

Abstract
The ability of a ketogenic diet to treat seizures and render a neuronal network more resistant to strong electrical activity has been observed for a century in clinics and for decades in research laboratories. Alongside ongoing efforts to understand how this therapy works to stop seizures, metabolic health is increasingly appreciated as critical buffer to resisting and recovering from acute and chronic disease. Accordingly, links between metabolism and health, and the broader emerging impact of the ketogenic diet in improving diverse metabolic, immunological and neurological conditions, have served to intensify the search for its key and/or common mechanisms. Here we review diverse evidence for increased levels of NAD+, and thus an altered ratio of NAD+/NADH, during metabolic therapy with a ketogenic diet. We propose this as a potential unifying mechanism, and highlight some of the evidence linking altered NAD+/NADH with reduced seizures and with a range of short and long-term changes associated with the beneficial effects of a ketogenic diet. An increase in NAD+/NADH is consistent with multiple lines of evidence and hypotheses, and therefore we suggest that increased NAD+ may be a common mechanism underlying beneficial effects of ketogenic diet therapy.
Keywords: Ketogenic diet, Seizures, Epigenetic changes, Adenosine, Mitochondria, Metabolism
1. Introduction
Effective energy production and consumption is essential, and as one example carbohydrates are converted to glucose via digestive enzymes, absorbed in the intestine, and released in the blood stream to provide the main reactant for energy production (Hewitt, 1924). When dietary carbohydrates are insufficient, an alternative mechanism to maintain ATP generation is increased production and metabolism of the ketone bodies acetoacetate (AcAc) and β-hydroxybutyrate (β-OHB) (Laffel, 1999). It has long been known that decreased carbohydrates accompanied by increased fat will shift the dependence of energy production from glucose to ketone bodies (Wigglesworth, 1924) and promote the metabolic changes observed during what is now termed a ketogenic diet (KD).
Since the early 1900s, the KD has been used to decrease seizures (Pulford, 1927), although its use declined for many decades due to the development of anticonvulsant drugs. Recently, the diet regained popularity due to wider public awareness and additional clinical data affirming its ability to treat refractory epilepsy (Kossoff et al., 2009; Lefevre and Aronson, 2000; Neal et al., 2008). While it is most popular in pediatric epilepsy, more recent evidence indicates significant anti-seizure effects in adult patients regardless of age or seizure type (Cervenka et al., 2017; Sirven et al., 1999), and establishes beneficial effects in a range of neurological conditions (Camberos-Luna and Massieu, 2020).
To date, a fundamental mechanism that explains its diverse beneficial effects remains elusive. It may be that there is none: the KD mobilizes multiple mechanisms, and a different cohort may benefit different disorders (Masino and Rho, 2019). Alternatively, there may be a fundamental mechanism that underlies these multiple mechanisms, and could be a powerful pan-disease therapeutic target. Identifying this mechanism and its accessibility as a therapeutic target has far-reaching implications.
Based on differential utilization of nicotinamide adenine dinucleotide (NAD) during glucose-based versus ketone-based metabolism (Lodish et al., 2000), altered cellular levels of NAD+ during ketone-based ATP generation would be expected. NAD is an essential metabolic coenzyme, a signaling molecule, a marker for mitochondrial and cellular health, and a substrate for deacetylating (sirtuins) and ribosyltransferase (PARP) enzymes implicated in longevity and DNA damage repair (Belenky et al., 2007; Lin and Guarente, 2003; Rustin et al., 1996). In this review, we highlight the role of the oxidized form of NAD (NAD+) as a potential fundamental starting point for multiple mechanisms proposed to underly KD action in supporting metabolic health as well as treating epileptic seizures and potentially other disorders.
2. Glucose versus ketone body-based metabolism and NAD redox state
Consumption of a diet with sufficient carbohydrates will increase blood glucose (Hewitt, 1924), which readily crosses the blood-brain-barrier and enters cells through facilitated diffusion or active transport and serves as the main energy source in the central nervous system (Lodish et al., 2000; Maher et al., 1994). Generating cellular ATP from glucose starts with glycolysis, where a single 6-carbon glucose molecule is converted to two 3-carbon pyruvate molecules (Lodish et al., 2000). For each pyruvate produced, an ATP molecule is generated and a single molecule of NAD+ is used as a cofactor and released in its reduced form, NADH. NADH is then oxidized in the electron transport chain to generate three ATP molecules (Fig. 1, upper left). Pyruvate is converted to acetyl-CoA via oxidative decarboxylation, which reduces another NAD+ molecule to NADH. Each acetyl-CoA molecule enters the citric acid cycle to generate twelve additional ATPs (Fig. 1, lower left).
Fig. 1.
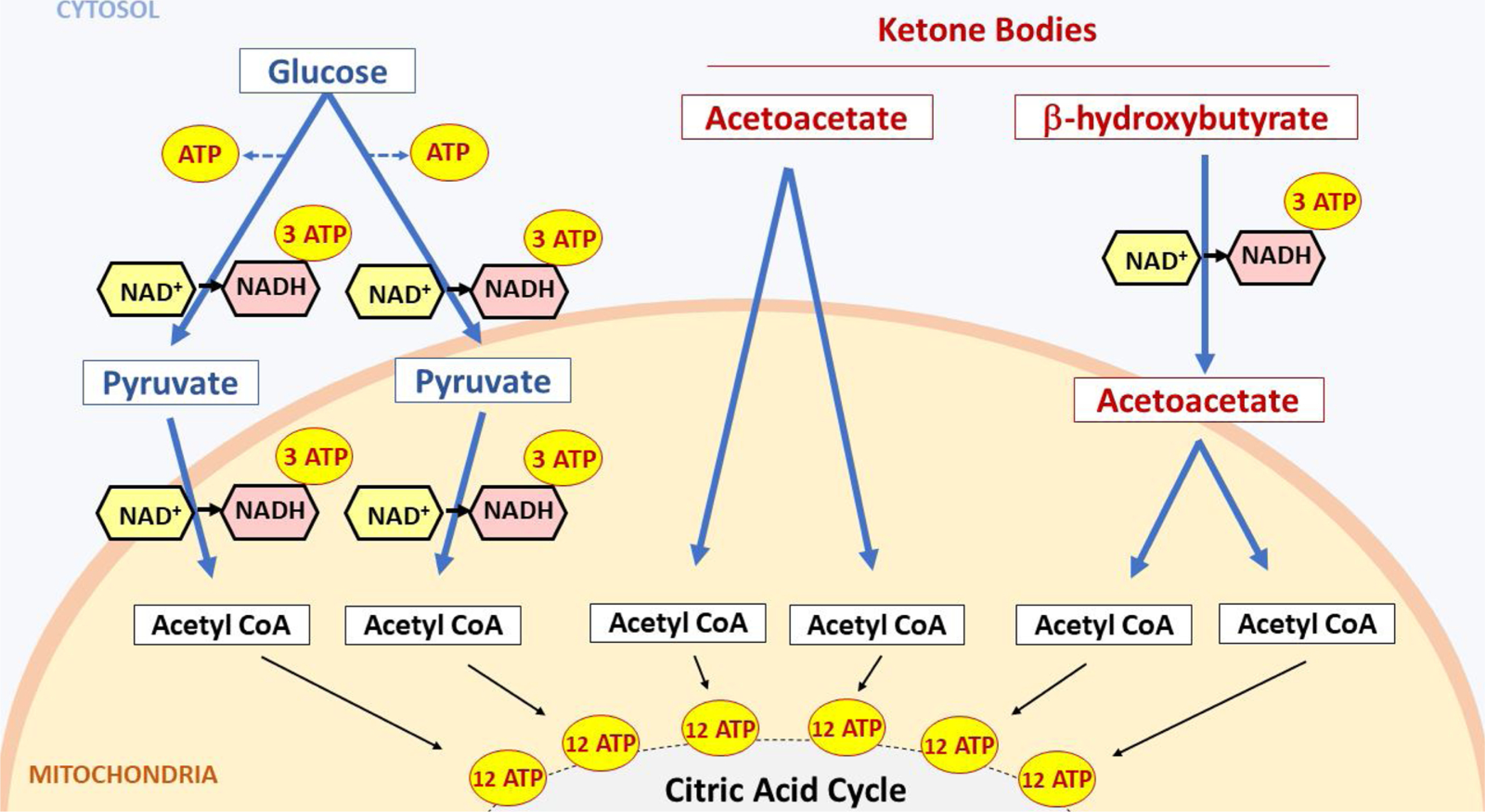
Alternate metabolic pathways whereby glucose (left) versus ketone bodies (right) are converted to ATP. Note decreased NAD+ consumption via ketone bodies versus glucose. See text for details.
In summary, when glucose is oxidized, the following changes in NADH and ATP occur: 1. during glycolysis, 8 ATP are produced (from 2 NADH + 2 ATP); 2. during oxidative decarboxylation, 6 ATP are produced (from 2 NADH) and 3. during the citric acid cycle, with two molecules of Acetyl CoA, 24 ATP are made from 12 NADH + 4 FADH2 + 4 GTP. In total 38 ATP molecules are produced.
Note that in the electron transport chain, NADH and FADH will produce approximately three and two ATP, respectively, while GTP will produce one ATP (Ahmad and Kahwaji, 2018; Lodish et al., 2000).
In contrast, fasting or consuming a KD causes the body to undergo lipolysis (fat breakdown) in order to generate fatty acids (FAs) as energy source in muscles and other tissues (Paoli, 2014). FAs are not able to cross the blood-brain-barrier: they are taken up by the liver to synthesize the ketone bodies AcAc and β-OHB which are then released into the bloodstream. Ketone bodies do cross the blood-brain barrier and enter the cells to undergo ketolysis to generate energy for the brain. Each ketone body molecule produces two molecules of acetyl-CoA (Fukao et al., 2004) (Fig. 1, right) which subsequently enter the citric acid cycle to produce ATP (Izuta et al., 2018; Krebs, 1970). When β-HB is converted to AcAc, it reduces one NAD+ molecule to NADH (Fig. 1, right), while AcAc molecules are converted to two acetyl-CoA molecules without the need to reduce NAD+ molecules. Therefore, the yield of ATP for a molecule of β-OHB is 27 (24 from Acetyl CoA and 3 from NADH produced during the conversion step to AcAc), and the net yield of ATP for AcAc is 24 (from Acetyl CoA) (Fig. 1, right).
At first glance, glucose is more efficient in ATP production. To produce 1000 ATP molecules only 28 glucose molecules are needed as compared to 37 β-OHB molecules and 42 AcAc molecules. But there are significant differences in NAD+ reduction: glucose reduces 112 molecules of NAD+, whereas β-OHB reduces 37 and AcAc reduces none. Accordingly, to produce comparable amounts of ATP, glucose-based metabolism reduces at least thrice the NAD+ molecules compared to ketone based-metabolism. Considering that a single cortical neuron can consume 4.7 billion ATPs per second at rest (Zhu et al., 2012), it is reasonable to assume that the above-mentioned differences in NAD+ utilization will have a significant effect on NAD redox state and the ratio of NAD+/NADH. It is worth noting that studies have already shown that ketones are a more efficient fuel compared to glucose. A study looking at the hydraulic work and oxygen consumption of hearts metabolizing glucose versus ketone bodies uncovered that ketones increased fuel efficiency by 25 % (Sato et al., 1995). Neurons grown in the presence of the ketone body β-OHB showed increased oxygen consumption and ATP production (Marosi et al., 2016). Multiple other studies also showed that metabolizing ketone bodies in lieu of glucose led to significant increases in ATP production (Kim et al., 2010; Murray et al., 2016; Nylen et al., 2009), further confirming the higher efficiency of ketones as energy substrates.
3. Key biological roles of NAD
Differential NAD+ utilization is significant because NAD plays a central role in two cellular processes:
Redox reactions, where it acts as an oxidizing agent – accepting electrons and converting to its reduced form, NADH. These redox reactions are important in cellular respiration, generation of ATP, and neutralization of microorganisms through the formation of free radicals (Imai and Johnson, 2018; Rustin et al., 1996).
Enzymatic reactions where it is required as a substrate. These NAD- dependent enzymes affect multiple cellular functions, ranging from gene expression and posttranslational modification of proteins to deacetylation and ADP-ribosylation reactions in the cells (Belenky et al., 2007). Two major enzymes that use NAD+ in the central nervous system are sirtuins and PARPs (Cantó et al., 2013 ).
In addition to its intracellular roles, NAD+ has been recognized as an extracellular signaling molecule released by multiple mammalian cell types, including astrocytes (Haag et al., 2007; Verderio et al., 2001). Multiple studies have also recognized NAD+ as a neurotransmitter released by neurons in both the central and peripheral nervous system (Breen et al., 2006; Durnin et al., 2012; Mutafova-Yambolieva et al., 2007; Smyth et al., 2004). Extracellular functions of NAD+ include – but are not limited to – modulating calcium signaling, purinergic signaling, immune cells function, and neurotransmitter release and postsynaptic signaling (Aarhus et al., 1995; Durnin et al., 2012; Haag et al., 2007; Kuzmin et al., 2016; Verderio et al., 2001).
Regarding a direct relationship between NAD+ and seizures, recent research shows that NAD+ can suppress the early stages of epileptogenesis in pilocarpine-treated rats, and also prevent hippocampal apoptosis (Liu et al., 2017). This is an important finding in a progressive seizure model that replicates the clinical relationship between a precipitating event and subsequent seizure development in patients. But if altered consumption of NAD+ by ketone-based metabolism is a starting point for the therapeutic benefits of a KD, there must be further credible links to key downstream processes that align with well-established clinical observations.
4. Ketogenic therapy in the management of epilepsy
Despite strong and consistent reports of clinical success for nearly a century, the first randomized controlled study utilizing KD in pediatric epilepsy was reported in 2008 (Neal et al., 2008). It demonstrated that refractory patients treated with KD improved significantly compared to those treated with a standard of care (who did not improve). Evidence of success continues to accumulate as more centers globally prescribe the diet and support families in its implementation as well as strive to collect long-term data (Bertoli et al., 2014; Gerges et al., 2019; Groleau et al., 2014; Heussinger et al., 2018; Tian et al., 2019). Correlations have been found between β-OHB levels in the blood and the CSF and the degree of seizure control in epileptic children treated with a ketogenic diet (Gilbert et al., 2000;Ruskin et al., 2017).
In the laboratory a variety of in vivo and in vitro models have been pursued, sometimes with mixed results. Overall it has been determined that the success of the diet in reducing neuronal excitability may depend on species, seizure model, and glucose concentration in vitro (Kawamura et al., 2016, 2014). In general, chronic seizure models in rats and mice that offer more clinically-relevant models show clear benefits of KD. Overall, the ketogenic diet does not have a marked effect on baseline activity but has a large effect in reducing high intensity (i.e. ATP-consuming) electrical activity (Blaise et al., 2015; Koranda et al., 2011; Ma et al., 2007; Viggiano et al., 2016).
5. Ketogenic therapy modulates NAD+ levels
As noted, the metabolic pathways of glucose versus ketone bodies exert differential effects on NAD+ reduction, and research has shown that a ketogenic treatment can modulate neuronal NAD redox state. In vitro, dissociated neurons from C57BL/6 mice incubated with β-OHB for 24 h show increased NAD+/NADH ratio and ATP production (Marosi et al., 2016). In vivo, KD increases hippocampal NAD+/NADH ratio in healthy wild-type rats, and this increase appears to be rapid (within two days) and persistent (lasting at least three weeks) (Elamin et al., 2018, 2017). Similarly, nutritional ketosis achieved via ingestion of medium chain triglyceride oils increases NAD+/NADH ratio in healthy human subjects (measured via 31P magnetic resonance spectroscopy) (Xin et al., 2018).
The relationship between KD and NAD+ is maintained under pathological conditions. After three months of subcutaneous injections of ketone bodies, fresh cortical and hippocampal tissues extracted from mice with an Alzheimer-linked gene variant showed a significant increase in NAD+/NADH ratio and ATP production (Yin et al., 2019). Moreover, treating hippocampal pyramidal neurons with ketone bodies resulted in an increase in NAD+/NADH ratio and oxygen consumption rate (Hasan-Olive et al., 2019), and application of the ketone bodies β-OHB and AcAc protected rat neocortical neurons from glutamate toxicity by increasing NAD+/NADH ratio (Maalouf et al., 2007), consistent with the ability of ketone bodies to modulate NAD redox state.
6. Common mechanisms mobilized by ketogenic therapy and altered NAD+
6.1. Enhanced mitochondrial function
NAD is an essential factor for mitochondrial ATP generation (Stein and Imai, 2012), and studies indicate NAD+ is a rate-limiting factor for several mitochondrial processes (Alano et al., 2004; Bai et al., 2011). Indeed, increasing NAD+ levels was found to enhance mitochondrial function and protect against oxidative stress damage (Kussmaul and Hirst, 2006; Pittelli et al., 2011). Cell lines and primary cultures exhibited an increase in mitochondrial ATP production and oxygen consumption after exogenous application of NAD (Kussmaul and Hirst, 2006; Lin and Guarente, 2003; Pittelli et al., 2011). Furthermore, circadian oscillations in mitochondrial NAD+ synthesis were shown to be associated with enhanced mitochondrial respiration and ATP production (Peek et al., 2013).
Regarding the role of NAD+ in ketogenic therapy, in vitro application of ketone bodies was shown to enhance mitochondrial respiration and protect against excitotoxicity via increasing NAD+/NADH ratio in isolated cortical mitochondria (Maalouf et al., 2007). Neurons showed similar results and the application of β-OHB increased the NAD+/NADH ratio while enhancing mitochondrial oxygen consumption and ATP production (Marosi et al., 2016). As noted, ketone-based metabolism is associated with enhanced mitochondrial functions (Bough et al., 2006; Sook Noh et al., 2004), which can play a significant role in controlling epileptic seizures (Rahman, 2012; Waldbaum and Patel, 2010).
Considering the ability of NAD+ to significantly enhance mitochondrial function in a manner similar to ketone-based metabolism (Kussmaul and Hirst, 2006; Maalouf et al., 2007; Marosi et al., 2016; Peek et al., 2013), it is reasonable to surmise that some of the beneficial effects of ketogenic therapy are mediated by NAD+. The rapidity through which NAD+ changes appear might partially explain the rapid efficacy of the KD observed in many patients after only a few days of KD treatment (Freeman and Vining, 1999). A rapid decrease in NAD+ availability and the subsequent effects on neuronal excitability could also be expected upon discontinuation of treatment, and indeed, 15 % of refractory epileptic patients experienced a rapid recurrence of seizures after discontinuation of the KD (Martinez et al., 2007). However, the inhibition of seizures persisted in other patients even after diet discontinuation (Martinez et al., 2007). This differential response among patients to treatment cessation indicates the existence of multiple downstream mechanisms and epigenetic changes implicated in seizure control or prevention (Lusardi et al., 2015; Masino and Rho, 2012). Upregulation of key ketogenic enzymes, mainly mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase, after longer periods of ketogenic treatment (Cullingford et al., 2002) might play a role in this maintenance of the beneficial effects after discontinuation of the diet.
6.2. Ketogenic diet, NAD+ and activation of sirtuin enzymes
Sirtuins are a group of deacetylase enzymes (SIRT1 - SIRT7) that use NAD+ as a main substrate (Michishita, 2005). Increasing the deacetylating activity of sirtuin enzymes can be achieved either by increasing gene expression levels, which subsequently increases the quantity of the enzymes, or by increasing cellular levels of NAD+ (Dali-Youcef et al., 2007; Landry et al., 2000; Revollo et al., 2004).
The most prominent and abundant member of this group is the nuclear SIRT1 whose main function is the deacetylation of several important targets that regulate apoptosis, inflammation, several growth factors, and transcription factors (Yang et al., 2006). SIRT2 is a cytosolic enzyme involved in the regulation of gluconeogenesis (Jiang et al., 2011) and central nervous system myelination (Beirowski et al., 2011), while SIRT4 is found to regulate fatty acid oxidation and mitochondrial gene expression (Nasrin et al., 2010). SIRT6 and SIRT7 are nuclear enzymes that are involved in decreasing age-associated DNA damage (McCord et al., 2009; Mostoslavsky et al., 2006; Vazquez et al., 2016). Hepatic SIRT6 is necessary for the ketogenic response to fasting and KD (Chen et al., 2019).
SIRT3 is the most abundant mitochondrial sirtuin (Michishita, 2005) that plays a pivotal role in the regulation of mitochondrial respiration and fatty acid oxidation. In fact, mice lacking SIRT3 were phenotypically normal at base line, however, they exhibited severe metabolic irregularities during fasting, highlighting the role of SIRT3 in maintaining energy homeostasis (Hirschey et al., 2010). β-OHB application to cultured hippocampal neurons elevated SIRT3 expression that had been lowered by oxidative stress, part of a pathway enhancing mitochondrial bioenergetics (Hasan-Olive et al., 2019). β-OHB treatment in vivo also elevates SIRT3 levels lowered by human apolipoprotein E4 expression in hippocampus and cortex of mice (Yin et al., 2019).
Taken together, sirtuins impact a wide range of functions. Increasing NAD+ levels will have a direct effect on sirtuins activity, and may mediate the beneficial effects associated with all seven sirtuin subtypes (Dali-Youcef et al., 2007; Landry et al., 2000; Revollo et al., 2004). SIRT1 was shown to mediate the seizure-suppressing effects of the micro RNA 199a-5p (Wang et al., 2016), highlighting the role of SIRT1 as a potential target for treating epilepsy. Administration of resveratrol, a well-known SIRT1 activator (Alcaín and Villalba, 2009), resulted in neuroprotection in a multitude of seizure models and showed synergistic effects with common anti-epileptic drugs (Pallàs et al., 2014). It has been hypothesized that one aspect of the neuroprotective effects of the KD is enhanced neuronal macroautophagy partially mediated by increased SIRT1 activity (McCarty et al., 2015).
In a temporal lobe epilepsy model, a decrease in NAD+ and a decrease in SIRT3 protein expression were uncovered in the acute and latent phases of epileptogenesis and thought to contribute to disease pathogenesis (Gano et al., 2018). Hippocampal pyramidal neurons in KD-fed mice exhibited upregulation of SIRT3, and treatment of the dissociated cells with ketone bodies increased NAD+/NADH ratio and oxygen consumption rates and improved mitochondrial biogenesis (Hasan-Olive et al., 2019).
It is important to note that sirtuin activity is not restricted to seizure inhibition but might also modulate neuronal death: in a model of acute acquired epilepsy, PARP-1 mediated neuronal death exposed the compromised enzymatic activity of SIRT1. NAD repletion was able to enhance SIRT1 activity and resulted in decreased neuronal death (Wang et al., 2013). Taken together these data affirm the positive role of sirtuin enzymes in the management of epilepsy, and support the idea that NAD+-driven activation of sirtuins is an important mechanism that may mediate the anti-seizure effects of ketogenic therapy.
6.3. Poly-ADP-ribosepolymerases (PARPs), DNA damage, and reactive oxygen species
The second NAD-dependent enzyme group is poly-ADP- ribosepolymerases (PARPs), with PARP-1 being the most abundantly expressed enzyme of the PARP family (Sodhi et al., 2010). This enzyme adds polymers of ADP-ribose into proteins, a process known as ADP-ribosylation and first recognized for its important role in cell survival and DNA damage repair (de Murcia and de Murcia, 1994; Grube and Burkle, 1992). A steady-state level of DNA damage exists as a result of DNA oxidation by reactive oxygen species (ROS), a by-product of normal cellular metabolism, and was reported to occur about 10,000 times per cell per day in humans and 50,000 times per cell per day in rats (Bernstein et al., 2013). PARP-1 enzyme was previously described as a molecular sensor for this type of oxidative, naturally occurring DNA damage (de Murcia and de Murcia, 1994), and it is considered an important marker for damage as levels of oxidative DNA damage were found to have an effect on both its activity (Dantzer et al., 2006) and protein levels (Shen et al., 2016). More recently, PARP-1 was found to play a role in gene transcription and programmed cell death (Kim et al., 2005).
PARP ribosylation reactions are major consumers of NAD+ in the brain. Despite the fact that PARP enzymes play a vital role in the DNA repair process, over-activation of PARP enzymes (and the subsequent depletion of NAD+) have been previously linked to several pathological conditions including MPTP-induced Parkinsonism, Alzheimer’s disease, ischemic brain injury, and metalloproteinase-mediated neuronal death (Endres et al., 1997; Kauppinen and Swanson, 2005; Love et al., 1999; Mandir et al., 1999).
With respect to epilepsy, PARP1 activation was shown to mediate neuronal death after status epilepticus, a type of severe continuous seizures that last more than 30 min (Kim et al., 2014). Inhibiting PARP enzymes was previously shown to increase the cellular availability of NAD+ (Hurtado-Bagès et al., 2020; Mendelsohn and Larrick, 2017). Animals treated with a KD for either two days or three weeks showed a significant decrease in hippocampal DNA damage and PARP-1 protein levels (Elamin et al., 2018). This inhibition of PARP-1 expression would be expected to augment the increase in NAD+ that results from utilizing ketone bodies as an energy source (Fig. 1) and further increase NAD+ bioavailability. Indeed, KD-induced PARP inhibition was associated with increased NAD+ (Elamin et al., 2018).
In a positive feedback manner, KD-induced increases in NAD+should also mobilize other NAD+-dependent mechanisms such as sirtuin enzymes to modify ROS generation and limit oxidative stress damage, which in turn should decrease PARP protein levels, making more NAD+ available. Sirtuins such as SIRT2 can decrease oxidative stress by deacetylating FOXO3a, a well-known pro-apoptotic protein (Wang et al., 2007) and SIRT3 was found to mediate the decrease in reactive oxygen species associated with caloric restriction (Qiu et al., 2010).
Ketogenic therapy decreased the formation of reactive oxygen radicals and reversed gene expression patterns of several genes that control ROS and oxidative stress in animal and cellular models (Maalouf et al., 2007; Stafford et al., 2010; Sullivan et al., 2004). Modification of cellular oxidative stress and ROS generation is important, since increased PARP1 activation and mitochondrial oxidative stress levels were shown to play a crucial role in epilepsy-associated neuronal death and contribute to epileptogenesis (Waldbaum and Patel, 2010; Wang et al., 2013). Moreover, inhibition of PARP protected epileptic neurons in the hippocampus from cell death and preserved NAD+ levels and mitochondrial respiration after status epilepticus (Lai et al., 2017; Yang et al., 2013). Hence, the collective paradigm of increasing NAD+, inhibiting PARP-1, and activating sirtuins might be a coherent strategy to protect neurons from epilepsy-induced cell death and a central part of the KD mechanism of action.
6.4. Ketogenic diet, NAD+ and adenosine
KD has been shown to increase activation of seizure-reducing adenosine A1 receptors and increase adenosine levels (Lusardi et al., 2015; Masino et al., 2011; Masino and Rho, 2012).
While there is no direct experimental evidence that we are aware of linking NAD+, adenosine and KD, NAD+ can be degraded into adenosine (Okuda et al., 2010; Zhang et al., 2018) – possibly contributing to the increased adenosine levels, activation of adenosine A1 receptors, and the seizure suppressing effects (Kawamura et al., 2010; Masino et al., 2009, 2012).
Research has shown that S-adenosylhomocysteine hydrolase, an enzyme involved in adenosine biosynthesis, contains an NAD-binding domain that modulates its activity (Kloor et al., 2003). The NAD+ precursor nicotinamide, in combination with adenosine, was shown to have strong protective effects against audiogenic seizures (Maitre et al., 1974), another potential link between NAD and adenosine that supports their role in working together to suppress seizures.
6.5. Ketogenic diet, NAD+ and modulation of ion channels
A plethora of evidence suggests that the KD acts directly or indirectly on ion channels. In particular, multiple lines of evidence suggest that the KD can increase activation of ATP-sensitive K+ channels (KATP channels) to decrease neuronal excitability (Giménez-Cassina et al., 2012; Kawamura et al., 2016, 2010; Tanner et al., 2011). In parallel, NAD is a known modulator of ionic transport, and several channels contain an NAD-binding domain (Kilfoil et al., 2013). Interestingly, NAD molecules can modulate KATP channels (Dukes et al., 1994) and directly interact with and bind to KATP channel subtypes (Dabrowski et al., 2003). Because of their ability to couple energy homeostasis to neuronal firing, KATP channels have been described as metabolic sensors (Olson and Terzic, 2010; Sun and Feng, 2013), and genetic defects of these channels can cause epilepsy among other neurological phenotypes (Olson and Terzic, 2010). Hence, it is not unreasonable to assume that NAD modulation of KATP is one of the mechanisms through which ketogenic therapy exerts its anti-epileptic effects.
Regarding other potassium channel subtypes, in vitro application of NAD+ increased open time and open probability of the voltage-gated potassium channels Kvα1.5 and Kvβ1.3, which play a role in setting the resting membrane potential and regulating neuronal firing (Tipparaju et al., 2005; Yellen, 2002). The effects on Kvα1.5 and Kvβ1.3 currents appear to be specific to NAD+, as application of NADH or NADPH did not achieve similar results (Tipparaju et al., 2005). Furthermore, physiological concentrations of NAD+, but not NADH, successfully modulated sodium-gated potassium channels (KNa), Slack channels, and SLO potassium channels through direct binding (Kilfoil et al., 2013).
Finally, NAD modulation of ion channels is not exclusive to potassium. Other channels that were shown to be directly modulated by NAD include the calcium-permeable TRPM2 cation channel, the ryanodine receptor calcium release channel, and Nav1.5 sodium channels (Hara et al., 2002; Kilfoil et al., 2013; Liu et al., 2009; Zima et al., 2004). All of this supports the idea that the observed modulation of ion channel function with ketones or KD could be mediated via intracellular NAD.
7. Summary and perspective
Whereas glucose or ketone bodies can each produce molecules of ATP, ketone-based metabolism is associated with diverse cellular benefits and requires the conversion of far fewer (õne-third as many) molecules of NAD+. Published research links ketone-based metabolism, altered levels of NAD+, and multiple postulated anti-seizure mechanisms mobilized by ketogenic therapies (Fig. 2). We suggest that a fundamental biochemical difference in NAD+ may be a common starting point for multiple downstream mechanisms. As such, it may explain the diverse beneficial effects of the KD in treating epilepsy and its emerging role in treating numerous additional disorders.
Fig. 2.
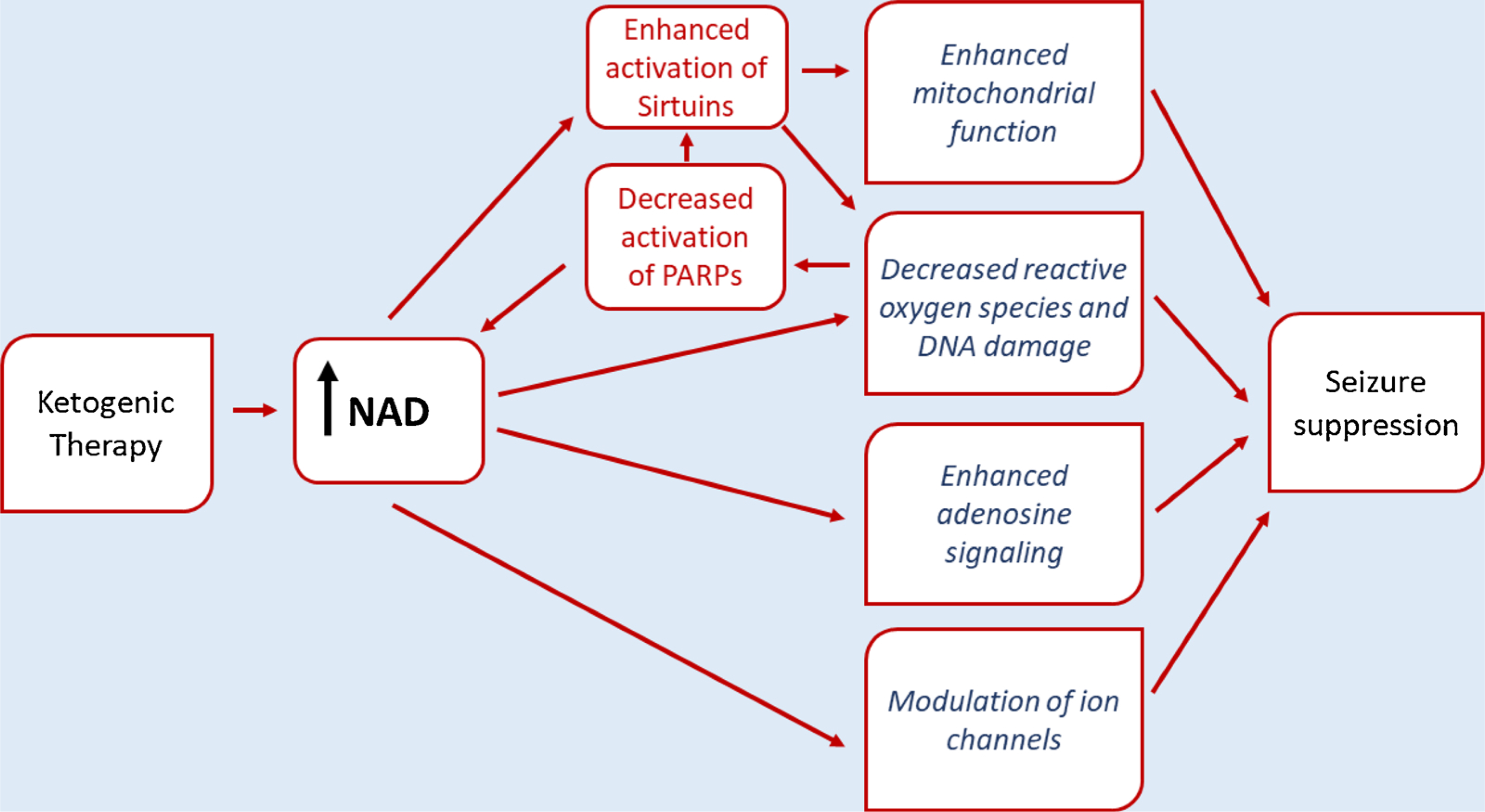
Summary of some of the proposed NAD-based links between ketogenic therapies and suppression of seizures. See text for details.
Funding
Schlumberger Foundation Faculty for the Future Award (ME), University of Hartford and Women’s Advancement Initiative (PS), NIH AT008742 (DNR), NIH NS065957 (SAM).
Footnotes
Declaration of Competing Interest
The authors report no declarations of interest.
References
- Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC, 1995. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem 270, 30327–30333. 10.1074/jbc.270.51.30327. [DOI] [PubMed] [Google Scholar]
- Ahmad M, Kahwaji CI, 2018. Biochemistry, electron transport Chain. StatPearls. StatPearls Publishing. [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA, 2004. Poly(ADP-ribose) Polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem 279, 18895–18902. 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alcaín FJ, Villalba JM, 2009. Sirtuin activators. Expert Opin. Ther. Pat 10.1517/13543770902762893. [DOI] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H,Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-De Murcia J, Auwerx J, 2011. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Gustin J, Armour SM, Yamamoto H, Viader A, North BJ, Michan S, Baloh RH, Golden JP, Schmidt RE, Sinclair DA, Auwerx J, Milbrandt J, 2011. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. U. S. A 108, E952–E961. 10.1073/pnas.1104969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C, 2007. NAD+ metabolism in health and disease. Trends Biochem. Sci 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Bernstein CRA, Nfonsam V, Bernstei H, 2013. DNA damage, DNA repair and cancer. New Research Directions in DNA Repair. InTech, pp. 413–466. 10.5772/53919. [DOI] [Google Scholar]
- Bertoli S, Trentani C, Ferraris C, De Giorgis V, Veggiotti P, Tagliabue A, 2014. Long-term effects of a ketogenic diet on body composition and bone mineralization in GLUT-1 deficiency syndrome: a case series. Nutrition 30, 726–728. 10.1016/j.nut.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Blaise JH, Ruskin DN, Koranda JL, Masino SA, 2015. Effects of a ketogenic diet on hippocampal plasticity in freely moving juvenile rats. Physiol. Rep 3 10.14814/phy2.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ, 2006. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol 60, 223–235. 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- Breen LT, Smyth LM, Yamboliev IA, Mutafova-Yambolieva VN, 2006. β-NAD is a novel nucleotide released on stimulation of nerve terminals in human urinary bladder detrusor muscle. Am. J. Physiol. - Ren. Physiol 290 10.1152/ajprenal.00314.2005. [DOI] [PubMed] [Google Scholar]
- Camberos-Luna L, Massieu L, 2020. Therapeutic strategies for ketosis induction and their potential efficacy for the treatment of acute brain injury and neurodegenerative diseases. Neurochem. Int 10.1016/j.neuint.2019.104614. [DOI] [PubMed] [Google Scholar]
- Cantó C, Sauve AA, Bai P, 2013. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka MC, Hocker S, Koenig M, Bar B, Henry-Barron B, Kossoff EH, Hartman AL, Probasco JC, Benavides DR, Venkatesan A, Hagen EC, Dittrich D, Stern T, Radzik B, Depew M, Caserta FM, Nyquist P, Kaplan PW, Geocadin RG, 2017. Phase I/II multicenter ketogenic diet study for adult superrefractory status epilepticus. Neurology 88, 938–943. 10.1212/WNL.0000000000003690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu Q, Tang Q, Kuang J, Li H, Pu S, Wu T, Yang X, Li R, Zhang J, Zhang Z, Huang Y, Li Y, Zou M, Jiang W, Li T, Gong M, Zhang L, Wang H, Qu A, Xie W, He J, 2019. Hepatocyte-specific Sirt6 deficiency impairs ketogenesis. J. Biol. Chem 294, 1579–1589. 10.1074/jbc.RA118.005309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullingford TE, Eagles DA, Sato H, 2002. The ketogenic diet upregulates expression of the gene encoding the key ketogenic enzyme mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in rat brain. Epilepsy Res. 49, 99–107. 10.1016/S0920-1211(02)00011-6. [DOI] [PubMed] [Google Scholar]
- Dabrowski M, Trapp S, Ashcroft FM, 2003. Pyridine nucleotide regulation of the KATP channel Kir6.2/SUR1 expressed in Xenopus oocytes. J. Physiol 550, 357–363. 10.1113/jphysiol.2003.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J, 2007. Sirtuins: the “magnificent seven”, function, metabolism and longevity. Ann. Med 39, 335–345. 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- Dantzer F, Amé J, Schreiber V, Nakamura J, Ménissier-de Murcia J, de Murcia G, ´ 2006. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 409, 493–510. 10.1016/S0076-6879(05)09029-4. [DOI] [PubMed] [Google Scholar]
- de Murcia G, de Murcia JM, 1994. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem. Sci 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- Dukes LD, McIntyre MS, Mertz RJ, Philipson LH, Roe MW, Spencer B, Worley JF, 1994. Dependence on NADH produced during glycolysis for β-cell glucose signaling. J. Biol. Chem 269, 10979–10982. [PubMed] [Google Scholar]
- Durnin L, Dai Y, Aiba I, Shuttleworth CW, Yamboliev IA, Mutafova-Yambolieva VN, 2012. Release, neuronal effects and removal of extracellular β-nicotinamide adenine dinucleotide (β-NAD +) in the rat brain. Eur. J. Neurosci 35, 423–435. 10.1111/j.1460-9568.2011.07957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin M, Ruskin DN, Masino SA, Sacchetti P, 2017. Ketone-based metabolic therapy: is increased NAD+ a primary mechanism? Front. J. Mol. Neurosci 10, 377. 10.3389/fnmol.2017.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin M, Ruskin DN, Masino SA, Sacchetti P, 2018. Ketogenic diet modulates NAD+-dependent enzymes and reduces DNA damage in hippocampus. Front. Cell. Neurosci 12 10.3389/fncel.2018.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA, 1997. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab 17, 1143–1151. 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Vining EPG, 1999. Seizures decrease rapidly after fasting: preliminary studies of the ketogenic diet. Arch. Pediatr. Adolesc. Med 153, 946–949. [DOI] [PubMed] [Google Scholar]
- Fukao T, Lopaschuk GD, Mitchell GA, 2004. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fat. Acids 70, 243–251. 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Gano LB, Liang LP, Ryan K, Michel CR, Gomez J, Vassilopoulos A, Reisdorph N, Fritz KS, Patel M, 2018. Altered mitochondrial acetylation profiles in a kainic acid model of temporal lobe epilepsy. Free Radic. Biol. Med 123, 116–124. 10.1016/j.freeradbiomed.2018.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges M, Selim L, Girgis M, El Ghannam A, Abdelghaffar H, El-Ayadi A, 2019. Implementation of ketogenic diet in children with drug-resistant epilepsy in a medium resources setting: egyptian experience. Epilepsy Behav. Case Reports 11, 35–38. 10.1016/j.ebcr.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DL, Pyzik PL, Freeman JM, 2000. The ketogenic diet: seizure control correlates better with serum -Hydroxybutyrate than with urine ketones. J. Child Neurol 15, 787–790. 10.1177/088307380001501203. [DOI] [PubMed] [Google Scholar]
- Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN, 2012. BAD-dependent regulation of fuel metabolism and KATP channel activity confers resistance to epileptic seizures. Neuron 74, 719–730. 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groleau V, Schall JI, Stallings VA, Bergqvist CA, 2014. Long-term impact of the ketogenic diet on growth and resting energy expenditure in children with intractable epilepsy. Dev. Med. Child Neurol. 56, 898–904. 10.1111/dmcn.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube K, Burkle A, 1992. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. U. S. A 89, 11759–11763. 10.1073/pnas.89.24.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag F, Adriouch S, Braß A, Jung C, Moller S, Scheuplein F, Bannas P, ¨ Seman M, Koch-Nolte F, 2007. Extracellular NAD and ATP: partners in immune cell modulation. Purinergic Signal 10.1007/s11302-006-9038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y, 2002. LTRPC2 Ca2+-Permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 9, 163–173. 10.1016/S1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- Hasan-Olive MM, Lauritzen KH, Ali M, Rasmussen LJ, Storm-Mathisen J, Bergersen LH, 2019. A ketogenic diet improves mitochondrial biogenesis and bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res 44, 22–37. 10.1007/s11064-018-2588-6. [DOI] [PubMed] [Google Scholar]
- Heussinger N, Della Marina A, Beyerlein A, Leiendecker B, Hermann-Alves S, Dalla Pozza R, Klepper J, 2018. 10 patients, 10 years – long term follow-up of cardiovascular risk factors in Glut1 deficiency treated with ketogenic diet therapies: a prospective, multicenter case series. Clin. Nutr 37, 2246–2251. 10.1016/j.clnu.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Hewitt JYJA, 1924. The Metabolism of Carbohydrates. Part III. The Absorption of Glucose, Fructose and Galactose From the Small Intestine. Biochem. J 18, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E, 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Bagès S, Knobloch G, Ladurner AG, Buschbeck M, 2020. The taming of PARP1 and its impact on NAD+ metabolism. Mol. Metab 10.1016/j.molmet.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Sichiro, Johnson S, 2018. NAD+ biosynthesis, aging, and disease. F1000Research 10.12688/f1000research.12120.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuta Y, Imada T, Hisamura R, Oonishi E, Nakamura S, Inagaki E, Ito M, Soga T, Tsubota K, 2018. Ketone body 3-hydroxybutyrate mimics calorie restriction via the Nrf2 activator, fumarate, in the retina. Aging Cell 17. 10.1111/acel.12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA, 2005. Poly(ADP-Ribose) Polymerase-1 promotes microglial activation, proliferation, and matrix Metalloproteinase-9-Mediated neuron death. J. Immunol 174, 2288–2296. 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Ruskin DN, Masino SA, 2010. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci 30, 3886–3895. 10.1523/JNEUROSCI.0055-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Ruskin DN, Geiger JD, Boison D, Masino SA, 2014. Ketogenic diet sensitizes glucose control of hippocampal excitability. J. Lipid Res 55, 2254–2260. 10.1194/jlr.M046755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Ruskin DN, Masino SA, 2016. Metabolic therapy for temporal lobe epilepsy in a dish: investigating mechanisms of ketogenic diet using electrophysiological recordings in hippocampal slices. Front. Mol. Neurosci 10.3389/fnmol.2016.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilfoil PJ, Tipparaju SM, Barski OA, Bhatnagar A, 2013. Regulation of ion channels by pyridine nucleotides. Circ. Res 10.1161/CIRCRESAHA.111.247940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL, 2005. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- Kim DY, Vallejo J, Rho JM, 2010. Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J. Neurochem 114 10.1111/j.1471-4159.2010.06728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Kim YJ, Kim JY, Kang TC, 2014. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic- independent manner. Cell Death Dis. 5, e1362. 10.1038/cddis.2014.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloor D, Lüdtke A, Stoeva S, Osswald H, 2003. Adenosine binding sites at S- adenosylhomocysteine hydrolase are controlled by the NAD+/NADH ratio of the enzyme. Biochem. Pharmacol 66, 2117–2123. 10.1016/S0006-2952(03)00581-1. [DOI] [PubMed] [Google Scholar]
- Koranda JL, Ruskin DN, Masino SA, Harry Blaise J, 2011. A ketogenic diet reduces long-term potentiation in the dentate gyrus of freely behaving rats. J.Neurophysiol 106, 662–666. 10.1152/jn.00001.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA, Rho JM, 2009. Ketogenic diets: an update for child neurologists. J. Child Neurol 24, 979–988. 10.1177/0883073809337162. [DOI] [PubMed] [Google Scholar]
- Krebs HA, 1970. Rate control of the tricarboxylic acid cycle. Adv. Enzyme Regul 8, 335–353. 10.1016/0065-2571(70)90028-2. [DOI] [PubMed] [Google Scholar]
- Kussmaul L, Hirst J, 2006. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc.Natl. Acad. Sci. U. S. A 103, 7607–7612. 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin VS, Pustovit KB, Abramochkin DV, 2016. Effects of exogenous nicotinamide adenine dinucleotide (NAD+) in the rat heart are mediated by P2 purine receptors. J. Biomed. Sci 23, 50. 10.1186/s12929-016-0267-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffel L, 1999. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev 15, 412–426. . [DOI] [PubMed] [Google Scholar]
- Lai YC, Scott Baker J, Donti T, Graham BH, Craigen WJ, Anderson AE, 2017. Mitochondrial dysfunction mediated by poly(ADP-ribose) polymerase-1 activation contributes to hippocampal neuronal damage following status epilepticus. Int. J. Mol. Sci 18 10.3390/ijms18071502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R, 2000. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U. S. A 97, 5807–5811. 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre F, Aronson N, 2000. Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics 105, e46. 10.1542/peds.105.4.e46. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Guarente L, 2003. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol 10.1016/S0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CLH, Grace A, London B, Dudley SC, 2009. Cardiac Na+ Current regulation by pyridine nucleotides. Circ. Res 105, 737–745. 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang B, Zhou P, Kong Y, Hu W, Zhu G, Ying W, Li W, Wang Y, Li S, 2017. Nicotinamide adenine dinucleotide suppresses epileptogenesis at an early stage. Sci. Rep 7, 7321. 10.1038/s41598-017-07343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Matsudaira P, Zipursky L, Baltimore D, Darnell J, 2000. Molecular Cell Biology. W.H. Freeman, New York. [Google Scholar]
- Love S, Barber R, Wilcock GK, 1999. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 122, 247–253. 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Akula KK, Coffman SQ, Ruskin DN, Masino SA, Boison D, 2015. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 99, 500–509. 10.1016/j.neuropharm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G, 2007. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J. Neurosci 27, 3618–3625. 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM, 2007. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher F, Vannucci SJ, Simpson IA, 1994. Glucose transporter proteins in brain. FASEB J. 8, 1003–1011. 10.1096/fasebj.8.13.7926364. [DOI] [PubMed] [Google Scholar]
- Maitre M, Ciesielski L, Lehmann A, Kempf E, Mandel P, 1974. Protective effect of adenosine and nicotinamide against audiogenic seizure. Biochem. Pharmacol 23, 2807–2816. 10.1016/0006-2952(74)90054-9. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM, 1999. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6- tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. U. S. A 96, 5774–5779. 10.1165/rcmb.2004-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, Camandola S, Mattson MP, 2016. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem 139, 769–781. 10.1111/jnc.13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez CC, Pyzik PL, Kossoff EH, 2007. Discontinuing the ketogenic diet in seizure-free children: recurrence and risk factors. Epilepsia 48, 187–190. 10.1111/j.1528-1167.2006.00911.x. [DOI] [PubMed] [Google Scholar]
- Masino SA, Rho JM, 2012. Mechanism of ketogenic diet action. Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information; (US), pp. 1003–1018. [PubMed] [Google Scholar]
- Masino SA, Rho JM, 2019. Metabolism and epilepsy: ketogenic diets as a homeostatic link. Brain Res 10.1016/j.brainres.2018.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino S, Kawamura M Jr., Wasser C, Pomeroy L, Ruskin D, 2009. Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Curr. Neuropharmacol 7, 257–268. 10.2174/157015909789152164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D, 2011. A ketogenic diet suppresses seizures in mice through adenosine A 1 receptors. J. Clin. Invest 121, 2679–2683. 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Ruskin DN, Geiger JD, Boison D, 2012. Purines and neuronal excitability: links to the ketogenic diet. Epilepsy Res. 100, 229–238. 10.1016/j.eplepsyres.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty MF, DiNicolantonio JJ, O’Keefe JH, 2015. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 85, 631–639. 10.1016/j.mehy.2015.08.002. [DOI] [PubMed] [Google Scholar]
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, Bohr VA, Chua KF, 2009. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany. NY) 1, 109–121. 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn AR, Larrick JW, 2017. The NAD+/PARP1/SIRT1 axis in aging. Rejuvenation Res 10.1089/rej.2017.1980. [DOI] [PubMed] [Google Scholar]
- Michishita E, 2005. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635. 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW, 2006. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, Pichulik T, Gulston MK, Atherton HJ, Schroeder MA, Deacon RMJ, Kashiwaya Y, King MT, Pawlosky R, Rawlins JNP, Tyler DJ, Griffin JL, Robertson J, Veech RL, Clarke K, 2016. Novel ketone diet enhances physical and cognitive performance. FASEB J. 30, 4021–4032. 10.1096/fj.201600773R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Sung JH, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM, 2007. β-Nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc. Natl. Acad. Sci. U. S. A 104, 16359–16364. 10.1073/pnas.0705510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrin N, Wu X, Fortier E, Feng Y, Bar OC, Chen S, Ren X, Wu Z, Streeper RS, Bordone L, 2010. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem 285, 31995–32002. 10.1074/jbc.M110.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH, 2008. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- Nylen K, Velazquez JLP, Sayed V, Gibson KM, Burnham WM, Snead OC, 2009. The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1−/− mice. Biochim. Biophys. Acta - Gen. Subj 1790, 208–212. 10.1016/j.bbagen.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H, Higashi Y, Nishida K, Fujimoto S, Nagasawa K, 2010. Contribution of P2X7 receptors to adenosine uptake by cultured mouse astrocytes. Glia 58, 1757–1765. 10.1002/glia.21046. [DOI] [PubMed] [Google Scholar]
- Olson TM, Terzic A, 2010. Human KATP channelopathies: diseases of metabolic homeostasis. Pflugers Arch. Eur. J. Physiol 10.1007/s00424-009-0771-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallàs M, Ortuno-Sahagún D, Anédrs-Benito P, Ponce-Regalado MD, Rojas- Mayorquín AE, 2014. Resveratrol in epilepsy: preventive or treatment opportunities? Front. Biosci. - Landmark 19, 1057–1064. 10.2741/4267. [DOI] [PubMed] [Google Scholar]
- Paoli A, 2014. Ketogenic diet for obesity: friend or foe? Int. J. Environ. Res. Public Health 10.3390/ijerph110202092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J, 2013. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 80, 342. 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A, 2011. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol 80, 1136–1146. 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- Pulford DS, 1927. Ketogenic diets for epileptics. Cal. West. Med 27, 50–56. [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D, 2010. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667. 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Rahman S, 2012. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol 10.1111/j.1469-8749.2011.04214.x. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai SI, 2004. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem 279, 50754–50763. 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Lindefeldt M, Freedgood NR, Dahlin M, Masino SA, 2017. Ketogenic diet shifts CSF metabolome and has differential effects in responsive vs. non- responsive pediatric epilepsy patients. Society for Neuroscience Washington, DC, 385.05. [Google Scholar]
- Rustin P, Parfait B, Chretien D, Bourgeron T, Djouadi F, Bastin J, RȌtig A, Munnich A, 1996. Fluxes of nicotinamide adenine dinucleotides through mitochondrial membranes in human cultured cells. J. Biol. Chem 271, 14785–14790. 10.1074/jbc.271.25.14785. [DOI] [PubMed] [Google Scholar]
- Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, Chance B, Clarke K, Veech RL, 1995. Insulin, Ketone-Bodies, and mitochondrial energy transduction. FASEB J. 9. [DOI] [PubMed] [Google Scholar]
- Shen Y, McMackin MZ, Shan Y, Raetz A, David S, Cortopassi G, 2016. Frataxin deficiency promotes excess microglial DNA damage and inflammation that is rescued by PJ34. PLoS One 11, e0151026. 10.1371/journal.pone.0151026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirven J, Whedon B, Caplan D, Liporace J, Glosser D, O’Dwyer J, Sperling MR, 1999. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 40, 1721–1726. 10.1111/j.1528-1157.1999.tb01589.x. [DOI] [PubMed] [Google Scholar]
- Smyth LM, Bobalova J, Mendoza MG, Lew C, Mutafova-Yambolieva VN, 2004. Release of β-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J. Biol. Chem 279, 48893–48903. 10.1074/jbc.M407266200. [DOI] [PubMed] [Google Scholar]
- Sodhi RK, Singh N, Jaggi AS, 2010. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul. Pharmacol 10.1016/j.vph.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Sook Noh H, Po Lee H, Wook Kim D, Soo Kang S, Jae Cho G, Rho JM, Sung Choi W, 2004. A cDNA microarray analysis of gene expression profiles in rat hippocampus following a ketogenic diet. Mol. Brain Res 129, 80–87. 10.1016/j.molbrainres.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Stafford P, Abdelwahab MG, Kim DY, Preul MC, Rho JM, Scheck AC, 2010. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. (Lond) 7, 74. 10.1186/1743-7075-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S, 2012. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab 23, 420. 10.1016/J.TEM.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM, 2004. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol 55, 576–580. 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, 2013. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol. Sin 10.1038/aps.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner GR, Lutas A, Martínez-François JR, Yellen G, 2011. Single KATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci 31, 8689–8696. 10.1523/JNEUROSCI.5951-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Chen J, Zhang J, Yang X, Ji T, Zhang Yao, Wu Y, Fang F, Wu X, Zhang Yuehua, 2019. The efficacy of ketogenic diet in 60 Chinese patients with dravet syndrome. Front. Neurol 10 10.3389/fneur.2019.00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Saxena N, Liu S-Q, Kumar R, Bhatnagar A, 2005. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am. J. Physiol., Cell Physiol 288, C366–C376. 10.1152/ajpcell.00354.2004. [DOI] [PubMed] [Google Scholar]
- Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T, Bunting S, Vaquero A, Tischfield JA, Serrano L, 2016. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 35, 1488–1503. 10.15252/embj.201593499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Bruzzone S, Zocchi E, Fedele E, Schenk U, De Flora A, Matteoli M, 2001. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem 78, 646–657. 10.1046/j.1471-4159.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- Viggiano A, Stoddard M, Pisano S, Operto FF, Iovane V, Monda M, Coppola G, 2016. Ketogenic diet prevents neuronal firing increase within the substantia nigra during pentylenetetrazole-induced seizure in rats. Brain Res. Bull 125, 168–172. 10.1016/j.brainresbull.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Waldbaum S, Patel M, 2010. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 10.1016/j.eplepsyres.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Nguyen M, Qin FXF, Tong Q, 2007. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514. 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- Wang S, Yang X, Lin Y, Qiu X, Li H, Zhao X, Cao L, Liu X, Pang Y, Wang X, Chi Z, 2013. Cellular NAD depletion and decline of SIRT1 activity play critical roles in PARP-1-mediated acute epileptic neuronal death in vitro. Brain Res. 1535, 14–23. 10.1016/j.brainres.2013.08.038. [DOI] [PubMed] [Google Scholar]
- Wang D, Li Z, Zhang Y, Wang G, Wei M, Hu Y, Ma S, Jiang Y, Che N, Wang X, Yao J, Yin J, 2016. Targeting of microRNA-199a-5p protects against pilocarpine-induced status epilepticus and seizure damage via SIRT1-p53 cascade. Epilepsia 57, 706–716. 10.1111/epi.13348. [DOI] [PubMed] [Google Scholar]
- Wigglesworth VB, 1924. Studies on ketosis: I. The relation between Alkalosis and ketosis. Biochem. J 18, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Ipek ö., Beaumont M, Shevlyakova M, Christinat N, Masoodi M, Greenberg N, Gruetter R, Cuenoud B, 2018. Nutritional ketosis increases NAD+/ NADH ratio in healthy human brain: an in vivo study by 31P-MRS. Front. Nutr 5 10.3389/fnut.2018.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Fu M, Pestell R, Sauve AA, 2006. SIRT1 and endocrine signaling. Trends Endocrinol. Metab 10.1016/j.tem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang S, Lin Y, Han Y, Qiu X, Zhao X, Cao L, Wang X, Chi Z, 2013. Poly(ADP-ribose) polymerase inhibition protects epileptic hippocampal neurons from apoptosis via suppressing Akt-mediated apoptosis-inducing factor translocation in vitro. Neuroscience 231, 353–362. 10.1016/j.neuroscience.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Yellen G, 2002. The voltage-gated potassium channels and their relatives. Nature. 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- Yin J, Nielsen M, Li S, Shi J, 2019. Ketones improves Apolipoprotein E4-related memory deficiency via sirtuin 3. Aging (Albany. NY) 11, 4579–4586. 10.18632/aging.102070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang C, Shi H, Wu D, Ying W, 2018. Extracellular degradation into adenosine and the activities of adenosine kinase and AMPK mediate extracellular NAD+-produced increases in the adenylate pool of BV2 microglia under basal conditions. Front. Cell. Neurosci 12 10.3389/fncel.2018.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W, 2012. Quantitative imaging of energy expenditure in human brain. Neuroimage 60, 2107–2117. 10.1016/j.neuroimage.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Copello JA, Blatter LA, 2004. Effects of cytosolic NADH/NAD+ levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J. Physiol 555, 727–741. 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]