

Abstract
Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) has caused substantially more infections, deaths, and economic disruptions than the 2002‐2003 SARS‐CoV. The key to understanding SARS‐CoV‐2's higher infectivity lies partly in its host receptor recognition mechanism. Experiments show that the human angiotensin converting enzyme 2 (ACE2) protein, which serves as the primary receptor for both CoVs, binds to the receptor binding domain (RBD) of CoV‐2's spike protein stronger than SARS‐CoV's spike RBD. The molecular basis for this difference in binding affinity, however, remains unexplained from X‐ray structures. To go beyond insights gained from X‐ray structures and investigate the role of thermal fluctuations in structure, we employ all‐atom molecular dynamics simulations. Microseconds‐long simulations reveal that while CoV and CoV‐2 spike‐ACE2 interfaces have similar conformational binding modes, CoV‐2 spike interacts with ACE2 via a larger combinatorics of polar contacts, and on average, makes 45% more polar contacts. Correlation analysis and thermodynamic calculations indicate that these differences in the density and dynamics of polar contacts arise from differences in spatial arrangements of interfacial residues, and dynamical coupling between interfacial and non‐interfacial residues. These results recommend that ongoing efforts to design spike‐ACE2 peptide blockers will benefit from incorporating dynamical information as well as allosteric coupling effects.
Keywords: allostery, COVID‐19, molecular dynamics, protein dynamics, protein‐protein interactions, SARS‐CoV, SARS‐CoV‐2, viral entry
1. INTRODUCTION
Within 10 months of its emergence, the severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) has caused more than 23 million confirmed infections and over 800 000 deaths globally, and these infections continue to grow rapidly. 1 In contrast, its genetic variant, SARS‐CoV, which caused the 2002 to 2003 outbreak, led to far fewer infections and was relatively easier to contain. 2
Several studies on coronaviruses show that their overall rates of replication, transmissibility and disease severity (in several species) correlate with the binding affinities of their spike proteins to the host receptor angiotensin converting enzyme 2 (ACE2). 3 , 4 , 5 , 6 Accordingly, calorimetric experiments, including surface plasmon resonance (SPR) and bilayer interferometry (BI), show that ACE2 does bind to the receptor binding domain (RBD) of CoV‐2 spike 5 to 20‐fold stronger compared to CoV spike RBD (Table S1 of supporting information). 7 , 8 , 9 , 10 Qualitative results from pull‐down binding assays are also consistent with this finding. 11 Note, however, that these findings do not necessarily imply that CoV‐2's full‐length spike will also have a higher affinity for ACE2. Experiments remain somewhat unclear about this issue. On the one hand, results from pull‐down assays indicate that ACE2 binds to full‐length spikes of CoV and CoV‐2 with comparable affinities. 11 This difference in relative binding affinities of spike RBD and full‐length spike to ACE2 is suggested to be due to differences in exposure rates of RBD in CoV and CoV‐2 spike. 11 Although the RBD of CoV binds ACE2 with lower affinity, it is suggested that its RBD has a higher exposure rate compared to the RBD of CoV‐2 spike, which compensates for its RBD's lower affinity. 11 These conformational states have also been suggested to be modulated by spike glycans. 12 On the other hand, SPR experiments indicate that inclusion of CoV‐2's spike ectodomain, which is expected to control RBD exposure, does not decrease its ACE2 affinity 8 (see also Table S1 of supporting information). Nevertheless, what is clear from all experiments is that ACE2 does have a much higher affinity for CoV‐2 spike RBD compared to CoV spike RBD.
Here we examine the molecular basis underlying the different binding affinities of CoV and CoV‐2 spike RBDs to ACE2. X‐ray structures are indispensable starting points to investigate this issue, however, as we discuss below, there is a clear disconnect between X‐ray structural data and experimental binding affinity data. The RBDs of the CoV and CoV‐2 spike have almost similar structures, and they interact with almost identical regions of the proteolytic domain (PD) of ACE2. 7 , 9 , 13 Out of the 43 amino acids that are different in the spike RBDs of CoV and CoV‐2 strains used for structure determination, 10 are at the ACE2 binding region. 7 These 10 amino acid differences do not alter the numbers of hydrogen bonds and salt bridges at the spike‐ACE2 interface. 7 They only make CoV‐2's spike‐ACE2 interface less hydrophobic compared to CoV's spike‐ACE2 interface. A smaller hydrophobicity implies a higher desolvation cost associated with protein‐protein binding and, therefore a lower binding affinity. 14 , 15 This, in principle, should have made ACE2 bind less strongly to CoV‐2's spike RBD, rather than the opposite.
Since the emergence of X‐ray structures, several research groups have performed molecular simulations on spike‐ACE2 complexes. 16 , 17 , 18 , 19 , 20 , 21 , 22 Some of these studies also address the issue of why ACE2 binds with different affinities to CoV and CoV‐2 spike RBDs. 16 , 19 , 20 , 21 , 22 In particular, MD simulations 21 reveal that CoV‐2's spike‐ACE2 interface consists of several high‐frequency polar contacts, which suggests that polar contacts may be important to CoV‐2's higher affinity. Alanine‐scanning free energy calculations by Zou et al 22 indicate that interfacial residues in both CoV and CoV‐2 RBDs contribute to their respective binding affinities with ACE2. Free energy calculations by Wang et al. 16 and Ghorbani et al. 20 indicate that individual substitutions of many of CoV's interfacial residues into CoV‐2 spike have a positive impact on spike‐ACE2 affinity, suggesting that the overall higher affinity of CoV‐2 spike perhaps results from a combination of multiple differences in sequences. However, these early simulation studies do not explain why CoV‐2's spike RBD binds more strongly to ACE2 despite making a smaller hydrophobic contact with ACE2 and despite making the same numbers of polar contacts compared to CoV spike.
A potentially important aspect of spike‐ACE2 interactions that is missing from literature is the characterization of thermal fluctuations in structure, and their role in relative binding. Our studies on paramyxoviruses, in fact, show that protein dynamics play a vital role in virus‐host protein‐protein interactions and also in regulation of viral entry, 23 , 24 and perhaps they may also explain why ACE2 binds more tightly to CoV‐2's spike RBD. To examine this and go beyond insights obtained from X‐ray structures and recent simulation studies, here we carry out a comparative analysis of spike‐ACE2 complexes using all‐atom MD simulations at physiological temperature and in explicit solvent. We use a protocol similar to that we have employed recently, 23 whose forward predictions on virus‐host protein‐protein interactions were also validated by targeted biophysical experiments. 25 Additionally, we carry out free energy calculations to examine the effect of mutating selected spike residues on spike‐ACE2 binding affinity. Results from these studies provide a physical basis for why ACE2 binds to CoV‐2 spike RBD more strongly compared to CoV spike RBD. This mechanistic, structural and energetic data is expected to directly benefit ongoing efforts to design small molecules, polymers, and antibodies targeting the spike‐ACE2 interaction.
2. RESULTS AND DISCUSSION
We first carry out 3 μs long MD trajectories of ACE2 PD complexed with spike RBDs of CoV and CoV‐2, and examine differences in binding modes, polar contacts, hydrophobic contacts and interfacial waters. Next, using conformations selected from MD simulations, we examine the effect of mutating selected CoV spike RBD residues on spike‐ACE2 binding free energy. Note first that here we do not simulate the entire spike protein—our goal here is understand why ACE2 binds more strongly to the RBD of CoV‐2's spike protein compared to the RBD of CoV's spike protein. This does not affect comparison to experimental structural and calorimetric binding data, as experiments were also conducted using the same truncated forms of spike proteins (Table S1 of Supporting Information). Note also that we do not model RBD and ACE2 glycans. This is because of the following reasons. Experiments on CoV have demonstrated that its spike glycans play no role in its spike binding to ACE2. 26 In the case of CoV‐2, there are two glycosylation sites on its spike RBD (residues 331 and 343), 27 both of which are far from the spike‐ACE2 interface. Importantly, both sites undergo complex glycosylations by the ER and Golgi‐resident processing enzymes, exhibiting substantial variability, 27 which suggests that whatever role they may have in directly affecting spike‐ACE2 interfaces is unlikely to be conserved. Finally, ACE2 glycosylation is also expected to exhibit large variations across tissues, 28 suggesting that its role in spike binding is also unlikely to be conserved. In fact, recent experiments show no effects of several types of glycosylations on spike binding, and only minor effects of a few types of glycosylations on spike binding. 29 Regardless, results from our work will provide the necessary baseline to isolate the specific effects that glycans may have on CoV‐2's spike‐ACE2 interface.
2.1. Comparison of MD ensembles
2.1.1. Binding modes
To characterize spike‐ACE2 interfacial binding modes, we extract from each simulation spike‐ACE2 conformations at one‐nanosecond intervals. This yields 3001 conformations for each complex. We calculate root mean square deviations (RMSDs) between each of the (3001 × 3000)/2 conformation pairs. For calculating RMSDs, we consider the backbone atoms of only those amino acids in ACE2 and spike that are part of the spike‐ACE2 interface. The interface is defined geometrically, and an amino acid is considered to part of the interface if any of its heavy atoms is within 5 Å from the complementary protein in any of the 3001 conformations. These pairwise RMSDs are shown in Figure 1. These RMSDs are then taken as a measure of similarity in the affinity propagation algorithm, 30 , 31 which clusters these conformations into five and six groups, respectively, in the CoV and CoV‐2 spike‐ACE2 systems. The advantage of the affinity propagation algorithm over traditional clustering approaches is that it does not assume a priori the number of clusters or a cutoff value for delineating clusters. We adopted this unsupervised machine learning algorithm previously to cluster correlations in structural fluctuations. 32
FIGURE 1.
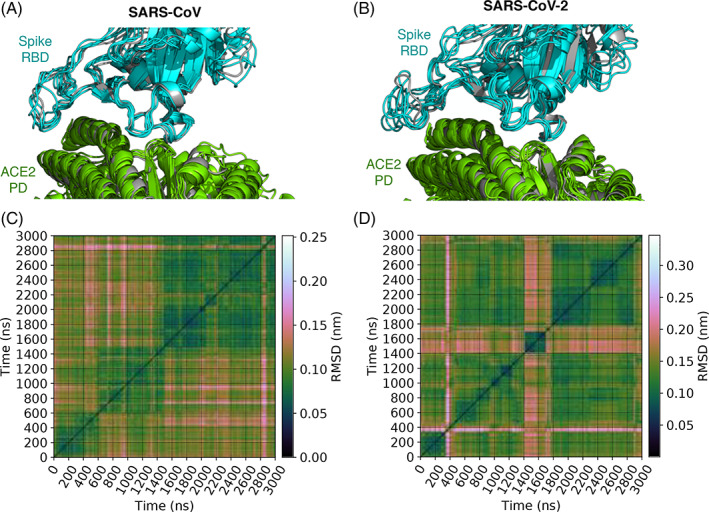
Binding modes of spike‐angiotensin converting enzyme 2 (ACE2) complexes in MD simulations. A and B show, respectively, the distinct binding mode conformations of the complexes containing spike proteins of CoV and CoV‐2. These conformations are superimposed over the X‐ray structures of spike‐ACE2 complexes (gray)—ACE2 bound with CoV spike is compared against 2AJF, 13 and ACE2 bound to CoV‐2 spike is compared against 6M0J. 7 They are the lowest energy conformations of the five and six binding modes identified, respectively, for the complexes involving CoV and CoV‐2 spike proteins. Binding modes are identified by clustering conformations extracted every nanosecond from MD. Conformational clustering is performed using affinity propagation 30 , 32 in which we take RMSD as an index of similarity between conformations. C and D show these pairwise RMSDs
The lowest energy conformation of each of the five and six clusters of CoV and CoV‐2 spike‐ACE2 complexes are shown in Figure 1. We note that the overall topologies of these cluster representatives, or binding modes, closely resemble their respective X‐ray structures. 7 , 13 The main variation between the different binding modes is in the structures of the spike RBD loops at the binding interface. Additionally, we find that the five binding mode representatives of the CoV spike‐ACE2 complex are similar to the six binding mode representatives of the CoV‐2 spike‐ACE2 complex, with RMSDs ranging between 2.2 and 3.4 Å (Figure S1 of Supporting Information).
2.1.2. Local spike‐ACE2 contacts
To examine differences in local interactions, we first determine hydrogen bonds and salt bridges between spike RBD and ACE2 PD. Hydrogen bonds are computed using the geometric definition proposed by Luzar and Chandler. 33 Salt bridges are defined using a 4 Å cutoff between carboxyl carbon and amine/guanidine nitrogen. The choice of this cutoff distance is discussed in Figure S2 of the supporting information. Figure 2A compares time evolutions of inter‐protein hydrogen bonds and salt bridges. The key observation we make is that, on average, ACE2 engages in discernibly greater numbers of hydrogen bonds and salt bridges with CoV‐2's spike RBD compared to CoV's spike RBD. On average, ACE2 engages in 9.9 ± 0.3 hydrogen bonds and 0.7 ± 0.1 salt bridges with CoV‐2's spike RBD, and 7.2 ± 0.8 hydrogen bonds and 0.07 ± 0.06 salt bridges with CoV's spike RBD. In relative terms, ACE2 makes 45% more polar contacts with CoV‐2 spike RBD, compared to CoV spike RBD. A breakdown of hydrogen bonds as a function of the interfacial binding modes observed in Figure 1 shows that all binding modes of CoV‐2 make greater numbers of hydrogen bonds than those of CoV (Table S2 of Supporting Information). We also note in Figure 2A that the hydrogen bond density in CoV's spike‐ACE2 interface shows long‐timescale fluctuations. This is also evident from the somewhat higher standard deviations in hydrogen bond numbers reported above. To examine the potential effect of these fluctuations on our conclusions, we analyze 10 μs long MD trajectories of spike‐ACE2 complexes made available by the DE Shaw research group. 34 We note similar long‐timescale fluctuations even in their trajectories (Figure S3 of Supporting Information), and importantly, the averages and standard deviations we find from our trajectories are quantitatively similar to those we obtain from their longer MD trajectories. Note that there is one key difference between our simulations and those conducted by the DE Shaw research group ‐ the spike RBDs in Shaw group's constructs include glycans, and ours do not, which tells us that spike glycans, if at all, play only a minor role in modulating polar contacts at the spike‐ACE2 interface.
FIGURE 2.
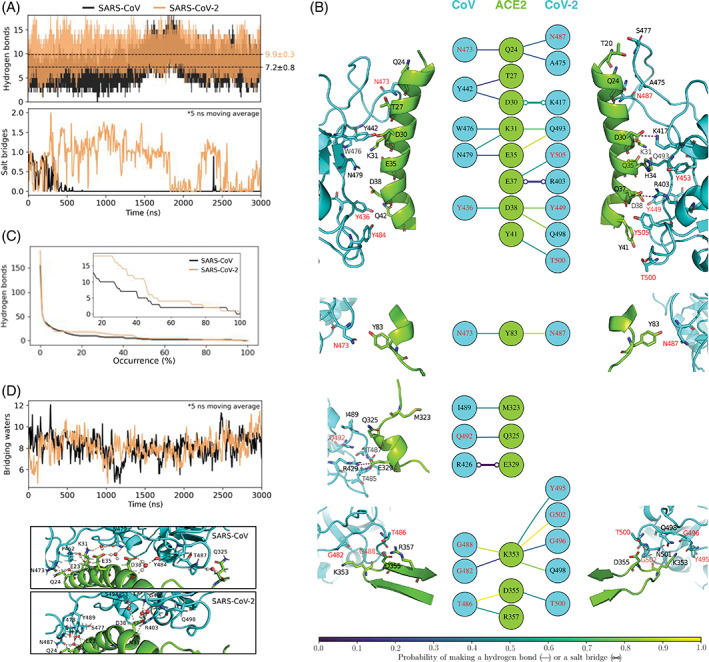
Polar contacts of angiotensin converting enzyme 2 (ACE2) with spike proteins of CoV and CoV‐2. A, Time evolution of hydrogen bonds and salt bridges between ACE2 and spike RBDs. Dashed lines indicate time‐averages, and statistical errors are obtained from block averaging (Figure S4 of Supporting Information). B, Structural map of hydrogen bonds and salt bridges between ACE2 and spike. ACE2 associates with spike at four regions that are non‐contiguous in its primary sequence. These four interfacial regions are shown separately. The colors of the lines connecting the residues in the central panel indicate their occurrence probabilities. Note that for the sake of clarity, only those hydrogen bonds and salt bridges are shown that are observed for at least 15% of the total simulated time. The amino acid of spike labeled in red are the ones that are conserved in the two spike RBDs. C, Numbers of unique hydrogen bonds as functions of their occurrence probabilities. The inset zooms in on the 15% to 100% probability region. D, Time evolution of waters that bridge interactions between ACE2 and spike by hydrogen bonding simultaneously with both proteins. The images below show the bridging waters in the 3 μs snapshots of the MD trajectories
To gain further insight into differences between CoV and CoV‐2 spike‐ACE2 interfacial polar contacts, Figure 2B compares the structural maps of the spike‐ACE2 hydrogen bonds and salt bridges. We note that the higher numbers of polar interactions in CoV‐2's spike‐ACE2 interface do not emanate from one specific region of the interface. In fact, almost all residues of CoV‐2 spike, including those that are conserved in CoV spike, have a higher probabilities of engaging in hydrogen bonds (and salt bridges) with ACE2. The probabilities here refer to the fractions of frames in which amino acids are found to be hydrogen bonded (or salt‐bridged).
To examine these hydrogen bond probabilities collectively, we plot in Figure 2C the numbers of unique hydrogen bonds as functions of their occurrence probabilities. We note that CoV‐2's spike‐ACE2 interface has 22% more possible combinations of hydrogen bonds compared to CoV's spike‐ACE2 interface. We also note that the more frequently observed hydrogen bonds (> 20% probability) are in greater numbers in CoV‐2's spike‐ACE2 interface, compared to CoV's spike‐ACE2 interface.
Polar interactions between spike and ACE2 can also be bridged by interstitial water molecules, that is, water molecules can hydrogen bond simultaneously with both proteins. These bridging waters are considered to stabilize protein‐protein interactions. 35 We note from Figure 2D that the spike‐ACE2 interfaces of CoV and CoV‐2 have similar numbers of bridging waters, which supports the possibility that bridging waters have comparable effects on the stabilities of both spike‐ACE2 complexes.
Finally, consistent with X‐ray structures of the spike‐ACE2 complexes, we note that ACE2 makes a more extensive hydrophobic contact with the spike RBD of CoV. Figure S6 of the Supporting Information shows that interfacial contacts break and form intermittently during the simulation, but at the same time there are about twice as many residue‐residue hydrophobic contact pairs in CoV's spike‐ACE2 interface compared to CoV‐2's spike‐ACE2 interface. A higher hydrophobic contact typically implies a higher binding affinity. 14 , 15 Therefore, this interaction should drive binding of ACE2 in favor of CoV, and not CoV‐2 spike RBD. Note, however, that this does not mean that CoV's spike interaction with ACE2 results in a larger burial of hydrophobic residues. In fact, CoV and CoV‐2 spike‐ACE2 interactions result in a very similar burial of hydrophobic residues (Table S3 of Supporting Information).
Overall, the analysis above shows that the density and dynamics of interfacial polar contacts are the primary differentiating factors that explains ACE2's relative affinity for CoV‐2/CoV spike RBDs. These differences, however, cannot be due to differences in interfacial charges or hydrophobic/polar ratios, as they are identical in the CoV and CoV‐2 spike‐ACE2 interfaces. Out of the 43 amino acid differences between CoV and CoV‐2 spike RBDs, 10 are at the ACE2 binding interface. 7 In CoV spike RBD, these interfacial residues are V404, R426, T433, Y442, L443, P462, L472, N479, Y484 and T487, and in CoV‐2 spike RBD the corresponding interfacial residues are K417, N439, G446, L455, F456, A475, F486, Q493, Q498 and N501.
The differences in the density and dynamics of interfacial polar contacts must, therefore, be due to (a) differences in the spatial arrangements of interfacial hydrophobic and polar groups, and/or (b) long‐ranged effects from amino acid differences in allosteric sites. We examine these below.
2.1.3. Long‐ranged coupling in conformational dynamics
Structural and thermodynamic experiments on model systems demonstrate that protein‐protein binding affinities can be modulated substantially by allosteric coupling in dynamics of interfacial and non‐interfacial residues. 36 Since dynamics is a key differentiating factor between CoV and CoV‐2 spike‐ACE2 interfaces, it raises the question of whether the dynamics of interfacial and non‐interfacial residues are coupled. To evaluate this quantitatively, we determine pairwise correlations in structural thermal fluctuations of amino acids, . In this expression, i and j refer to two amino acids. r(t) is the time‐dependent position of a residue's center‐of‐mass, is its time‐averaged position, and σ denotes its fluctuation. C ij is bounded between 0 and 1 and a value closer to 1 indicates a higher correlation.
Figure 3 shows that C ij decreases with distance between residues i and j, as also observed in model proteins. 32 However, even at distances greater than 20 Å the dynamics of interfacial and non‐interfacial residues are moderately correlated. There are also striking differences in the long‐distance correlations of spike RBDs of CoV and CoV‐2. (The pairwise correlation maps are provided in Figure S7 of the supporting information.) This means that the dynamics of polar interfacial residues are affected by non‐interfacial residues, and so non‐interfacial residues in spike RBD can also allosterically modulate ACE2 binding affinity. Note that allosteric signals can, in principle, transduce across protein domains. A comprehensive assessment of allosteric signals will, therefore, require modeling the full length spike. Nevertheless, what we find here is that there is dynamical allosteric coupling within RBDs of CoV and CoV‐2, and that there are discernible differences between them.
FIGURE 3.
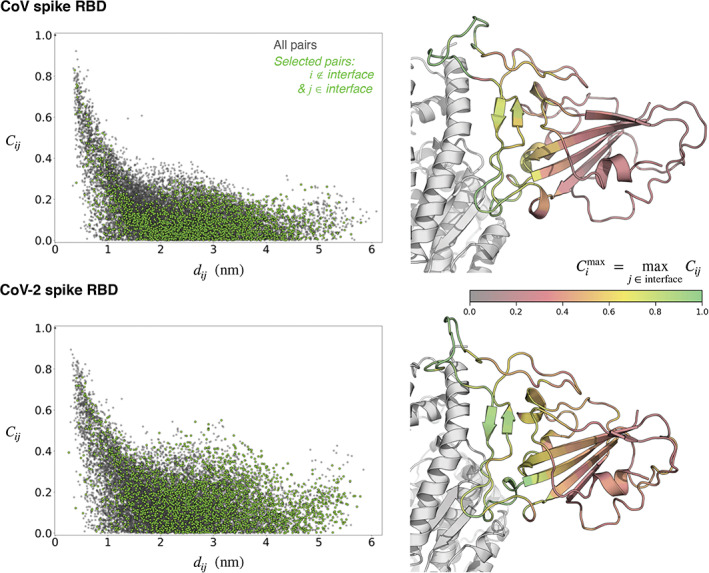
Distance dependence of structural fluctuation correlations in spike proteins. C ij are pairwise correlations in thermal structural fluctuations between the positions of the centers‐of‐masses (CoMs) of residues i and j, and d ij are the distances between the time‐averaged CoMs of these residues. The cartoons on the right show the highest correlation that each residue (in spike) has with an interfacial residue. The specific values of these highest correlations as well as the identities of the interfacial residues with which they have highest correlations are provided in Table S4 of the Supporting Information. The cartoon represented in gray is angiotensin converting enzyme 2 (ACE2)
2.2. Effect of interfacial spike mutations on binding free energy
As discussed above, the differences in the density and dynamics of interfacial polar contacts could be due to differences in the spatial arrangements of interfacial residues. Additionally, we observe from Figure 3 that non‐interfacial residues can also allosterically impact spike‐ACE2 affinity by altering the dynamics of interfacial residues. To examine these issues, we substitute CoV‐2's interfacial residues into CoV, and determine the effect of this substitution on spike‐ACE2 binding free energy. This substitution will only alter the spatial arrangement of hydrophobic and polar residues at the interface, but would not affect the net interfacial charge and the polar/hydrophobic ratio. Note that even after this substitution, there will be 30 + non‐interfacial residues in CoV RBD that will differ from CoV‐2 RBD, and since they could allosterically modulate spike‐ACE2 affinity, this substitution will not necessarily produce CoV‐2's experimental spike‐ACE2 affinity. Nonetheless, deviations from experiment could provide insight into the role of allosteric coupling.
Specifically, we engineer eight mutations in CoV spike RBD: V404K, R426N, T433G, Y442L, L472F, N479Q, Y484Q and T487N (Figure S8 of supporting information). We do not engineer the L443 → F456 mutation because neither L443 in CoV nor its corresponding residue F456 in CoV‐2 is found to make hydrophobic contact with ACE2 (Figure S6 of Supporting Information). The mutation P462 → A475 is also not engineered due to the current unavailability of established transition pathways. 37
Free energy calculations show that this substitution increases the binding affinity of CoV spike RBD, with a ΔG = −5.4 ± 0.4 kcal/mol (Figure S8 of supporting information). A recent study on force field accuracy in predicting mutational free energies 37 suggests that our prediction could contain an error of about 1 kcal/mol. Note, however, that this is an average error estimate, and there are cases for which free energy predictions have been off by several kcal/mol. 37 A general issue associated with all force fields is that they do not perform well at predicting non‐covalent interactions at high electric fields, 38 , 39 , 40 and we suspect that this may also be true for charged amino acids. Nevertheless, going by the average error in that study, 37 we note that our ΔG estimate for interfacial spike substitutions is overestimated with respect to experiments by 2 to 3 kcal/mol. 7 , 8 , 9 , 10 This difference between predicted and experimental estimates could be due to three reasons other than that related to force fields. First, due to technical reasons, we did not engineer the P462 → A475 substitution at the interface. This substitution can be expected to increase the flexibility at the interface, which, based on experiments conducted on other systems, 41 can potentially negatively impact spike‐ACE2 affinity by about a kcal/mol. Second, while all biophysical experiments concur that ACE2 binds to spike RBD of CoV‐2 more strongly than the spike RBD of CoV, we note that the experimental dissociation constants (k d ) for the same truncated spike systems and employing the same methodology (SPR) do differ by an order in magnitude (Table S1 of Supporting Information). Consequently, some of the mismatch could be due to experimental uncertainties. Finally, the enhanced effect of this substitution could be due to the fact that we did not modify any of the 30+ non‐interfacial residues in CoV that differ in CoV‐2. Thermodynamic experiments on other systems have shown that altering non‐interfacial residues can affect protein‐protein binding affinities by several kcal/mol through dynamical allosteric coupling with interfacial residues. 36
3. CONCLUSIONS
Analysis of MD simulations reveals three main differences between CoV and CoV‐2 spike‐ACE2 interfaces. Firstly, consistent with observations from X‐ray structures, 7 , 9 ACE2 makes a more extensive hydrophobic contact with CoV's spike. Secondly, there are 45% greater numbers of hydrogen bonds and salt bridges in CoV‐2's spike‐ACE2 interface. Thirdly, CoV‐2's spike‐ACE2 interface has 22% more possible combinations of hydrogen bonds compared to CoV's spike‐ACE2 interface. While the first difference will drive ACE2 binding in favor of CoV, the latter two will drive ACE2 binding in favor of CoV‐2. These observations lead to the conclusion that the higher affinity of ACE2 for CoV‐2's spike is due to the higher density and combinatorics of polar contacts, which also compensate for the smaller hydrophobic contacts in CoV‐2's spike‐ACE2 interface.
These observations bear important correspondence to those made from X‐ray structures. First, all of the polar contacts observed in X‐ray structures 7 , 9 , 13 are also observed in MD (Figure S5 of supporting information), but MD yields an additional set of possible combinations of polar contacts between spike and ACE2 that were not observed in X‐ray structures. Second, conformational sampling in MD assigns a probability to each polar contact, which cannot be obtained from X‐ray structures. Third, while X‐ray structures indicate that CoV and CoV‐2 spike‐ACE2 interfaces consist of equal numbers of polar contacts, 7 we find in MD that there are substantially more polar contacts in CoV‐2's spike‐ACE2 interface. These differences between MD and X‐ray structures could be due to structural thermal fluctuations present in MD, and due to the dynamics of interfacial water molecules, 42 which is present in MD, but absent in X‐ray structures. Taken together, it appears that differences in polar contacts between CoV and CoV‐2 spike‐ACE2 interfaces emerge when polar contacts are determined after accounting for their thermal fluctuations, combinatorics and probabilities.
The differences in density and dynamics of interfacial polar contacts between CoV and CoV‐2 spike‐ACE2 interfaces cannot be due to differences in interfacial charges or hydrophobic/polar ratios, as they are identical in the two interfaces. The spatial arrangement of interfacial residues, however, matters. Free energy calculations show that substitution of CoV‐2's interfacial residues into CoV spike positively impacts spike‐ACE2 affinity. The predicted effect is, however, greater than ACE2's relative CoV‐2/CoV spike affinity measured in experiments. This could be due to several reasons, as discussed in detail in an earlier section. These include potential errors from force fields, a missing P462 → A475 substitution at the interface, potential errors in experiments, and allosteric coupling from 30+ non‐interfacial residues in CoV that were not mutated in the chimera to their corresponding chemical types in CoV‐2. The potential role of allosteric coupling is also supported by the observation in equilibrium MD simulations that the dynamics of interfacial residues are distinctly influenced by non‐interfacial residues. In fact, results from Wang et al.'s free energy calculations on the spike‐ACE2 interface 16 also support the presence of allosteric coupling effects in binding. In their study, eight interfacial residues in CoV spike were individually/pair substituted into corresponding residues present in CoV‐2 spike. The net effect of the these individual/pair substitutions (− 6.8 ± 1.4 kcal/mol) is actually also greater than ACE2's relative CoV‐2/CoV spike affinity. Further insights into allostery can be obtained by engineering and studying the effects of allosteric mutations using several different computational and experimental techniques (reviewed recently in references 32 , 43 ). A reasonable starting set of target mutations could be the non‐interfacial residues that are found to be coupled strongly to interfacial residues (Figure 3 and Table S4 of Supporting Information). Mutational targets can also be obtained using structure‐based methods, 44 where our clustered binding modes in Figure 1 can be used as starting points for network analysis and hot‐spot prediction.
Given existing evidence on the general reliability of employed MD methods, 32 , 37 , 45 , 46 and our recent work 23 that yielded validated predictions on virus‐host protein‐protein interactions, 25 we consider our conclusions to be robust. In fact, MD simulations 21 performed using a potential energy function different from ours reveals a set of high‐frequency polar contacts in CoV‐2's spike‐ACE2 interface that is very similar to those found in our simulation (Figure 2). Nevertheless, from the perspective of intermolecular interaction theory, the underlying potential energy functions that we employ do rely on describing interactions using point charges, no polarization, and only pairwise vdW interactions. These approximations should be properly scrutinized in future studies of complex protein‐protein interactions. Higher‐level electronic structure calculations 47 are in progress to assess the role of multipole electrostatics, induced polarization, and many‐body dispersion interactions on spike‐ACE2 binding.
Overall, the molecular understanding that our work provides on the relative binding affinities of CoV and CoV‐2 spike to ACE2 is important for understanding their different infectivity rates. The proposed model and our quantitative findings can be validated by NMR and binding kinetics experiments on wild type CoV/CoV‐2 and its mutational variants. Our mechanistic model is, in fact, timely as it lays the foundation to understand the molecular bases for the new set of highly infectious strains that are beginning to emerge and spread rapidly across the globe. 48 Based on high‐throughput calorimetric experiments, 49 their higher infectivity is suspected to be due to increased affinities of their spike proteins from mutations at and near their ACE2 binding sites, 48 however, that still remains to be demonstrated.
These results are also expected to lend direct insight into designing spike‐ACE2 blockers, which, like in the cases of HIV and Influenza, can serve as antivirals. 50 As such, recent experiments 51 show that ACE2‐derived peptides can serve as spike‐ACE2 blockers, although their binding affinities to spike are three‐orders in magnitude weaker compared to ACE2. We anticipate that results from these studies will directly serve to rationally improve their binding affinities from the micromolar to the nanomolar range for them to serve as effective competitive inhibitors of spike‐ACE2 interactions. Specifically, our results suggest that the efficacy of candidate peptides can be improved by considering not only structural, but also dynamical information. MD based free energy calculations, just as we conduct here, can be carried out to examine the specific roles of ACE2 interfacial residues and their targeted mutations on spike binding. Virtual screening has been employed to accelerate search for potential compounds for experimental testing, 52 and our results suggest incorporation of an additional filtration step that examines the role of protein dynamics to avoid discarding candidates solely based on structural information. Our results also strongly recommend considering allosteric affects in design of longer peptides. In fact, a recent study shows that mini‐proteins designed by computational modeling can serve as high‐affinity spike‐ACE2 blockers, 53 and allosteric effects from non‐interfacial residues can be explored in these mini‐proteins to reduce their sizes for effective delivery, while maintaining their high affinities and stabilities.
4. METHODS
4.1. Molecular dynamics
All MD simulations are performed using Gromacs 2020. 54 Protein and water bonds are restrained, 55 , 56 and consequently an integration time step of 2 fs is employed. Simulations are conducted under isobaric‐isothermal boundary conditions. Pressure is regulated at 1 bar using a coupling constant of 1 ps and a compressibility of 4.5 × 10−5 bar−1. Temperature is maintained at 310 K. Extended ensemble approaches are used for maintaining both temperature and pressure. 57 , 58 , 59 Electrostatic interactions beyond 10 Å are computed using the particle mesh Ewald Scheme 60 with a Fourier grid spacing of 1.5 Å, a fourth‐order interpolation. Van der Waals interactions are computed explicitly for interatomic distances smaller than 10 Å. We use Amber99sb‐ILDN parameters to describe protein and ions, 45 and SPC/E parameters to describe water molecules. 61 This force field has been demonstrated to perform well in reproducing structural data from X‐ray diffraction and NMR spectroscopy, 45 , 46 and also dynamics data from NMR spectroscopy. 32 The ACE2 protein contains a Zn2+ ion in its catalytic core, which is about 20 Å away from the spike‐ACE2 interface. In line with earlier work on modeling Zn2+ ions in proteins, 62 the coordination of the Zn2+ ion in ACE2 is maintained through application of distance restraints. Specifically, flat‐bottomed quadratic potentials are assigned to distances between the ion and three atoms of ACE2, H374/NE2, H378/NE2 and E402/OE1, that are observed to coordinate it in X‐ray structures. In MD simulations, the average restraining energies for the CoV and the CoV‐2 complexes are found to be small, that is, 0.14 ± 0.08 and 0.11 ± 0.06 kcal/mol, respectively.
Starting coordinates of CoV's spike‐ACE2 complex are taken from its X‐ray structure (PDB ID: 2AJF), 13 and those of CoV‐2's spike‐ACE2 complex are taken from its cryo‐EM structure (PDB ID: 6 M17). 63 Note that a higher resolution X‐ray structure of CoV‐2's spike‐ACE2 complex is now also available, 7 and is used in our analysis, but was unavailable at the start of this project. Nevertheless, there is only little difference between the X‐ray and cryo‐EM structures ‐ the RMSD between all heavy atoms is <1 Å, which is less than the RMSDs between the different interfacial binding modes observed in MD simulations. The carbohydrate groups in spike are removed. The missing loops in the ACE2 protein in 2AJF are built using MODELER. 64 To make ACE2 PD sequences identical in CoV and CoV‐2 constructs, the N‐terminal residues 19 and 20 in ACE2 of 6 M17 are built, and the C‐terminal of ACE2 in 6 M17 is truncated at the last C‐terminal residue resolved in 2AJF. The N‐ and C‐termini of both spike RBD and ACE2 PD are capped with ACE and NME, respectively. Hydrogen atom positions and histidine types are determined using the PDB2PQR algorithm. 65 Each of the two complexes is initially placed in a cubic unit cell of length 160 Å, and then energy minimized using the steepest descent algorithm implemented in Gromacs. The vacant space in the box is then filled with water, and the system is again subjected to energy minimization. The unit cells containing CoV and CoV‐2 spike‐ACE2 complexes contain, respectively, 131 995 and 131 897 waters. Note that the crystallographically resolved waters are retained. Na+ and Cl− ions are added by randomly substituting non‐crystallographic waters. NaCl concentration is set at 50 mM with extra Na+ ions to compensate for the charge of the complex. Specifically, the CoV spike‐ACE2 unit cell contains 144 Na+ and 120 Cl− ions, the CoV‐2 spike‐ACE2 unit cell contains 143 Na+ and 120 Cl− ions. After adding salt, the system is energy minimized a final time. Each of these two complexes is then subjected to 3 μs long MD simulations.
In addition to carrying out MD simulations of spike‐ACE2 complexes, we also carry out 0.5 μs long MD simulations of isolated spike RBDs of CoV and CoV‐2 in solution. These simulations are used for getting starting conformations for free energy calculations. For these simulations, the starting coordinates of spike RBDs are taken from final snapshots of the MD simulations of the spike‐ACE2 complexes. Each of the two spike RBDs is initially placed in a cubic unit cell of length 90 Å, and the protocol described above is followed for adding waters and salt. The unit cell containing CoV's spike RBD contains 23 247 waters, 21 Na+ and 22 Cl− ions, and the unit cell containing CoV‐2's spike RBD contains 23 240 waters, 21 Na+ and 23 Cl− ions.
4.2. Free energy calculations
The effect of mutating spike residues on its binding free energy with ACE2 is determined as
| (1) |
Here, ΔG spike−ACE2 is the effect of mutations on the free energy of the spike‐ACE2 complex in solution, and ΔG spike is the effect of mutations on the free energy of isolated spike in solution. ΔG spike and ΔG spike−ACE2 are computed using thermodynamic integration, and using the 5‐point Gauss‐Quadrature rule, that is,
| (2) |
where the weights w i = {0.237, 0.479, 0.559, 0.479, 0.237} and λ i = {0.047, 0.231, 0.5, 0.769, 0.953}.
The starting conformations for engineering mutations are taken from MD simulations. Hybrid topology files, which contain coordinates and force field parameters for both states of the amino acids (natural and mutated) are constructed using the PMX module. 37 To avoid singularities and numerical instabilities that may arise due to particle appearance and annihilation, we use a modified form of the “soft core” potentials suggested by Beutler et al. 66 implemented in Gromacs. In these soft core potentials, the distances between particles 'i' and “j” in state A (λ = 0) are modified as and those between particles in state B (λ = 1) are modified as . In these expressions, σ = (C 12/C 6)1/6 is the ratio of the LJ parameters, and if either C 12 or C 6 is zero, then we take σ = 3 Å. We set the soft core parameters to be α = 1 and P = 1. Sampling is conducted using stochastic dynamics and under NVT conditions. For each λ i , ∂H/∂λ is averaged for 250 ns (Figure S8 of Supporting Information), and statistical errors are determined from the final 50 ns using block averaging.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
This research used resources of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE‐AC05‐00OR22725.
Delgado JM, Duro N, Rogers DM, Tkatchenko A, Pandit SA, Varma S. Molecular basis for higher affinity of SARS‐CoV‐2 spike RBD for human ACE2 receptor. Proteins. 2021;89:1134–1144. 10.1002/prot.26086
This manuscript has been authored in collaboration with UT‐Battelle, LLC, under contract DE‐AC05‐00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid‐up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Funding information Office of Science of the U.S. Department of Energy, Grant/Award Number: DE‐AC05‐00OR22725
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track covid‐19 in real time. Lancet Infect Dis. 2020;20:533‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petersen E, Koopmans M, Go U, et al. Comparing sars‐cov‐2 with sars‐cov and influenza pandemics. Lancet Infect Dis. 2020;20(9):e238‐e244. 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guan Y, Zheng BJ, He YQ, et al. Isolation and characterization of viruses related to the sars coronavirus from animals in southern China. Science. 2003;302:276‐278. [DOI] [PubMed] [Google Scholar]
- 4. Li W, Greenough TC, Moore MJ, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J Virol. 2004;78:11429‐11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li W, Zhang C, Sui J, et al. Receptor and viral determinants of sars‐coronavirus adaptation to human ace2. EMBO J. 2005;24:1634‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade‐long structural studies of sars coronavirus. J Virol. 2020;94(7):e00127‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lan J, Jiwan G, Jinfang Y, et al. Structure of the sars‐cov‐2 spike receptor‐binding domain bound to the ace2 receptor. Nature. 2020;581:215‐220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 8. Wrapp D, Wang N, Corbett K, et al. Cryo‐em structure of the 2019‐ncov spike in the prefusion conformation. Science. 2020;367:1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by sars‐cov‐2. Nature. 2020;581:221‐224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the sars‐cov‐2 spike glycoprotein. Cell. 2020;181:281‐292.e6. http://www.sciencedirect.com/science/article/pii/S0092867420302622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shang J, Wan Y, Chuming L, et al. Cell entry mechanisms of sars‐cov‐2. Proc Natl Acad Sci U S A. 2020;117:11727‐11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Casalino L, Gaieb Z, Goldsmith JA, et al. Beyond shielding: the roles of glycans in the sars‐cov‐2 spike protein. ACS Central Sci. 2020;6:1722‐1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li F, Li W, Farzan M, Harrison SC. Structure of sars coronavirus spike receptor‐binding domain complexed with receptor. Science. 2005;309:1864‐1868. [DOI] [PubMed] [Google Scholar]
- 14. Kastritis PL, Bonvin AMJJ. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J Royal Soc Interface. 2013;10:20120835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rahman S, Wineman‐Fisher V, Al‐Hamdani Y, Tkatchenko A, Varma S. Predictive QM/MM modeling of the effect of lysine methylation on protein‐protein binding. J Mol Biol. 2021;433:166745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y, Liu M, Gao J. Enhanced receptor binding of sars‐cov‐2 through networks of hydrogen‐bonding and hydrophobic interactions. Proc Natl Acad Sci U S A. 2020;117:13967‐13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laurini E, Marson D, Aulic S, Fermeglia M, Pricl S. Computational alanine scanning and structural analysis of the sars‐cov‐2 spike protein/angiotensin‐converting enzyme 2 complex. ACS Nano. 2020;14:11821‐11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah M, Ahmad B, Choi S, Woo HG. Mutations in the sars‐cov‐2 spike rbd are responsible for stronger ace2 binding and poor anti‐sars‐cov mabs cross‐neutralization. Comput Struct Biotechnol J. 2020;18:3402‐3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Andrade J, Goncalves PFB, Netz PA. Why does the novel coronavirus spike protein interact so strongly with the human ace2? A thermodynamic answer. ChemBioChem. 2020;22(5):865‐875. [DOI] [PubMed] [Google Scholar]
- 20. Ghorbani M, Brooks BR, Klauda JB. Critical sequence hotspots for binding of novel coronavirus to angiotensin converter enzyme as evaluated by molecular simulations. J Phys Chem B. 2020;124:10034‐10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ali A, Vijayan R. Dynamics of the ace2–sars‐cov‐2/sars‐cov spike protein interface reveal unique mechanisms. Sci Rep. 2020;10:14214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zou J, Yin J, Fang L, et al. Computational prediction of mutational effects on sars‐cov‐2 binding by relative free energy calculations. J Chem Inform Model. 2020;60:5794‐5802. 10.1021/acs.jcim.0c00679. [DOI] [PubMed] [Google Scholar]
- 23. Dutta P, Siddiqui A, Botlani M, Varma S. Stimulation of nipah fusion: small intradomain changes trigger extensive interdomain rearrangements. Biophys J. 2016;111:1621‐1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duro N, Varma S. Role of structural fluctuations in allosteric stimulation of paramyxovirus hemagglutinin‐neuraminidase. Structure. 2019;27:1601‐1611. [DOI] [PubMed] [Google Scholar]
- 25. Wong JJ, Young TA, Zhang J, et al. Monomeric ephrinb2 binding induces allosteric changes in nipah virus g that precede its full activation. Nat Commun. 2017;8:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chakraborti S, Prabakaran P, Xiao X, Dimitrov DS. The sars coronavirus s glycoprotein receptor binding domain: fine mapping and functional characterization. Virol J. 2005;2:73. 10.1186/1743-422X-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site‐specific glycan analysis of the sars‐cov‐2 spike. Science. 2020;369:330‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parekh RB, Tse AG, Dwek RA, Williams AF, Rademacher TW. Tissue‐specific n‐glycosylation, site‐specific oligosaccharide patterns and lentil lectin recognition of rat thy‐1. EMBO J. 1987;6:1233‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen JD, Watanabe Y, Chawla H, Newby ML, Crispin M. Subtle influence of ace2 glycan processing on sars‐cov‐2 recognition. J Mol Biol. 2021;433:166762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315:972‐976. [DOI] [PubMed] [Google Scholar]
- 31. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit‐learn: machine learning in python. J Mach Learning Res. 2011;12:2825‐2830. [Google Scholar]
- 32. Botlani M, Siddiqui A, Varma S. Machine learning approaches to evaluate correlation patterns in allosteric signaling: a case study of the pdz2 domain. J Chem Phys. 2018;148:241726. 10.1063/1.5022469. [DOI] [PubMed] [Google Scholar]
- 33. Luzar A, Chandler D. Hydrogen‐bond kinetics in liquid water. Nature. 1996;379:55‐57. 10.1038/379055a0. [DOI] [Google Scholar]
- 34. Molecular dynamics simulations related to sars‐cov‐2. D. E. Shaw Research Technical Data (2020). http://www.deshawresearch.com/resources\_sarscov2.html.
- 35. Janin J. Wet and dry interfaces: the role of solvent in protein‐protein and protein‐dna recognition. Structure. 1999;7:R277‐R279. [DOI] [PubMed] [Google Scholar]
- 36. Fuentes EJ, Gilmore SA, Mauldin RV, Lee AL. Evaluation of energetic and dynamic coupling networks in a pdz domain protein. J Mol Biol. 2006;364:337‐351. [DOI] [PubMed] [Google Scholar]
- 37. Gapsys V, Michielssens S, Seeliger D, de Groot BL. Accurate and rigorous prediction of the changes in protein free energies in a large‐scale mutation scan. Angew Chem, Int Ed. 2016;55:7364‐7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wineman‐Fisher V, Al‐Hamdani Y, Addou I, Tkatchenko A, Varma S. Ion‐hydroxyl interactions: from high‐level quantum benchmarks to transferable polarizable force fields. J Chem Theory Comput. 2019;15:2444‐2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wineman‐Fisher V, Al‐Hamdani Y, Nagy PR, Tkatchenko A, Varma S. Improved description of ligand polarization enhances transferability of ion‐ligand interactions. J Chem Phys. 2020;153:094115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wineman‐Fisher V, Melendez J, Nagy PR, et al. Transferable interactions of li+ and mg2+ ions in polarizable models. J Chem Phys. 2020;153:104113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borcherds W, Theillet FX, Katzer A, et al. Disorder and residual helicity alter p53‐mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10:1000‐1002. [DOI] [PubMed] [Google Scholar]
- 42. Dutta P, Botlani M, Varma S. Water dynamics at protein‐protein interfaces: molecular dynamics study of virus‐host receptor complexes. J Phys Chem B. 2014;118:14795‐14807. [DOI] [PubMed] [Google Scholar]
- 43. Wodak SJ, Paci E, Dokholyan NV, et al. Allostery in its many disguises: from theory to applications. Structure. 2019;27:566‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J, Jain A, McDonald LR, et al. Mapping allosteric communications within individual proteins. Nat Commun. 2020;11:3862. 10.1038/s41467-020-17618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lindorff‐Larsen K, Piana S, Palmo K, et al. Improved side‐chain torsion potentials for the amber ff99sb protein force field. Proteins. 2010;78:1950‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beauchamp KA, Lin Y‐S, Das R, Pande VS. Are protein force fields getting better? A systematic benchmark on 524 diverse nmr measurements. J Chem Theory Comput. 2012;8:1409‐1414. 10.1021/ct2007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stöhr M, Tkatchenko A. Quantum mechanics of proteins in explicit water: the role of plasmon‐like solute‐solvent interactions. Sci Adv. 2019;5(12):eaax0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baric RS. Emergence of a highly fit sars‐cov‐2 variant. N Engl J Med. 2020;383:2684‐2686. [DOI] [PubMed] [Google Scholar]
- 49. Starr TN, Greaney AJ, Hilton SK, et al. Deep mutational scanning of sars‐cov‐2 receptor binding domain reveals constraints on folding and ace2 binding. Cell. 2020;182:1295‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho Neves N, Franco OL. Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci. 2019;76:3525‐3542. 10.1007/s00018-019-03138-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang J, Petitjean SJL, Koehler M, et al. Molecular interaction and inhibition of sars‐cov‐2 binding to the ace2 receptor. Nat Commun. 2020;11:4541. 10.1038/s41467-020-18319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Panda PK, Arul MN, Patel P, et al. Structure‐based drug designing and immunoinformatics approach for sars‐cov‐2. Sci Adv. 2020;6(28):eabb8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cao L, Goreshnik I, Coventry B, et al. De novo design of picomolar sars‐cov‐2 miniprotein inhibitors. Science. 2020;370:426‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abraham MJ, Murtola T, Schulz R, et al. Gromacs: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19‐25. [Google Scholar]
- 55. Hess B. P‐lincs: a parallel linear constraint solver for molecular simulation. Journal of Chemical Theory and Computation. 2008;4:116‐122. [DOI] [PubMed] [Google Scholar]
- 56. Miyamoto S, Kollman PA. Settle: an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13:952‐962. [Google Scholar]
- 57. Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52:255‐268. [Google Scholar]
- 58. Hoover WG. Canonical dynamics: equilibrium phase‐space distributions. Phys Rev A. 1985;31:1695. [DOI] [PubMed] [Google Scholar]
- 59. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:14101. [DOI] [PubMed] [Google Scholar]
- 60. Darden T, York D, Pedersen L. Particle mesh ewald: an n log (n) method for ewald sums in large systems. J Chem Phys. 1993;98:10089‐10092. [Google Scholar]
- 61. Berendsen H, Grigera J, Straatsma T. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269‐6271. [Google Scholar]
- 62. Peters MB, Yang Y, Wang B, et al. Structural survey of zinc‐containing proteins and development of the zinc amber force field (zaff). J Chem Theory Comput. 2010;6:2935‐2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of sars‐cov‐2 by full‐length human ace2. Science. 2020;367:1444‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Webb B, Sali A. Comparative protein structure modeling using modeller. Curr Protocols Bioinform. 2016;54:5.6.1‐5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. Pdb2pqr: an automated pipeline for the setup of poisson–boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665‐W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett. 1994;222:529‐539. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.