind slightly more strongly with the hACE2 than the SARS‐CoV sRBD; therefore, the N501Y variant with an even higher sRBD binding affinity with hACE2 is more transmissible. Note that, so far, it remains unknown whether the N501Y mutation works synergistically with other changes (such as the deletion of amino acids 69/70 and the P681H mutation) to result in a more contagious variant.
Here, we were motivated to investigate the molecular mechanism underlying the enhanced hACE2‐sRBD binding, induced by the N501Y mutation. Using the all‐atom molecular dynamics (MD) simulation as a computational microscope, we directly imaged hACE2‐sRBD binding at the atomic level, and explored how the N501Y mutation can cause conformational changes for residues residing at the hACE2‐sRBD interface. Furthermore, we employed the rigorous free energy perturbation (FEP) method to predict the binding affinity difference caused by the N501Y mutation detected in lineages B.1.1.7 and B.1.351 of SARS‐CoV‐2.
Materials and methods
MD simulations
All‐atom MD simulations were carried out for both the bound (the complex of hACE2 and sRBD) and free (stand‐alone sRBD) states using the namd2.13 [16] running on an IBM Power Cluster. To model the hACE2‐sRBD complex (a bound state), we first obtained the previously resolved crystal structure (Protein Data Bank code: 6VW1) [17] and then solvated the complex (with a bound Zn2+) in a rectangular water box measuring approximately 95 × 75×133 Å3. 104 Na+ and 79 Cl− were added into the system to neutralize the entire simulation system, setting the ion concentration at 0.15 m (Fig. 1A). The final system containing 96 897 atoms was first minimized for 10 ps and further equilibrated for 1000 ps in the NPT ensemble (P ~ 1 bar and T ~ 300 K), with atoms in the backbones harmonically constrained (spring constant k = 1 kcal·mol−1·Å−2). During the production run in the NVT ensemble, only atoms in the backbones of hACE2 that are far away from the sRBD (residues 110–290, 430–510 and 580–615) were constrained, preventing the whole complex from rotating out of the water box. We also performed MD simulation for the sRBD alone in the 0.15 m NaCl electrolyte (a free state) using the same protocol.
Fig. 1.
MD simulation of the hACE2‐sRBD complex. (A) Cartoon illustration of hACE2 (orange) bound with sRBD (gray). The residue N501 on sRBD is in the van der Waals sphere representation. Na+ and Cl− are shown as tan and cyan balls, respectively, and, for clarity, water is not shown. (B) RMSFs for residues in sRBD in MD simulation. (C) Time‐dependent interfacial contact areas between hACE2 and sRBD. (D) Equilibrated atomic structure for N501 in sRBD coordinating with Y41 and K353 in hACE2.
With equilibrated structures in bound and free states, we carried out FEP calculations [18]. In the perturbation method, many intermediate stages (denoted by λ) for which Hamiltonian H(λ) = λHf + (1 − λ)Hi are inserted between initial and final states to yield a high accuracy. With the softcore potential enabled, λ in each FEP calculation for ΔG A or ΔG B varies from 0 to 1.0 in 20 perturbation windows (lasting 0.3 ns in each window), yielding gradual annihilation and exnihilation processes for N501 and Y501, respectively. We followed our protocol (used in previous mutagenesis studies for optimizing a neutralizing antibody targeting SARS‐CoV‐2 [19]) to obtain the mean and error for ΔG A and ΔG B.
We applied the CHARMM36 force field [20] for proteins, the TIP3P model [21, 22] for water and the standard force field [23] for ions. The periodic boundary conditions were used in all three dimensions. Long‐range Coulomb interactions were calculated using particle‐mesh Ewald full electrostatics with a grid size of approximately 1 Å in each dimension. The van der Waals energies between atoms were calculated using a smooth (10–12 Å) cut‐off. The temperature T was maintained at 300 K by applying the Langevin thermostat [24], whereas the pressure was kept constant at 1 bar using the Nosé–Hoover method [25]. With the SETTLE algorithm [26] allowing all bonds to be kept rigid, the simulation time‐step was set at 2 fs for bonded and non‐bonded (including van der Waals, angle, improper and dihedral) interactions, and electric interactions were calculated every 4 fs with the multiple time‐step algorithm [27].
Results and Discussion
Figure 1 illustrates the simulation system for modeling the interaction between the hACE2 and sRBD, with the focus on the interfacial region. Detailed simulation protocols are provided in the Materials and methods. Briefly, atomic coordinates for the complex of hACE2 and sRBD were taken from the crystal structure (Protein Data Bank code: 6VW1). The protein complex was further solvated in a 0.15 NaCl electrolyte. The residue N501 in sRBD locates at the peripheral contact between hACE2 and sRBD (Fig. 1A). During the 185‐ns MD simulation, the hACE2‐sRBD complex originating from the crystal environment was properly equilibrated in the physiology‐like environment. Figure 1B shows the root mean square fluctuations (RMSF) of alpha carbon atoms in the backbone of sRBD. RMSF values for most residues in sRBD are less than or approximately 1.0 Å, indicating that the secondary structure of sRBD was stable. In addition to the disordered N‐terminal (residues 334–337) and C‐terminal (residues 519–527), residues from A475 to N487 (located in a long turn between two short anti‐parallel β‐sheets near the hACE2‐sRDB interface) fluctuated significantly, as manifested through their large RMSF values. However, these residues are not in contact with hACE2 and their fluctuations barely affected the binding stability between hACE2 and sRBD.
We then quantified the binding stability by calculating the time‐dependent contact area S between the hACE2 and sRBD (Fig. 1C). When analyzing the MD trajectory, we first calculated the solvent accessible surface area (SASA) for both the hACE2 (S A) and sRBD (S B) and then calculated the SASA for the entire complex (S AB). Thus, the contact area S between the hACE2 and sRBD can be estimated as (S A + S B − S AB)/2. Figure 1C shows that, during the MD simulation, the contact areas were almost constant (~ 8 nm2), indicating a stable binding between hACE2 and sRBD.
By analyzing residues located at the interface, we found that both Y41 and K353 in the hACE2 were within 3.5 Å from N501 in sRBD. Figure 1D illustrates the atomic coordinations among these interfacial residues. During most of the simulation time, the hydrophilic N501 was in the proximity of the hydrophobic benzene ring of Y41 and the hydrophobic alkane chain in K353. Therefore, it is concluded that these interfacial interactions can be improved by N501 being mutated into a hydrophobic residue. Consistently, through experimental screening of all possible mutations in sRBD, it was found that mutating N501 into V, F, W or Y can enhance sRBD binding with hACE2 [14]. In the emergent SARS‐CoV‐2 variant, the presence of the N501Y mutation in sRBD indeed caused the disease to become more contagious, a consequence of the enhanced binding between the hACE2 and sRBD.
To unveil the underlying molecular mechanism of the N501Y mutation, we performed FEP calculations [18]. As required in FEP calculations, 70‐ns long MD simulations of sRBD alone in a 0.15 m NaCl electrolyte (a free state) were also carried out. After obtaining protein structures for both bound and free states in respective MD simulations, we employed the FEP method to calculate the binding free energy difference for the N501Y mutation on the sRBD, using the thermodynamic cycle shown in Fig. 2. The changes in sRBD's binding free energies induced by the N501Y mutation can be calculated as ΔΔG = ΔG 2 − ΔG 1 = ΔG A − ΔG B. In practice, direct calculations of ΔG 1 and ΔG 2 (Fig. 2) are challenging, although this can be circumvented by computing ΔG A and ΔG B instead. Through the ensemble average [18], ΔG A and ΔG B can be calculated theoretically as ΔG A,B = −k B Tln < exp(Hf − Hi/k B T) > i, where k B is the Boltzmann constant; T is the temperature; and Hi and Hf are the Hamiltonians for the initial (i) and final (f) stages respectively. As shown in Fig. 2, for the N501Y mutation in bound/free states, the original sRBD with N501 is present in the initial stage and, in the final stage, the same residue becomes Y501 through the alchemical process in the FEP calculation.
Fig. 2.
Illustration of a thermodynamic cycle used in the FEP calculations with the mutation N501Y. (A) The bound state between the original sRBD and hACE2. (B) The bound state between the mutated sRBD (N501Y) and hACE2. (C) The free state of the original sRBD (with N501) in water. (D) The free state of the mutated sRBD (with Y501) in water. Protein segments (in cartoon representation) are colored the same as those in Fig. 1A.
Table 1 summarizes the results from the FEP calculations. In the bound state (Fig. 2A,B), the N501Y mutation resulted in an increase in free energies (i.e. ΔG A = 67.19 kcal·mol−1). In the free state (Figs. 2C,D), the same mutation yielded the free energy change ΔG B of approximately 68.00 kcal·mol−1. Note that these energy values for ΔG A and ΔG B include the interaction energy change between a residue and its environment, as well as the internal energy change within the residue. The latter cancels out when calculating ΔΔG. Overall, the value of ΔΔG is −0.81 kcal·mol−1 (with an error of 0.67 kcal·mol−1), suggesting that the N501Y mutation increases the binding affinity between hACE2 and sRBD (consistent with previous experimental results [13, 14]).
Table 1.
Values of for the N501Y mutation with respect to sRBD binding with hACE2 and CB6 (mAb).
| N501Y | ΔG A,B (kcal·mol−1) | ΔΔG (kcal·mol−1) |
|---|---|---|
| sRBD only | 68.00 ± 0.18 | – |
| sRBD‐hACE2 | 67.19 ± 0.65 | −0.81 |
| sRBD‐CB6 | 68.62 ± 0.43 | 0.62 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
We further explored the molecular mechanism of the N501Y mutation by analyzing the interfacial atomic structures in the simulation trajectory. As mentioned above, in the wild‐type S‐protein, N501 is actually unfavorably coordinated by hydrophobic fragments in Y41 and K353 of hACE2. With the mutation, Fig. 3 demonstrates that the hydrophobic pocket formed by Y41 and K353 (in hACE2) can be fit well by Y501 (in sRBD). Evidently, the edge of the Y501 benzene ring is in contact with the surface of the Y41 benzene ring (Fig. 3), forming the perpendicular T‐shaped contact, which is a well‐known hydrophobic interaction in addition to parallel π–π stacking. Additionally, the surface of the Y501 benzene ring interacts hydrophobically with the alkane chain of the amphipathic K353 (Fig. 3). We note that, with fluctuations, it is also possible that the surface of the Y501 benzene ring forms the so‐called cation‐π interaction with the charged group in K353. Therefore, Y501 in sRBD coordinates very well with both Y41 and K353 in hACE2. In Movie S1, we show how, during the alchemy FEP calculation, the exnihilated Y501 (in sRBD) gradually makes a good coordination with Y41 and K353 (in hACE2).
Fig. 3.
Enhanced interfacial coordinations between Y501 in sRBD and key residues (Y41 and K353) in hACE2.
The above results suggest that the N501Y mutation is favorable in the bound state. On the other hand, in the free state, the hydrophilic N501 is better than the hydrophobic Y501 at coordinating the surrounding water molecules. Thus, the N501Y mutation in the free state is unfavorable. Taken together, we found that Y501 in mutant sRBD energetically favors the bound state and can enhance the binding affinity between hACE2 and sRBD (i.e. ΔΔG < 0). We further verified the stability of new interfacial coordinations between Y501 in sRBD and residues Y41 and K353 in hACE2 by performing an additional large‐scale MD simulation (Fig. S1 and Movie S2 in Supporting Information).
Currently, neutralizing mAbs hold the promise of being both therapeutic and prophylactic for COVID‐19. It is still unknown how the N501Y mutation affects the binding between the S‐protein of SARS‐CoV‐2 and neutralizing antibodies. Apparently, this mutation cannot affect the binding of mAbs that target domains other than sRBD, such as the N‐terminal domain of S‐protein [28]. Additionally, even on targeting sRBD, some mAbs (e.g. CR3022 [29], S309 [30] and REGN10987 [31]) bind different epitopes and thus the N501Y mutation has little effect on such binding. However, many other mAbs (e.g. CB6 [32], P2B‐2F6 [33], B38 [34] and REGN10933 [19]) bind the epitope site in sRBD that overlaps with the binding site of hACE2. Therefore, the N501Y mutation is likely to affect the binding of these mAbs to sRBD. Below, we focus on how the N501Y mutation can affect the binding between sRBD and the mAb CB6.
The bound state for the complex of sRBD and the CB6 Fab (Fig. 4A) was obtained from our previous 200‐ns long MD simulation [19]. The complex highlights that sRBD mainly interacts with the heavy chain (blue in Fig. 4A) in the Fab. Although not as important as the heavy chain, the light chain (orange in Fig. 4A) also contacts the sRBD and the residue N501 is inside this contact (Fig. 4A). From FEP calculations, we found that ΔG A = 68.62 kcal·mol−1 for the N501Y mutation in the bound state. With respect to the value of ΔG B for the N501Y mutation in the free state (Table 1), ΔΔG = 0.62 kcal·mol−1 (with an error of 0.47 kcal·mol−1). The positive value of ΔΔG indicates that the N501Y mutation can weaken the binding between sRBD and the CB6 Fab. It is worth noting that, as a result of multiple epitopes on the spike protein targeted by various mAbs, the single N501Y mutation might not allow the virus to evade the human immune system.
Fig. 4.
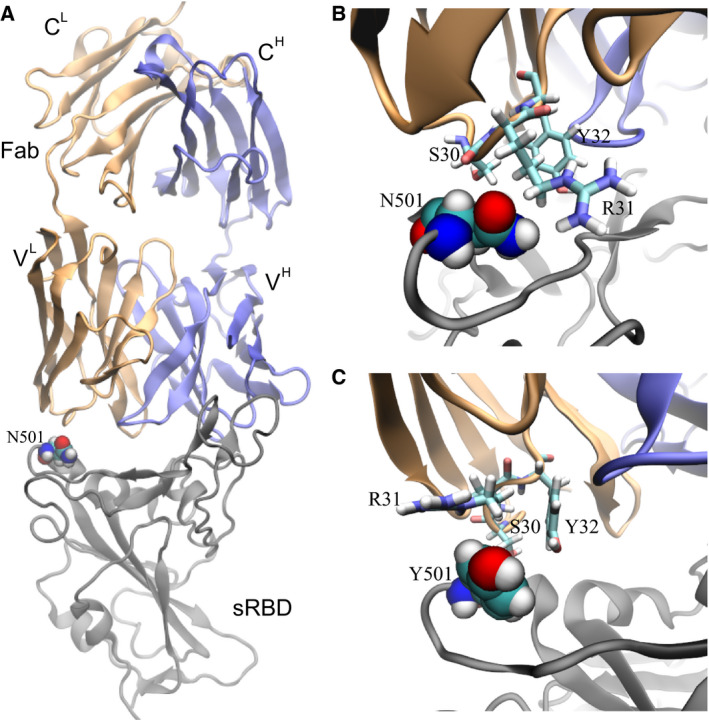
Effects of the N501Y mutation on the binding between the neutralizing mAb CB6 and sRBD. (A) The complex of sRBD and the Fab (in CB6) equilibrated in MD simulation. The Fab comprises one heavy chain (fragment) and one light chain, colored in blue and orange respectively; the sRBD is in gray. The heavy (light) chain contains a variable region VH (VL) and a constant region CH (CL). (B) Coordinations between N501 in sRBD and its surrounding residues (S30, R31 and Y32) in Fab, at the beginning of the FEP calculation. (C) Coordinations between Y501 in sRBD and surrounding residues (S30, R31 and Y32) in Fab, at the end of the FEP calculation.
However, it is essential to investigate the molecular mechanism with the aim of designing more efficacious therapeutic mAbs. Figure 4B illustrates the atomic coordination between N501 in sRBD and the surrounding residues (S30, R31 and Y32) in the Fab. Residues S30 and R31 are closer to N501 than Y32, forming more direct interactions. Despite being hydrophilic, N501, S30 and R31 failed to form stable hydrogen bonds among them. Occasionally, we observed the hydrogen bond between the ‐NH atoms in the guanidino group in R31 and the oxygen atom in the carboxamide side‐chain in N501, indicating a weak attraction. In the final stage of FEP calculations for the N501Y mutation, Fig. 4C illustrates that hydrophobic Y501 repels the charged guanidino group in R31 and is not close to the hydrophobic hydrocarbons in R31. Y32 and Y510 are spatially distant from each other, forming neither T‐shaped, nor π–π interactions. Additionally, S30 is not in close contact with Y501. Overall, Y501 cannot coordinate well with nearby residues from the CB6 Fab, which accounts for the positive value of ΔΔG.
As a biologics drug, CB6 can be modified accordingly to accommodate the N501Y mutation in sRBD. According to Fig. 4C, it would appear to be a promising strategy to mutate residue 31 in the light chain of the CB6 Fab from R to F, Y or W, which increases the hydrophobic interaction across the binding interface.
Conclusions
In summary, our in silico studies suggest that the N501Y mutation can enhance sRBD binding affinity with hACE2 and potentially cause the virus to evade antibody neutralization. These results are consistent with the fact that N501Y is a naturally occurring and selected mutation [13]. Prior to July, 2020, it had been already discovered that the N501Y mutation could be generated de novo from an adaptive murine model using only the parental wild‐type virus strain (IMEBJ05) first isolated in Beijing [13]. It only took the parental strain one passage to start exhibiting and accumulating the N501Y variant and, by the third passage, the N501Y strain took over 93% of the viral population, suggesting a much more favorable adaptation to the host than the wild‐type strain, presumably as a result of enhanced sRBD interaction with hACE2. In a more recent study [14], it was further validated that N501Y together with N501F, N501W and N501V exhibited enhanced binding affinity between sRBD and hACE2 in vitro. The N501Y mutation only requires one mutation (A23063T [6]), whereas the codon requirement for N501 converting to F, W or V requires at least two bases to mutate simultaneously, which is much less likely to occur directly from the wild‐type strain. Therefore, it is much easier for N501Y to occur naturally by evolution and selection. As a result of the hydrophobic nature of Y501, we conjectured that the more hydrophobic Y501 (than N501) might promote the closed prefusion form of the spike protein (i.e. all three sRBDs down), evading human antibodies (targeting the sRBD) before binding with hACE2.
We discovered that, after the N501Y mutation, Y501 in sRBD can simultaneously form hydrophobic interactions with Y41 and K353 in hACE2, yielding an enhanced interfacial binding. Namely, the benzene ring edge of Y501 forms the T‐shaped interaction with the benzene ring surface of Y41, and the benzene ring surface of Y501 interacts hydrophobically with the alkane chain in K353 (Fig. 3). Regarding the polymorphism of hACE2, two mutations, K26R and I468V, in hACE2 (and close to the bound sRBD) can occur frequently in non‐Finnish Europeans and East Asians [35] respectively; however, these residues (26 and 468) in hACE2 are not close to the residue 501 in sRBD. In a more complete study of different human ACE2 allelic variants [36], all of the identified mutations were also not close to the side‐chain of Y501 in the bound sRBD. Therefore, the molecular mechanism of the N501Y mutation revealed above is not altered by the known polymorphisms of hACE2.
Based on the molecular mechanism revealed in the present study for sRBD‐hACE2 binding (Fig. 3), we hypothesized that mutations of R31 in the light chain of CB6 Fab into hydrophobic F, Y or W might stabilize and improve the interfacial binding between sRBD and the modified CB6 antibody. This modified mAB specifically targeting the SARS‐CoV‐2 variant could be added into an antibody cocktail for treating all COVID‐19 patients. Overall, our results complement the experimental findings, provide structural insights to assist in evaluating the functional impact of this mutation and shed light on the design of more efficacious antibodies. Apparently, the N501Y mutation in sRBD provides significant edges for the virus to proliferate; therefore, continued studies on the interaction between sRBD and hACE2 are warranted for the development of treatments against COVID‐19.
Author Contributions
BL designed the study; BL and TH carried out simulations and data analysis; BL and HW discussed the results; BL, HW and TH rote paper together.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Supporting information
Fig. S1. MD simulation of the sRBD‐hACE2 complex with the N501Y mutation in the sRBD.
Movie S1. Showing how, during the alchemical process (FEP calculations), Y501 in sRBD gradually shows up and coordinates Y41 and K353 in hACE2. For clarity, the simultaneous gradual annihilation of N501 is not shown.
Movie S2. Showing the stability of the N501Y‐induced interfacial coordinations.
Acknowledgements
T.H and B.L. gratefully acknowledge the computing resources of the IBM Cognitive Computing Program.
Edited by Michael Bubb
References
- 1. Zhou P, Yang X‐L, Wang X‐G, Hu B, Zhang L, Zhang W, Si H‐R, Zhu Y, Li B, Huang C‐L et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R et al. (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yurkovetskiy L, Wang X, Pascal KE, Tomkins‐Tinch C, Nyalile TP, Wang Y, Baum A, Diehl WE, Dauphin A, Carbone C et al. (2020) Structural and functional analysis of the D614G SARS‐CoV‐2 spike protein variant. Cell 183, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, Zhang X, Muruato AE, Zou J, Fontes‐Garfias CR et al. (2021) Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature 592, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hou YJ, Chiba S, Halfmann P, Ehre C, Kuroda M, Dinnon KH, Leist SR, Schäfer A, Nakajima N, Takahashi K et al. (2020) SARS‐CoV‐2 D614G variant exhibits efficient replication ex vivo and transmission in vivo . Science 370, 1464–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rambaut A, Loman N, Pybus O, Barclay W, Barrett J and Carabelli AEA (2020) Preliminary genomic characterisation of an emergent SARS‐CoV‐2 lineage in the UK defined by a novel set of spike mutations. https://virological.org/
- 7. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N‐H, Nitsche A et al. (2020) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT and Veesler D (2020) Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang Y, Yang C, Xu X‐F, Xu W and Liu S‐W (2020) Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacol Sin 41, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turonová B, Sikora M, Schürmann C, Hagen WJ, Welsch S, Blanc FE, von Bülow S, Gecht M, Bagola K, Hörner C et al. (2020) In situ structural analysis of SARS‐CoV‐2 spike reveals flexibility mediated by three hinges. Science 370, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan R, Zhang Y, Li Y, Xia L, Guo Y and Zhou Q (2020) Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A and Li F (2020) Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci USA 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu H, Chen Q, Yang G, He L, Fan H, Deng Y‐Q, Wang Y, Teng Y, Zhao Z, Cui Y et al. (2020) Adaptation of SARS‐CoV‐2 in BALB/c mice for testing vaccine efficacy. Science 369, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC et al. (2020) Deep mutational scanning of SARS‐CoV‐2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nguyen HL, Lan PD, Thai NQ, Nissley DA, O’Brien EP and Li MS (2020) Does SARS‐CoV‐2 bind to human ACE2 more strongly than does SARS‐CoV? J Phys Chem B 124, 7336–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L and Schulten K (2005) Scalable molecular dynamics with NAMD. J Comp Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A and Li F (2020) Structural basis of receptor recognition by SARS‐CoV‐2. Nature 581, 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chipot C and Pohorille A (2007) Free Energy Calculations. Springer, New York, NY. [Google Scholar]
- 19. Luan B and Huynh T (2020) In silico antibody mutagenesis for optimizing its binding to spike protein of severe acute respiratory syndrome coronavirus 2. J Phys Chem Lett 11, 9781–9787. [DOI] [PubMed] [Google Scholar]
- 20. MacKerell A Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S et al. (1998) All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 21. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW and Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79, 926–935. [Google Scholar]
- 22. Neria E, Fischer S and Karplus M (1996) Simulation of activation free energies in molecular systems. J Chem Phys 105, 1902–1921. [Google Scholar]
- 23. Beglov D and Roux B (1994) Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J Chem Phys 100, 9050–9063. [Google Scholar]
- 24. Allen MP and Tildesley DJ (1987) Computer Simulation of Liquids. Oxford University Press, New York, NY. [Google Scholar]
- 25. Martinetz T and Schulten K (1994) Topology representing networks. Neural Netw 7, 507–522. [Google Scholar]
- 26. Miyamoto S and Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water molecules. J Comp Chem 13, 952–962. [Google Scholar]
- 27. Tuckerman M, Berne BJ and Martyna GJ (1992) Reversible multiple time scale molecular dynamics. J Chem Phys 97, 1990–2001. [Google Scholar]
- 28. Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, Zhang Z, Fan P, Dong Y, Yang Y et al. (2020) A neutralizing human antibody binds to the N‐terminal domain of the Spike protein of SARS‐CoV‐2. Science 369, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan M, Wu NC, Zhu X, Lee C‐CD, So RT, Lv H, Mok CK and Wilson IA (2020) A highly conserved cryptic epitope in the receptor binding domains of SARS‐CoV‐2 and SARS‐CoV. Science 368, 630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinto D, Park Y‐J, Beltramello M, Walls AC, Tortorici MA, Bianchi S, Jaconi S, Culap K, Zatta F, De Marco A et al. (2020) Cross‐neutralization of SARS‐CoV‐2 by a human monoclonal SARS‐CoV antibody. Nature 583, 290–295. [DOI] [PubMed] [Google Scholar]
- 31. Hansen J, Baum A, Pascal KE, Russo V, Giordano S, Wloga E, Fulton BO, Yan Y, Koon K, Patel K et al. (2020) Studies in humanized mice and convalescent humans yield a SARS‐CoV‐2 antibody cocktail. Science 369, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi R, Shan C, Duan X, Chen Z, Liu P, Song J, Song T, Bi X, Han C, Wu L et al. (2020) A human neutralizing antibody targets the receptor binding site of SARS‐CoV‐2. Nature 584, 120–124. [DOI] [PubMed] [Google Scholar]
- 33. Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S et al. (2020) Human neutralizing antibodies elicited by SARS‐CoV‐2 infection. Nature 584, 115–119. [DOI] [PubMed] [Google Scholar]
- 34. Wu Y, Wang F, Shen C, Peng W, Li D, Zhao C, Li Z, Li S, Bi Y, Yang Y et al. (2020) A noncompeting pair of human neutralizing antibodies block COVID‐19 virus binding to its receptor ACE2. Science 368, 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Q, Cao Z and Rahman P (2020) Genetic variability of human angiotensin‐converting enzyme 2 (hACE2) among various ethnic populations. Mol Genet Genomic Med 8, e1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A and Aziz B (2020) Structural variations in human ACE2 may influence its binding with SARS‐CoV‐2 spike protein. J Med Virol 92, 1580–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. MD simulation of the sRBD‐hACE2 complex with the N501Y mutation in the sRBD.
Movie S1. Showing how, during the alchemical process (FEP calculations), Y501 in sRBD gradually shows up and coordinates Y41 and K353 in hACE2. For clarity, the simultaneous gradual annihilation of N501 is not shown.
Movie S2. Showing the stability of the N501Y‐induced interfacial coordinations.