

Abstract
Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) pandemic spread rapidly and this scenario is concerning in South America, mainly in Brazil with more than seven million cases of infection. Three major pandemic lineages/clades could be identified along with SARS‐CoV‐2 dissemination (G, GR, and GH) in the Americas. These clades differ according to their genomic characteristics, virulence, and spreading times. The present study describes the main clades and the respective temporal spreading analyses based on SARS‐CoV‐2 whole‐genome sequences (WGS) from South America, obtained in the early pandemic phase (from March 1 to May 31 in 2020). SARS‐CoV‐2 WGSs with available information from country and year of sampling were obtained from different countries and the main clades were identified and analyzed independently with a Bayesian approach. The results demonstrated the prevalence of clades GR (n = 842; 54.6%), G (n = 529; 34.3%), and GH (n = 171; 11.1%). The frequencies of the clades were significantly different between South American countries. Clade G was the most prevalent in Ecuador, Suriname, and Uruguay, clade GR in Argentina, Brazil, and Peru, and clade GH in Colombia. The phylodynamic analysis indicated that all these main lineages increased viral spreading from February to early March and after an evolutionary stationary phase was observed. The decrease observed in the virus dissemination was directly associated to the reduction of social movement after March. In conclusion, these data demonstrated the current predominance of clades G, GR, and GH in South America because of the early dissemination of them in the first pandemic phase in South America.
Keywords: dissemination, pandemic, SARS coronavirus
Research Highlights
Three major pandemic clades could be identified along with SARS‐CoV‐2 dissemination (G, GR, and GH) in South America.
The prevalence of clades/lineages GR, G, and GH in South America were: 54.6%, 34.3%, and 11.1%.
These lineages increased viral spreading from February to early March 2020.
The decrease observed in the virus dissemination was directly associated to the reduction of social movement after March 2020.
In South America, the clades G, GR, and GH are predominant because of the early dissemination of them in the first pandemic phase.
1. INTRODUCTION
Several patients with pneumonia of unknown causes were identified in Wuhan, Hubei Province, China in December 2019. 1 , 2 , 3 Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) was rapidly associated to this new disease (coronavirus disease 2019 [COVID‐19]) that spread worldwide, accounting to more than 50 million confirmed cases and one million deaths in the first eight months from the pandemic. 4 In the American continent, 20,131,365 (48.6% in South America, 44.3% in North America, and 7.1% in Central America) cases were detected at 12 November 2020, and the five countries reporting most cases are United States (10,516,513), Brazil (5,747,660), Argentina (1,273,356), Colombia (1,165,326) and Peru (928,006). Death case records reach 614,626 deaths and the five countries reporting most deaths are United States (242,557), Brazil (163,368), Mexico (96,430), Peru (35,031), and Argentina (34,351). 4
A difference in case fatality rates of SARS‐CoV‐2 infection across countries was observed, possibly related to the diverse demographic composition around the World and the control measures adopted in different countries to reduce viral spreading. 5 , 6 , 7 According to the public database of the GISAID, three major SARS‐CoV‐2 lineages/clades could be initially identified and they were named as G (variant of the spike protein S‐D614G), V (variant of the ORF3a coding protein NS3‐G251), and S (variant ORF8‐L84S). 8 Afterwards, two other clades (GR and GH) emerged and rapidly spread together with G clade in the Western World. All these three clades are genetically similar and they differ in few single nucleotide polymorphisms (SNPs), some of them altering amino acid sequences in the viral proteins. Clade G has four main SNPs (C241, C3037T, C14408T, A23403G) that affect 5′‐untranslated region, NSP3 (F106F), NSP12b (P314L), and the spike protein (D614G) genes. Clade GR presents these same SNPs with an additional modification at the codon GGG28881AAC that generates the modification in the nucleocapsid protein (RG203KR). Finally, clade GH has another modification (G25563T) that affects the ORF3a (Q57H). 9 The high predominance of the clades G, GR, and GH are mainly associated with the occurrence of the amino acid substitution D614G that improved viral fitness. 10 , 11
Clades G, GR, and GH gradually predominated in the Americas, Africa, and Europe. 12 The extremely rapid viral spread raised questions about the way their evolution was driven during the pandemic. In South America, clades G, GR, and GH are the most frequent and their spreading was very pronounced. 12 The main objective was to study the introduction, the frequencies, and the temporal and geographic spreading of these three main clades in South America, mainly in Brazil, through a Bayesian approach with SARS‐CoV‐2 WGSs obtained from March 1 to May 31 in 2020.
2. METHODS
2.1. Data collection
SARS‐CoV‐2 WGSs with available information from country and year of sampling were obtained from GISAID 13 from March 1 to May 31, 2020. Five datasets were assembled: (I) with 414 sequences of clade G from South American countries (Table S1); (II) with 791 sequences of clade GR from South American countries (Table S2); (III) with 165 sequences of clade GH from South American countries (Table S3); (IV) with 207 sequences of clade G from Brazil (Table S4); (V) with 614 sequences of clade GR from Brazil (Table S5); GH clade WGSs from Brazil were not evaluated in a specific dataset because of the reduced number (n = 18).
Putative recombination events were verified using the Recombination Detection Program version 4 (RDP4) software 14 with the default settings using the algorithms RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq, and LARD. The beginning and end breakpoints of the potential recombinant sequences were also defined by the RDP4 software. Putative recombinant events were considered significant when p ≤ .01 was observed for the same event using four or more algorithms. Sequences that presented putative recombination events, clonal dissemination, and missing record of the collection were excluded from the evolutionary analysis.
2.2. Root‐to‐tip regression
Previous alignments were performed for each genotype by MAFFT v7 15 and visually inspected with AliView v1.26. The best‐fitting nucleotide substitution (HKY) model was selected using a hierarchical likelihood ratio, Akaike information criterion, and Bayesian information criterion tests with Model Finder in IQ‐TREE webserver. 16 SARS‐CoV‐2 sequences maximum likelihood phylogenetic tree were inferred according to the best‐fitting model using IQ‐TREE web server (http://iqtree.cibiv.univie.ac.at/). Statistical supports for internal branches in the phylogeny were assessed by bootstrapping (1000 replicates) and the approximate likelihood ratio test (aLRT). 17 The resulting tree was used to further obtain root‐to‐tip regressions in TempEst v1.5 18 by selecting the root position that maximized the correlation coefficient. The root‐to‐tip versus divergence plot of the full datasets showed a correlation between sampling time and genetic distance to the root of the ML tree of the available sequences (R 2 = 0.65 for dataset I, R 2 = 0.71 for dataset II, R 2 = 0.58 for dataset III, R 2 = 0.67 for dataset IV, and R 2 = 0.56 for dataset V), suggesting moderate temporal signals and the possibility to calibrate a reliable molecular clock.
2.3. Bayesian coalescent inference
Time‐scaled phylogenetic tree estimation was performed using BEAST/BEAGLE v 2.5 software. 19 The best‐fitting nucleotide substitution (HKY) model with gamma site distribution was selected using a hierarchical likelihood ratio, Akaike information criterion, and Bayesian information criterion tests with Model Finder in IQ‐TRE web server (http://iqtree.cibiv.univie.ac.at/). For each run of 250 million of Monte Carlo Markov Chains (MCMC), the marginal likelihood was estimated via path sampling (PS) and stepping stone (SS) methods and the resulting Bayes factors (BF) (ratio of marginal likelihoods) used to select the best‐fitting clock/demographic model. The models can be compared to evaluate the strength of evidence against the null hypothesis (H0) defined in the following way: 2lnBF < 2 indicates no evidence against H0; 2–6, weak evidence; 6–10: strong evidence, and greater than 10 very strong evidence. 20 Both SS and PS estimators indicated the uncorrelated lognormal molecular clock as the best‐fitted model to the datasets under analysis (BF > 10 for all datasets). MCMC analysis was performed and the maximum likelihood estimations of the obtained trees were compared using a BF to select the best model and parameter values. BF analysis showed that the uncorrelated lognormal clock fitted the data significantly better than other clocks (2lnBF > 500 for all datasets). BF analysis showed that the Bayesian skyline plot (BSP) was better than other models (2lnBF > 100 for both datasets). MCMC was run for 500 million generations to ensure stationary and adequate effective sample size for all statistical parameters. Tracer v.1.6 software 21 was used to diagnose MCMC, adjust initial burn‐in, and to perform the Skyline demographic reconstruction. Uncertainty in parameter estimates was evaluated in the 95% highest posterior density (HPD 95%) interval. TreeAnnotator v1.8.2 was used to summarize the maximum clade credibility (MCC) tree from the posterior distribution of trees and the MCC tree was visualized and edited in FigTree v.1.4.4 (available at http://tree.bio.ed.ac.uk/software/figtree/).
2.4. Statistical analysis
Statistical analyses were performed using the SPSS (Statistical Package for Social Sciences, version 23.0) software. The frequencies of SARS‐CoV‐2 clades were compared between South American countries and Brazilian regions using the Pearson chi‐square test. All estimates were bilateral with a significance level of 5% (p < .05).
3. RESULTS
3.1. SARS‐CoV‐2 clades in South America
A total of 1542 SARS‐CoV‐2 genomes classified as clades G, GR, and GH were identified from GISAID database (GISAID, 2020c). The prevalence of clades G, GR, and GH in South America were 34.3% (n = 529), 54.6% (n = 842), and 11.1% (n = 171) respectively. The frequencies of these three clades were significantly different between South American countries (p < .01) (Table 1). Clade G had a frequency of 23.7% in Argentina, 25.8% in Brazil, 34.8% in Chile, 54.3% in Ecuador, 34.0% in Peru, 94.4% in Suriname and 69.8% in Uruguay. The clade GR had frequencies of 52.6% in Argentina, 72.1% in Brazil, 31.9% in Chile, 11.9% in Colombia, 38.6% in Ecuador, 60.4% in Peru, 27.9% in Uruguay and 33.3% in Venezuela. The clade GH was detected in 23.7% cases in Argentina, 2.1% in Brazil, 33.3% in Chile, 50.0% in Colombia, 7.1% in Ecuador, 5.7% in Peru, 2.3% in Uruguay and 66.7% in Venezuela. Clade G was the most frequent in Suriname (94.4%), Uruguay (69.8%), Ecuador (54.3%), and Chile (34.8%). Clade GR was the most frequent in Brazil (72.1%), Peru (60.4%), and Argentina (52.6%). Finally, GH was frequent in Colombia (50.0%) and Venezuela (66.7%). Clade G was the most prevalent in Ecuador, Suriname, and Uruguay (p < .01); clade GR Argentina, Brazil, and Peru (p < .01); and clade GH in Colombia (p < .01) (Table 1; Figure 1A).
Table 1.
Distribution of SARS‐CoV‐2 clades G, GR, and GH in South American countries
| South American Country | G | GR | GH | Total | p Value a | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | ||
| Argentina | 9 | 23.7 | 20 | 52.6 | 9 | 23.7 | 38 | 2.5 | <.01 |
| Brazil | 221 | 25.8 | 618 | 72.1 | 18 | 2.1 | 857 | 55.6 | <.01 |
| Chile | 49 | 34.8 | 45 | 31.9 | 47 | 33.3 | 141 | 9.1 | .53 |
| Colombia | 61 | 38.1 | 19 | 11.9 | 80 | 50.0 | 160 | 10.4 | <.01 |
| Ecuador | 38 | 54.3 | 27 | 38.6 | 5 | 7.1 | 70 | 4.5 | <.01 |
| Peru | 54 | 34.0 | 96 | 60.4 | 9 | 5.7 | 159 | 10.3 | <.01 |
| Suriname | 67 | 94.4 | 4 | 5.6 | 0 | 0.0 | 71 | 4.6 | <.01 |
| Uruguay | 30 | 69.8 | 12 | 27.9 | 1 | 2.3 | 43 | 2.8 | <.01 |
| Venezuela | 0 | 0.0 | 1 | 33.3 | 2 | 66.7 | 3 | 0.2 | .51 |
| Total | 529 | 34.3 | 842 | 54.6 | 171 | 11.1 | 1542 | 100.0 | <.01 |
Abbreviation: SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Pearson's χ 2 test was used to analyze the differences between the frequencies of clades G, GR, and GH between each country specifically. Values of p < .05 were significant.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 1.
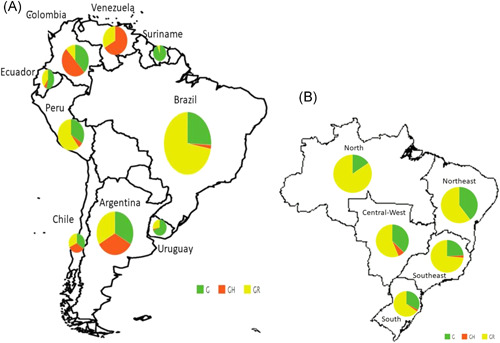
Distribution of severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) clades G, GR, and GH in (A) South American countries and in (B) Brazilian regions
The prevalence of clades G, GR, and H in Brazil were 25.8%, 72.1%, and 2.1% (p < .01) respectively (Table 2). Clade GR was the most prevalent in all Brazilian regions (65.0% in the South; 73.5% in the Southeast; 56.3% in the Central‐West; 84.6% in the North; 60.2% in the Northeast). Clade G had a frequency of 32.5% in the South, 24.0% in Southeast, 37.5% in Central‐West, 15.4% in the North, and 39.8% in Northeast. Clade GH was detected in 2.5% cases in the South, 2.4% in the Southeast, and 6.3% in Central‐West (Table 2; Figure 1B).
Table 2.
Distribution of SARS‐CoV‐2 clades G, GR, and GH according to Brazilian regions
| Brazilian region | G | GR | GH | Total | p Value a | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | ||
| South | 13 | 32.5 | 26 | 65.0 | 1 | 2.5 | 40 | 4.7 | <.01 |
| Southeast | 157 | 24.0 | 481 | 73.5 | 16 | 2.4 | 654 | 76.3 | <.01 |
| Central West | 6 | 37.5 | 9 | 56.3 | 1 | 6.3 | 16 | 1.9 | .03 |
| North | 8 | 15.4 | 44 | 84.6 | 0 | 0 | 52 | 6.1 | <.01 |
| Northeast | 37 | 39.8 | 56 | 60.2 | 0 | 0 | 93 | 10.9 | <.01 |
| Not available | 0 | 0 | 2 | 100 | 0 | 0 | 2 | 0.2 | .67 |
| Total | 221 | 25.8 | 618 | 72.1 | 18 | 2.1 | 857 | 100 | <.01 |
Abbreviation: SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Pearson's χ 2 test was used to analyze the differences between the frequencies of clades G, GR, and GH between Brazilian regions. Values of p < .05 were significant.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2. Clade G phylodynamic in South America
SARS‐CoV‐2 clade G phylogenetic tree demonstrated five clusters clades (I–V). In cluster G I (n = 19) were detected sequences only from Peru in basal branches from the whole phylogenetic tree. Cluster G II (n = 6) presented sequences from Peru (66.7%), Chile (16.7%), and Colombia (16.7%). Cluster G III (n = 95) demonstrated sequences from Brazil (29.5%), Uruguay (27.4%), Chile (13.7%), Colombia (12.6%), Peru (11.6%), and Ecuador (5.3%). Cluster G IV (n = 99) showed sequences from Colombia (35.4%), Chile (32.3%), Peru (10.1%), Brazil (9.1%), Argentina (8.1%), Uruguay (4.0%), Ecuador (1.0%), and Uruguay (4.0%). Finally, cluster G V (n = 195) showed sequences from Brazil (87.7%), Ecuador (5.6%), Suriname (2.6%), Chile (1.5%), Colombia (1.5%), Argentina (0.5%), and Peru (0.5%) (Figure 2A).
Figure 2.
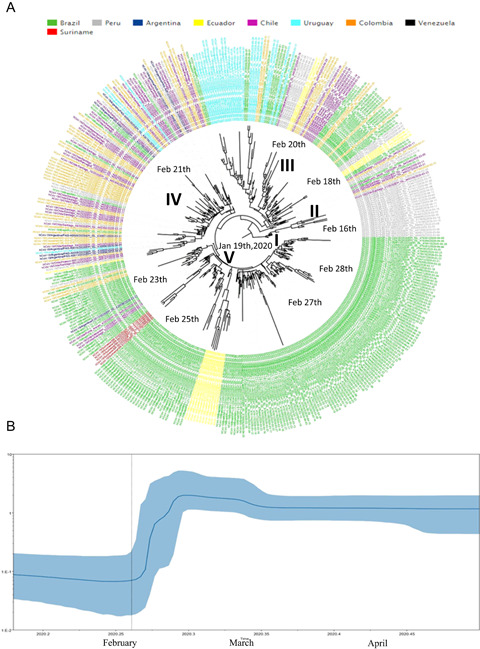
(A) Time‐scaled maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 clade G whole‐genome sequences from South American countries available in GISAID (from March 1 to May 31, 2020). The time of the most recent common ancestor (tMRCA) are demonstrated in the nodes with significant posterior probabilities (≥0.95). (B) Bayesian skyline plot (BSP) of SARS‐CoV‐2 clade G whole‐genome sequences obtained from GISAID. The effective number of infections is reported on the Y‐axis. The timeline is reported on the X‐axis. The colored area corresponds to the 95% credibility intervals of highest probability density (95% HPD). The vertical line indicate the 95% lower HPD (dotted) of the tree root. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2
Brazilian sequences were branched in three clusters: G III (n = 28; 75.0% from Southeast, 17.9% from North, 3.6% from Central‐West, and 3.6% from South), G IV (n = 9; 55.6% from Southeast, 22.2% from South, and 22.2% from Northeast), and G V (n = 171; 69.6% from Southeast, 19.9% form Northeast, 5.8% from South, 2.9% from Central‐West, and 1.8% from North) (Figure 2A).
Clade G root of the phylogenetic tree dated back to 19 January 2020 (HPD 95%: December 23, 2019– February 6, 2020) and sequences from Peru clustered in the more basal branches from the phylogenetic tree. Clade G substitution rates was 2.26E10‐3 (HPD 95%: 1.02E10‐3 to 3.55E10‐3) nucleotides per site per year. Clade G seems to be disseminated between Peru, Chile, and Colombia. Afterward, strains of this clade were disseminated between Argentina, Brazil, Chile, Ecuador, Suriname, and Uruguay. The BSP analysis of Clade G genomes showed that the dissemination grew between February 16 and 28, 2020. After March 2020, a stationary growth phase was observed (Figure 2B).
3.3. Clade GR phylodynamic in South America
SARS‐CoV‐2 clade GR phylogenetic tree demonstrated six main clusters (I–VI). In cluster GR I (n = 57) were detected sequences from Peru (73.7%), Argentina (8.8%), Uruguay (7.0%), Chile (5.3%), Brazil (3.5%), and Ecuador (1.8%) in basal branches from the whole phylogenetic tree. Cluster GR II (n = 15) presented sequences from Brazil (53.3%), Argentina (13.3%), Chile (13.3%), Uruguay (13.3%), and Peru (6.7%). Cluster GR III (n = 106) demonstrated sequences from Brazil (40.6%), Colombia (17.9%), Chile (17.0%), Peru (11.3%), Ecuador (7.5%), Argentina (4.7%), and Venezuela (0.9%). Cluster GR IV (n = 28) showed sequences from Peru (53.6%), Chile (42.9%), and Brazil (3.2%). Cluster GR V (n = 244) showed sequences from Brazil (99.6%) and Argentina (0.4%). Finally, Clade GR VI (n = 337) showed sequences from Brazil (93.8%) (Figure 3A).
Figure 3.
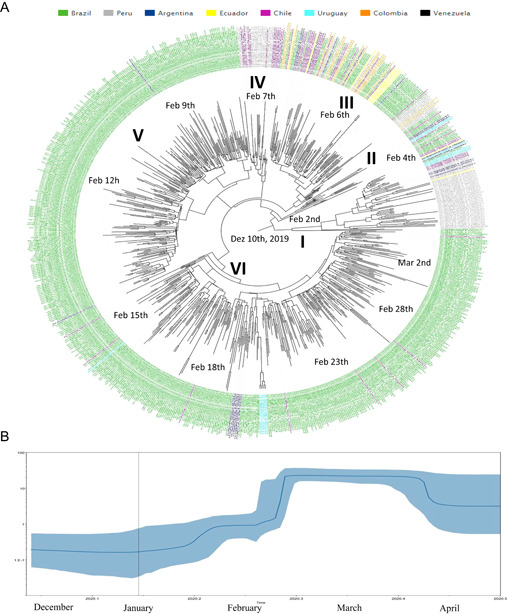
(A) Time‐scaled maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 clade GR whole‐genome sequences from South American countries available in GISAID (from 1 March to 31 May 2020). The tMRCA are demonstrated in the nodes with significant posterior probabilities (≥0.95). (B) BSP of SARS‐CoV‐2 clade GR whole‐genome sequences obtained from GISAID. The effective number of infections is reported on the Y‐axis. The timeline is reported on the X‐axis. The colored area corresponds to the 95% credibility intervals of HPD. The vertical line indicate the 95% lower HPD (dotted) of the tree root. BSP, Bayesian skyline plot; HPD, highest probability density; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; tMRCA, time of the most recent common ancestor
Brazilian sequences were branched in six clusters: GR I (n = 2; 50% from Southeast and 50% from North), GR II (n = 8; 75.0% from Northeast and 25.0% from Southeast), GR III (n = 43; 69.8% from North, 25.6% from Southeast, and 4.7% from South), GR IV (n = 1; 100% from Southeast), GR V (n = 243; 87.9% from Southeast, 6.2% from North, 4.9% from South, and 2.1% from North), and GR VI (n = 315; 79.7% from Southeast, 8.9% from Northeast, 4.7% from North, 3.8% from South, and 2.8% from Central‐West) (Figure 3A).
Clade GR root of the phylogenetic tree dated back to January 2, 2020 (HPD 95%: December 21, 2019–February 14, 2020) and sequences from Argentina, Uruguay, Chile, Brazil, Peru, and Ecuador clustered in the more basal branches from the phylogenetic tree, highlighting that these countries were the probable geographic origins of this clade. Clade GR substitution rates was 2.89E10‐3 (HPD 95%: 1.77E10‐3 to 3.87E10‐3) nucleotides per site per year. Clade GR strains possibly were disseminated in Brazil, Peru, Argentina, Chile, Ecuador, Peru, Uruguay, and Venezuela. The BSP analysis of Clade G genomes showed that the dissemination grew between February 2 and March 1, 2020. After March 2020, an evolutionary stationary phase was observed (Figure 3B).
3.4. Clade GH phylodynamic in South America
SARS‐CoV‐2 clade GH phylogenetic tree demonstrated six main clusters (I–III). Sequences from Brazil and Colombia were observed in basal branches from the whole phylogenetic tree. Cluster GH I (n = 19) presented sequences from Argentina (36.8%), Colombia (36.8%), Chile (10.5%), Brazil (10.5%), and Uruguay (5.3%). Cluster GH II (n = 39) demonstrated sequences from Colombia (64.1%), Chile (25.6%), Brazil (5.1%), Argentina (2.6%), and Peru (2.6%). Finally, in cluster GH III (n = 100) were observed sequences from Colombia (44.0%), Chile (34.0%), Brazil (11.0%), Peru (7.0%), Venezuela (2.0%), Argentina (1.0%), and Ecuador (1.0%). Brazilian sequences were branched in three clusters: GH I (n = 3; 100% from Southeast), GH II (n = 2; 100% from Southeast), and GH III (n = 11; 90.9% from Southeast and 9.1% from Central‐West) (Figure 4A).
Figure 4.
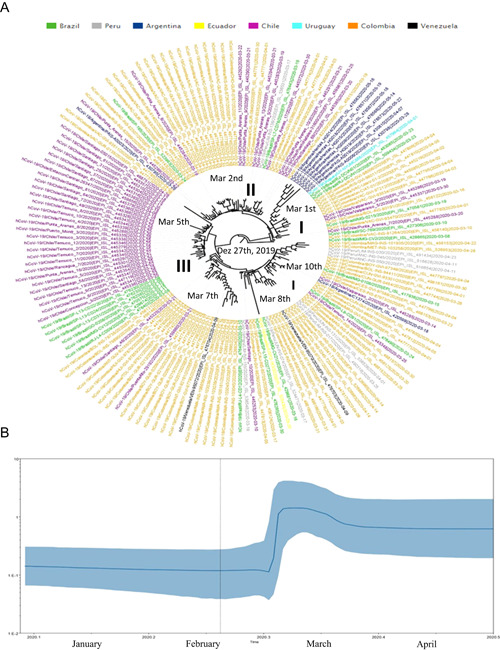
(A) Time‐scaled maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 clade GH whole‐genome sequences from South American countries available in GISAID (from March 1 to May 31, 2020). The tMRCA are demonstrated in the nodes with significant posterior probabilities (≥0.95). (B) BSP of SARS‐CoV‐2 clade GH whole‐genome sequences obtained from GISAID. The effective number of infections is reported on the Y‐axis. The timeline is reported on the X‐axis. The colored area corresponds to the 95% credibility intervals of HPD. The vertical line indicate the 95% lower HPD (dotted) of the tree root. BSP, Bayesian skyline plot; HPD, highest probability density; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; tMRCA, time of the most recent common ancestor
Clade GH root of the phylogenetic tree dated back to December 27, 2019 (HPD 95%: December 12, 2019–February 7, 2020) and sequences from Brazil (Southeast) clustered in the more basal branches from the phylogenetic tree, highlighting that this country was the probable geographic origin of this clade. Clade GH substitution rates was 2.68E10‐3 (HPD 95%: 1.03E10‐3 to 3.31E10‐3) nucleotides per site per year. This clade seems to be disseminated in Brazil, Peru, Argentina, Chile, Colombia, Ecuador, Peru, Uruguay, and Venezuela. The BSP analysis of Clade G genomes showed that the dissemination grew between March 1 and 6, 2020. After March 2020, an evolutionary stationary phase was observed (Figure 4B).
3.5. Clades G and GR phylodynamic in Brazil
In an attempt to understand the evolutionary history of the G and GR clades of SARS‐CoV‐2 in Brazil, we estimate these phylodynamic processes using datasets with sequences from Brazil. Clade G root of the phylogenetic tree dated back to December 10, 2019 (HPD 95%: December 5 2019–February 19, 2020) and sequences from Southeast clustered in the more basal branches from the phylogenetic tree, highlighting that this region was the probable geographic origin of this clade in Brazil. Clade G substitution rates was 2.33E10‐3 (HPD 95%: 1.13E10‐3 to 3.74E10‐3) nucleotides per site per year. This clade seems to be disseminated from the Southeast to other regions after February 26, 2020 (Figure 5A).
Figure 5.
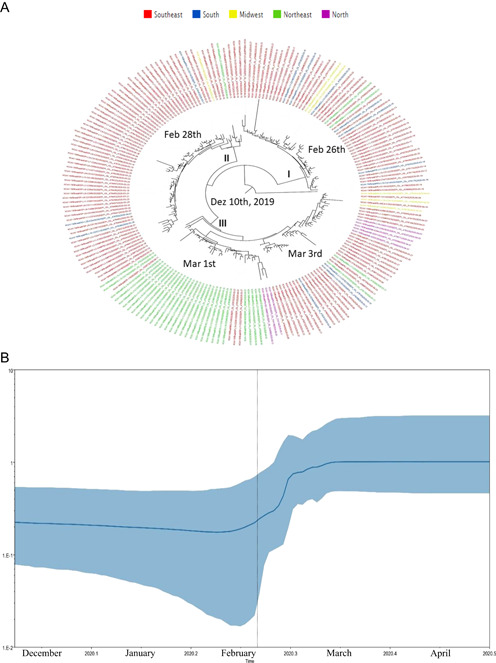
Maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 clade G (Panel A) whole‐genome sequences from Brazilian regions available in GISAID (from March 1 to May 31, 2020). The tMRCA are demonstrated in the nodes with significant posterior probabilities (≥0.95). Panel B showed BSP of SARS‐CoV‐2 whole‐genome sequences obtained from GISAID of clade G. The effective number of infections is reported on the Y‐axis. The timeline is reported on the X‐axis. The colored area corresponds to the 95% credibility intervals of HPD. The vertical line indicate the 95% lower HPD (dotted) of the tree root. BSP, Bayesian skyline plot; HPD, highest probability density; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; tMRCA, time of the most recent common ancestor
Clade GR root of the phylogenetic tree dated back to January 20, 2020 (HPD 95%: December 21, 2019–February 13, 2020) and sequences from Southeast clustered in the more basal branches from the phylogenetic tree, highlighting that this region was the probable geographic origin of this clade in Brazil. Clade GR substitution rates was 2.48E10‐3 (HPD 95%: 1.51E10‐3 to 3.47E10‐3) nucleotides per site per year. This clade seems to be disseminated from the Southeast to other regions after February 15, 2020 (Figure 6A).
Figure 6.
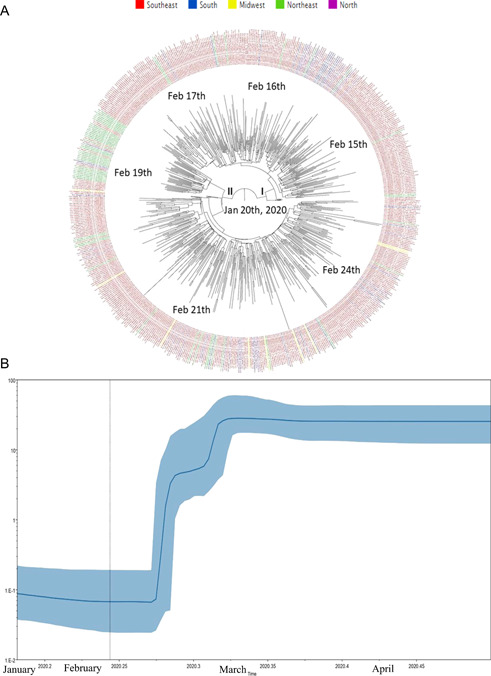
Maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 clade GR (Panel A) whole‐genome sequences from Brazilian regions available in GISAID (from March 1 to May 31, 2020). The tMRCA are demonstrated in the nodes with significant posterior probabilities (≥0.95). Panel B showed BSP of SARS‐CoV‐2 whole‐genome sequences obtained from GISAID of clade GR. The effective number of infections is reported on the Y‐axis. The timeline is reported on the X‐axis. The colored area corresponds to the 95% credibility intervals of HPD. The vertical line indicate the 95% lower HPD (dotted) of the tree root. BSP, Bayesian skyline plot; HPD, highest probability density; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; tMRCA, time of the most recent common ancestor
The BSP analysis of Clade G genomes showed that the dissemination grew from February 26 to March 10. After mid‐March 2020, a stationary growth phase was observed (Figure 5B). Also, the BSP analysis of Clade GR genomes showed that the dissemination grew from February 18 to March 13. After mid‐March 2020, a stationary growth phase was observed (Figure 6B).
4. DISCUSSION
The phylogenetic characterization of an emerging viral infection can help in understanding and monitoring the pandemic progression. Currently, there is a very large amount of WGSs data to study the recent SARS‐CoV‐2 spreading. More evolutionary and dissemination studies are necessary to understand the SARS‐CoV‐2 genetic diversity and to identify the main epidemic findings. These data are essential to define public health measures to control the current pandemic. In South America, most countries presented massive SARS‐CoV‐2 dissemination from March to May 2020. 22 Reports showed that the first COVID‐19 cases were identified in late February and early March 2020. 9 , 22 , 23 , 24 Previous studies reported that growing epidemics in Brazil, Peru, Mexico, Chile, Colombia, Panama, and possibly Venezuela and Nicaragua at this time. 23 , 24 , 25 , 26 , 27 , 28
Clades L, O, S, and V were more frequent at the beginning of the pandemic, but clades G, GR, and GH gradually predominated in most western countries. These clades are widely prevalent in the World, especially in South America, North America, Africa, and Europe. 12 , 13 Historically, clade L seems to be the common ancestor of all SARS‐CoV‐2 strains disseminated later in the western world countries. After mid‐February, clades G, GR, and GH were originated due to specific mutations, mainly D614G that improved viral fitness. 10 , 12 , 13 The clade G is characterized by the spike protein D614G mutation which has been suggested to increase transmissibility but not pathogenicity. After this initial stage, the clade GR increased rapidly, stabilized around 30% between March and May 2020, and increased further to become the most frequent clade in June 2020. Clade GH showed a peak of cases in May 2020 (30%) and has rapidly declined since then. 12 , 13
In the present study, the evolution and spread of the main clades circulating in South America (G, GR, and GH) were evaluated by a Bayesian coalescent approach at the beginning of the high dissemination in the Americas (March–May, 2020). Another study had already demonstrated that clades G and GR became the most prevalent in the Americas. 12 Here it was highlighted that clade GR was the predominant one followed by G and GH in South America. Possibly, all these clades have become predominant because of the emergence of the D614G mutation that increased viral fitness. 10 , 11 Our results demonstrate that the prevalence of clades G, GR, and GH in South America were 34.3%, 54.6%, and 11.1%, respectively. Also, all these clades presented a clear geographic distribution. Clade G was the most frequent in Suriname, Uruguay, Ecuador, and Chile; clade GR was the most frequent in Brazil, Peru, and Argentina; clade GH predominates in Colombia and Venezuela. The prevalence of clades G, GR, and GH demonstrated here agree with the results observed in other studies that evaluated South American sequences (≅ 30.0% for G, ≅ 60.0% for GR, and ≅ 10.0% for GH). 9 , 12
In Brazil, all three clades were detected in different geographic regions because of the several introductions into the country. 22 , 23 , 24 The predominance of clades G, GR, and GH in Brazil and South America are also in accordance with the data previously reported. 9 The greatest spread of these clades seems to have occurred during early March 2020 worldwide, and high dissemination was evident in South America and Brazil. 9 , 22 , 23 , 24 These findings indicate that these clades were extremely important in the establishment of the beginning of the SARS‐CoV‐2 pandemic in South America.
Here we also demonstrated that all these clades (G, GR, and GH) were possibly disseminated between February and March 2020 in South America. This temporal pattern of dissemination was also similar to that observed specifically for Brazil. In this sense, there is evidence that more than 100 international introductions of SARS‐CoV‐2 occurred in Brazil. It was observed a rapid spread of COVID‐19 through the country, with more populated and better‐connected districts being affected prior, and less populated districts being affected at a later stage of the epidemic. Brazil announced COVID‐19 as a national public health crisis on February 3 2020. 22 , 29 After the development of a national crisis plan and the early establishment of molecular diagnostic offices over Brazil's network of public health laboratories, the country detailed its first confirmed COVID‐19 case on February 25, 2020, in a traveler returning to São Paulo from northern Italy. 22 , 30
Currently, Brazil has one of the fastest‐growing COVID‐19 epidemics in the world, comprising over 55% of the overall number of reported cases in Latin America and the Caribbean. 31 SARS‐CoV‐2 was mainly disseminated between February and March 2020, a period that includes the carnival, which was possibly the trigger for the initial dissemination in the country. Further, the Brazilian Southeast region was fundamental for the fast spread of SARS‐CoV‐2, since it was one of the epicenters for the dissemination to other regions of the country. 23 , 32 After March, we can observe the evolutionary stability phase in the spread of clades G, GR, and GH. We can understand that with the beginning of the quarantine, social isolation measures were adopted, the use of masks was mandatory, so limiting the movement of people and reducing service opening hours (i.e., grocery, workplaces, transit station, parks, retail, and recreation) and resulting in a first control of the spread of SARS‐CoV‐2 strains. In this sense, Figure S1 represents the number of visitors change since the beginning of the pandemic in Brazil, demonstrating the reduction of social movement activities in different areas (grocery, pharmacy stories, workplaces, transit stations, parks, and retail and recreations) after March 2020.
Constant monitoring of mutations will also be pivotal in tracking the movement of the virus between individuals and across geographical areas. For example, our descriptive analysis of clade prevalence over time (Figures 2, 3, and 4A) shows the possibly common ancestor for clades G, GR, and GH in South America between December 2019 and March 2020 with mainly spread events between February and March 2020. These clades possibly have then reached the American continent between February and March 2020 and are currently the fastest‐evolutionary viral subpopulation worldwide. 33 , 34
SARS‐CoV‐2 genome phylogeny investigation reveals that this D614G mutation appeared to emerge from an ancestral D residue, in a glycosylated region of the viral spike protein. It has been hypothesized that mutations in this region change the intensely glycosylated viral spike (S) and improve the membrane fusion capabilities between the SARS‐CoV‐2 and the host cell, increasing viral transmissibility, and pathogenicity. 10 , 35 , 36 Noteworthy, D614G is highly conserved in clades G, GR, and GH of SARS‐CoV‐2 and it has been strongly associated with the high prevalence of these clades worldwide today. This mutation affects viral fitness, increasing the transmissibility and pathogenicity of the virus, allowing strains of clades G, GR, and GH to have an advantage over others by the natural selection process. 10 , 11 , 37 Recent studies have reported other mutations in the SARS‐CoV‐2 spike gene improving, even more, the viral fitness, resulting in novel strains and lineages with very high transmissibility and pathogenicity in different continents of the World, such as Europe (as B.1.1.7 from the United Kingdom), Africa (as B.1.351 from South Africa) and also South America (as P.1 from Brazil). 38 , 39 , 40
Competition among viral strains of changing virulence is being evidenced during the current SARS‐CoV‐2 dissemination and COVID‐19 pandemic. The continuous monitoring of the most frequent SARS‐CoV‐2 clades, lineages, and strains as well as their specific dynamic evolution processes are now imperative for epidemiologists to define public health measures, such as to limit or to relax the social movement. Furthermore, this information will be necessary to develop more appropriate diagnostic tests (as the molecular biology methods) and vaccines for the circulating SARS‐CoV‐2 strains. Deeply surveillance of viral transmission at local and global scales and the evaluation of the effect of the different control measures on COVID‐19 transmission will offer assistance to decide an ideal mitigation procedure to minimize infections and decrease public healthcare demand. Therefore, continued monitoring of the SARS‐CoV‐2 genetic and antigenic diversity is already essential for public human health in the World.
5. CONCLUSION
Three SARS‐CoV‐2 clades were disseminated in the early pandemic phase in South America: G (mainly in Suriname, Uruguay, Ecuador, and Chile), GR (mainly in Brazil, Peru, and Argentina), GH (mainly in Colombia and Venezuela). The strains of these three clades had D614G amino acid modification and spread in the continent mainly from February to early March. The statistical results suggested that strains from clade G spread mainly between February 16 and 28, 2020, clade GR between February 2 and March 1, and clade GH between March 1 and March 6, 2020. The continuous monitoring of the most frequent SARS‐CoV‐2 clades, lineages, and strains as well as their specific dynamic evolution processes are now imperative for epidemiologists to define public health measures, diagnostic tests, and vaccines.
CONFLICT OF INTERESTS
The authors declare that htere are no conflicts of interests.
AUTHOR CONTRIBUTIONS
Jonas Michel Wolf and Vagner Ricardo Lunge designed the study. Jonas Michel Wolf performed the bioinformatics analyses. Jonas Michel Wolf, André Felipe Streck, André Fonseca, Nilo Ikuta, Daniel Simon, and Vagner Ricardo Lunge wrote the first draft of the manuscript and contributed to the literature review and discussion of results. All authors contributed to and have approved the final manuscript.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
This study was funded by ULBRA, Simbios Biotecnologia, and Financier of Studies and Projects (COVID—07/2020 Program FINEP—TECNOVA/RS II—2nd Edition—Economic Subsidy to Innovation, process numbers: 48080.599.26791.12062020). Vagner Ricardo Lunge were also financially supported by the National Council for Scientific and Technological Development from Brazil (CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico; process numbers 313564/2014‐0; 313304/2014‐9). Jonas Michel Wolf was further supported by the Coordination for the Improvement of Higher Education Personnel from Brazil (CAPES—Coordenação de Aperfeiçoamento de Pessoal de Nível Superior; Finance Code 001).
Wolf JM, Streck AF, Fonseca A, Ikuta N, Simon D, Lunge VR. Dissemination and evolution of SARS‐CoV‐2 in the early pandemic phase in South America. J Med Virol. 2021;93:4496–4507. 10.1002/jmv.26967
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Zhu N, Zhang D, Wang W, et al. China Novel Coronavirus Investigating and Research Team . A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727‐733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019‐nCoV. J Med Virol. 2020;92(5):501‐511. 10.1002/jmv.25701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus‐infected pneumonia. N Engl J Med. 2020;382(13):1199‐1207. 10.1056/NEJMoa2001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.GISAID Initiative. COVID‐19 dashboard by the Center for Systems Science and Engineering (CSSE) at John Hopkins University. 2020. https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6. Accessed November 12, 2020.
- 5. Castells M, Lopez‐Tort F, Colina R, Cristina J. Evidence of increasing diversification of emerging severe acute respiratory syndrome coronavirus 2 strains. J Med Virol. 2020;92(10):2165‐2172. 10.1002/jmv.26018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dowd JB, Andriano L, Brazel DM, et al. Demographic science aids in understanding the spread and fatality rates of COVID‐19. Proc Natl Acad Sci USA. 2020;117(18):9696‐9698. 10.1073/pnas.2004911117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai A, Bergna A, Acciarri C, Galli M, Zehender G. Early phylogenetic estimate of the effective reproduction number of SARS‐CoV‐2. J Med Virol. 2020;92(6):675‐679. 10.1002/jmv.25723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS‐CoV‐2 genomes. Proc Natl Acad Sci USA. 2020;117(17):9241‐9243. 10.1073/pnas.2004999117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mercatelli D, Giorgi FM. Geographic and Genomic Distribution of SARS‐CoV‐2 Mutations. Front Microbiol. 2020;11:1800. 10.3389/fmicb.2020.01800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brufsky A. Hyperglycemia, hydroxychloroquine, and the COVID‐19 pandemic. J Med Virol. 2020;92(7):770‐775. 10.1002/jmv.25887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plante JA, Liu Y, Liu J, et al. Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature. 2020. 10.1038/s41586-020-2895-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. GISAID . Latest update. 2020. https://www.gisaid.org/hcov-19-analysis-update/. Accessed November 12, 2020.
- 13. GISAID . GISAID initiative. 2020. https://www.epicov.org/epi3/frontend#502179. Accessed November 12, 2020.
- 14. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003. 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32(1):268‐274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minh BQ, Nguyen MA, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30(5):1188‐1195. 10.1093/molbev/mst024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evol. 2016;2(1):vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bouckaert R, Vaughan TG, Barido‐Sottani J, et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLOS Comput Biol. 2019;15(4):e1006650. 10.1371/journal.pcbi.1006650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773‐795. [Google Scholar]
- 21. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol. 2018;67(5):901‐904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poterico JA, Mestanza O. Genetic variants and source of introduction of SARS‐CoV‐2 in South America. J Med Virol. 2020;92(10):2139‐2145. 10.1002/jmv.26001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Candido DDS, Watts A, Abade L, et al. Routes for COVID‐19 importation in Brazil. J Travel Med. 2020;27(3):taaa042. 10.1093/jtm/taaa042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paiva MHS, Guedes DRD, Docena C, et al. Multiple introductions followed by ongoing community spread of SARS‐CoV‐2 at one of the largest metropolitan areas of Northeast Brazil. Viruses. 2020;12(12):E1414. 10.3390/v12121414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burki T. COVID‐19 in Latin America. Lancet Infect Dis. 2020;20(5):547‐548. 10.1016/S1473-3099(20)30303-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cimerman S, Chebabo A, Cunha CAD, Rodríguez‐Morales AJ. Deep impact of COVID‐19 in the healthcare of Latin America: the case of Brazil. Braz J Infect Dis. 2020;24(2):93‐95. 10.1016/j.bjid.2020.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ezequiel G, Jafet A, Hugo A, et al. The COVID‐19 pandemic: a call to action for health systems in Latin America to strengthen quality of care. Int J Qual Health Care. 2020:mzaa062. 10.1093/intqhc/mzaa062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller MJ, Loaiza JR, Takyar A, Gilman RH. COVID‐19 in Latin America: novel transmission dynamics for a global pandemic? PLOS Negl Trop Dis. 2020;14(5):e0008265. 10.1371/journal.pntd.0008265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Croda J, Oliveira WK, Frutuoso RL, et al. COVID‐19 in Brazil: advantages of a socialized unified health system and preparation to contain cases. Rev Soc Bras Med Trop. 2020;53:e20200167. 10.1590/0037-8682-0167-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jesus JG, Sacchi C, Candido DDS, et al. Importation and early local transmission of COVID‐19 in Brazil, 2020. Rev Inst Med Trop Sao Paulo. 2020;62:e30. 10.1590/s1678-9946202062030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. World Health Organization . Coronavirus disease (COVID‐2019) situation reports. 2020. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Accessed November 12, 2020.
- 32. de Souza WM, Buss LF, Candido DDS, et al. Epidemiological and clinical characteristics of the COVID‐19 epidemic in Brazil. Nat Hum Behav. 2020;4(8):856‐865. 10.1038/s41562-020-0928-4 [DOI] [PubMed] [Google Scholar]
- 33. Hufsky F, Lamkiewicz K, Almeida A, et al. Computational strategies to combat COVID‐19: useful tools to accelerate SARS‐CoV‐2 and coronavirus research. Brief Bioinform. 2020:bbaa232. 10.1093/bib/bbaa232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercatelli D, Triboli L, Fornasari E, Ray F, Giorgi FM. Coronapp: A web application to annotate and monitor SARS‐CoV‐2 mutations. J Med Virol. 2020:jmv.26678. 10.1002/jmv.26678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260‐1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292.e6. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182(4):812‐827.e19. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volz E, Mishra S, Chand M, et al. Transmission of SARS‐CoV‐2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2020. 10.1101/2020.12.30.20249034 [DOI] [Google Scholar]
- 39. Wibmer CK, Ayres F, Hermanus T, et al. SARS‐CoV‐2 501Y.V2 escapes neutralization by South African COVID‐19 donor plasma. Nat Med. 2021. 10.1038/s41591-021-01285-x [DOI] [PubMed] [Google Scholar]
- 40. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of a novel SARS‐CoV‐2 lineage in Manaus, Brazil. medRxiv. 2021. 10.1101/2021.02.26.21252554 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.