

Abstract
Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) is the causative agent of coronavirus disease 2019 (COVID‐19) infection, which has emerged as a global pandemic causing serious concerns. Lack of specific and effective therapeutics for the treatment of COVID‐19 is a major concern and the development of vaccines is another important aspect in managing the infection effectively. The first step in the SARS‐CoV‐2 pathogenesis is the viral entry and it is mediated by its densely glycosylated spike protein (S‐protein). Similar to the SARS‐CoV, SARS‐CoV‐2 also engages angiotensin‐converting enzyme 2 (ACE2) as the host cell entry receptor. In addition to ACE2, several recent studies have implicated the crucial role of cell surface heparan sulfate (HS) as a necessary assisting cofactor for ACE2‐mediated SARS‐CoV‐2 entry. Furthermore, SARS‐CoV‐2 was also identified to use both endosomal cysteine proteases cathepsin B and L (CatB/L) and the transmembrane serine protease 2 (TMPRSS2) for the pivotal role of S‐protein priming mediating viral entry. As the entry of SARS‐CoV‐2 into host cells is mandatory for viral infection, it becomes an extremely attractive therapeutic intervention point. In this regard, this review will focus on the therapeutic targeting of the crucial steps of SARS‐CoV‐2 viral entry like S‐protein/ACE2 interaction and S‐protein priming by host cell proteases. In addition, this review will also give insights to the readers on several therapeutic opportunities, pharmacological targeting of the viral‐entry facilitators like S‐Protein, ACE2, cell surface HS, TMPRSS2, and CatB/L and evidence for those drugs currently ongoing clinical studies.
Keywords: ACE2, CatB/L, clinical trials, heparan sulfate, SARS‐CoV‐2, S‐protein, TMPRSS2
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) is the recent viral pathogen found to be responsible for the emergence of global pandemic coronavirus disease 2019 (COVID‐19). Originated from Wuhan, China, SARS‐CoV‐2 infection has drastically spread to almost all the countries and territories in the world. Started with just 6 fatalities and 282 confirmed cases, SARS‐CoV‐2 has paved its path to 2,703,780 fatalities and 122,536,880 confirmed cases as of March 21, 2021.1, 2 Over the past two decades, seven coronaviruses (CoVs) have emerged and caused respiratory diseases in humans. Most of these CoVs, including SARS‐CoV‐2, are found to cause lung injuries and even multiorgan failure in patients. Interestingly, the SARS‐CoV‐2 was found to be a novel virus of the beta‐coronavirus (β‐CoV) genus, which shares 80% genome identity with severe acute respiratory syndrome coronavirus (SARS‐CoV) and 96.2% genome identity with bat coronavirus BatCoV RaTG13.3, 4, 5, 6, 7
Human coronaviruses (HCoVs) like HcoV‐OC43, HCoV‐229E, HCoV‐NL63, and HCoV‐HKU1 are some previously identified CoVs that are known for its circulation among the population. Most of these HCoVs have only caused seasonal and mild respiratory tract infections that are commonly associated with symptoms of “common cold.” Contradictive to these HCoVs, severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle‐East respiratory coronavirus (MERS‐CoV), and SARS‐CoV‐2, which have emerged in the global population over the past 20 years, are notably highly pathogenic and caused serious concern in the human population. Through their rigorous infections in human bronchial epithelial cells, upper respiratory tract, and pneumocytes, these three high pathogenic CoVs (SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2) can lead to life‐threatening lung injuries and respiratory pathologies. 8 As of date, specific and effective therapeutics are not available for the treatment of COVID‐19 and the current management is solely dependent on social distancing, patient isolation, travel restrictions, and supportive medical care. 9
SARS‐CoV‐2 makes use of its densely glycosylated spike protein (S‐protein) to gain entry into the host cells. Similar to the SARS‐CoV, SARS‐CoV‐2 also engages angiotensin‐converting enzyme 2 (ACE2) as the host cell entry receptor. SARS‐CoV‐2 initiates its infection by binding to the ACE2 receptor, which is found abundant in the epithelial cells of the oral and nasal mucosa. The viral infection further precipitates a decreased sense of taste and smell as it moves down to the lungs (the primary site of infection). The eventual process of infection includes endocytosis, exploitation of host cell machinery for genome replication, transcription, assembly, and viral egress. Followed by the viral egress, SARS‐CoV‐2 initiates the infection of neighboring cells and subsequent infiltration of organ systems that ultimately results in a multiple‐organ failure. 10
Several recent studies on cell surface heparan sulfate (HS) have implicated its role as a crucial viral‐recruiting co‐factor that promotes the ACE2‐dependant viral entry of SARS‐CoV‐2. Furthermore, SARS‐CoV‐2 was also identified to use both endosomal cysteine proteases cathepsin B and L (CatB/L) and the transmembrane serine protease 2 (TMPRSS2) for the pivotal role of S‐protein priming mediating viral entry.6, 11, 12, 13 This review will focus on the therapeutic targeting of the crucial steps of SARS‐CoV‐2 viral entry like S‐protein/ACE2 interaction and S‐protein priming by host cell proteases. In addition, this review will also give insights to the readers on several therapeutic opportunities and pharmacological targeting of the viral‐entry facilitators like S‐Protein, ACE2, cell surface HS, TMPRSS2, and CatB/L.
2. SARS‐COV‐2 VIRAL ENTRY
Structurally, CoVs comprise the largest known RNA genome of 26–32 kilo‐bases length. CoVs are enveloped, positive sensed, non‐segmented, single‐stranded RNA viruses, which encode for four vital structural proteins like nucleocapsid protein (N‐protein), membrane protein (M‐protein), an envelope protein (E‐protein), and spike protein (S‐protein). 14 Viral entry into the host cell is an essential factor for cross‐species transmission, particularly for the β‐CoVs. Recently, the release of the SARS‐CoV‐2 sequence has predicted the presence of a cleavage site for the cellular proteases. As hypothesized, the S‐protein of SARS‐CoV‐2 contains a polybasic cleavage motif present at the S1/S2 cleavage site, which is absent in other variants of CoVs. This motif is responsible for the cleavage of the full‐length S‐protein into S1 and S2 subunits, mediated by the host proteases furin. 15 Thus, CoVs have an encoded S‐protein with two subunits, namely S1‐subunit (responsible for binding to host cell receptor) and S2‐subunit (responsible for the fusion of viral and cellular membranes).16, 17 The receptor‐binding domain (RBD) of the S1 subunit directly interacts and initiates direct‐binding with the peptidase domain (PD) of the ACE2 receptor. Further, S1‐subunit with RBD also stabilizes the prefusion state of S2‐subunit, which contains the fusion machinery and other basic components necessary for membrane fusion.14, 17, 18, 19, 20 The cleaved S‐proteins are incorporated into the viral capsid during the assembly, enabling efficient entry into the cell. Proteolytic cleavage of the S‐protein by furin or other cellular proteases, including TMPRSS2 can help to progress the infection as the cleavage provides two distinct functions for the subunits. It also plays an important role in the selection of host species and infection. Hence, the presence of a furin cleavage site confirms the transmission of the virus from the bat to humans.15, 21
Further, it has been reported that the cleavage of spike protein at the S1/S2 site is crucial for the entry of the virus into the human lung cells. In addition, it was also found that the spike protein of SARS‐CoV‐2 contains the optimal cleavage site and follows a specific activity for different hosts, thus being an important virulent factor. The plentiful source of furin proteases in the respiratory tract, the S‐protein may get cleaved upon exit from the epithelial cells, resulting in the efficient infection of the neighboring cells. 22 For the complete activation of S‐protein, SARS‐CoV‐2 displays a dibasic cleavage site recognized not only by furin but also by other proteases, making them more pathogenic unlike other strains of CoVs. Further, acquisition of the furin cleavage site by SARS‐CoV‐2 is considered as the functional gain, which enables the transfer of virus from bats to humans and epidemic spread. As a consequence, the cleavage motif at the S1/S2 site has gained significant interest in analyzing the structural loops and cleavage residues for treatment purposes.15, 22, 23
Recently, Zhou et al., with the help of HeLa cells, confirmed that SARS‐CoV‐2 uses the same ACE2 receptor‐like SARS‐CoV. Furthermore, the same group also confirmed that conventional CoV entry receptors like aminopeptidase‐N (APN) dipeptidyl peptidase‐4 (DPP4) are not being engaged by SARS‐CoV‐2. Subsequent to receptor binding, CoV must gain access to the host cell cytosol for further pathogenesis. This could be attained by any of the two distinct viral‐entry pathways, which include the CatL‐mediated‐endosomal pathway (where the S‐protein activation is mediated by the pH‐dependent endosomal protease CatL) and TMPRSS2‐mediated pathway (where the S‐protein activation is mediated by TMPRSS2 for subsequent entry via host plasma membrane). Several evidence have been reported on the roles of these protease activators, indicating that both TMPRSS2 and endosomal cysteine protease CatB/L are crucial for SARS‐CoV‐2 entry.4, 11, 24, 25, 26 Even though the high expression of both TMPRSS2 and cathepsins have been confirmed in the lung tissues, 27 the commonly used cell lines for performing viral assays may exhibit varying expression levels of both TMPRSS2 and cathepsins, which can potentially result in a dramatic impact on the viral‐entry mechanism of SARS‐CoV‐2. 28 Since animal models of SARS‐CoV‐2 are still under optimization, the controversy on the expression levels of TMPRSS2 and cathepsins in cell lines and their ability to accurately mimic aspects of the human infection still warrants investigation. Altogether, strategic selection of cell lines for the purpose of antiviral testing plays a crucial aspect in the excluding selection and screening of drugs, which could be efficiently tackled by SARS‐CoV‐2 by its redundant viral‐entry pathways. 28 A detailed pictorial representation of the SARS‐CoV‐2 life cycle is given in Figure 1.
Figure 1.

Life cycle of SARS‐CoV‐2. ACE2, angiotensin‐converting enzyme 2; CatL, cathepsin L; E‐protein, envelope protein; HSPG, heparan sulfate proteoglycans; M‐protein, membrane glycoprotein; N‐protein, nucleocapsid protein; ORF1a, open‐reading frame 1a; ORF1ab, open‐reading frame 1ab; RER, rough endoplasmic reticulum; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; S‐protein, spike protein; TMPRSS2, transmembrane serine protease 2
3. TARGETING THE VIRAL‐ENTRY FACILITATORS
3.1. S‐protein
S‐protein is a crucial structural protein of CoV, which is assembled into a unique corolla structure and exists as a trimer on the surface of the virus. As mentioned earlier, S‐protein plays a pivotal role in interactive binding to host cell receptors for facilitating the viral invasion and also acts as a determinant factor for host tropism. Since the structural integrity and cleavage activation of S‐protein are key factors for both virulence and viral invasion, therapeutic strategies targeting S‐protein can result in the development of effective antivirals and vaccines.29, 30 Among the two subunits of S‐protein (S1 and S2), S1‐subunit has diverged in sequence whereas S2‐subunit acts as the most conserved region of the protein. C‐terminal domain (CTD) and N‐terminal domain (NTD) are the two subdomains of S‐protein where both the subdomains can function as RBD. 31 In this approach of targeting S‐protein, a pivotal target for neutralizing antibodies is the RBD. Apart from the high homology of the S‐protein of the SARS‐CoV‐2 to that of SARS‐CoV, remarkable alterations (>85%) have been identified in the RBD antibody epitopes of SARS‐CoV‐2 when compared to SARS‐CoV. This variation in the RBD antibody epitopes of SARS‐CoV‐2 necessitates the development of new monoclonal antibodies against SARS‐CoV‐2.32, 33
3.1.1. Pharmacological treatment
In the line of antibodies against S‐protein, recombinant human angiotensin‐converting enzyme 2 (rhACE2), also referred to as APN01, may block the S‐protein interaction with host cell receptor ACE2 and thereby could potentially block the viral entry of SARS‐CoV‐2. Recent studies conducted on cellular and embryonic stem cell‐derived organoids, rhACE2 reported to show blocking effects against SARS‐CoV‐2 and was found to potentially inhibit the SARS‐CoV‐2 replication by a factor of 1000–5000 times.34, 35 Studies have also reported that the renin–angiotensin system (RAS) was found to be negatively regulated by ACE2, and further, angiotensin II (Ang II) receptor (AT2) and ACE reduces the sepsis/acid aspiration induced lung damage in the mouse. CoV also downregulates the resveratrol (RES; experimentally deactivated RAS), which can subsequently lead to an elevated level of ACE2.36, 37 Administration of rhACE2 can potentially reduce the serum levels of AT2 by circumventing the contact between the substrate and the related enzyme (angiotensin‐converting enzyme [ACE]). This activity of rhACE2 could possibly preserve and prevent the pulmonary vascular integrity and acute respiratory distress syndrome (ARDS), respectively, by preventing the activation of host cell receptor ACE2.34, 38
Several other specific neutralizing antibodies for SARS‐CoV (m396, S109.8, and CR3022) and SARS‐CoV‐2 (311 monoclonal antibodies (mab)−32D4 and 311mab‐31B5) were also reported for targeting S‐protein. Further, recombinant ACE2‐Ig fusion protein has been reported to show neutralizing activity on both pseudotyped SARS‐CoV and SARS‐CoV‐2 with an inhibitory concentration (IC50) of 0.8 and 0.1 μg/ml, respectively. Also, recent studies on 47D11, a cross‐neutralizing human antibody, showed binding efficacy toward the full‐length S‐protein expressed on cells. 47D11 could potentially inhibit the pseudotyped SARS‐CoV and SARS‐CoV‐2 in Vero E6 cells by exhibiting an IC50 of 0.06 and 0.08 μg/ml, respectively, by targeting conserved epitope in the RBD region of the S‐protein.39, 40, 41, 42 Apart from neutralizing antibodies, viral fusion inhibitors are focused on numerous research for its blocking/disruption of the viral fusion to host cell membranes.
As mentioned earlier, the S2 subunit plays a key role in the process of membrane fusion. SARS‐CoV‐2 and SARS‐CoV have about 89% of homology between their S2 segments. Initial interaction between the RBD of S1 domain and PD of ACE2 initiates the process of fusion, which is followed by heptad repeat 1 (HR1) domain's interaction with heptad repeat 2 (HR2) domains to form a six‐helix bundle (6HB) fusion core, ultimately resulting in viral fusion with the host cell membrane.4, 43, 44, 45 Most of the research on fusion inhibitors is primarily focused on peptide drugs. Recent reports on pan‐CoV (pan‐coronavirus) peptide fusion inhibitors (OC43‐HR2P, EK1, and EK1C4) showed potential inhibitory activity against CoV fusion by targeting the HR1 domain. Interestingly, EK1 has a broad‐spectrum neutralization and antiviral activity on both bat‐SARSr‐CoVs (like WIV1, pseudotyped Rs3367, and SHC014) and human‐CoVs (like pseudotyped SARS‐CoV, MERS‐CoV, NL63, 229E, and OC43) in vitro with a range of 0.26–6.02 μM of IC50. More interestingly, EK1C4, a lipopeptide resultant from the conjugation of cholesterol to the C‐terminus of EK1, was found to be the most effective fusion inhibitor against S‐protein‐mediated membrane fusion in SARS‐CoV‐2 infection with a 150‐ and 240‐fold enhanced anti‐CoV activity compared to EK1.42, 46, 47, 48
Furthermore, in recent computational modeling, it was suggested that nelfinavir can also target the S‐protein, thereby inhibiting viral entry into the cell. 49 Nelfinavir is an antiretroviral protease inhibitor used in the treatment and prevention of human immunodeficiency virus (HIV) infection. Docking studies have revealed that nelfinavir is a potential multi‐target agent. In silico approaches have confirmed the binding of nelfinavir to SARS‐CoV‐2 at Glu166 position and the interaction is stronger than the potent drugs lopinavir/ritonavir. Further to substantiate computational approaches, its antiviral activity was demonstrated in vitro by multiple research groups. 50 It was found that nelfinavir was three to five times more potent than the combination of lopinavir/ritonavir. Recent studies have reported that nelfinavir mesylate has significantly inhibited both SARS‐CoV and SARS‐CoV‐2 S‐protein‐mediated membrane fusion in a dose‐dependent manner (complete inhibition observed at a lowest effective concentration of 10 µM). Further in silico docking experiments revealed that nelfinavir may possibly bind to the S2 amino terminus of S trimer and thereby inhibit the heptad‐repeat complex formation that is responsible for S‐protein‐mediated membrane fusion. 51
Recently Rory et al., have conducted experimental in vitro and in vivo studies on a dimeric form of SARS‐CoV‐2 S‐specific lipopeptide that acts as a potential fusion inhibitor of S‐protein‐mediated viral entry. The intranasal administration of the dimeric lipopeptide used in this study has successfully prevented direct‐contact transmission of SARS‐CoV‐2 ferrets. 52 The same group has designed dimeric cholesterol‐conjugated peptide ([SARSHRC‐PEG4]2‐chol) that shows significant in vitro and in vivo efficacy. The designed [SARSHRC‐PEG4]2‐chol dimeric lipopeptide has inhibited live virus entry in both VeroE6 cells (IC50 ~300 nM, IC90 ~1 µM) and TMPRSS2 overexpressing VeroE6‐TMPRSS2 cells (IC50 ~5 nM). Also, the group has measured the efficacy of [SARSHRC‐PEG4]2‐chol dissolved in sucrose solution (as a substitute for dimethylsulfoxide) that would support the translational potential for human use. The designed [SARSHRC‐PEG4]2‐chol dimeric lipopeptide has maintained its potency in the new formulation (dissolved in sucrose solution instead of dimethylsulfoxide solution) on both VeroE6 cells (IC50 ~300 nM) and VeroE6‐TMPRSS2 cells (IC50 ~5 nM). Further, the group has also conducted a cellular toxicity (MTT) assay and found that [SARSHRC‐PEG4]2‐chol exhibited no toxicity even at its IC90 entry inhibitory concentration (~350 nM). 52
Further, the same research group has performed in vivo experiments using ferrets to assess the efficacy of [SARSHRC‐PEG4]2‐chol dimeric lipopeptide. Two noninfected ferrets were treated prophylactically with the [SARSHRC‐PEG4]2‐chol dimeric lipopeptide before their co‐housing with SARS‐CoV‐2‐infected ferrets. Initially, one SARS‐CoV‐2‐infected ferret was co‐housed with a group of four naive ferrets (two [SARSHRC‐PEG4]2‐chol‐treated and two mock‐treated). Co‐housing was stopped after 24 h of transmission period and the three groups (infected, mock‐treated, and [SARSHRC‐PEG4]2‐chol‐treated) of ferrets were separated and each was observed for their presence of viral load. SARS‐CoV‐2‐infected ferrets showed productive infection in both throat and nose swabs, and the infected ferret had also efficiently transmitted SARS‐CoV‐2 to the two mock‐treated ferrets. Interestingly, the third group, [SARSHRC‐PEG4]2‐chol‐treated ferrets, was not detected with productive SARS‐CoV‐2 infection in both throat and nose swabs. These in vitro and in vivo studies of [SARSHRC‐PEG4]2‐chol dimeric lipopeptide can be a promising antiviral candidate with potential efficacy in the prevention of SARS‐CoV‐2 transmission and fusion‐mediated viral entry even in intense direct contact for a period of 24 hours. Additionally, [SARSHRC‐PEG4]2‐chol has a long shelf life, does not require refrigeration, and inexpensive to produce. 52 Thus, with increasing evidence and reports on S‐protein as a crucial target in the treatment of COVID‐19, pharmacological interventions targeting S‐protein is gaining a large interest among global researchers and may act as an effective treatment strategy against SARS‐CoV‐2 infection.
3.2. ACE2 and cellular HS
ACE2 (homolog of ACE), a multifunctional zinc metalloprotease, is divided into two domains (amino‐ and carboxy‐terminal domain). With several imperative roles, the activities of ACE2 are associated with RAS, which is responsible for fluid/salt balance and maintenance of blood pressure homeostasis. Cleavage of angiotensinogen by renin generates angiotensin (Ang) I and further formation of Ang II are catalyzed by ACE. Ang II reduces hypoxia by the induction of vascular permeability and pulmonary vasoconstriction. Further, Ang II also mediates the extravasation of cytokines directing to the site of inflammation, where if this inflammatory response gets exacerbated, it would lead to detrimental conditions like respiratory distress and edema.53, 54, 55, 56
Although, both SARS‐CoV and SARS‐CoV‐2 share the same host cell receptor (ACE2) for their host entry, recent studies proved that the S‐protein ectodomain of the emergent virus (SARS‐CoV‐2) has 10‐ to 20‐fold higher ACE2‐binding affinity when compared to the S‐protein of SARS‐CoV. This stronger binding affinity could be a possible result of the several alterations in the amino acid (AA) residues leading to an enhanced salt‐bridge formation and hydrophobic interaction of SARS‐CoV‐2.6, 57, 58 ACE2 receptors can be found and observed in the epithelial cells of the lung, liver, testis, respiratory tract, and mouth, respectively. A noteworthy observation to mention is that the enhanced expression of ACE2 is associated with age, which partially leads to the severity of symptoms and higher viral load detected in the elderly patients of COVID‐19.9, 56, 59, 60, 61
Glycocalyx is generally composed of complex mixtures of glycoconjugates and glycans. Depending upon its location, several infectious organisms and viruses must get passed through the glycocalyx to engage with the receptors that mediate viral entry into the host cells. Most of the viral pathogens like the influenza virus, human immunodeficiency virus, herpes simplex virus, SARS‐CoV‐1, and MERS‐CoV have evolved to exploit glycans as an effective attachment factor. This exploitation of host glycans further facilitates the initial viral–host interaction.13, 62, 63, 64 Cell surface HS are linear polysaccharides that are highly negatively charged and are generally attached to extracellular matrix proteoglycans. Recent studies have shown that the S‐protein of SARS‐CoV‐2 interacts with the cell surface HS via RBD present in the S1 subunit. Further, the binding of heparin to the S‐protein of SARS‐CoV‐2 facilitates and favors the open conformation of RBD that binds to ACE2. Imaging studies by the same group have also inferred that heparin may increase the proportion of S‐protein bound to ACE2 and the occupancy of individual S‐protein through its stabilizing effects on ACE2 interactions. 13 These studies have shown that the binding of S‐protein to the host cells requires the dual engagement of cell surface HS and ACE2. Wherein the binding of S‐protein to cell surface HS/heparin enhances the recruitment of SARS‐CoV‐2 to the cell surface and thereby intensifies its local concentration for an effective engagement with the host cell receptor ACE2. Altogether, this pathogenic mechanism of exploiting host cell surface HS for an effective infection suggests that cell surface HS/S‐protein interactions of SARS‐CoV‐2 can be a potential novel therapeutic target.12, 13
3.2.1. Pharmacological treatment
Use of small soluble‐RBD (s‐RBD), a key engineered neutralizing fragment based on the S‐protein of SARS‐CoV is a considerable approach to accomplish the blockade of the ACE2 receptor. The administration of this key domain (s‐RBD), 193 AA in size, was found to occupy the host cell receptor ACE2 and thereby effectively blocks the viral entry of SARS in cell cultures. The existence of similar binding sites on ACE2 for both SARS‐CoV and SARS‐CoV‐2 provides a potential therapeutic opportunity against SARS‐CoV‐2 infection. Further, there has been a key focus for clinical interventions targeting ACE because of its unaltered structure that provides a key advantage on high affinity in the binding of therapeutic agents.65, 66, 67 A similar strategy targeting ACE2 is the administration of antibodies that are capable of binding to the ACE2 receptor. This strategy was reported to have efficient blockage of SARS‐CoV entry and replication in the in vitro experiments. While designing an anti‐ACE2 antibody, it is mandatory to remove the effector functions from the fragment crystallizable (Fc) domain so that the detrimental inflammatory response would be averted in the tissues expressing ACE2. Further, this modification would also preserve the long half‐life equipped by the Fc domain.67, 68, 69
Other small molecules with antiviral activities like interferon‐inducible transmembrane (IFITM) proteins and ACE2 inhibitors can also be considered for targeting ACE2. A specific peptide inhibitor, N‐(2‐aminoethyl)−1‐aziridine‐ethanamine, a known small‐molecule inhibitor, showed inhibitory activity against ACE2 of SARS‐CoV with an IC50 of 57 ± 7 μM, resulting in efficient inhibition of S‐protein‐mediated membrane fusion in vitro. Further, IFITM can potentially promote the buildup of cholesterol in the endosomes and thereby obstruct the viral entry of several enveloped viruses, including SARS‐CoV, MERS‐CoV, HCoV‐NL63, and HCoV‐229E. A broad‐spectrum small molecule inhibitor for an efficient anti‐CoV activity is still lacking and remains unexplored.42, 70, 71, 72, 73 Apart from the above pharmacological treatments, an investigational drug Umifenovir (also known as Arbidol), is also considered as an efficient repurposed antiviral drug with a promising mechanism of action against the interaction of S‐protein/ACE2. 74 With multifunctional roles in both normal and detrimental conditions, ACE2 is a promising therapeutic target for the treatment of SARS‐CoV‐2 infection.
Lactoferrin (LF) is a nontoxic, naturally occurring iron‐binding glycoprotein found in numerous mucosal secretions, which plays a crucial role in the first line of defense against infectious microbes. LF is known for its broad‐spectrum antiviral activity against several RNA and DNA viruses that infects both humans and animals. This antiviral activity of LF and its mechanism of action against these viruses are found to be mediated via the binding of LF to heparan sulfate proteoglycans (HSPGs; an abundant expression form of cell surface HS). Recent in vitro study has experimentally shown that LF can bind to the cell surface HSPGs, which, in turn, disrupts the SARS‐CoV‐2 interaction with HSPGs and thereby prevents the viral attachment to the host cells. Taken together, the study concluded that LF has considerable antiviral activity against most common HCoV, including SARS‐CoV‐2, indicating that LF can be a potential promising antiviral drug candidate for treating SARS‐CoV‐2 infection. 75 Tilorone, an established pan‐antiviral agent is also identified as a potential drug that prevents SARS‐CoV‐2 infection in vitro through the inhibition of cell surface HS‐dependent endocytosis. Interestingly, Raloxifene, a heparin/cell surface HS‐binding drug was found to enhance the antiviral activity of Tilorone and this combinational administration of Tilorone and Raloxifene are convenient for oral administration and was also well tolerated even at highest concentrations. However, further studies on the synergistic effects of Tilorone and Raloxifene and their potential antiviral activity still warrants investigation. Further, Mitoxantrone, a DNA intercalator known for its inhibition of type II DNA topoisomerase has shown promising blockade effects on cell surface HS‐dependent endocytosis. Experimental evidence has shown the inhibitory activity of Mitoxantrone in pseudoviral particles (PP) treated ACE2‐GFP HEK293 cells, where Mitoxantrone strongly inhibited the viral entry. Collective results suggest that Mitoxantrone directly binds to cell surface HS and might be capable of influencing the mode of cell surface HS interaction with the spike and thereby blocks the viral entry.12, 76, 77, 78
3.3. TMPRSS2 and cathepsin B/L
Subsequent to the receptor interaction, host cell proteases play an essential role in the virus–host cell membrane fusion, which ultimately leads to the release of the viral genome. TMPRSS2, an androgen‐responsive, type II transmembrane serine protease located on the surface of the host cell, is responsible for the activation of the S‐protein of highly pathogenic HCoV like MERS‐CoV and SARS‐CoV. TMPRSS2 is normally expressed in the human epithelia of the urogenital, gastrointestinal, and respiratory tracts. Recent studies have confirmed that TMPRSS2 facilitates the viral entry of SARS‐CoV‐2. More precisely, the cleavage of the S‐protein of SARS‐CoV‐2 is mediated by TMPRSS2 resulting in viral activation, which is one of the crucial host factors for the pathogenicity of SARS‐CoV‐2. Despite the activation of S‐protein by other proteases like TMPRSS4, TMPRSS11A, TMPRSS11D, TMPRSS11E1, and endosomal cathepsin, the activity of TMPRSS2 is found to be most crucial for the viral entry and pathogenesis.11, 56, 79, 80, 81, 82, 83
Apart from TMPRSS2, Cathepsin L (CatL) also mediates the S1 subunit cleavage, which is adequate for the viral‐entry of CoV and virus–host cell endosome membrane fusion followed by the release of viral RNA. Several studies have reported that both TMPRSS2 (facilitates the fusion of viral and host membrane) and CatB/L (facilitates the fusion of viral and endosomal membrane) are disparate in nature and either of the two proteases works independently, which leaves two independent pathways for the viral entry. A recent study predicted a complete blockade of viral entry with a combined usage of both TMPRSS2 and CatB/L inhibitors.11, 84, 85, 86
3.3.1. Pharmacological treatment
The absence of TMPRSS2 in the TMPRSS2 knockout mice has been confirmed to lack both CoV and influenza virus infection, which pose a new useful drug target for the treatment of COVID‐19. Even though the protease inhibitors like camostat mesylate, nafamostat mesylate, and gabexate mesylate were originally not developed for targeting TMPRSS2 specifically, these protease inhibitors are found to attenuate the protease activity of TMPRSS2.42, 56, 79, 87, 88 Camostat mesylate (CM), originally used for chronic pancreatitis, was found to subdue the activity of TMPRSS2 of SARS‐CoV‐2. CM could potentially inhibit the pseudotyped SARS‐CoV‐2 infection in the Calu‐3 cells with an EC50 (half‐maximal effective concentration) of 1 μM and EC90 (concentration for 90% of maximal effect) of 5 μM, which showed 80% inhibition of pseudotyped SARS‐CoV‐2 entry in the airway epithelial cells of humans. Similar to camostat mesylate, nafamostat mesylate (NM) was also observed for its inhibitory activity on S‐protein‐mediated entry of SARS‐CoV‐2 in Calu‐3 cells. Interestingly, when comparing the EC50 of CM (87 nM) and NM (5 nM) against the S‐protein‐mediated SARS‐CoV‐2 entry, the EC50 of NM was found to be 15‐fold greater than that of CM. In addition to the above two protease inhibitors, gabexate mesylate (FOY), was found to be less potent on both SARS‐CoV (EC50 = 1.2 M) and SARS‐CoV‐2 (EC50 = 115 μM) S‐protein‐mediated viral entry into Calu‐3 cells.11, 42, 89, 90
The entry of CoVs through endocytosis has relied on different lysosomal cathepsins where a recent study has highlighted that the entry of SARS‐CoV‐2 via endocytosis is solely dependent on CatL and not on cathepsin B (CatB). Treating a CatL selective inhibitor SID26681509 on HEK293/hACE2 cells reduced the viral entry of SARS‐CoV‐2 pseudovirus by >76%.11, 24, 56, 91 K11777, a small molecule CatL inhibitor, showed inhibition of the pseudotyped SARS‐CoV (IC50 = 0.68 nM), MERS‐CoV (IC50 = 46.12 nM), HCoV‐229E (IC50 = 1.48 nM), and HCoV‐NL63 (IC50 = 6.78 nM). However, this inhibitory activity of K11777 can only be observed in the absence of activating serine proteases. To overcome this scenario, a combination of K11777 with a serine protease inhibitor could be used. Further, K11777 being an irreversible covalent inhibitor of cysteine protease CatL often involves undesirable toxicity. Whereas, oxocarbazate, a reversible CatL inhibitor may act as a selective, potent, and low toxic candidate for the treatment of COVID‐19 in humans and needs relevant studies and animal experiments for its activity against SARS‐CoV‐2 infection.42, 56, 85, 91, 92, 93
Teicoplanin, an antimicrobial drug with potential activity on inhibiting CatL in the late endosome that ultimately leads to the blockade of CoV entry. While it has been already found that teicoplanin can inhibit the pseudotyped SARS‐CoV (IC50 = 3760 nM) and MERS‐CoV (IC50 = 630 nM) entry, a recent study has been reported that teicoplanin with an IC50 value of 1660 nM can inhibit the viral entry of pseudotyped SARS‐CoV‐2. Additionally, E64d, a broad spectrum cysteine protease inhibitor, was reported to inhibit 92.5% of SARS‐CoV‐2 pseudovirion entry. Further, a combined synergistic effect of both camostat (TMPRSS2 inhibitor) and E64d (CatL inhibitor) may result in a complete blockade of the SARS‐CoV‐2 viral entry.42, 56, 84, 85, 91, 94 Even though cathepsin inhibitors like K11777 and E64d shows efficient blockade of viral entry in some cell lines, their antiviral activity can be compromised upon the expression of activating serine proteases like TMPRSS2. 28 K11777 was found to completely inhibit CoV infection only when target cells lack the presence of activating serine proteases, and in case of the TMPRSS2 expressed cells, K11777 in combination with CM were required for complete inhibition. Further, only the inhibition of drugs targeting serine proteases has attenuated the SARS‐CoV‐2 pathogenesis in vivo. Altogether, future developments on targeting TMPRSS2 and CatL must consider the fact that SARS‐CoV‐2 is capable of utilizing redundant viral‐entry pathways, that is either endosomal‐mediated (in presence of CatL) or TMPRSS2‐mediated (in absence of CatL) viral‐entry. Further CatL inhibitor drugs that show efficient blockade of SARS‐CoV‐2 in cell lines that do not express TMPRSS2 can likely lose their antiviral activity upon the expression of TMPRSS2 in other in vivo models.24, 28 Thus, the translational potential of existing CatL inhibitors in the presence of TMPRSS2 remains to be validated and further warrants investigation.
Inhibitors targeting SARS‐CoV‐2 main protease (Mpro) was recently identified using FRET‐based assays and these Mpro inhibitors include boceprevir, GC‐376, calpain inhibitors II and XII. The significant discovery of calpain inhibitors II and XII suggest that it could be a feasible option to design and develop a dual inhibitor of SARS‐CoV‐2 Mpro and the host calpains/cathepsins. 95 Further, the X‐ray crystal structures of Mpro with calpain inhibitors II and XII revealed that calpain XII bound to Mpro in an unexpected binding mode with an inverted, semi‐helical conformation where the P1′ pyridine ring has been placed in S1 pocket instead of the conventional P1 norvaline side chain. This complex structure of calpain II and XII divulges that the S1 pocket of Mpro is capable of accommodating both hydrophobic and hydrophilic substitutions which in turn paves way for the design of dual inhibitors targeting viral Mpro and host CatL. 96 This hypothesis of dual inhibition of calpain inhibitors II and XII was further studied experimentally Hu et al., where the group conducted pseudovirus neutralization assay and drug time‐of‐addition assay. The SARS‐CoV‐2 pseudovirus entry (mediated by CatL) was inhibited by the inhibitory activity of both calpain inhibitor II (IC50 values of 9.26 ± 1.35 μM) and XII (IC50 values of 5.28 ± 0.74 μM) in VeroE6 cells (with minimal levels of TMPRSS2, and thus viral entry relied on CatL). Thus, calpain inhibitors II and XII have a potential novel mechanism of action that acts as a dual‐inhibitor by targeting both viral Mpro and host CatL‐mediated viral entry. 97 Extensive research on these crucial host‐proteases that are responsible for viral entry through independent mechanisms, may pose a possibility for the development of novel and effective therapy against the SARS‐CoV‐2 infection. A pictorial summarization of the abovementioned strategies targeting the viral‐entry facilitators is given in Figure 2.
Figure 2.
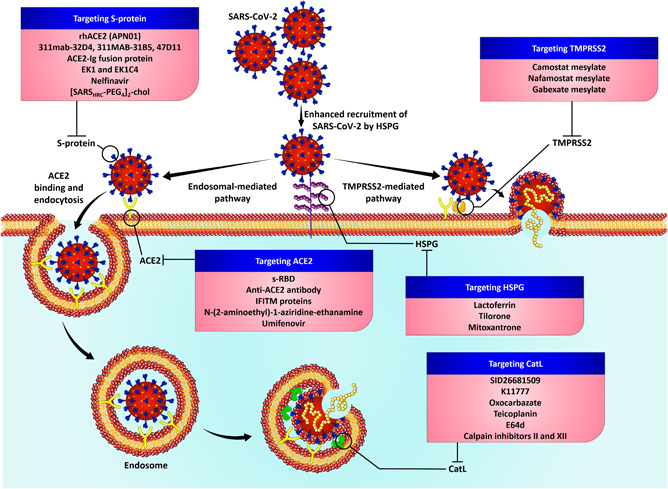
Targeting the viral‐entry facilitators of SARS‐CoV‐2 infection. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2
4. A GLANCE AT THE IN‐LINE PHARMACOLOGICAL TREATMENT AND THEIR ONGOING CLINICAL TRIALS
While several antiviral drugs against SARS‐CoV‐2 infection are in rigorous study and research their ongoing clinical trials play a pivotal role in the testing and development of the same drug for their indispensable need in treating the current pandemic. Recent findings using the publicly available human host gene expression profiles of SARS‐CoV‐2, eight pro‐viral factors have been identified that mediates the COVID‐19 infection. The study concluded by identifying 12 repurposed drugs for the potential treatment of SARS‐CoV‐2 infection. In addition to pro‐viral factors, six drugs targeting the prostaglandin‐endoperoxide synthase 2 (PTGS2) or cyclo‐oxygenase2 (Cox‐2) were also found to be of potential use, 98 in treating SARS‐CoV‐2 infection. A data‐driven drug repositioning framework study recently identified a PARP‐1 inhibitor CVL218 as a potential therapeutic agent for the treatment of COVID‐19. 99
Another study using a network‐based methodology involving the drug–target networks, HCoV–host interactions, HCoV‐induced transcriptome in human cell lines, and human protein–protein interactome network revealed 16 repurposable drugs and 3 potential drug combinations targeting SARS‐CoV‐2 infection. 100 A study involving 332 high‐confidence SARS‐CoV‐2 protein–human–protein interactions found 69 ligands that can interact with these interactions, of which 29 found to be FDA‐approved drugs, 28 compounds in preclinical, and 12 are in clinical trials. 101 The outcome of this study revealed that many of these drugs need to be tested for COVID‐19 and few of them have been tested in the following studies.102, 103 Such computational studies represent a drug repurposing approach and further experimental studies using multidisciplinary approaches will pave way for repurposing FDA‐approved drugs for targeting COVID‐19. A summarized glance on the potential drug candidates and their clinical trial status on COVID‐19 treatment are tabulated in Table 1.
Table 1.
An overview of potential drug candidates targeting viral‐entry facilitators and their clinical trial status
| Target | Role in the viral entry of SARS‐CoV‐2 | Pharmacological treatment | Mechanism of action | Ongoing clinical trials | References | ||||
|---|---|---|---|---|---|---|---|---|---|
| ClinicalTrials. gov identifier | Study type | Intervention mode | Phase | Recruitment status | |||||
| S‐protein | Facilitates the viral entry through interactive binding with the host‐cell receptor ACE2 | rhACE2 (APN01) | May inhibit the attachment of the virus to the cells | NCT04335136 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Recruiting | 34, 35 |
| NCT04287686 | Interventional (Clinical Trial) | Parallel Assignment | NA | Withdrawn (Without CDE Approval) | |||||
| 311mab‐32D4, 311mab‐31B5 | Specifically bind to RBD. May block the SARS‐CoV‐2 RBD‐human ACE2 interaction | No clinical trials have been initiated | 40, 42 | ||||||
| ACE2‐Ig fusion protein | Binds to RBD with high affinity, and may possess neutralizing effects on SARS‐CoV‐2 | No clinical trials have been initiated | 41, 42 | ||||||
| Human monoclonal antibody 47D11 | Can potentially to full‐length S‐protein by targeting conserved epitope in the RBD | No clinical trials have been initiated | 39, 42 | ||||||
| EK1 | May block S‐protein‐mediated cell–cell fusion by competitively inhibiting the 6HB formation | No clinical trials have been initiated | 42, 47, 104 | ||||||
| EK1C4 | May exhibit potent inhibitory activity against S‐protein‐mediated membrane fusion | No clinical trials have been initiated | 42, 48 | ||||||
| Nelfinavir | May exhibit inhibitory activity on S‐protein‐mediated membrane fusion | No clinical trials have been initiated | 49, 50, 51 | ||||||
| [SARSHRC‐PEG4]2‐chol dimeric lipopeptide | May act as a potential fusion inhibitor that inhibits S‐protein‐mediated membrane fusion | No clinical trials have been initiated | 52 | ||||||
| ACE2 | Acts as the host‐cell receptor for SARS‐CoV‐2 entry | s‐RBD | Can potentially occupy the ACE2 and may avoid the S‐protein interaction with ACE2 | No clinical trials have been initiated | 65, 66, 67 | ||||
| Anti‐ACE2 antibody | May potentially bind to the ACE2 receptor and interfere with the S‐protein/ACE2 interaction | No clinical trials have been initiated | 67, 68, 69 | ||||||
| IFITM proteins | Obstructs the entry of several CoVs by building up cholesterol in endosomes | No clinical trials have been initiated | 42, 73 | ||||||
| N‐(2‐aminoethyl)‐1‐aziridine‐ethanamine | May block the S‐protein‐mediated membrane fusion via targeting ACE2 receptor | No clinical trials have been initiated | 42, 70, 71, 72 | ||||||
| Umifenovir (Arbidol) | May potentially disrupts the viral entry of SARS‐CoV‐2 by targeting S‐protein/ACE2 interaction | NCT04350684 | Interventional (Clinical Trial) | Parallel Assignment | 4 | Enrolling by invitation | 74, 105, 106 | ||
| NCT04260594 | Interventional (Clinical Trial) | Parallel Assignment | 4 | Not yet recruiting | |||||
| NCT04273763 | Interventional (Clinical Trial) | Sequential Assignment | NA | Active, not recruiting | |||||
| NCT04476719 | Interventional (Clinical Trial) | Crossover Assignment | 1 | Active, not recruiting | |||||
| Cell surface HS | Facilitates and favors the open conformation of RBD that binds to ACE2 and thereby enhances the recruitment of SARS‐CoV‐2 | Lactoferrin | May potentially bind to HSPG and thereby prevent the viral attachment to the cell surface | NCT04421534 | Interventional (Clinical Trial) | Parallel Assignment | 2, 3 | Not yet recruiting | 75 |
| NCT04427865 | Interventional (Clinical Trial) | Parallel Assignment | 2, 3 | Not yet recruiting | |||||
| NCT04526821 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Not yet recruiting | |||||
| NCT04475120 | Interventional (Clinical Trial) | Parallel Assignment | 2, 3 | Completed | |||||
| NCT04412395 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Not yet recruiting | |||||
| Tilorone | May prevent SARS‐CoV‐2 infection through the process of inhibiting cell surface HS | No clinical trials have been initiated | 12, 76 | ||||||
| Mitoxantrone | May block the cell surface HS‐dependent endocytosis | No clinical trials have been initiated | 12, 77, 78 | ||||||
| TMPRSS2 | Facilitates the viral‐entry of SARS‐CoV‐2 by S‐protein priming | Camostat mesylate | Acts as a potential TMPRSS2 inhibitor, and may possess inhibitory activity against S‐protein‐mediated viral‐entry | NCT04353284 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Active, not recruiting | 11, 42 |
| NCT04455815 | Interventional (Clinical Trial) | Sequential Assignment | 2, 3 | Not yet recruiting | |||||
| NCT04338906 | Interventional (Clinical Trial) | Parallel Assignment | 4 | Not yet recruiting | |||||
| NCT04435015 | Interventional (Clinical Trial) | Parallel Assignment | 1 | Not yet recruiting | |||||
| NCT04321096 | Interventional (Clinical Trial) | Parallel Assignment | 1 | Recruiting | |||||
| NCT04355052 | Interventional (Clinical Trial) | Single Group Assignment | 3 | Recruiting | |||||
| NCT04470544 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Not yet recruiting | |||||
| NCT04374019 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Recruiting | |||||
| Nafamostat mesylate | Similar to the function of camostat, but much more efficient in the blockade of S‐protein‐mediated SARS‐CoV‐2 entry | NCT04418128 | Interventional (Clinical Trial) | Parallel Assignment | 2, 3 | Not yet recruiting | 42, 90 | ||
| NCT04352400 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Not yet recruiting | |||||
| NCT04473053 | Interventional (Clinical Trial) | Parallel Assignment | 2 | Recruiting | |||||
| Gabexate mesylate | May block the S‐protein‐mediated viral entry | No clinical trials have been initiated | 42, 90 | ||||||
| CatL/B | Responsible for the viral and endosomal membrane fusion | SID26681509 | Can selectively inhibit CatL and thereby disrupts the viral–endosome fusion | No clinical trials have been initiated | 11, 24, 46, 56, 91, 107 | ||||
| K11777 | Can potentially possess inhibitory activity against the CatL | No clinical trials have been initiated | 42, 56, 85, 91, 92, 93, 108 | ||||||
| Oxocarbazate | Shows selective, potent, less‐toxic inhibitory activity against CatL | No clinical trials have been initiated | 85, 92 | ||||||
| Teicoplanin | Can potentially block the viral entry by targeting CatL in the late endosome | No clinical trials have been initiated | 42, 85, 94, 109 | ||||||
| E64d | May potentially block SARS‐CoV‐2 entry by inhibitory activity against CatL | No clinical trials have been initiated | 42, 84, 85, 91 | ||||||
| Calpain inhibitors II and XII | May act as a dual‐inhibitor targeting both Mpro and CatL | No clinical trials have been initiated | 95, 96, 97 | ||||||
Abbreviations: ACE2, angiotensin‐converting enzyme 2; CatL/B, cathepsin L/B; CDE, Center for Drug Evaluation; HS, heparan sulfate; IFITM, interferon‐inducible transmembrane; rhACE2, recombinant human angiotensin‐converting enzyme 2; RBD, receptor‐binding domain; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; TMPRSS2, transmembrane serine protease 2.
5. CONCLUSION
The COVID‐19 outbreak is declared as a public health emergency worldwide by the WHO. The incidence of infections continues to rise even though extensive quarantine and control measures are implemented. SARS‐CoV‐2 continues to spread rapidly across the world raising serious global concerns. Though several research groups have set their goals prioritized on the identifications and development of new therapeutic strategies, there are no effective treatments as of date. The viral entry being the first step in viral pathogenesis is considered to be a promising therapeutic intervention point where the inhibition of receptor interaction and virus–host cell membrane fusion efficiently enhances the process of evading viral genome delivery. Our survey on the ongoing clinical trials targeting the viral‐entry facilitators has shown the lack of clinical trial initiation in many potent drugs that showed efficient blockade of SARS‐CoV‐2 in the experimental in vivo and in vitro cultures. This gap on drugs targeting the crucial viral‐entry facilitators needs further investigation and clinical trials for effective management of the emerged pandemic.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Shibi Muralidar: Writing—original draft; Writing—review & editing; collection of data. Gayathri Gopal: Writing—review & editing; collection of data. Senthil V. Ambi: Conceptualization, visualization, supervision, and writing—review & editing.
ACKNOWLEDGMENTS
The authors express their gratitude to SASTRA‐Deemed‐to‐be‐University, Tamil Nadu, India for infrastructure and financial support. They also extend their appreciation for the contribution of Biopharmaceutical research lab members, SASTRA‐Deemed‐to‐be‐University.
Muralidar S, Gopal G, Visaga Ambi S. Targeting the viral‐entry facilitators of SARS‐CoV‐2 as a therapeutic strategy in COVID‐19. J Med Virol. 2021;93:5260–5276. 10.1002/jmv.27019
REFERENCES
- 1. World Health Organization. (WHO) . Novel Coronavirus (2019‐nCoV) Situation Report—1, 21 January 2020. WHO Bull. 2020;(January):1‐7.
- 2. World Health Organization . COVID‐19 Weekly Epidemiological Update 22. World Health Organization. 2020;(December):1‐3. https://www.who.int/docs/default-source/coronaviruse/situation-reports/weekly_epidemiological_update_22.pdf
- 3. Muralidar S, Ambi SV, Sekaran S, Krishnan UM. The emergence of COVID‐19 as a global pandemic: understanding the epidemiology, immune response and potential therapeutic targets of SARS‐CoV‐2. Biochimie. 2020;179:85‐100. 10.1016/j.biochi.2020.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu C, Liu Y, Yang Y, et al. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science (80‐). 2020;3(3):1‐8. 10.20944/preprints202003.0226.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science (80‐). 2020;367(6483):1260‐1263. 10.1126/science.aax0902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Devaux CA, Rolain JM, Raoult D. ACE2 receptor polymorphism: Susceptibility to SARS‐CoV‐2, hypertension, multi‐organ failure, and COVID‐19 disease outcome. J Microbiol Immunol Infect. 2020;53:425‐435. 10.1016/j.jmii.2020.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. V′kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol. 2021;19:155‐170. 10.1038/s41579-020-00468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin‐converting enzyme 2 (ACE2) as a SARS‐CoV‐2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586‐590. 10.1007/s00134-020-05985-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poduri R, Joshi G, Jagadeesh G. Drugs targeting various stages of the SARS‐CoV‐2 life cycle: exploring promising drugs for the treatment of Covid‐19. Cell Signal. 2020;74(July):109721. 10.1016/j.cellsig.2020.109721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Q, Chen CZ, Swaroop M, et al. Heparan sulfate assists SARS‐CoV‐2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020;6(1):80. 10.1038/s41421-020-00222-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clausen TM, Sandoval DR, Spliid CB, et al. SARS‐CoV‐2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043‐1057. 10.1016/j.cell.2020.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baig MS, Alagumuthu M, Rajpoot S, Saqib U. Identification of a potential peptide inhibitor of SARS‐CoV‐2 targeting its entry into the host cells. Drugs R&D. 2020;20:161‐169. 10.1007/s40268-020-00312-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Racaniello V. Furin cleavage site in the SARS‐CoV‐2 coronavirus glycoprotein. Virol Blog Viruses Viral Dis. 2020;February:1‐34. [Google Scholar]
- 16. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562‐569. 10.1038/s41564-020-0688-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tai W, He L, Zhang X, et al. Characterization of the receptor‐binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17:613‐620. 10.1038/s41423-020-0400-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan R, Zhang Y, Guo Y, Xia L, Zhou Q. Structural basis for the recognition of the 2019‐nCoV by human ACE2. bioRxiv. 2020;(March):2762. 10.1101/2020.02.19.956946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176(February):104742. 10.1016/j.antiviral.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang T, Jaimes JA, Bidon MK, Straus MR, Daniel S, Whittaker GR. Proteolytic activation of SARS‐CoV‐2 spike at the S1/S2 boundary: potential role of proteases beyond furin. ACS Infect Dis. 2021;7:264‐272. 10.1021/acsinfecdis.0c00701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xia S, Lan Q, Su S, et al. The role of furin cleavage site in SARS‐CoV‐2 spike protein‐mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020;5(1):92. 10.1038/s41392-020-0184-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Örd M, Faustova I, Loog M. The sequence at spike S1/S2 site enables cleavage by furin and phospho‐regulation in SARS‐CoV2 but not in SARS‐CoV1 or MERS‐CoV. Sci Rep. 2020;10(1):1‐10. 10.1038/s41598-020-74101-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76‐84. 10.1016/j.antiviral.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maier HJ, Bickerton E, Britton P. Coronaviruses: Methods and protocols. Vol. 1282. Springer Protocols; 2015:1‐282. 10.1007/978-1-4939-2438-7 [DOI] [Google Scholar]
- 26. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci U S A. 2020;117(21):11727‐11734. 10.1073/pnas.2003138117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lukassen S, Chua RL, Trefzer T, et al. SARS‐CoV‐2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;39(10):1‐15. 10.15252/embj.20105114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steuten K, Kim H, Widen JC, et al. Challenges for targeting SARS‐CoV‐2 proteases as a therapeutic strategy for COVID‐19. ACS Infect Dis. 2021:acsinfecdis.0c00815. 10.1021/acsinfecdis.0c00815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pang J, Wang MX, Ang IYH, et al. Potential rapid diagnostics, vaccine and therapeutics for 2019 novel coronavirus (2019‐nCoV): a systematic review. J Clin Med. 2020;9(3):623. 10.3390/jcm9030623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS‐CoV‐2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766‐788. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spike Protein/S Protein . https://www.sinobiological.com/research/virus/hcov-spike-protein-overview. Accessed July 25, 2020.
- 32. Zheng M, Song L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS‐CoV‐2 compared to SARS‐CoV. Cell Mol Immunol. 2020;17(5):536‐538. 10.1038/s41423-020-0385-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H, Zhou Y, Zhang M, Wang H, Updated Approaches against SARS‐CoV‐2. Antimicrobial Agents and Chemotherapy. 2020;(64 (6):1‐7. https://aac.asm.org/content/64/6/e00483-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tu YF, Chien CS, Yarmishyn AA, et al. A review of SARS‐CoV‐2 and the ongoing clinical trials. Int J Mol Sci. 2020;21(7):2657. 10.3390/ijms21072657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181(4):905‐913. 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Magrone T, Magrone M, Jirillo E. Focus on receptors for coronaviruses with special reference to angiotensin‐converting enzyme 2 as a potential drug target—a perspective. Endocrine Metab Immune Disord—Drug Targets. 2020;20(6):807‐811. 10.2174/1871530320666200427112902 [DOI] [PubMed] [Google Scholar]
- 37. Imai Y, Kuba K, Rao S, et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112‐116. 10.1038/nature03712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khan A, Benthin C, Zeno B, et al. A pilot clinical trial of recombinant human angiotensin‐converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21(1):1‐9. 10.1186/s13054-017-1823-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS‐CoV‐2 infection. Nat Commun. 2020;11(1):1‐6. 10.1038/s41467-020-16256-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen X, Li R, Pan Z, et al. Human monoclonal antibodies block the binding of SARS‐CoV‐2 spike protein to angiotensin converting enzyme 2 receptor. Cell Mol Immunol. 2020;17(6):647‐649. 10.1038/s41423-020-0426-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lei C, Qian K, Li T, et al. Neutralization of SARS‐CoV‐2 spike pseudotyped virus by recombinant ACE2‐Ig. Nat Commun. 2020;11(1):1‐5. 10.1038/s41467-020-16048-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang X, Xia S, Wang Q, et al. Broad‐spectrum coronavirus fusion inhibitors to combat COVID‐19 and other emerging coronavirus diseases. Int J Mol Sci. 2020;21(11):3843. 10.3390/ijms21113843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bosch BJ, Martina BEE, Van Der Zee R, et al. Severe acute respiratory syndrome coronavirus (SARS‐CoV) infection inhibition using spike protein heptad repeat‐derived peptides. Proc Natl Acad Sci U S A. 2004;101(22):8455‐8460. 10.1073/pnas.0400576101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kirchdoerfer RN, Cottrell CA, Wang N, et al. Pre‐fusion structure of a human coronavirus spike protein. Nature. 2016;531(7592):118‐121. 10.1038/nature17200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar N, Kanchan T, Unnikrishnan B, et al. Drug targets for corona virus: a systematic review. Indian J Pharmacol. 2020;49(5):344‐347. 10.4103/ijp.IJP [DOI] [Google Scholar]
- 46. Joshi S, Joshi M, Degani MS. Tackling SARS‐CoV‐2: proposed targets and repurposed drugs. Future Med Chem. 2020;12(17). 10.4155/fmc-2020-0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xia S, Yan L, Xu W, et al. A pan‐coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci Adv. 2019;5(4):eaav4580. 10.1126/sciadv.aav4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xia S, Liu M, Wang C, et al. Inhibition of SARS‐CoV‐2 (previously 2019‐nCoV) infection by a highly potent pan‐coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343‐355. 10.1038/s41422-020-0305-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Z, Yao H, Shen J, et al. Nelfinavir is active against SARS‐CoV‐2 in Vero E6 cells. ChemRxiv. 2020:0‐3. 10.26434/chemrxiv.12039888.v1 [DOI] [Google Scholar]
- 50. Lee PC. Nelfinavir for COVID19: summary of basic science data and initial clinical experience. Res Square. 2020:1‐6. 10.21203/rs.3.rs-27346/v1 [DOI] [Google Scholar]
- 51. Musarrat F, Chouljenko V, Dahal A, et al. The anti‐HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV‐2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID‐19 infections. J Med Virol. 2020;92(10):2087‐2095. 10.1002/jmv.25985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Vries RD, Schmitz KS, Bovier FT, et al. Intranasal fusion inhibitory lipopeptide prevents direct contact SARS‐CoV‐2 transmission in ferrets. bioRxiv. 2020. 10.1101/2020.11.04.361154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Serfozo P, Wysocki J, Gulua G, et al. Ang II (angiotensin II) conversion to angiotensin‐(1‐7) in the circulation is POP (prolyloligopeptidase)‐dependent and ACE2 (angiotensin‐converting enzyme 2)‐independent. Hypertens (Dallas, Tex 1979). 2020;75(1):173‐182. 10.1161/HYPERTENSIONAHA.119.14071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Offringa A, Montijn R, Singh S, Paul M, Pinto YM, Pinto‐Sietsma S‐J. The mechanistic overview of SARS‐CoV‐2 using angiotensin‐converting enzyme 2 to enter the cell for replication: possible treatment options related to the renin–angiotensin system. Eur Hear J—Cardiovasc Pharmacother. 2020;6:317‐325. 10.1093/ehjcvp/pvaa053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuba K, Imai Y, Ohto‐Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin‐angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128(1):119‐128. 10.1016/j.pharmthera.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gil C, Ginex T, Maestro I, et al. COVID‐19: drug targets and potential treatments. J Med Chem. 2020;63:12359‐12386. 10.1021/acs.jmedchem.0c00606 [DOI] [PubMed] [Google Scholar]
- 57. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin‐converting enzyme 2: SARS‐CoV‐2 receptor and regulator of the renin–angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126:1456‐1474. 10.1161/CIRCRESAHA.120.317015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID‐19 virus targeting the CNS: tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995‐998. 10.1021/acschemneuro.0c00122 [DOI] [PubMed] [Google Scholar]
- 59. Xiao L, Sakagami H, Miwa N. ACE2: the key molecule for understanding the pathophysiology of severe and critical conditions of COVID‐19: demon or angel? Viruses. 2020;12(5):2002‐2003. 10.3390/v12050491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Y, Li L. SARS‐CoV‐2: virus dynamics and host response. Lancet Infect Dis. 2020;20(5):515‐516. 10.1016/S1473-3099(20)30235-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu C, Zheng M. Single‐cell RNA expression profiling shows that ACE2, the putative receptor of Wuhan 2019‐nCoV, has significant expression in the nasal, mouth, lung and colon tissues, and tends to be co‐expressed with HLA‐DRB1 in the four tissues. Res Square. 2020;(February):1‐3. 10.21203/rs.3.rs-16992/v1 [DOI] [Google Scholar]
- 62. Koehler M, Delguste M, Sieben C, Gillet L, Alsteens D. Initial step of virus entry: virion binding to cell‐surface glycans. Annu Rev Virol. 2020;7:143‐165. 10.1146/annurev-virology-122019-070025 [DOI] [PubMed] [Google Scholar]
- 63. Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses. 2019;11(7):1‐24. 10.3390/v11070596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stencel‐Baerenwald JE, Reiss K, Reiter DM, Stehle T, Dermody TS. The sweet spot: defining virus–sialic acid interactions. Nat Rev Microbiol. 2014;12(11):739‐749. 10.1038/nrmicro3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shetty R, Ghosh A, Honavar SG, Khamar P, Sethu S. Review article therapeutic opportunities to manage COVID‐19/SARS‐CoV‐2 infection: present and future. Indian J Ophthalmol. 2020;68(5):693‐702. 10.4103/ijo.IJO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193‐amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin‐converting enzyme 2. J Biol Chem. 2004;279(5):3197‐3201. 10.1074/jbc.C300520200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kruse RL. Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research. 2020;9:72. 10.12688/f1000research.22211.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol. 2016;40:78‐87. 10.1016/j.coi.2016.03.005 [DOI] [PubMed] [Google Scholar]
- 69. Tanonaka K, Marunouchi T. Angiotensin‐converting enzyme 2. Folia Pharmacol Jpn. 2016;147(2):120‐121. 10.1254/fpj.147.120 [DOI] [PubMed] [Google Scholar]
- 70. Aggarwal A, Mehta S, Gupta D, et al. Clinical & immunological erythematosus patients characteristics in systemic lupus Maryam. J Dent Educ. 2012;76(11):1532‐1539. 10.4103/ijmr.IJMR [DOI] [PubMed] [Google Scholar]
- 71. Han DP, Penn‐Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS‐CoV entry and development of a potent entry inhibitor. Virology. 2006;350(1):15‐25. 10.1016/j.virol.2006.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Huentelman MJ, Zubcevic J, Hernández Prada JA, et al. Sructure‐based discovery of a novel angiotensin‐converting enzyme 2 inhibitor. Hypertension. 2004;44(6):903‐906. 10.1161/01.HYP.0000146120.29648.36 [DOI] [PubMed] [Google Scholar]
- 73. Wrensch F, Winkler M, Pöhlmann S. IFITM proteins inhibit entry driven by the MERS‐coronavirus spike protein: evidence for cholesterol‐independent mechanisms. Viruses. 2014;6(9):3683‐3698. 10.3390/v6093683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID‐19): a review. JAMA. 2020;323(18):1824‐1836. 10.1001/jama.2020.6019 [DOI] [PubMed] [Google Scholar]
- 75. Hu Y, Meng X, Zhang F, Xiang Y, Wang J. The in vitro antiviral activity of lactoferrin against common human coronaviruses and SARS‐CoV‐2 is mediated by targeting the heparan sulfate co‐receptor. Emerg Microbes Infect. 2021;10(1):1‐32. 10.1080/22221751.2021.1888660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Drugs F, Jeon S, Ko M, et al. Identification of antiviral drug candidates against SARS‐CoV‐2 from FDA‐approved drugs. Antimicrob Agents Chemother. 2020;64(7):e00819‐e00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Crespi D, Ivanier E, Genovese J, Baldi A. Mitoxantrone affects topoisomerase activities in human breast cancer cells. Biochem Biophys Res Commun. 1986;2:521‐528. [DOI] [PubMed] [Google Scholar]
- 78. Kapuscinski J, Darzynkiewicz Z. Interactions of antitumor agents ametantrone and mitoxantrone (novatrone) with double‐stranded DNA. Biochem Pharmacol. 1985;34(24):4203‐4213. [DOI] [PubMed] [Google Scholar]
- 79. Iwata‐Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. 2019;93(6):1‐15. 10.1128/jvi.01815-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vaarala MH, Porvari KS, Kellokumpu S, Kyllönen AP, Vihko PT. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J Pathol. 2001;193(1):134‐140. [DOI] [PubMed] [Google Scholar]
- 81. Shirato K, Kawase M, Matsuyama S. Wild‐type human coronaviruses prefer cell‐surface TMPRSS2 to endosomal cathepsins for cell entry. Virology. 2018;517:9‐15. 10.1016/j.virol.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mollica V, Rizzo A, Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020;16(27):1‐5. 10.2217/fon-2020-0571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Strope JD, PharmD CHC, Figg WD. TMPRSS2: potential biomarker for COVID‐19 outcomes. J Clin Pharmacol. 2020;60(7):801‐807. 10.1002/jcph.1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pranesh P, Rajat D, Narendra D. Targeting TMPRSS2 and cathepsin B/L together may be synergistic against SARS‐CoV‐2 infection. chemRxiv. 2020:1‐41. 10.26434/chemrxiv.12213125.v1 [DOI] [Google Scholar]
- 85. Liu T, Luo S, Libby P, Shi GP Cathepsin L‐selective inhibitors: a potentially promising treatment for COVID‐19 patients. Pharmacol Ther. 2020;213(February):107587. 10.1016/j.pharmthera.2020.107587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kawase M, Shirato K, Van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86(12):6537‐6545. 10.1128/jvi.00094-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW. TMPRSS2 and COVID‐19: serendipity or opportunity for intervention? Cancer Discov. 2020;10(6):779‐782. 10.1158/2159-8290.CD-20-0451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen KY. Coronaviruses‐drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327‐347. 10.1038/nrd.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bryce AH, Fonseca R. Targeting TMPRSS2 in SARS‐CoV‐2 infection. Mayo Clin Proc. 2020;95(9):1989‐1999. 10.1016/j.mayocp.2020.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hoffmann M, Schroeder S, Kleine‐Weber H, Müller MA, Drosten C, Pöhlmann S. Nafamostat mesylate blocks activation of SARS‐CoV‐2: new treatment option for COVID‐19. Antimicrob Agents Chemother. 2020;64(6):19‐21. 10.1128/AAC.00754-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun. 2020;11(1):1620. 10.1038/s41467-020-15562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shah PP, Wang T, Kaletsky RL, et al. A small‐molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol Pharmacol. 2010;78(2):319‐324. 10.1124/mol.110.064261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Engel BJC, Doyle PS, Hsieh I, Mckerrow JH. Cysteine protease inhibitors cure an experimental. Survival (Lond). 1998;188(4):725‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhang J, Ma X, Yu F, et al. Teicoplanin potently blocks the cell entry of 2019‐nCoV. bioRxiv. January 2020. 10.1101/2020.02.05.935387 [DOI] [Google Scholar]
- 95. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC‐376, and calpain inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678‐692. 10.1038/s41422-020-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS‐CoV‐2 main protease reveals strategy for developing dual inhibitors against Mpro and cathepsin L. bioRxiv. 2020;(December). 10.1101/2020.07.27.223727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hu Y, Ma C, Szeto T, Hurst B, Tarbet B, Wang J. Boceprevir, calpain inhibitors II and XII, and GC‐376 have broad‐spectrum antiviral activity against coronaviruses in cell culture. bioRxiv. 2020. 10.1101/2020.10.30.362335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Loganathan T, Ramachandran S, Shankaran P, Nagarajan D, Suma Mohan S. Host transcriptome‐guided drug repurposing for COVID‐19 treatment: a meta‐analysis based approach. PeerJ. 2020;2020(6):1‐27. 10.7717/peerj.9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ge Y, Tian T, Huang S, et al. A data‐driven drug repositioning framework discovered a potential therapeutic agent targeting COVID‐19. bioRXiv. 2020:1‐62. 10.1101/2020.03.11.986836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network‐based drug repurposing for novel coronavirus 2019‐nCoV/SARS‐CoV‐2. Cell Discov. 2020;6(1):14. 10.1038/s41421-020-0153-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459‐468. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Riva L, Yuan S, Yin X, et al. A large‐scale drug repositioning survey for SARS‐CoV‐2 antivirals. bioRxiv Prepr Serv Biol. 2020. 10.1101/2020.04.16.044016 [DOI] [Google Scholar]
- 103. Si L, Bai H, Rodas M, et al. Human organs‐on‐chips as tools for repurposing approved drugs as potential influenza and COVID19 therapeutics in viral pandemics. bioRxiv. 2020. 2020.04.13.039917. 10.1101/2020.04.13.039917 [DOI] [Google Scholar]
- 104. Xia S, Zhu Y, Liu M, et al. Fusion mechanism of 2019‐nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020;17(7):765‐767. 10.1038/s41423-020-0374-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kadam RU, Wilson IA. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci U S A. 2017;114(2):206‐214. 10.1073/pnas.1617020114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect Dis. 2020;71(15):769‐777. 10.1093/cid/ciaa272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mahmoud IS, Jarrar YB, Alshaer W, Ismail S. SARS‐CoV‐2 entry in host cells‐multiple targets for treatment and prevention. Biochimie. 2020;175:93‐98. 10.1016/j.biochi.2020.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xiu S, Dick A, Ju H, et al. Inhibitors of SARS‐CoV‐2 entry: current and future opportunities. J Med Chem. 2020;63(21):12256‐12274. 10.1021/acs.jmedchem.0c00502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhou N, Pan T, Zhang J, et al. Glycopeptide antibiotics potently inhibit cathepsin l in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS‐CoV), and severe acute respiratory syndrome coronavirus (SARS‐CoV). J Biol Chem. 2016;291(17):9218‐9232. 10.1074/jbc.M116.716100 [DOI] [PMC free article] [PubMed] [Google Scholar]