

Abstract
The evolution of the SARS‐CoV‐2 new variants reported to be 70% more contagious than the earlier one is now spreading fast worldwide. There is an instant need to discover how the new variants interact with the host receptor (ACE2). Among the reported mutations in the Spike glycoprotein of the new variants, three are specific to the receptor‐binding domain (RBD) and required insightful scrutiny for new therapeutic options. These structural evolutions in the RBD domain may impart a critical role to the unique pathogenicity of the SARS‐CoV‐2 new variants. Herein, using structural and biophysical approaches, we explored that the specific mutations in the UK (N501Y), South African (K417N‐E484K‐N501Y), Brazilian (K417T‐E484K‐N501Y), and hypothetical (N501Y‐E484K) variants alter the binding affinity, create new inter‐protein contacts and changes the internal structural dynamics thereby increases the binding and eventually the infectivity. Our investigation highlighted that the South African (K417N‐E484K‐N501Y), Brazilian (K417T‐E484K‐N501Y) variants are more lethal than the UK variant (N501Y). The behavior of the wild type and N501Y is comparable. Free energy calculations further confirmed that increased binding of the spike RBD to the ACE2 is mainly due to the electrostatic contribution. Further, we find that the unusual virulence of this virus is potentially the consequence of Darwinian selection‐driven epistasis in protein evolution. The triple mutants (South African and Brazilian) may pose a serious threat to the efficacy of the already developed vaccine. Our analysis would help to understand the binding and structural dynamics of the new mutations in the RBD domain of the Spike protein and demand further investigation in in vitro and in vivo models to design potential therapeutics against the new variants.
Keywords: KD (dissociation constant), MD simulation, new variants, protein–protein docking, SARS‐CoV‐2
This study precisely explored the mechanism of the interaction of the spike RBD with the host ACE2 and revealed the differences in the binding of the reference and new variants. The systematic investigation revealed that the South African and Brazilian variants are more lethal than the others due to inter‐protein contacts specifically the electrostatic while the N501Y is comparable with the wild type. We hypothesized that the residue at 501Y is continuously subjected to positive selection pressure. We further demonstrated the dynamic behavior is also changed with the protein evolution. Conclusively, this study provides strong basis for structure and rationale‐based drug designing against the new variant by exploring the noticeable differences.
1. INTRODUCTION
During the 21st century, Asia has remained the epicenter of coronavirus caused epidemics such as SARS and MERS. Recently the novel β‐coronavirus named SARS‐CoV‐2 emerged in Wuhan (China; Xydakis et al., 2020) that has devastated human health across the globe by causing upper respiratory complexities resulting in severe pneumonia and bronchiolitis (Huang et al., 2020). Rapid human‐to‐human transmission is the most striking feature of SARS‐CoV‐2, which enabled its worldwide penetrations (Wang et al., 2020b). Consequently, the SARS‐CoV‐2 caused the coronavirus disease‐19 (COVID‐19) was announced as a pandemic by the world health organization (Rothan & Byrareddy, 2020).
The SARS‐CoV‐2 possessed the positive‐sense single‐stranded ~30 kb RNA genome, which codes for structural envelope (E), spike (S), nucleocapsid (N), and membrane (M), nonstructural and accessory proteins (Wu et al., 2020). The bat coronavirus RaTG13 is considered as the evolutionarily closest relative of SARS‐CoV‐2 and genomes of these two viruses that codes for structural protein depict ~96% sequence similarity (Fehr & Perlman, 2015; Hussain et al., 2020).
Mechanisms of SARS‐CoV‐2 transmission and pathogenesis involve the binding of the virus to the host cellular angiotensin‐converting enzyme (ACE2) receptors through the surface S‐protein (Spike protein) which is composed of S1 and S2 subunit (Li, 2016). The RBD (receptor binding domain) of S1 facilitates the binding of the virus to ACE2; however, the S2 subunit is responsible for membrane fusion which permits the entry of viral genome into host cytoplasm (Lan et al., 2020; Li, 2016). The binding between viral S‐protein and ACE2 receptor triggers the host antibody production against the RBD domain of S‐protein, leading to host immunization (Abraham et al., 1990; Hoffmann et al., 2020). Therefore, blocking ACE2‐RBD interaction has been recognized as an essential way to inhibit SARS‐CoV‐2 transmission and infection (Walls et al., 2020). Consequently, the SARS‐CoV‐2 RBD domain can serve as an important target for designing the anti‐COVID‐19 therapeutic strategies (Lan et al., 2020)
Recent reports from England regarding the origin of novel contagious strain (B.1.1.7) of SARS‐CoV‐2 have further exacerbated the situation (Leung et al., 2021). Novel mutations in the spike protein of B.1.1.7 (deletion 69–70, 144 and substitution K417N, K417T, E484K, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H, and many others) might have altered the SARS‐CoV‐2 ability to transmit and infect. Consequently, the currently available vaccines against COVID‐19 might not be effective against B.1.1.7 (Harrington et al., 2021). The substitution mutations N501Y, E484K, and others) within RBD of the UK and South African SARS‐CoV‐2 strains are now spreading unchecked. The number of 501Y mutations associated cases has raised from 0.1 percent in October to 49.7% in the UK as estimated in late November. The mutation N501Y co‐occurs with other mutations in the N, orf8, orf1a, and S glycoprotein in 501, involving two deletions Δ69 and Δ70. The new variant of South Africa carries K417N‐E484K‐N501Y mutations amongst others (Kirby, 2021; Koyama et al., 2020). The new variants of both UK and South Africa seem to be more contagious; however, mutations in the UK variants are unlikely to impede the effectiveness of the developed vaccines, though the South African variants may interfere with it to some extent (Davies et al., 2020). In this regard, the lack of empirical data is the major challenge to predict which one of the newly emerged strains of SARS‐CoV‐2 is more lethal.
To relieve this challenge and diminish the dread of the current pandemic, many researchers worldwide with molecular and computational expertise are using different strategies and approaches like the repurposing of available antiviral drugs, vaccine designing, and mutational analysis. A more focused and detailed analysis is crucial for understanding the effect of these novel amino acid substitutions on the structure, function, and binding of the ACE2 receptor. Therefore, in the present study, we employed the protein‐protein docking methods with a biophysical investigation to examine the effect of K417N‐E484K‐N501Y, K417T‐E484K‐N501Y, E484K, N501Y, and E484K‐N501Y mutations on the structure and binding of the ACE2 receptor and their correlation with infectivity of newly emerged strains of SARS‐CoV‐2. Our analysis would help to understand the structural dynamics of the new mutations in the RBD domain of the Spike protein.
2. METHODOLOGY
2.1. Data retrieval and variants modeling
Recently the novel strains of SARS‐CoV‐2 were reported in England, South Africa, and Brazil that has a high transmission speed and claimed to be very contagious. The predicted mutations in novel strains were found in Spike protein, which plays an indispensable role in the binding of the virus to the host cell. Keeping the importance of Spike protein in the viral infection, we retrieved the latest submitted amino acid sequence of SARS‐CoV‐2 Spike protein from UniProt (Magrane, 2011) available under accession number: P0DTC2 to identify the exact location of newly emerged mutations. Finally, the reported wild‐type structure (6M0J) of SARS‐CoV‐2 (Lan et al., 2020) Spike protein was obtained from a protein data bank (Rose et al., 2010) and used for variant modeling by Chimera software (Goddard et al., 2005).
2.2. Protein–protein docking and determination of dissociation constant (KD)
High ambiguity‐driven protein–protein docking (HADDOCK) algorithm which uses biophysical and biochemical interaction data such as mutagenesis, ambiguous interaction restraints (AIRs), and chemical shift perturbation from NMR titration experiments was used to perform the protein–protein docking process (Dominguez et al., 2003). To predict docking, we used the Guru interface, which is known to be the strongest interface between all four interfaces owned by the HADDOCK server. Approximately 500 features are used by Guru interface for protein–protein/DNA/RNA docking. Furthermore, to give a convincible insight into the KD (dissociation constant) was calculated using PROtein binDIng enerGY prediction (PRODIGY) which is an online server to compute the binding affinity and KD for different biological complexes (Xue et al., 2016).
2.3. Molecular dynamics simulation of the top complexes
The dynamic behavior of complex wild type, K417N‐E484K‐N501Y, K417T‐ E484K‐N501Y, E484K, N501Y, and E484K‐N501Y was checked by MD simulation performed on Amber20 (Salomon‐Ferrer et al., 2013) using FF14SB force field. The system solvation was performed in a TIP3P water box, and the system was neutralized by the addition of counter ions (Price & Brooks, 2004). Energy minimization protocol was used for the removal of the bad clashes in the system. The steepest descent algorithm (Meza, 2010) and the conjugate gradient algorithm were used for 6000 and 3000 cycles, respectively (Watowich et al., 1988). After heating up to 300 K the system was equilibrated at constant pressure 1 atm with weak restraint and then equilibrated without any restraint. Finally, the production step was run for 100 ns. The long‐range electrostatic interaction was treated with particle mesh Ewald algorithm (Salomon‐Ferrer et al., 2013) with a cutoff distance of 10.0 Å. The SHAKE algorithm was used to treat a covalent bond (Kräutler et al., 2001). The production step of MD simulation was performed on PMEMD.CUDA and trajectories were processed usingAmber20 CPPTRAJ package (Roe & Cheatham, 2013).
2.4. Binding free energy calculations
To estimate the real binding energy calculations of the wild type, K417N‐E484K‐N501Y, K417T‐ E484K‐N501Y, E484K, N501Y, and E484K‐N501Y, MMGBSA approach was used. This method is the best approach used by different studies to estimate the real binding energy of different biological complexes, such as Spike protein–ligand, protein–protein, and protein–DNA/RNA (Ali et al., 2019; Khan et al., 2018; Khan et al., 2019; Khan et al., 2020a; 2020b; 2020c; 2020d). The MMGBSA.py (Hou et al., 2011) script was used to estimate the total binding free energy of the top ligand complexes. Each energy term such as vdW, electrostatic, GB, and SA was calculated as a part of the total binding energy.
For free energy calculation, the following equation was used:
Each component of the total free energy was estimated using the following equation:
Where Gbond, Gele, and GvdW denote bonded, electrostatic, and van der Waals interactions, respectively. G‐pol and Gnpol are polar and nonpolar solvated free energies. The Gpol and Gnpol are calculated by the generalized born (GB) implicit solvent method with the solvent‐accessible surface area SASA term.
3. RESULTS AND DISCUSSION
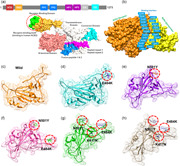
A distressing situation has been created by the new variants of the SARS‐CoV‐2 that has ascended in the UK, South Africa, and Brazil. The emergence of such novel strain suggests that the earlier strain that emerged in Wuhan has passed through further genetic pressure and acquired genetic variations. The variations caused by these genetic alterations may have led to notable differences in the transmission, infectivity, and available treatment options. The scientist also speculated the available vaccine developed recently against the Wuhan strain may work against the UK (N501Y; VOC 202012/01) variant but not against the South African variant (K417N‐E484K‐N501Y; 501Y.V2 Variant) and the Brazil variant (K417T‐E484K‐N501Y) formally known as P.1 (or B.1.1.248). This speculation prompted us to characterize the new variants further and explore their uniqueness. Comparative genomics inspection of genomes obtained from the UK, South Africa, Brazil, and other parts of the world revealed that the spike protein had acquired multiple crucial mutations including (K417N, K417T, E484K, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H) and deletions (69–70, 144). The Spike protein contains receptor‐binding domain (RBD), N‐terminal domain (NTD), the hepated peptides 1 and 2, the connector domain (CD), the transmembrane, and the central helix (Figure 1a). The spike glycoprotein is a homotrimeric protein that uses the RBD domain to bind to ACE2 protein. Such binding induces a cascade of instances that contribute to the fusion for cell entry between the host cell and viral membranes. This binding stabilizes the spike protein from premetastable state to the poststable state. These deletions and mutations may further aggravate the severity of SARS‐CoV‐2 and potentially hinder the efficacy of new vaccines. Among the given mutations, three are specific to the RBD domain and required insightful scrutiny for new therapeutic options. These substitutions may be associated with functional uniqueness and may distantly use a different approach for infection. Given the importance of the Spike RBD in the virulence and pathogenesis, the RBD of the SARS‐CoV‐2 was subjected to comparative binding and biophysical investigation upon the interaction with ACE2. The structure of the Spike‐RBD complex retrieved from RCSB (PDB ID:6M0J). Among these residues in the binding interface, only eight are conserved in SARS‐CoV‐2. Among the evolved residues Leu455, Phe456, Phe486, Gln493, Gln498, and Asn501 (from SARs‐CoV) in SARS‐CoV‐2 also involved Asn501 (Figure 1b). This residue Asn501 (SARS‐CoV‐2 Wuhan isolate) is changed to Tyr501 in the SARS‐CoV‐2 (B.1.1.7) strain, which shows that this residue is continuously subjected to positive selection pressure(Lan et al., 2020; Wang et al., 2020a). Previously studies demonstrated that these five residues (given above) are responsible for the stronger binding of spike RBD to ACE2 in SARS‐CoV‐2 than the SARs‐CoV (Li et al., 2005). Looking into the significant role of these substitutions in the RBD domain of the spike glycoprotein, we generated K417N‐E484K‐N501Y, K417T‐ E484K‐N501Y, E484K, N501Y, and E484K‐N501Y double mutant to perform comparative structural and binding analysis. The mutants were generated by using Chimera and are given in Figure 1c (wild type), Figure 1d (E484K), Figure 1e (N501Y), Figure 1f (E484K‐N501Y), Figure 1g (K417N‐E484K‐N501Y), Figure 1h (K417T‐ E484K‐N501Y).
Figure 1.
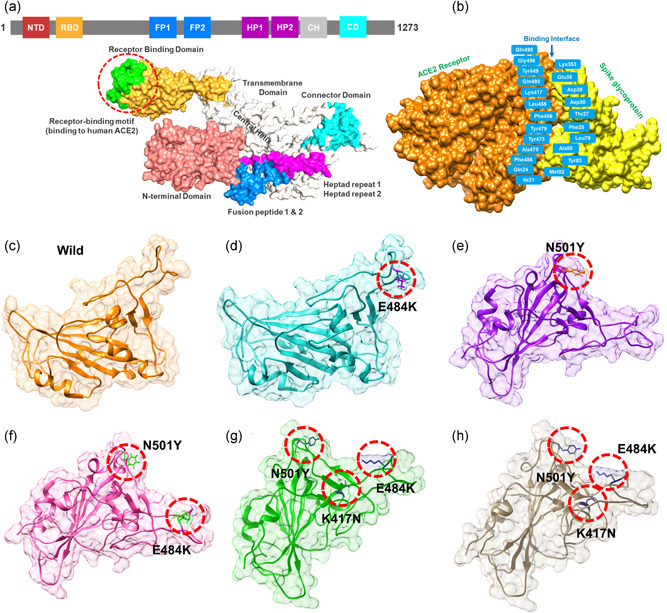
Structural representation of the spike glycoprotein (PDB ID:6M0J) and the receptor‐binding domain of the SARS‐CoV‐2. (a) shows the distribution of different domains differentiated with different colors. The RBD domain is shown as yellow specifically. (B) shows the binding interface of the ACE2 and spike RBD. (c) wild type, (d) E484K, (e) N501Y, and (f) E484K‐N501Y, (g) K417N‐E484K‐N501Y, and (h) shows the K417T‐E484K‐N501Y structure of the spike RBD
Given the role of Spike protein in many important processes including attachment and pathogenesis next, we performed the binding analysis of the wild type and mutants (Spike RBD). Nearly all biological processes inside the cells are regulated by the interactions among different proteins. Variations in these interactions are liable for several disorders, making protein‐protein complexes a key target for the advancement of therapeutics (Ray, 2014). In this case, the identification of the structural determinants of these interactions and their binding energy is a crucial step towards a deeper understanding and regulation of these processes. Notably, the binding affinity, which determines whether or not complex formation occurs under particular circumstances, holds the key to regulating molecular interactions (e.g., engineering high‐affinity interactions), developing novel therapeutics (e.g., guiding rational drug design), or predicting the effect of variations on protein interfaces (Smith & Sternberg, 2002). The binding affinity has been calculated for decades by different methodologies ranging from exact approaches (e.g., free energy perturbation), that are precise however computationally expensive compared to empirical methods (e.g., scoring functions in docking, various regression models), which are fast and accurate (Sprinzak et al., 2003). Therefore, we used HADDOCK to perform the protein–protein docking of ACE2 with the wild spike‐RBD, E484K spike‐RBD, N501Y spike‐RBD, double mutant (E484K‐N501Y) spike‐RBD, K417N‐E484K‐N501Y spike‐RBD, and K417T‐E484K‐N501Y spike‐RBD domain to explore the structural mechanism behind the higher infectivity of the SARS‐CoV‐2 variants. HADDOCK predicted the docking score −122.6 ± 0.7 for the ACE2‐wild (spike‐RBD). HADDOCK cluster the binding conformations which revealed that 64 structures docked at the same place formed the best cluster (cluster 1) which was selected for the analysis. Interaction analysis explored through PDBsum reported that 37 residues form the interface among which 18 residues are contributed by the ACE2 while 15 residues by the spike‐RBD. The interaction analysis revealed that both the structures form 1 salt bridge, 11 hydrogen bonds, and 125 nonbonded interactions. The hydrogen bonds formed by the ACE2‐spike(RBD) wild include Glu30‐Lys417, Glu35‐Gln493, Glu38‐Tyr449, Glu38‐Gly496, Tyr41‐Thr500, Tyr41‐Thr500, Gln42‐Gln498, Asn330‐Thr500, Lys353‐Gly502, Lys353‐Gly496, and Lys353‐Gln498 (Figure 2a,c). The only salt bridge was reported between Glu30 (ACE2) and Lys417 (spike RBD). The E484K (ACE2‐spike RBD) complex possesses a higher docking score than the wild type. The HADDOCK docking score for E484K (ACE2‐spike RBD) was reported to be −128.8 ± 2.6 kcal/mol. This difference was further explored through the molecular interaction of the two protein structures. With an extra salt bridge and an additional hydrogen bond formed by the substituted residue Lys484 increased the binding ACE2‐E484K mutant complex. A total of two salt bridges, 12 hydrogen bonds, and 144 nonbonded contacts were reported (Figure 2b,d). The key residues Glu30‐Lys417 and Glu35‐Lys484 formed the salt bridges. Among the hydrogen bonds Ser19‐Ala475, Glu35‐Lys484, Glu38‐Tyr449, Glu38‐Gln498, Tyr41‐Thr500, Tyr41‐Thr500, Gln42‐Gly446, Gln42‐Tyr449, Tyr83‐Asn487, Lys353‐Gly502, Lys353‐Gly496, and Lys353‐Gln498 residues are involved. The E484K substitution as mentioned earlier is responsible for the fast spread and high infectivity of the SARS‐CoV‐2 variant is actively involved in both salt bridges and hydrogen bonding interaction and consequently enhanced the binding affinity and infectivity. Scientists have speculated that the available vaccines against COVID‐19 might not be effective against it the E484K variant and hence, therefore, E484K along with other interface residues are key hotspots for the drug discovery against the SARS‐CoV‐2 variants.
Figure 2.
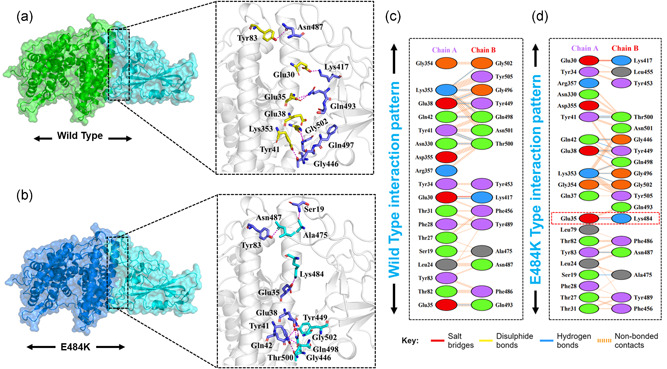
Docking representation of the Wild type and E484K mutant complexes. (a) represent the binding interface of the wild‐type complex along with its stick representation of the key hydrogen interactions. (b) shows the binding interface and stick representation of the key hydrogen bonding interactions of the E484K mutant. (c, d) represent the 2D interactions representation including hydrogen, salt bridges, and nonbonded interactions in wild type and E484K complex
Furthermore, the novel mutation N501Y reported in the UK variant is also reported to increases the spread and thus responsible for increased coronavirus cases (Leung et al., 2021). To determine how the N501Y impacts the binding of the ACE2‐spike RBD domain, herein structural insight was provided. Employing a similar approach, HADDOCK has predicted the docking score similar to wild type −122.4 ± 1.1. However, significant variation was found in the interaction pattern. With a single salt bridge formed by the Glu30‐Lys417 twelve hydrogen bonds and 139 nonbonded contacts are also detected. The substituted residue Tyr501 formed an extra hydrogen bond with the ACE2 receptor justifying that the substitution forms extra interaction and increases the binding affinity and infectivity. Among the hydrogen bonding interactions Glu30‐Lys417, Tyr34‐Ser494, Glu35‐Gln493, Glu38‐Tyr449, Tyr41‐Thr500, Tyr41‐Thr500, Gln42‐Gln498, Gln42‐Gly446, Tyr83‐Asn487, Asn330‐Thr500, Lys353‐Gly502, and Lys353‐Tyr501 residues are involved (Figure 3a,c). From this data, it is evident that the N501Y possess similar behavior to the wild type; hence it can be inferred that the already developed vaccines might work against the new N501Y variant. Since only two variants E484K and N501Y are reported alone but not simultaneously. Herein to understand the impact of double substitution on the binding of ACE2‐spike RBD, we also generated the E484K‐N501Y mutant. HADDOCK predicted the docking score −126.5 ± 3.0 for the double mutant. The E484K‐N501Y mutant complex formed 1 salt bridge, 11 hydrogen bonds, and 121 nonbonded interactions. The only salt bridge was formed between Glu30 and Lys417. The hydrogen bonding residues include Glu30‐Lys417, Glu35‐Gln493, Glu38‐Tyr449, Glu38‐Gln498, Tyr41‐Thr500, Tyr41‐Thr500, Gln42‐Gln498, Gln42‐Gly446, Gln42‐Tyr449, Lys353‐Gly502, and Lys353‐Tyr501(Figure 3b,d). The triple variants exhibiting K417N‐E484K‐N501Y (reported in South Africa) and K417T‐E484K‐N501Y (reported in Brazil) mutations were also subjected to molecular docking investigation. A docking score of −127.6 ± 1.1 kcal/mol for South African variants with triple mutations was reported. Intriguingly the electrostatic energy (−212.2 ± 39.0 kcal/mol) remained the major contributing factor in the tighter binding. With one salt bridge only, 11 hydrogen bonds and nonbonded interactions, 118 in total, were also involved in the binding. Among the key hydrogen bonding interactions Ser19‐Lys444, Glu30‐Gln493, Glu30‐Lys484, Thr31‐Ser494, Tyr34‐Phe490, Glu35‐Asn417, Gln37‐Tyr489, Glu75‐Tyr501, Glu75‐Tyr505, Tyr83‐Tyr449, and Tyr83‐Gln498 residues are involved. It can be seen that the mutated residues, such as K417N, E484K, and N501Y, are involved primarily in interactions. Along with the hydrogen bond, the only salt bridge is formed by Lys484 with Glu30. This shows that the binding of K417N‐E484K‐N501Y (reported in South Africa) is due to the substitutions which formed essential extra interactions and thus signifies the higher infectivity. However, the docking scores of South African and Brazilian variants are comparable. The docking score for the Brazilian variant carrying K417T mutation with the E484K and N501Y was reported to be −127.0 ± 1.4 kcal/mol. One salt bridge, 13 hydrogen bonds, and 133 nonbonded interactions were recorded. Among the key residues involved in hydrogen bonding includes Ser19‐Ala475, Tyr34‐Ser494, Glu35‐Gln493, Glu35‐Lys484, Glu38‐Ser494, Tyr41‐Thr500, Tyr41‐Thr500, Gln42‐Gln498, Gln42‐Tyr449, Tyr83‐Asn487, Asn330‐Thr500, Lys353‐Gly502, and Lys353‐Tyr501. The only salt bridge was formed between Glu35 and Lys483. Similarly, the electrostatic interactions substantially increased the binding.
Figure 3.
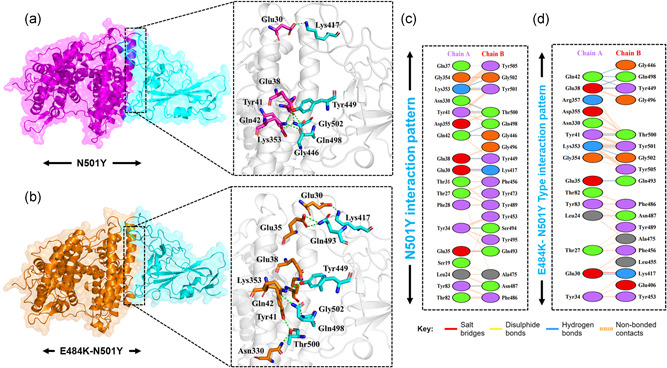
Docking representation of the N501Y and E484K‐ N501Y mutant complexes. (a) represent the binding interface of the N501Y complex along with its stick representation of the key hydrogen interactions. (b) shows the binding interface and stick representation of the key hydrogen bonding interactions of the E484K‐ N501Y mutant. (c, d) represent the 2D interactions representation including hydrogen, salt bridges and nonbonded interactions in N501Y and E484K‐ N501Y complex
Additionally, as reported that the salt‐bridges and other interactions are increased in the mutant complexes than the wild type, which is possible due to the increase in the electrostatic energy, specifically in the E484K complex. Among these, some interactions are strongly conserved between the wild type and mutant complexes. Among the key hotspots, Gln498 formed an interaction with the Lys353 of ACE2. This interaction is previously reported to be sustained during the molecular dynamics simulation (Ali & Vijayan, 2020). Tyr449, a conserved residue, form key interaction with Glu30 is an important interaction for the ACE2 recognition. A previous study also reported sustained interactions between Gln93‐Glu35 and Gln498‐Glu38 (Ali & Vijayan, 2020). This shows our consistent results of all the complexes. In addition, Lys417 forms an important interaction with the Asp30, which is also preserved here. For the wild type and the mutants, the Van der Waals energy was reported to be −59.6 ± 2.3 (wild complex), −53.6 ± 5.3 (E484K), −55.2 ±1.9 (N501Y), and −59.2 ± 7.5 for the double mutants. The electrostatic energy for the wild type reported here to be −181.4 ± 15.5, for the E484K complex it was reported to be −274.1 ± 10.0, for the N501Y it was reported to be −205.8 ± 10.3 for E484K‐N501Y complex the electrostatic energy was found to be −182.4 ± 24.3, for K417N‐E484K‐N501Y the electrostatic contribution was −212.2 ± 39.0 while for K417T‐E484K‐N501Y it was −207.7 ± 15.2 kcal/mol. This notion of more electrostatic energy is supported by previous studies state that evaluation of the binding of SARS‐CoV‐2 and SARS‐CoV RBDs with ACE2 have revealed more hydrogen bonds and electrostatic interactions in SARS‐CoV‐2 (Lan et al., 2020; Wang et al., 2020a). Conclusively, this justifies that the stronger binding of the mutant complexes is mainly due to the electrostatic contribution. These findings further suggest that the key differences in the interaction pattern are noteworthy for higher infectivity. The interaction pattern of South African and Brazilian triple variants are shown in Figure 4a,b while the 2D interactions are shown in Figure 4c,d.
Figure 4.
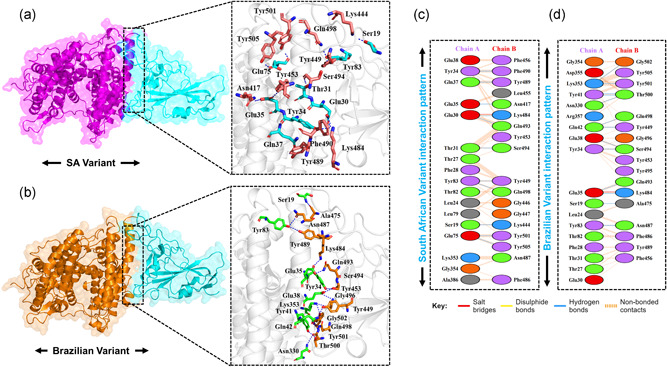
Docking representation of the South African and Brazilian mutant complexes. (a) represent the binding interface of the South African complex along with its stick representation of the key hydrogen interactions. (b) shows the binding interface and stick representation of the key hydrogen bonding interactions of the Brazilian mutant. (c, d) represent the 2D interactions representation including hydrogen, salt bridges, and nonbonded interactions in South African and Brazilian complex
Additionally, to provide a convincible insight into the binding differences, we also calculated the KD (dissociation constant) of the wild and mutant complexes. KD is used to evaluate and rank order strengths of biomolecular interactions (Landry et al., 2012). The KD kinetics is widely practiced for the antigen–antibodies binding, protein–ligand interaction, and large biological macromolecule interactions affinity prediction. The lowest KD the value, the stronger the interaction (Landry et al., 2011). This method has been previously used to determine the KD value for different macromolecules associations (Landry et al., 2008). To further provide a deep insight into the real‐time binding of these wild and mutant complexes, we used PRODIGY (PROtein binDIng enerGY prediction), an online server to compute the binding affinity KD for different biological complexes. Herein the wild type the binding affinity ΔG was predicted to be −13.2 and the KD value for the wild complex was reported to be 5.2E−10, for the E484K the ΔG was predicted to be −13.2 while the KD was 3.0E−10, for the N501Y the ΔG was predicted to be −13.1 while the KD was 6.6E−10. In case of the double mutant (E484K‐N501Y), the predicted ΔG value was −12.8 kcal/mol while the KD 9.4E−10 was reported. For the K417N‐ E484K‐N501Y variant the KD was reported to be 4.2E−10 which reflects the tighter binding of the new variant than wild type. While for the K417T‐E484K‐N501Y the KD value was reported to be 4.2E−08. Hence our analysis shows that the South African variant and Brazilian variant possess similar behavior even there are variations in the binding pattern of both variants. These findings are consistent with a significantly lower equilibrium dissociation constant (KD) obtained in in vitro binding assays of SARS‐CoV‐2 compared to SARS‐CoV (Tian et al., 2020; Walls et al., 2020). All the parameters including HADDOCK docking scores, cluster size, vdW energy, electrostatic energy, and KD of all the complexes are given in Table 1.
Table 1.
The HADDOCK predicted docking score for all the mutant complexes and additional parameters including cluster size, vdW energy, electrostatic energy, and Z‐score are given. The table also tabulate the KD (dissociation constant) for each complex predicted by PRODIGY (PROtein binDIng enerGY prediction) is also given
| Parameters | Wild | E484K | N501Y | E484K‐N501Y | K417N‐ E484K‐N501Y | K417T‐ E484K‐N501Y |
| HADDOCK score | −122.6 ± /−0.7 | −128.8 ± 2.6 | −122.4 ± /− 1.1 | −126.5 ± 3.0 | −127.6 ± 1.1 | −127.0 ± 1.4 |
| Cluster size | 64 | 61 | 71 | 92 | 86 | 64 |
| RMSD | 1.7 ± 1.0 | 0.5 ± /− 0.5 | 0.5 ± 0.4 | 1.1 ± 0.7 | 5.8 ± 0.6 | 0.8 ± 0.5 |
| Van der Waals energy | −59.6 ± 2.3 | −53.6 ± /− 5.3 | −55.2 ± 1.9 | −59.2 ± 7.5 | −58.2 ± 8.1 | −61.6 ± 2.6 |
| Electrostatic energy | −181.4 ± 15.5 | −274.1 ± /− 10.0 | −205.8 ± 10.3 | −182.4 ± 24.3 | −212.2 ± 39.0 | −207.7 ± 15.2 |
| Desolvation energy | −27.1 ± 3.4 | −20.8 ± /− 4.7 | −28.2 ± 2.0 | −33.0 ± 1.4 | −30.1 ± 3.3 | −26.0 ± 2.0 |
| Restraints violation energy | 4.7 ± 3.8 | 4.0 ± /− 1.4 | 20.8 ± 18.1 | 21.7 ± 11.0 | 31.6 ± 21.4 | 20.4 ± 13.5 |
| Buried Surface Area | 1965.3 ± 120.6 | 1899.9 ± /− 93.2 | 1819.3 ± 36.0 | 1755.9 ± 61.8 | 1894.9 ± 97.5 | 1938.6 ± 67.1 |
| Z‐score | −1.9 | −1.4 | −1.4 | −1.2 | −1.4 | −1.6 |
| Prodigy binding score | −13.2 | −13.6 | −13.1 | −12.8 | −12.8 | −13.3 |
| KD (dissociation constant) | 5.2E−10 | 3.0E−10 | 6.6E−10 | 9.4E−10 | 4.2E−10 | 4.2E−08 |
Next, we further determined the effect of the fixed amino‐acid substitutions reported within the receptor‐binding domain (RBD) of spike glycoprotein of SARS‐CoV‐2 by predicting the thermodynamic state function (root mean square deviation [RMSD]). RMSD is primarily practiced for quantifying the variance between the backbone of a protein from its initial structural conformation to its final position. The dynamics stability of a biological molecule relative to its conformation can be estimated by the deviations experienced during the simulation time. The stability of a protein is associated with the deviation it faces during the simulation. Smaller deviation means the structure is more stable and may not meet the convergence. Herein, to calculate the stability (RMSD) from the Cα backbone was estimated for the 100 ns trajectory of each protein–protein complex. Figure 5 shows that the wild‐type system equilibrated at 5 ns and reached stability at 2.0 Å. The deviation was conserved, and any ample convergence was not observed during the simulation time except the RMSD deviated between 50 and 60 ns. At this point the RMSD increased sharply up to 4.0 Å; however, the RMSD value decreased then to 2.0 Å. Afterward, the RMSD remained uninformed and maintained at a level of 2.0 Å for the wild‐type complex. The E484K (ACE2‐spike RBD) complex initially converged largely between 0 and 18 ns reaching at 5.0 Å. The RMSD remained higher; however, then the RMSD decreased to 2.0 Å followed the same pattern over the course of the simulation. After 18 ns the RMSD remained uniform maintaining 2.2 Å until the end. This shows a more rigid binding of the ACE2 and E484K‐spike RBD. Though the average RMSD remained higher, not significantly, still this mutant did not face any convergence after reaching the equilibrium point at 18 ns. This shows that the fixed amino acid substitution (E484K) has helped the viral protein to evolve stably and thus binds to the host receptor with stronger affinity. Next, we calculated the stability index for the N501Y (ACE2‐spike RBD) complex to reveal the dynamic behavior. It can be seen from Figure 4 that N501Y gained stability at 2.0 Å. However, after reaching 20 ns the RMSD significantly diverged from its initial position and increased up to 4.0 Å. This trend remained uniform until 40 ns. Later the RMSD reached the equilibration and maintained 2.5 Å until the end. At some intervals, the N501Y (ACE2‐spike RBD) possesses similar behavior as the wild type over the course of the simulation. Finally, we calculated the stability of the E484K‐N501Y (ACE2‐spike RBD) complex, which shows that the system RMSD increased continuously during the 100 ns simulation. The RMSD did not converge substantially but increased gradually. It can be seen that during the 0–8 ns, the RMSD was reported to be 2.0 Å; however, the RMSD remained higher between 9 and 38 ns. The RMSD between 9 and 38 ns was observed to be 2.2Å. Afterward, the system converged more, and the RMSD continues to increase until 100 ns. For the rest of the simulation 39–100 ns the average RMSD was observed to be 3.0 Å. As previously reported that the oscillating RMSD of these complexes during the course of simulation is associated with the opening or closing motion of the claw‐like structure in the ACE2 (Yan et al., 2020). A higher RMSD at different intervals during simulation is probably associated with the binding and unbinding of some destabilized interaction in the ACE2‐spike interface. Structural‐dynamics features of the K417N‐E484K‐N501Y variant were further characterized. The RMSD results show that the K417N‐E484K‐N501Y system initially converged until 30 ns but afterward the complex attained stability and did not face substantial perturbation. The average RMSD remained 2.5 Å until 100 ns with RMSD convergence at different intervals. In the case of the K417T‐E484K‐N501Y variant, a more radical behavior was observed during the simulation. Substantial convergence at different intervals was experienced. Until 40 ns the system remained uniform however significant perturbation between 41 and 60 ns was reported. Further, the RMSD value also increased over the simulation time. The average RMSD for the Brazilian variant was reported to be 3.0 Å. Longer simulation is needed for a deep understanding of the dynamic behavior of the Brazilian variant. This shows that the naturally reported K417N, K417T, E484K, and N501Y mutations have stably evolved than the hypothetical E484K‐N501Y mutations. Since these mutations, K417N, K417T, E484K, and N501Y modulates the binding pattern of the spike protein and supports their adaptive significance. Consequently, the unusual virulence of this virus is potentially the consequence of Darwinian selection‐driven epistasis in protein evolution (Hussain et al., 2020).
Figure 5.
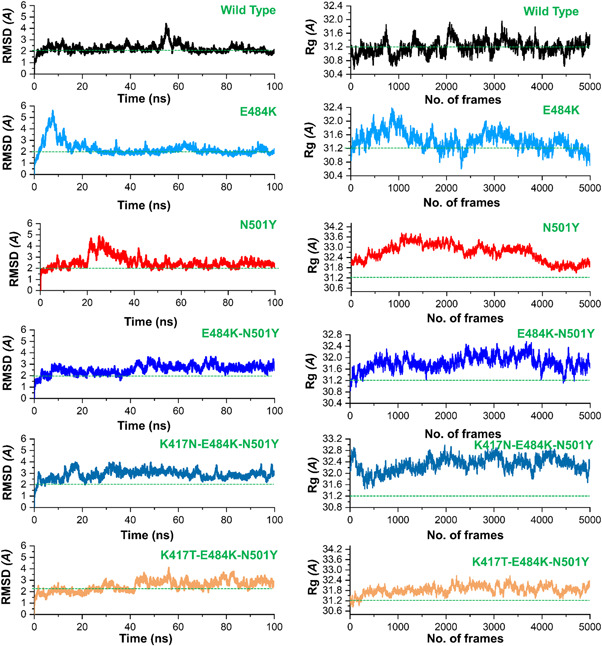
The figure represents the RMSDs and Rg(s) of all the complexes. The RMSD and Rg of the wild type is shown in black color while the other mutants are given in different colors. RMSDs, root mean square deviations
Next, we calculated the radius of gyration (Rg), which is an indicator of protein structure compactness during the simulation. As given in Figure 5, all the systems possess the same pattern of Rg. All the systems faced increased and decreased in the Rg value during the simulation. The average Rg value for the wild type was observed to be (31.2 Å), for E484K (31.6 Å), for N501Y (31.8 Å) while for the E484K‐N501Y the average Rg value was observed to be 31.6 Å. In the case of the K417N‐E484K‐N501Y mutant, the Rg value remained higher comparatively. The average Rg value was observed to be 32.0 Å. Similarly, the Rg value for K417T‐E484K‐N501Y increased during the simulation. The average Rg value for the K417T‐E484K‐N501Y variant was observed to the same as K417N‐E484K‐N501Y which shows a similar behavior of the two variants. The average Rg value was observed to be 32.0 Å which is similar to that of the K417N‐E484K‐N501Y variant. Increased or decreased in the Rg value at different intervals in all the systems is due to the binding and unbinding of one or another end of the spike RBD domain.
Moreover, to provide noteworthy insights into the dynamics–function relationship due to the evolutionary divergence of protein motions, RMSFs of backbone C‐alpha were calculated and compared. In different biological processes such as biological manufacturing, binding, and unbinding of biological molecules, catalysis, and molecular recognition residual flexibility, or rigidity play an essential role. The higher root mean square fluctuation (RMSF) value indicates a more flexible region, whereas the low RMSF value shows minimal movements about its average position during the simulation. In the case of South African and Brazilian variants, a higher fluctuation between 125 and 200 residues was observed. The wild type exhibited minimal fluctuation as compared to the reported mutants. Figure 6a shows that the region between 40 and 50 in N501Y displayed considerably higher fluctuation than the others. Intriguingly, the four mutant complexes but not the wild type showed a higher fluctuation between 100 and 200. Fluctuation in this region is considerably higher than the others, which is due to the distribution of three important loops vital for the binding with ACE2 thus implicating the functional relevance of the mutant complexes for better binding and essential conformational optimization during Darwinian evolution and amino acids fixation. In addition, the higher fluctuation was observed between 300 and 400 (ACE2) essential for the binding. Overall these results show that the spike protein has passed through critical structural evolution and considerable adjustment for better binding and ultimately leads to the increased infectivity.
Figure 6.
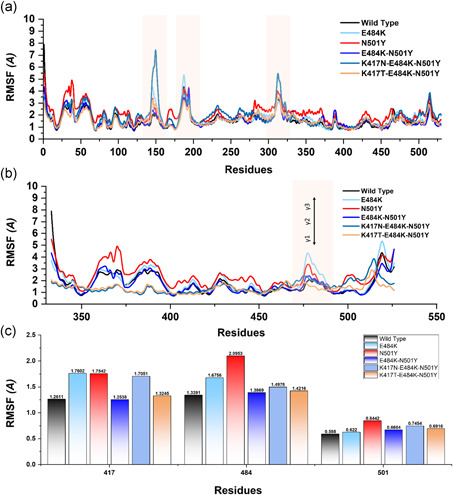
This figure represents the residual flexibility index of the wild‐type and mutant complexes. (a) shows the RMSFs of the complexes while the fluctuated regions are highlighted in light orange color. (b) represent the residual flexibility index of the wild and mutant spike RBD only. The three important loops required for interaction with ACE2 are represented with γ1, γ2, and γ3. These regions γ1 (474–485), γ2 (488–490), and γ3 (494–505) are crucial for binding. (c) show the individual residue flexibility, that is, K417N, K417T, E48K, and N501Y. RMSFs, root mean square fluctuations
Subsequently, we also calculated the RMSF for the Spike RBD domain of the wild and mutants apo states (Figure 6b). Three loops in the spike RBD domain γ1 (474–485), γ2 (488–490), and γ3 (494–505) are crucial for binding with ACE2 possesses higher fluctuation in the mutant systems but not in the wild type. The residue Lys417 is an essential residue for the binding with ACE2, which showed limited fluctuation. These findings are similar to the previous study reported ACE2‐RBD dynamics. Our findings also showed that region 469–505 possess higher fluctuation. In E484K, the region where the mutated residue (E484K) reside possesses higher fluctuation. In comparison, in the case of N501Y, the regions 500–505 have higher fluctuation but not others (Figure 6C). Moreover, no noticeable difference was observed in other regions.
To further connect protein conformational changes with the binding of the receptor‐binding domain (RBD), we used the MM‐GBSA approach to compute the binding energy of spike RBD to ACE2 (Figure 7). Binding comparison discovered that there is a noticeable divergence in spike RBD binding between wild type, E484K, N501Y, E484K‐N501Y, K417N‐E484K‐N501Y, and K417T‐E484K‐N501Y complexes. Intriguingly, the total binding energy of the wild type (−73.11 kcal/mol) and N501Y (−73.00 kcal/mol) was comparable. The binding of spike RBD with the E484K to the ACE2 was more efficient than any other. The total binding energy of the ACE2‐spike RBD (E484K) complex was reported to be the highest (−79.19 kcal/mol). Furthermore, the total binding energy for the E484K‐N501Y (ACE2‐spike RBD) was observed to be −77.53 kcal/mol. Interestingly the electrostatic energy significantly increased in the ACE2‐spike RBD (E484K) and E484K‐N501Y (ACE2‐spike RBD) complexes. To characterize the binding of the South African variant (K417N‐ E484K‐N501Y) the total binding energy remained higher than any other variant. The predicted binding energy for the South African variant with triple mutants was reported to be −91.54 kcal/mol. For the Brazilian variant (K417T‐ E484K‐N501Y) the total binding energy was reported to be −90.99 kcal/mol. The vdW and electrostatic energies for the wild type and N501Y are comparable thus justify this notion that the already developed vaccine may work against the UK mutant but not against the variants that emerged in South Africa and Brazil. The closet similarity in the binding of South African and Brazilian variants determines that it may enforce similar strategies to induce infection. We speculate the higher affinity of spike RBD South African and Brazilian variants for ACE2 is due to the structural evolution with an adaptive extraordinary binding affinity that enables the South African and Brazilian variants variant to be more contagious than the others and the already developed vaccine may not work against it. This shows that protein conformational epistasis evolution may play a significant role in the binding of ACE2 to the spike RBD in the wild type and mutants. In addition, the mutants with the hypothetical mutant (E484K‐N501Y) were observed to be radical, thus implying the biological significance of the designed mutant, which may result in relatively higher infectivity than the wild type. The electrostatic energy significantly improved than the wild type.
Figure 7.
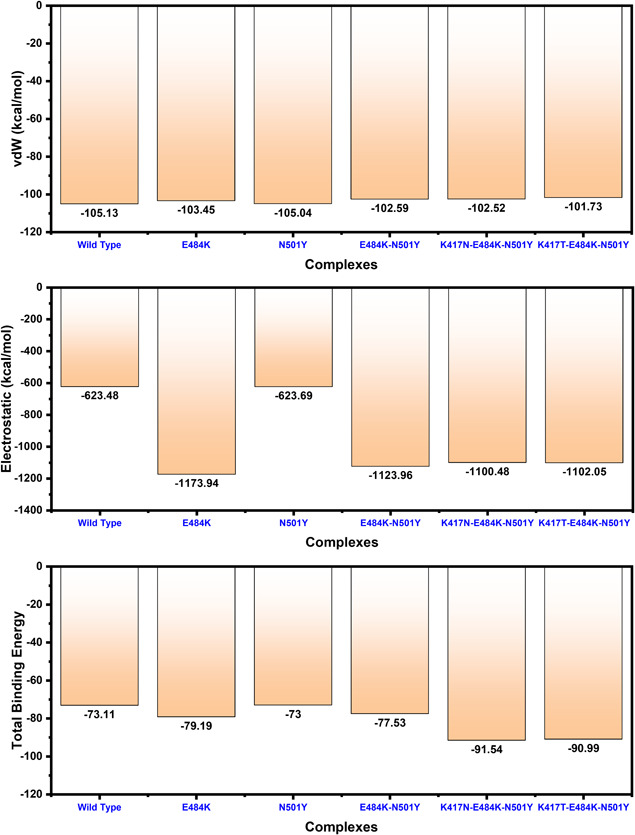
Free energy calculation results obtained from MD simulation trajectory of the wild type and mutant complexes. The top bar graph shows the vdW contribution by each complex, the 2nd bar graph shows the electrostatic energy while the bottom bar graph shows the total binding energy. All the energies given here are calculated in kcal/mol
In conclusion, this study precisely explored the mechanism of the interaction of the spike RBD with the host ACE2 and revealed the differences in the binding of the reference and new variants. The systematic investigation revealed that the South African and Brazilian variants are more lethal than the others due to interprotein contacts specifically the electrostatic while the N501Y is comparable with the wild type. We hypothesized that the residue at 501Y is continuously subjected to positive selection pressure. We further demonstrated the dynamic behavior is also changed with the protein evolution. Conclusively, this study provides a strong basis for structure and rationale‐based drug designing against the new variant by exploring the noticeable differences.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENTS
Dong‐Qing Wei is supported by grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Science Foundation of China (Grant No. 32070662, 61832019, 32030063), the Science and Technology Commission of Shanghai Municipality (Grant No.: 19430750600), as well as SJTU JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2021ZD02). The computations were partially performed at the Pengcheng Lab. and the Center for High‐Performance Computing, Shanghai Jiao Tong University.
Khan, A. , Zia, T. , Suleman, M. , Khan, T. , Ali, S. S. , Abbasi, A. A. , Mohammad, A. , & Wei, D. (2021). Higher infectivity of the SARS‐CoV‐2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J Cell Physiol, 236, 7045–7057. 10.1002/jcp.30367
REFERENCES
- Abraham, S. , Kienzle, T. E. , Lapps, W. , & Brian, D. A. (1990). Deduced sequence of the bovine coronavirus spike protein and identification of the internal proteolytic cleavage site. Virology, 176(1), 296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, A. , Khan, A. , Kaushik, A. C. , Wang, Y. , Ali, S. S. , Junaid, M. , Saleem, S. , Cho, W. C. , Mao, X. , & Wei, D.‐Q. (2019). Immunoinformatic and systems biology approaches to predict and validate peptide vaccines against Epstein–Barr virus (EBV). Scientific Reports, 9(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, A. , & Vijayan, R. (2020). Dynamics of the ACE2–SARS‐CoV‐2/SARS‐CoV spike protein interface reveal unique mechanisms. Scientific Reports, 10(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, N. G. , Barnard, R. C. , Jarvis, C. I. , Kucharski, A. J. , Munday, J. , Pearson, C. A. , Russell, T. W. , Tully, D. C. , Abbott, S. , & Gimma, A. (2020). Estimated transmissibility and severity of novel SARS‐CoV‐2 Variant of Concern 202012/01 in England. medRxiv. [Google Scholar]
- Dominguez, C. , Boelens, R. , & Bonvin, A. M. (2003). HADDOCK: A protein−protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society, 125(7), 1731–1737. [DOI] [PubMed] [Google Scholar]
- Fehr, A. R. , & Perlman, S. (2015). Coronaviruses: An overview of their replication and pathogenesis. Coronaviruses: Springer, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard, T. D. , Huang, C. C. , & Ferrin, T. E. (2005). Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure, 13(3), 473–482. [DOI] [PubMed] [Google Scholar]
- Harrington, D. , Kele, B. , Pereira, S. , Couto‐Parada, X. , Riddell, A. , Forbes, S. , Dobbie, H. , & Cutino‐Moguel, T. (2021). Confirmed Reinfection with SARS‐CoV‐2 Variant VOC‐202012/01. Clinical Infectious Diseases, 72, ciab014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , & Pöhlmann, S. (2020). A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Molecular Cell, 78, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, T. , Wang, J. , Li, Y. , & Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling, 51(1), 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Wang, Y. , Li, X. , Ren, L. , Zhao, J. , Hu, Y. , Zhang, L. , Fan, G. , Xu, J. , & Gu, X. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The lancet, 395(10223), 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain, I. , Pervaiz, N. , Khan, A. , Saleem, S. , Shireen, H. , Wei, D.‐Q. , Labrie, V. , Bao, Y. , & Abbasi, A. A. (2020). Evolutionary and structural analysis of SARS‐CoV‐2 specific evasion of host immunity. Genes & Immunity, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Ali, S. S. , Khan, M. T. , Saleem, S. , Ali, A. , Suleman, M. , Babar, Z. , Shafiq, A. , Khan, M. , & Wei, D.‐Q. (2020a). Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS‐CoV‐2 main protease (3CLpro). Journal of Biomolecular Structure and Dynamics, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Junaid, M. , Kaushik, A. C. , Ali, A. , Ali, S. S. , Mehmood, A. , & Wei, D.‐Q. (2018). Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS One, 13(5), e0196484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Junaid, M. , Li, C.‐D. , Saleem, S. , Humayun, F. , Shamas, S. , Ali, S. S. , Babar, Z. , & Wei, D.‐Q. (2020b). Dynamics insights into the gain of flexibility by Helix‐12 in ESR1 as a mechanism of resistance to drugs in breast cancer cell lines. Frontiers in Molecular Biosciences, 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Khan, M. , Saleem, S. , Babar, Z. , Ali, A. , Khan, A. A. , Sardar, Z. , Hamayun, F. , Ali, S. S. , & Wei, D.‐Q. (2020c). Phylogenetic analysis and structural perspectives of RNA‐dependent RNA‐polymerase inhibition from SARs‐CoV‐2 with natural products. Interdisciplinary Sciences: Computational. Life Sciences, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Khan, M. T. , Saleem, S. , Junaid, M. , Ali, A. , Ali, S. S. , Khan, M. , & Wei, D.‐Q. (2020d). Structural Insights into the mechanism of RNA recognition by the N‐terminal RNA‐binding domain of the SARS‐CoV‐2 nucleocapsid phosphoprotein. Computational and Structural Biotechnology Journal, 18, 2174–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. T. , Khan, A. , Rehman, A. U. , Wang, Y. , Akhtar, K. , Malik, S. I. , & Wei, D.‐Q. (2019). Structural and free energy landscape of novel mutations in ribosomal protein S1 (rpsA) associated with pyrazinamide resistance. Scientific Reports, 9(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby, T. (2021). New variant of SARS‐CoV‐2 in UK causes surge of COVID‐19. The Lancet. Respiratory Medicine, 9(2), e20–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama, T. , Platt, D. , & Parida, L. (2020). Variant analysis of SARS‐CoV‐2 genomes. Bulletin of the World Health Organization, 98(7), 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kräutler, V. , Van Gunsteren, W. F. , & Hünenberger, P. H. (2001). A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry, 22(5), 501–508. [Google Scholar]
- Lan, J. , Ge, J. , Yu, J. , Shan, S. , Zhou, H. , Fan, S. , Zhang, Q. , Shi, X. , Wang, Q. , & Zhang, L. (2020). Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature, 581(7807), 215–220. [DOI] [PubMed] [Google Scholar]
- Landry, J. , Sun, Y. , Guo, X. , & Zhu, X. (2008). Protein reactions with surface‐bound molecular targets detected by oblique‐incidence reflectivity difference microscopes. Applied Optics, 47(18), 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry, J. P. , Fei, Y. , & Zhu, X. (2011). High throughput, label‐free screening small molecule compound libraries for protein‐ligands using combination of small molecule microarrays and a special ellipsometry‐based optical scanner. International Drug Discovery, 8. [PMC free article] [PubMed] [Google Scholar]
- Landry, J. P. , Fei, Y. , & Zhu, X. (2012). Simultaneous measurement of 10,000 protein‐ligand affinity constants using microarray‐based kinetic constant assays. ASSAY and Drug Development Technologies, 10(3), 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, K. , Shum, M. H. , Leung, G. M. , Lam, T. T. , & Wu, J. T. (2021). Early transmissibility assessment of the N501Y mutant strains of SARS‐CoV‐2 in the United Kingdom. October to November 2020. Eurosurveillance, 26(1), 2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology, 3, 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Li, W. , Farzan, M. , & Harrison, S. C. (2005). Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science, 309(5742), 1864–1868. [DOI] [PubMed] [Google Scholar]
- Magrane, M. (2011). UniProt Knowledgebase: A hub of integrated protein data. Database, 2011, 2011–bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza, J. C. (2010). Steepest descent. Wiley Interdisciplinary Reviews: Computational Statistics, 2(6), 719–722. [Google Scholar]
- Price, D. J. , Brooks III, & C. L. (2004). A modified TIP3P water potential for simulation with Ewald summation. The. Journal of Chemical Physics, 121(20), 10096–10103. [DOI] [PubMed] [Google Scholar]
- Ray, S. (2014). The Cell: A molecular approach. The Yale Journal of Biology and Medicine, 87(4), 603. [Google Scholar]
- Roe, D. R. , Cheatham III, & T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. [DOI] [PubMed] [Google Scholar]
- Rose, P. W. , Beran, B. , Bi, C. , Bluhm, W. F. , Dimitropoulos, D. , Goodsell, D. S. , Prlić, A. , Quesada, M. , Quinn, G. B. , & Westbrook, J. D. (2010). The RCSB protein data bank: Redesigned website and web services. Nucleic Acids Research, 39(suppl_1), D392–D401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan, H. A. , & Byrareddy, S. N. (2020). The epidemiology and pathogenesis of coronavirus disease (COVID‐19) outbreak. Journal of Autoimmunity, 109, 102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon‐Ferrer, R. , Case, D. A. , & Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3(2), 198–210. [Google Scholar]
- Salomon‐Ferrer, R. , Götz, A. W. , Poole, D. , Le Grand, S. , & Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. Journal of Chemical Theory and Computation, 9(9), 3878–3888. [DOI] [PubMed] [Google Scholar]
- Smith, G. R. , & Sternberg, M. J. (2002). Prediction of protein–protein interactions by docking methods. Current Opinion in Structural Biology, 12(1), 28–35. [DOI] [PubMed] [Google Scholar]
- Sprinzak, E. , Sattath, S. , & Margalit, H. (2003). How reliable are experimental protein–protein interaction data? Journal of Molecular Biology, 327(5), 919–923. [DOI] [PubMed] [Google Scholar]
- Tian, X. , Li, C. , Huang, A. , Xia, S. , Lu, S. , Shi, Z. , Lu, L. , Jiang, S. , Yang, Z. , & Wu, Y. (2020). Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus‐specific human monoclonal antibody. Emerging Microbes & Infections, 9(1), 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y.‐J. , Tortorici, M. A. , Wall, A. , McGuire, A. T. , & Veesler, D. 2020. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. [DOI] [PMC free article] [PubMed]
- Wang, Q. , Zhang, Y. , Wu, L. , Niu, S. , Song, C. , Zhang, Z. , Lu, G. , Qiao, C. , Hu, Y. , & Yuen, K.‐Y. 2020a. Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell. [DOI] [PMC free article] [PubMed]
- Wang, W. , Tang, J. , & Wei, F. (2020b). Updated understanding of the outbreak of 2019 novel coronavirus (2019‐nCoV) in Wuhan, China. Journal of Medical Virology, 92(4), 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watowich, S. J. , Meyer, E. S. , Hagstrom, R. , & Josephs, R. (1988). A stable, rapidly converging conjugate gradient method for energy minimization. Journal of Computational Chemistry, 9(6), 650–661. [Google Scholar]
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y.‐M. , Wang, W. , Hu, Y. , Song, Z.‐G. , Tao, Z.‐W. , Tian, J.‐H. , & Pei, Y.‐Y. 2020. Complete genome characterisation of a novel coronavirus associated with severe human respiratory disease in Wuhan, China. bioRxiv.
- Xue, L. C. , Rodrigues, J. P. , Kastritis, P. L. , Bonvin, A. M. , & Vangone, A. (2016). PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics, 32(23), 3676–3678. [DOI] [PubMed] [Google Scholar]
- Xydakis, M. S. , Dehgani‐Mobaraki, P. , Holbrook, E. H. , Geisthoff, U. W. , Bauer, C. , Hautefort, C. , Herman, P. , Manley, G. T. , Lyon, D. M. , & Hopkins, C. 2020. Smell and taste dysfunction in patients with COVID‐19. The Lancet Infectious Diseases. [DOI] [PMC free article] [PubMed]
- Yan, R. , Zhang, Y. , Li, Y. , Xia, L. , Guo, Y. , & Zhou, Q. (2020). Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science, 367(6485), 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]