

Abstract
The SARS‐CoV‐2 virus has been rapidly spreading globally since December 2019, triggering a pandemic, soon after its emergence. While Iran was among the first countries confronted with rapid spread of virus in February 2020, no real‐time SARS‐CoV‐2 whole‐genome tracking in early phase of outbreak was performed in the country. To address this issue, we provided 50 whole‐genome sequences of viral isolates ascertained from different geographical locations in Iran during March–July 2020. The corresponding analysis on origins, transmission dynamics and genetic diversity of SARS‐CoV‐2 virus, represented at least two introductions of the virus into the country, constructing two major clusters defined as B.4 and B.1*. The first entry of the virus might have occurred around very late 2019/early 2020, as suggested by the time to the most recent common ancestor, followed by a rapid community transmission that led to dominancy of B.4 lineage in early epidemic till the end of June. Gradually, reduction in dominancy of B.4 occurred possibly as a result of other entries of the virus, followed by surge of B.1* lineages, as of mid‐May. Remarkably, variation tracking of the virus indicated the increase in frequency of D614G mutation, along with B.1* lineages, which showed continuity till October 2020. The increase in frequency of D614G mutation and B.1* lineages from mid‐May onwards predicts a rapid viral transmission that may push the country into a critical health situation followed by a considerable change in composition of viral lineages circulating in the country.
Keywords: COVID‐19, Iran, phylogenetic study, SARS‐CoV‐2, whole genome sequencing
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) pandemic caused by SARS‐CoV‐2 (Zhu et al., 2020) has, as of 23 October 2020, exceeded one million deaths, while infecting 42,334,976 people worldwide. Real‐time whole‐genome sequencing of this emerging virus was commenced at the early phase of outbreak globally, as it can accurately evaluate the magnitude of transmission, offering insights for management of the epidemic (Oude Munnink et al., 2020). Therefore, an increasing number of sequences are being deposited in the global initiative on sharing all influenza data (GISAID) (Shu & McCauley, 2017).
Iran was among the first countries confronting the rapid virus spread. The first COVID‐19 confirmed patient and death was reported on 19 February 2020, from Qom city. Local transmission to neighbouring provinces was reported just a day after and then the disease spread shortly Iran wide. The first outbreak peak dropped in April, but soon after relaxing the lockdown, the country experienced another notable increase in SARS‐CoV‐2 cases in May, which is still sustained. As of 23 October, 556,891 infected cases and 31,985 deaths have been reported officially and there is a concern about the increase in mortality rate in autumn and winter (https://www.worldometers.info/coronavirus/country/iran/).
Although the outbreak in Iran initiated early in February along with Italy and South Korea, no real‐time SARS‐CoV‐2 whole‐genome tracking was performed in the first months of epidemic. The first virus sequence from Iran was deposited in GISAID (EPI_ISL_424349) on April 4. In the same month, the study by Eden et al. revealed three major substitutions of G1397A, T28688C and G29742T in genomes of patients with travel history to Iran, which constitute a distinct clade representative of the specific viral diversity present in Iran at that time (Eden et al., 2020).
As of 23 October 2020, there were only eight complete genomes available in GISAID, not sufficient for tracking the virus in the country, and the only epidemiologic study of Iranian outbreak used the genomic sequences ascertained from travellers to Iran, which estimated 21/01/2020 (95% HPD: 05/12/2019–14/02/2020) as the start of epidemic in Iran with a doubling time of 3 days (95% HPD: 1.68–16.27) (Ghafari et al., 2020). To address this issue, we performed genome sequencing of 50 SARS‐CoV‐2 samples ascertained from different geographical locations and especially in early time intervals of the epidemic in Iran. We aimed at improving the understanding of the origins and transmission dynamics, circulating lineages and variation tracking of SARS‐CoV‐2 outbreak at early phase of Iranian epidemic, using molecular and phylogenetic methods.
2. MATERIALS AND METHODS
2.1. Specimen recruitment
We recruited 50 SARS‐CoV‐2 RNA samples, obtained as part of clinical testing in different referral centres of the following provinces: Alborz (n = 2), Ardabil (n = 4), Gilan (n = 6), Tehran (n = 27), Khuzestan (n = 3), Qom (n = 1), Sistan and Baluchestan (n = 3) and South Khorasan (n = 4). All patients were referred between March and July 2020, with clinical presentations of COVID‐19 disease, confirmed by real‐time RT‐PCR assay at those corresponding local centres.
2.2. Sequencing and Genome assembly
Whole‐genome sequencing of SARS‐CoV‐2 RNA samples was performed by targeted enrichment using CleanPlex® SARS‐CoV‐2 Research and Surveillance Panel (Paragon Genomics, Inc.). All samples were paired‐end sequenced on Illumina MiSeq instrument using 300‐cycle MiSeq v2 reagent kits (Illumina, Inc.), generating 5.4 Gb of data (94.5% of bases > = Q30). Initially, FASTQ files were assessed by FastQC (Andrews, 2010) and then pre‐processed using Fastp (Chen et al., 2018). The sequences were aligned to the SARS‐CoV‐2 reference genome (NC_045512.2) using Bowtie2 (Langmead & Salzberg, 2012) and keeping the reads mapped in proper pair. The resultant filtered BAM files were used for assembly of consensus SARS‐CoV‐2 sequences with Samtools mpileup and Bcftools (Li et al., 2009.). Finally, the consensus FASTQ files were converted into FASTA format by Seqtk (https://github.com/lh3/seqtk), masking bases with quality lower than 20 to ambiguous nucleotides (N).
2.3. Lineage assignment
In addition to 50 sequenced samples in this project, eight other SARS‐CoV‐2 Iranian sequences from GISAID were subjected to lineage assignment. We applied Pangolin v2.0.7 (Rambaut et al., 2020), CoV‐GLUE (Singer et al., 2020) and NextClade v.0.6.0 (Hadfield et al., 2018) to assign the global lineages present in Iranian SARS‐CoV‐2 outbreak.
2.4. Phylogenetic analysis
BEAST v1.10.4 was used to construct a phylogenetic tree and to estimate the most recent common ancestor (TMRCA) (Drummond & Rambaut, 2007). First, consensus sequences were evaluated by NextClade (Hadfield et al., 2018), and sequences containing >5% ambiguous nucleotides and bearing private mutations above the threshold were excluded from downstream analysis. To explore the temporal signal, high‐quality FASTA files were aligned by MAFFT v7.407 using the FFT‐NS‐2 algorithm (Katoh & Standley, 2013), and then, a maximum‐likelihood phylogenetic tree was built applying IQ‐TREE v2.1.1 with GTR + gamma model (Minh et al., 2020) and temporal signal was then explored by TempEst (Rambaut et al., 2016).
Eventually, BEAST was used to estimate TMRCA and construct a Bayesian phylogenetic tree of 45 sequences, plus Wuhan‐1 patient (EPI_ISL_402125) as outgroup, using a simple model consisting of HKYγ codon partitioned 1 + 2, 3 substitution model, strict clock and coalescent exponential growth tree prior. Maximum clade credibility (MCC) tree was then made with 10% burn‐in from two separate Markov chain‐Monte Carlo runs (Drummond and Rambaut, 2007).
To trace possible sources of SARS‐CoV‐2 entry into Iran, an additional phylogenetic tree was constructed based on a total of 261 samples including a list of high‐quality genomes in GISAID (Rambaut, 2020) from the start of epidemic till the end of February, and random subsets of samples in GISAID during March–June interval, that were selected using mothur (Schloss et al., 2009).
2.5. Variant analysis
Variant analysis was performed by CoV‐GLUE, relative to the reference sequence (NC_045512.1) (Singer et al., 2020). In total, 53 samples were investigated after excluding the sequences bearing private mutations above the threshold. Moreover, to track the renowned D614G mutation frequency after July 2020, sanger sequencing of additional 67 SARS‐CoV2 positive samples collected during July–October, was performed using the following primer pairs designed by ARTIC network (https://github.com/artic‐network/artic‐ncov2019/tree/master/primer_schemes/nCoV‐2019/V3); 5ʹ‐CCAGCAACTGTTTGTGGACCTA‐3ʹ, 5ʹ ‐CAGCCCCTATTAAACAGCCTGC‐3ʹ.
3. RESULTS
3.1. Genome assembly and data availability
In this study, we obtained 44 high‐quality SARS‐CoV‐2 sequences (>98% of the genome is complete) from the Iranian outbreak. The remaining six samples covered >82% of the NC_045512.2 genome sequence. The metadata information including the geographical location, collection date, age, gender, CT value, specimen source, percentage of ambiguous nucleotides (N) for each genome, percentage of genome coverage compared to NC_045512.2 and the exact lineages defined for each sample are provided in Table S1.
3.2. Lineage assignment
Lineage assignment with Pangolin and CoV‐GLUE, although slightly different for some samples, both yielded B.4 as the dominant lineage circulating in Iranian SARS‐CoV‐2 outbreak; comprising 75.9% and 74%, respectively (see Figure 1a for spectrum of circulating lineages and Table S1 for exact lineage of each sample).
FIGURE 1.

(a) Lineages assignment of 58 SARS‐CoV‐2 sequences from the Iranian outbreak. Abundance of SARS‐CoV‐2 lineages over time from March to the end of June 2020 indicates a reduction in dominancy of the B.4 lineage. Trend of circulating lineages assigned by (b) Pangolin v2.0.7, C. CoV‐GLUE and D. NextClade v0.6.0
This is consistent with previous reports (Eden et al., 2020), as also the majority (83%) of SARS‐CoV‐2 sequences in GISAID that were primarily exposed in Iran, belonged to the B.4 lineage (Table S2). Additionally, the allocated lineages provided by NextClade showed a dominancy of 19A major clade (77.6%), one of the most prevalent clades during the early phase of outbreak, especially in Asia. The dominancy of this clade in the Iranian outbreak is consistent with Iran being one of the first countries infected by the virus.
Therefore, our results confirm B.4 as the dominant lineage in Iranian outbreak during the February–June 2020 interval and introduce B, B.1, B.1.* and B.4 as the circulating lineages in the country.
Sequencing more SARS‐CoV‐2 samples from the end of June onwards is required to evaluate whether B.4 still persists as the dominant lineage. However, as shown in Figure 1, the current data already exhibit a trend towards the appearance of other SARS‐CoV‐2 lineages and reduction in dominancy of B.4. As of May, the 20A clade, being the dominant European clade in early 2020, started to appear and more B.1* lineages can be observed in Iranian epidemic. This is explicable by new sources of virus introductions to the country at that time interval. Its appearance could also be due to SARS‐CoV‐2 genome mutations in the existing Iranian B lineages, which were circulating alongside the B.4 lineage in early phase of the epidemic, although with a lower proportion.
3.3. Phylogenetic analysis
3.3.1. SARS‐CoV‐2 entry and circulating lineages
The phylogenetic tree of SARS‐CoV‐2 genomes from the Iranian outbreak clearly revealed two different circulating clusters (Figure 2), suggesting at least two separate introductions into the country.
FIGURE 2.
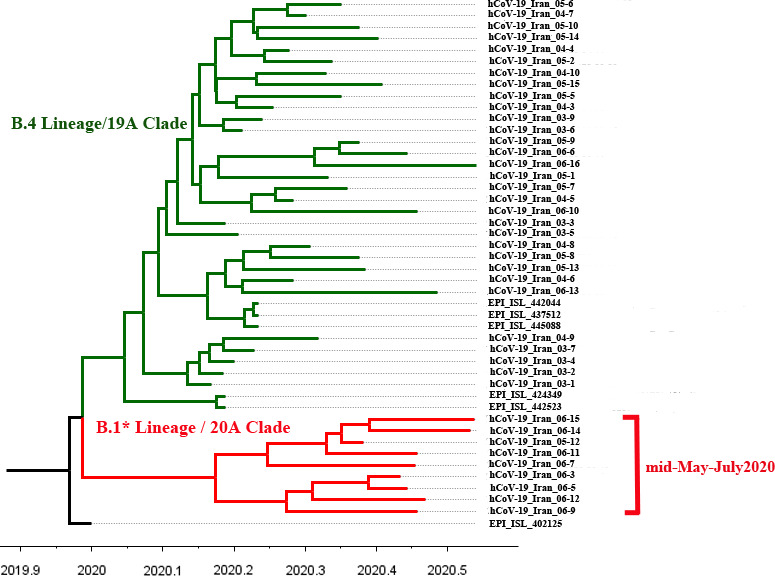
Tempo‐spatial phylogenetic tree of SARS‐CoV‐2 emergence in Iran
The older green cluster is comprised of 36 genomes almost all of the B.4 lineage [B.4/19A], carrying [G1397A‐T28688C‐G29742T] substitutions (Eden et al., 2020). These genomes were spread across different geographical regions including as follows: Alborz (n = 2), Gilan (n = 4), Khuzestan (n = 3), Qom (n = 1), Sistan and Baluchestan (n = 3), Semnan (n = 1), South Khorasan (n = 1), Tehran (n = 18) and unknown (n = 3). This indicates that the [B.4/19A] cluster originated very early in 2020 the latest and began circulating around the country thereafter, reflecting multiple local transmissions. Additionally, there are two samples with [B/19A] lineage in this older cluster. These samples were collected back in early March, showing that in the first phase of pandemic; at least two different lineages entered into the country. The [B/19A] samples also carry the G1397A and G29742T substitutions but not T28688C.
The red cluster is comprised of nine genomes the entire [B.1.*/20A] lineage and collected after May 15, compatible with the pattern shown in Figure 1. The [B.1.*/20A] samples did not show [G1397A‐T28688C‐G29742T] substitutions but instead harboured [C241T‐C3037T‐C14408T‐A23403G] or [C241T‐C3037T‐C14408T‐A23403G‐G25563T], which are the common patterns of variant co‐occurrence of B.1 and B.1.* lineages in Europe and North America (Mercatelli & Giorgi, 2020).
3.3.2. TMRCA estimates
The TMRCA of B4 clade was estimated as 29–12–2019 with 95% highest posterior density (HPD) intervals of [03–11–2019 to 06–02–2020], considering Wuhan‐1 sample (EPI_ISL_402125) as an outgroup. Additionally, to track the appearance of [B.1.*/20A] cluster in the country, the TMRCA of the nine B.1 samples, placing all the other genomic samples in this study as outgroup was estimated. The TMRCA was 22–02–2020, with HPD intervals of [12–01–2020 to 29–03–020]. Clearly, more high‐quality B.1* genomes are required to narrow the credible interval and predict a more precise TMRCA for entry of this lineage. However, the above results still indicate that the new lineage might have been introduced separately, after the entry of B.4, and then gradually increased in the population, becoming detectable in our cohort since mid‐May.
3.3.3. Sources of SARS‐CoV‐2 entries
Subsequent analysis in the context of 216 genomes from around the world clarified the location of two main clusters among the global samples (Figure 3).
FIGURE 3.

Radial phylogenetic tree of SARS‐CoV‐2 genomes from the Iranian outbreak in the context of 216 set of genomes from around the world. The major B.4 cluster in the Iranian SARS‐CoV‐2 outbreak is highlighted in light green. The [B.1.*/20A] cluster is highlighted in light red. The red circles denote the two B.4 samples from China located near the major Iranian cluster. The green circles denote the five B.4 samples from Australia, Canada and New Zealand located within the major Iranian cluster
As expected, the B.4 cluster was linked to the very early samples collected in January–February and mostly in China. This supports the hypothesis of an early virus introduction to Iran and most likely from China, which is consistent with Iran's health ministry statements that the virus was brought from China by travellers (https://en.wikipedia.org/wiki/COVID‐19_pandemic_in_Iran). Interestingly, the Iranian B.4 cluster is closely linked to the two B.4 samples collected on mid‐January (19–01–2020 and 18–01–2020) in Hubei/Wuhan and Shandong/Qingdao in China (EPI_ISL_408482 and EPI_ISL_412981). This suggests that the three [G1397A‐T28688C‐G29742T] substitutions might have occurred before their introduction to Iran, subsequently becoming the major lineage and driving the epidemic in the country. Afterwards, the virus was transferred to the other countries, such as Canada, Australia and New Zealand, by travellers (Eden et al., 2020). This is now confirmed by locating five samples from these countries within the major Iranian cluster (EPI_ISL_412965, EPI_ISL_413213, EPI_ISL_412975, EPI_ISL_413214 and EPI_ISL_413490). All these five samples were collected in late February having a travel history to Iran (Figure 3, Figure 4).
FIGURE 4.
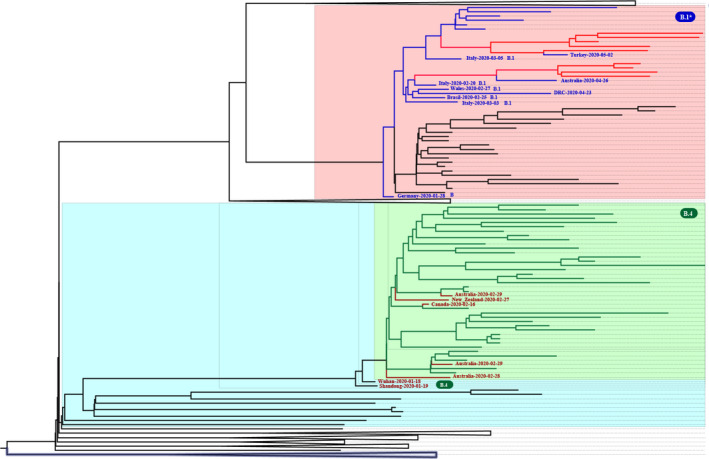
The zoomed and collapsed phylogenetic tree of SARS‐CoV‐2 genomes from the Iranian outbreak in the context of 216 set of genomes from around the world. The two main clusters circulating in Iran are zoomed. The major B.4 cluster in SARS‐CoV‐2 Iranian outbreak is highlighted in light green and the lines corresponding to Iranian samples within this cluster are also shown in green while other global samples are shown in red lines. The [B.1.*/20A] cluster is highlighted in light red, and the lines corresponding to Iranian samples within this cluster are also shown in red, while other global samples are shown in blue lines
The [B.1*/20A] cluster localizes in a completely different position, namely among the samples from various parts of the world, prominently Europe. This corroborates the hypothesis of new sources of virus introduction to the country (probably in late February as suggested by TMRCA of B.1* samples) before suspension of air flights; as the international travel lockdown was started at 23 February 2020 for some neighbouring countries and expanded until 8 March 2020 by more countries. Furthermore, important foreign and Iranian airlines transferring passengers between Iran, Europe and North America were suspended on February 25 and March 8, respectively.
3.4. Variant analysis of SARS‐CoV‐2 genomes from Iranian outbreak
3.4.1. Common variants
We detected 14 different variants with >10% frequency in SARS‐CoV‐2 genomes from Iranian outbreak (Table 1). Notably, just four variants, namely G1397A, T28688C, G29742T and G11083T, contributed to >70% of samples; comprising the common co‐occurrence of variants, [G1397A‐T28688C‐G29742T] in B.4 lineage.
TABLE 1.
Common and novel variants observed in SARS‐CoV‐2 genomes of the Iranian outbreak
| No | Genomic change | Type of mutation | Gene/protein | Amino acid change | No. of samples | Sample lineages | Description |
|---|---|---|---|---|---|---|---|
| Common SARS‐CoV−2 variants observed in early phase of Iranian outbreak | |||||||
| 1 | G1397A | Non‐synonymous | nsp2 | V198I | 43 (81%) | B.4, B |
Known coexistence of variants, constituting B.4 lineage, as suggested by Eden et al., 2020. |
| 2 | T28688C | Synonymous | N | L139L | 41 (77%) | B.4 | |
| 3 | G29742T | Non‐coding | 3ʹUTR | NA | 43 (81%) | B.4, B | |
| 4 | G11083T | Non‐synonymous | nsp6 | L37F | 39 (74%) | B.4, B | The most common mutation in Asia, during December 2019‐March 2020. |
| 5 | C241T | Non‐coding | 5ʹ‐UTR | NA | 10 (19%) | B.1* |
Known coexistence of variants, constituting B.1 (G) and B.1.* (GH) clades, according to Mercatelli & Giorgi, 2020. |
| 6 | C3037T | Synonymous | nsp3 | F106F | 10 (19%) | B.1* | |
| 7 | C14408T | Non‐synonymous | nsp12 (RdRp) | P323L | 10 (19%) | B.1* | |
| 8 | A23403G | Non‐synonymous | S | D614G | 13 (24.5%) | B.1* (n = 10), B.4 (n = 3) | |
| 9 | G25563T | Non‐synonymous | ORF3a | Q57H | 10 (19%) | B.1*, B.4, B | |
| Unique SARS‐CoV−2 haplotypes observed in early phase of Iranian outbreak | |||||||
| 10 | G20887A | Non‐synonymous | nsp16 | G77R | 9 (17%) | B.4 |
New coexistence of variants observed in same nine samples also carrying B.4 common variants. |
| 11 | C28830T | Non‐synonymous | N | S186F | 9 (17%) | B.4 | |
| 12 | C21627T | Non‐synonymous | S | T22I | 9 (17%) | B.4 | |
| 13 | G8653T | Non‐synonymous | nsp4 | M33I | 6 (11%) | B.4 |
New coexistence of variants observed in same six samples also carrying B.4 common variants. |
| 14 | C884T | Non‐synonymous | nsp2 | R27C | 6 (11%) | B.4 | |
| Unique SARS‐CoV−2 variants observed in early phase of Iranian outbreak | |||||||
| 15 | C28388G | Non‐synonymous | N | Q39E | 1 | B.1.1/20B | Known variants located at the same position: Q39L/ Q39H/ Q39R/ Q39* |
| 16 | G18712A | Non‐synonymous | nsp14 | A225T | 1 | B.4/19A | Known variants located at the same position: A225D / A225S |
| 17 | T3926C | Non‐synonymous | nsp3 | S403P | 1 | B.4/19A | Known variants located at the same position: S403L / S403A |
| 18 | G6461A | Non‐synonymous | nsp3 | V1248M | 1 | B.4/19A | Known variants located at the same position: V1248G / V1248L |
Abbreviations: N, nucleocapsid phosphoprotein; nsp14, 3'‐to‐5' exonuclease; nsp16, 2'‐O‐ribose methyltransferase; nsp2, Non‐Structural protein 2; nsp3, Predicted phosphoesterase, papain‐like proteinase; nsp4, Transmembrane protein; nsp6, Transmembrane protein; ORF, open reading frame; RdRp, RNA‐dependent RNA polymerase; S, Spike glycoprotein.
G11083T is one of the most frequent mutations observed in Asia from December 2019 to March 2020 (Koyama et al., 2020; Mercatelli and Giorgi, 2020) and, not surprisingly, is observed mostly along with B.4 substitutions.
Other less frequent variants were observed in ~19% of samples constituting the known co‐occurrence of variants; [C241T‐C3037T‐C14408T‐A23403G] and [C241T‐C3037T‐C14408T‐A23403G‐G25563T], occurring in clade G (B.1), which is prevalent in Europe, Oceania, South America and Africa and clade GH (B.1.*), prevalent in North America (Mercatelli and Giorgi, 2020).
3.4.2. Novel variants
Remarkably, we observed two specific haplotypes; the co‐occurrence of [G20887A‐C28830T‐C21627T] and [G8653T‐C884T] variants in 17% and 11% of samples, respectively. Both groups also carried B.4 [G1397A‐T28688C‐G29742T] variants. These haplotypes are less frequent in CoV‐GLU and 48,635 genomes investigated by Mercatelli & Giorgi (Mercatelli and Giorgi, 2020; Singer et al., 2020). However, further investigations are required to assess their extent of significance into SARS‐CoV2 genetic diversity in Iran. Analysis of viral isolates also revealed four samples harbouring unique variants (Table 1), not detected in sequences from SARS‐CoV‐2 pandemic (Mercatelli and Giorgi, 2020; Singer et al., 2020). Although the exact variants were unique, different missense variants at the same location were identified in other viral sequences around the world.
3.4.3. Variants located in spike protein
Spike is the key glycoprotein mediating entry of the virus to the cell and, therefore, the target of most vaccine strategies (Korber et al., 2020). We thus focussed on variants located in spike protein of viral isolates from Iran and identified spike variants in 28 samples (53%), in which D614G and T22I were occurring at higher frequencies of 24.5% and 17%, respectively (Table 2). T22I mutation was only observed in the [B.4/19A] cluster. D614G, the most prevalent mutation globally, was also the most prevalent spike mutation in viral isolates from Iran, while showing an increasing trend from mid‐May, observed mostly within the [B.1.*/20A] cluster. Remarkably, co‐occurrence of D614G mutation with B.4 [G1397A‐T28688C‐G29742T] and G11083T variants was also observed in three samples.
TABLE 2.
Variants located in the spike, observed in SARS‐CoV‐2 genomes of the Iranian outbreak
| Genomic change | Type of mutation | Gene/protein | Amino acid change | No. of samples | Sample lineages |
|---|---|---|---|---|---|
| A23403G | Non‐synonymous | S (S1) | D614G | 13 (24.5%) | B.1*/20A, B.1*/20B, B.4/19A |
| C21627T | Non‐synonymous | S (S1) | T22I | 9 (17%) | B.4/19A |
| G22100A | Non‐synonymous | S (S1) | E180K | 2 (4%) | B.4/19A |
| G22592T | Non‐synonymous | S (S1‐RBD Domain) | A344S | 1 (2%) | B.4/19A |
| C23679T | Non‐synonymous | S (S2) | A706V | 1 (2%) | B.4/19A |
| G24348T | Non‐synonymous | S (S2) | S929I | 1 (2%) | B.4/19A |
| G25249T | Non‐synonymous | S (S2) | M1229I | 1 (2%) | B.4/19A |
Abbreviation: S, spike glycoprotein.
Moreover, Sanger sequencing of additional 67 SARS‐CoV2 positive samples confirmed the increase in D614G frequency till October, becoming the dominant mutation in the Iranian outbreak (Figure 5).
FIGURE 5.

The frequency of D614G mutation during March–October interval in Iranian SARS‐CoV‐2 outbreak
4. DISCUSSION
The current study is the first comprehensive analysis of SARS‐CoV‐2 full genomes obtained from Iranian outbreak. Regarding the importance of real‐time sequencing of emerging viruses, and lack of full genomes from Iran, this study was designed to provide 50 SARS‐CoV‐2 full genome sequences of the early time interval of epidemic in Iran.
Lineage assignment and phylogenetic analysis of these sequences clarified the origins and transmission dynamics of SARS‐CoV‐2 outbreak in Iran, in which two major introductions into the country were detected, constructing two major clusters of the virus in different times, followed by rapid community transmission throughout the country.
These data confirm the B.4 as the dominant lineage in Iran, from the start of epidemic till the end of June. This lineage was primarily introduced as a prevalent distinct clade in Australian travellers from Iran in early April (Eden et al., 2020). Since then, it is recognized as the Iranian epidemic. Nonetheless, no specific study on Iranian SARS‐CoV‐2 samples was performed so far, which is addressed in this study.
Furthermore, the B.4 clade TMRCA in this study along with the previous epidemiologic study that used genomic samples ascertained from travellers to Iran (Ghafari et al., 2020) may propose the possibility of unrecognized transmission of the virus in the country due to presence of asymptomatic or misdiagnosed patients or limited testing capacities for more than 1 month prior to official report of first COVID‐19 patients in the country.
Therefore, the B.4 lineage originated first in late 2019/early 2020 followed by multiple local transmissions, developing the major SARS‐CoV‐2 clade in the beginning of outbreak. Our data suggest a reduction in B.4 dominancy, followed by a surge of B.1.* lineages, which has been exported from Europe globally. This new cluster might be explained by new sources of virus entries before suspension of flights in late February or being the mutant product of B lineage in early phase.
In addition to outbreak tracing, these full genome sequences can provide beneficial information in understanding the genome diversity of viral isolates in Iran, helpful in adapting more specific diagnostic tests, therapeutic approaches and vaccines.
Generally, RNA viruses are known to have a high mutation rate, explained by lack of proofreading activity of RNA‐dependent RNA polymerase (RdRp). However, coronaviruses are among the exceptions, with lower mutation rates due to presence of RdRp‐independent proofreading activity (Ahmadpour et al., 2020; Peck and Lauring, 2018). The mutation rate of 1.12 × 10–3 mutations per site year (Koyama et al., 2020) and the average 7.23 mutations per sample (Mercatelli and Giorgi, 2020) are in support of moderate mutation rate of SARS‐CoV‐2. Variation tracking of SARS‐CoV‐2 has shown that some mutations—such as P323L and D614G—are distributed globally, while some others are accumulated in specific geographical regions (Kannan et al., 2020).
Investigation of prevalent mutations in early phase of outbreak in Iran indicated the co‐occurrence of some widespread mutations consistent with the two main lineages in the country. Furthermore, the unique mutations and also haplotypes of [G20887A‐C28830T‐C21627T] or [G8653T‐C884T] with [G1397A‐T28688C‐G29742T] were identified in low proportion of samples. Therefore, none of those could be considered as adapted geographically in Iranian SARS‐CoV‐2 samples. Indeed, massive sequencing of a larger cohort is a requisite for investigating the significance of these variants. Moreover, the impact of these country‐specific variants (yet to be defined) on the behaviour of virus (replication efficiency, virulence. etc.) needs further investigation.
Despite the relatively low mutation rate of SARS‐CoV‐2, still a total of 353,341 mutations were identified in 48,635 SARS‐CoV‐2 genomes (Mercatelli and Giorgi, 2020). Among these, studying the mutations in spike protein is necessary, as this immunogenic structural protein mediates the virus entry to the host cells via interacting with cellular receptors such as angiotensin‐converting enzyme 2 (ACE2) and as it plays a key role in induction of neutralizing antibodies (Dearlove et al., 2020 ; Franco‐Muñoz et al., 2020). This glycoprotein is composed of two functional subunits, S1 and S2.
S1 contains a receptor‐binding domain (RBD) through which SARS‐Co‐2 binds to the ACE2 receptor, while S2 is responsible for virus‐host membrane fusion (Yang et al., 2020). Furthermore, the spike protein is the target for many vaccine candidates currently in development, and therefore, tracking the mutations in this protein (especially RBD region) is crucial (Dearlove et al., 2020 ).
Monitoring the spike mutations in Iranian SARS‐CoV‐2 genomes revealed no novel mutations in this genomic region, while determined two commonly known mutations, T22I and D614G. Both of these variants are outside RBD region, suggesting no negative effect for efficacy of the future vaccines on the viral lineages currently circulating in Iran (Dearlove et al., 2020 ).
While the T22I mutation is less frequent in other regions, the D614G variant is now the most prevalent mutation in COVID‐19 pandemic (Ahmadpour et al., 2020; Korber et al., 2020; Mercatelli and Giorgi, 2020). Recent studies suggested a fitness advantage for G614, rapidly making it the dominant form in each geographical location (Korber et al., 2020). A significant conformational change in spike protein may lead to more feasible virus‐host cell membrane fusion. As a consequence, increased infectivity, transmission and replication fitness is reported for G614 (Hu et al., 2020), but there are still some debates about its transmission effect (Grubaugh et al., 2020 , van Dorp et al., 2020). Hopefully, the mutation is not related to disease severity and does not reduce the effect of neutralizing antibodies (Korber et al., 2020; Fau et al., 2021.).
Notably, in accordance with the increasing [B.1*/20A] clade among the viral isolates of the Iranian epidemic as of mid‐May, the D614G variant is dominating in this population. This can partly explain the accelerated transmission of the virus in recent months, although the negative effect of relaxed quarantine policies should not be overlooked. Due to the strong fitness of G614, we could also observe its co‐occurrence with common B.4 variants, while the mutation is known as always co‐occurring with variants defining the G clade (Mercatelli and Giorgi, 2020).
In conclusion, genomic sequencing and phylogenetic analysis suggested that SARS‐CoV‐2 entered in very late 2019/early 2020 in Iran and circulated among vulnerable patients. The increase in frequency of D614G mutation and B.1* lineages from mid‐May onwards predicts a rapid viral transmission followed by considerable change in the composition of viral lineages circulating in the country.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
ETHICS STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received (Institutional ethical approval number of IR.USWR.REC.1399.094).
Supporting information
Table S1
Table S2
ACKNOWLEDGMENTS
Iranian Network for Research in Viral Diseases (INRVD) is acknowledged for help with the sample and data collection through its collaborative research centres and hospitals. This study was funded by Iran Vice deputy for Research and Technology at Iran Ministry of Health and Medical Education, grant number: 99/801/A/6/7586.
Fattahi Z, Mohseni M, Jalalvand K, et al. SARS‐CoV‐2 outbreak in Iran: The dynamics of the epidemic and evidence on two independent introductions. Transbound Emerg Dis. 2022;69:1375–1386. 10.1111/tbed.14104
Joint first authors: Zohreh Fattahi and Marzieh Mohseni.
Seyed Mohammad Jazayeri and Hossein Najmabadi contributed equally to this manuscript.
Contributor Information
Seyed Mohammad Jazayeri, Email: jazayeri42@gmail.com.
Hossein Najmabadi, Email: hnajm12@yahoo.com.
REFERENCES
- Ahmadpour, D. , Ahmadpoor, P. , & Rostaing, L. (2020). Impact of Circulating SARS‐CoV‐2 Mutant G614 on the COVID‐19 Pandemic. Iranian journal of kidney diseases, 14(5), 331–334. [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Chen, S. , Zhou, Y. , Chen, Y. , & Gu, J. (2018). fastp: An ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34(17), i884–i890. 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearlove, B. , Lewitus, E. , Bai, H. , Li, Y. , Reeves, D. B. , Joyce, M. G. , Scott, P. T. , Amare, M. F. , Vasan, S. , Michael, N. L. , Modjarrad, K. , & Rolland, M. (2020). A SARS‐CoV‐2 vaccine candidate would likely match all currently circulating variants. PNAS, 117(38), 23652–23662. 10.1073/pnas.2008281117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden, J.‐S. , Rockett, R. , Carter, I. , Rahman, H. , de Ligt, J. , Hadfield, J. , Storey, M. , Ren, X. , Tulloch, R. , Basile, K. , Wells, J. , Byun, R. , Gilroy, N. , O’Sullivan, M. V. , Sintchenko, V. , Chen, S. C. , Maddocks, S. , Sorrell, T. C. , Holmes, E. C. , … Arnott, A. (2020). An emergent clade of SARS‐CoV‐2 linked to returned travellers from Iran. Virus Evol, 6(1), veaa027. 10.1093/ve/veaa027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco‐Muñoz, C. , Álvarez‐Díaz, D. A. , Laiton‐Donato, K. , Wiesner, M. , Escandón, P. , Usme‐Ciro, J. A. , Franco‐Sierra, N. D. , Flórez‐Sánchez, A. C. , Gómez‐Rangel, S. , Rodríguez‐Calderon, L. D. , Barbosa‐Ramirez, J. , Ospitia‐Baez, E. , Walteros, D. M. , Ospina‐Martinez, M. L. , & Mercado‐Reyes, M. (2020). Substitutions in Spike and Nucleocapsid proteins of SARS‐CoV‐2 circulating in South America. Infection, Genetics and Evolution, 85, 104557. 10.1016/j.meegid.2020.104557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafari, M. , Hejazi, B. , Karshenas, A. , Dascalu, S. , Ferretti, L. , Ledda, A. , & Katzourakis, A. (2020). Ongoing outbreak of COVID‐19 in Iran: Challenges and signs of concern with under‐reporting of prevalence and deaths: Research square. Research square. 10.21203/rs.3.rs-32500/v2 [DOI] [Google Scholar]
- Grubaugh, N. D. , Hanage, W. P. , & Rasmussen, A. L. (2020). Making Sense of Mutation: What D614G Means for the COVID‐19 Pandemic Remains Unclear. Cell, 182(4), 794–795. 10.1016/j.cell.2020.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield, J. , Megill, C. , Bell, S. M. , Huddleston, J. , Potter, B. , Callender, C. , Sagulenko, P. , Bedford, T. , & Neher, R. A. (2018). Nextstrain: Real‐time tracking of pathogen evolution. Bioinformatics, 34(23), 4121–4123. 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , He, C. L. , Gao, Q. Z. , Zhang, G. J. , Cao, X. X. , Long, Q. X. , … Huang, A. L. (2020). The D614G mutation of SARS‐CoV‐2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. bioRxiv. 10.1101/2020.06.20.161323 [DOI] [Google Scholar]
- Kannan, S. R. , Spratt, A. N. , Quinn, T. P. , Heng, X. , Lorson, C. L. , Sönnerborg, A. , Byrareddy, S. N. , & Singh, K. (2020). Infectivity of SARS‐CoV‐2: There is something more than D614G? Journal of Neuroimmune Pharmacology, 15(4), 574–577. 10.1007/s11481-020-09954-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber, B. , Fischer, W. M. , Gnanakaran, S. , Yoon, H. , Theiler, J. , Abfalterer, W. , Hengartner, N. , Giorgi, E. E. , Bhattacharya, T. , Foley, B. , Hastie, K. M. , Parker, M. D. , Partridge, D. G. , Evans, C. M. , Freeman, T. M. , de Silva, T. I. , McDanal, C. , Perez, L. G. , Tang, H. , … Wyles, M. D. (2020). Tracking changes in SARS‐CoV‐2 spike: Evidence that D614G increases infectivity of the COVID‐19 virus. Cell, 182(4), 812–827.e819. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama, T. , Platt, D. , & Parida, L. (2020). Variant analysis of SARS‐CoV‐2 genomes. Bulletin of the World Health Organization, 98(7), 495–504. 10.2471/blt.20.253591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The Sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercatelli, D. , & Giorgi, F. M. (2020). Geographic and Genomic Distribution of SARS‐CoV‐2 Mutations. Frontiers in Microbiology, 11, 1800. 10.3389/fmicb.2020.01800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B. Q. , Schmidt, H. A. , Chernomor, O. , Schrempf, D. , Woodhams, M. D. , von Haeseler, A. , & Lanfear, R. (2020). IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530–1534. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink, B. B. , Nieuwenhuijse, D. F. , Stein, M. , O’Toole, Á. , Haverkate, M. , Mollers, M. , Kamga, S. K. , Schapendonk, C. , Pronk, M. , Lexmond, P. , van der Linden, A. , Bestebroer, T. , Chestakova, I. , Overmars, R. J. , van Nieuwkoop, S. , Molenkamp, R. , van der Eijk, A. A. , GeurtsvanKessel, C. , Vennema, H. , … Koopmans, M. (2020). Rapid SARS‐CoV‐2 whole‐genome sequencing and analysis for informed public health decision‐making in the Netherlands. Nature Medicine, 26(9), 1405–1410. 10.1038/s41591-020-0997-y [DOI] [PubMed] [Google Scholar]
- Peck, K. M. , & Lauring, A. S. (2018). Complexities of viral mutation rates. Journal of virology, 92(14), e01031‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante, J. A. , Liu, Y. , Liu, J. , Xia, H. , Johnson, B. A. , Lokugamage, K. G. , Zhang, X. , Muruato, A. E. , Zou, J. , Fontes‐Garfias, C. R. , Mirchandani, D. , Scharton, D. , Bilello, J. P. , Ku, Z. , An, Z. , Kalveram, B. , Freiberg, A. N. , Menachery, V. D. , Xie, X. , Plante, K. S. , Weaver, S. C. , & Shi, P. Y. (2021). Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature, 592, 116–121. 10.1101/2020.09.01.278689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. (2020). Phylodynamic analysis | 176 genomes | 6 Mar 2020. https://virological.org/t/phylodynamic‐analysis‐176‐genomes‐6‐mar‐2020/356
- Rambaut, A. , Holmes, E. C. , O’Toole, Á. , Hill, V. , McCrone, J. T. , Ruis, C. , du Plessis, L. , & Pybus, O. G. (2020). A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nature Microbiology, 5(11), 1403–1407. 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Lam, T. T. , Max Carvalho, L. , & Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evolution, 2(1), vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , Lesniewski, R. A. , Oakley, B. B. , Parks, D. H. , Robinson, C. J. , Sahl, J. W. , Stres, B. , Thallinger, G. G. , Van Horn, D. J. , & Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environment Microbiology, 75(23), 7537–7541. 10.1128/aem.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y. , & McCauley, J. (2017). GISAID: Global initiative on sharing all influenza data ‐ from vision to reality. Eurosurveillance Weekly, 22(13), 30494–30494. 10.2807/1560-7917.es.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, J. G. R. , Cotten, M. , & Robertson, D. (2020). CoV‐GLUE: A Web Application for Tracking SARS‐CoV‐2 Genomic Variation. Preprints, 2020060225, 10.20944/preprints202006.0225.v1 [DOI] [Google Scholar]
- van Dorp, L. , Richard, D. , Tan, C. C. , Shaw, L. P. , Acman, M. , & Balloux, F. (2020). No evidence for increased transmissibility from recurrent mutations in SARS‐CoV‐2. Nature Communications, 11(1), 5986. 10.1101/2020.05.21.108506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Petitjean, S. J. L. , Koehler, M. , Zhang, Q. , Dumitru, A. C. , Chen, W. , Derclaye, S. , Vincent, S. P. , Soumillion, P. , & Alsteens, D. (2020). Molecular interaction and inhibition of SARS‐CoV‐2 binding to the ACE2 receptor. Nature communications, 11(1), 4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N. A. , Zhang, D. , Wang, W. , Li, X. , Yang, B. O. , Song, J. , Zhao, X. , Huang, B. , Shi, W. , Lu, R. , Niu, P. , Zhan, F. , Ma, X. , Wang, D. , Xu, W. , Wu, G. , Gao, G. F. , & Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2