

Summary
The global propagation of SARS‐CoV‐2 and the detection of a large number of variants, some of which have replaced the original clade to become dominant, underscores the fact that the virus is actively exploring its evolutionary space. The longer high levels of viral multiplication occur – permitted by high levels of transmission –, the more the virus can adapt to the human host and find ways to success. The third wave of the COVID‐19 pandemic is starting in different parts of the world, emphasizing that transmission containment measures that are being imposed are not adequate. Part of the consideration in determining containment measures is the rationale that vaccination will soon stop transmission and allow a return to normality. However, vaccines themselves represent a selection pressure for evolution of vaccine‐resistant variants, so the coupling of a policy of permitting high levels of transmission/virus multiplication during vaccine roll‐out with the expectation that vaccines will deal with the pandemic, is unrealistic. In the absence of effective antivirals, it is not improbable that SARS‐CoV‐2 infection prophylaxis will involve an annual vaccination campaign against ‘dominant’ viral variants, similar to influenza prophylaxis. Living with COVID‐19 will be an issue of SARS‐CoV‐2 variants and evolution. It is therefore crucial to understand how SARS‐CoV‐2 evolves and what constrains its evolution, in order to anticipate the variants that will emerge. Thus far, the focus has been on the receptor‐binding spike protein, but the virus is complex, encoding 26 proteins which interact with a large number of host factors, so the possibilities for evolution are manifold and not predictable a priori. However, if we are to mount the best defence against COVID‐19, we must mount it against the variants, and to do this, we must have knowledge about the evolutionary possibilities of the virus. In addition to the generic cellular interactions of the virus, there are extensive polymorphisms in humans (e.g. Lewis, HLA, etc.), some distributed within most or all populations, some restricted to specific ethnic populations and these variations pose additional opportunities for/constraints on viral evolution. We now have the wherewithal – viral genome sequencing, protein structure determination/modelling, protein interaction analysis – to functionally characterize viral variants, but access to comprehensive genome data is extremely uneven. Yet, to develop an understanding of the impacts of such evolution on transmission and disease, we must link it to transmission (viral epidemiology) and disease data (patient clinical data), and the population granularities of these. In this editorial, we explore key facets of viral biology and the influence of relevant aspects of human polymorphisms, human behaviour, geography and climate and, based on this, derive a series of recommendations to monitor viral evolution and predict the types of variants that are likely to arise.
Introduction
The on‐going COVID‐19 pandemic is an authentic real‐time experiment in molecular evolution. It unveils the behaviour of a virus from the time when it entered a naïve population. This « experiment » spans almost the totality of the planet with a host population of 7.8 billions individuals of Homo sapiens that displays a huge environmental and genetic polymorphism. The origins of the SARS‐CoV‐2 betacoronavirus remains somewhat of a mystery – despite a not‐so‐distant origin in a bat species (Ji et al., 2020; Makarenkov et al., 2021) – but its evolution can be now traced with some accuracy as it spreads in different populations and countries. As of 2nd March 2021, the number of mutations with at least one SNP identified in 193,687 SARS‐CoV‐2 full genomes available from the Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org) compared with the initial sequenced isolate named « Wuhan‐Hu‐1 » [INSDC AccNum MN908947.3 (H. Wang et al., 2020b; Wu et al., 2020; Zhu et al., 2020)] reaches 19,794.
While providing us with a valuable tool to highlight important features of the evolution of the epidemic, vigilance is required as some of these SNPs may not express genuine viral diversity but rather stem from data of uneven quality. In addition, a biased or lop‐sided geographical distribution of sequences collection may over/under‐represent specific SNPs. Identifying viral variants with modified behaviour – virulence, tropism, transmission – is a key objective of viral genome surveillance. While increased transmission is expectingly selected over time, some of the mutations also modify virulence (Oulas et al., 2021). However, because of the wide spectrum of virus variants, variation in geographical conditions and extensive human polymorphisms, this must certainly be taken with caution, despite the importance of identifying either more virulent or attenuated strains of the virus (see comments in Fig. S1 and Table S1). This difficulty is highlighted by the enormous number of publications, of evident uneven quality, that discuss SARS‐CoV‐2 and COVID‐19 (109,000 publications listed at PubMed on 3rd March 2021) and the lack of systematic metadata sharing in the sequencing databases. In this Editorial, we attempt to anticipate some of the features of the evolution of the virus and pandemic, taking as much as possible an ‘out of the box’ approach, in order to avoid the ruts of fashion and bibliometric biases. For this, we revisit the constraints of evolution in the light of basic virus biology (which is geared to its primordial function of propagation/persistence over time) through an analysis of diverse functions of the pathogen, and their corresponding interactions with the host, that may impact development of an epidemic. This allows to highlight some of the less obvious features we are witnessing that might help us anticipate what may happen as the pandemic progresses.
Key facets of evolution
Evolution, by definition, is witnessed after the fact. Involving time implies that there is a huge difference when we anticipate short term and long term evolution of a biological entity. Furthermore, making predictions is hazardous, as evolution is myopic. It cannot have any grand design. However, in the long term, the very fact that an organism is still extant will highlight functions that allowed it to keep reproducing during that span of time. Things are different in the short term, with only a limited set of descent‐related functions, possibly missing those that allow propagation in the distant future, for example. Moving to a new host from a host with which it has interacted for a long time, will suddenly expose the virus to an unfamiliar environment. Yet, it still follows the program of functions that allowed it to thrive in its usual host. In parallel, the new host is also the result of long‐term evolution. While naïve for this specific invader, it has been shaped by natural selection that retained a variety of generic responses to react against that kind of invasion. In the case of viruses, natural innate immunity has been selected for functions that recognize the presence of viral features, as well as prevent, or at least control viral development (Nan et al., 2014; Chen et al., 2017; Hur, 2019). Many of these functions are shared by animal and even plant families. This is witnessed, for example, by the discovery – unexpected at the time – of the role of Toll receptors in Drosophila (Belvin and Anderson, 1996).
Evolution cannot decide beforehand whether a virus will be able to beget progeny in the long term In the short term, this implies that a successful virus will have maximized its descent without any direct feedback from the survival of its host, unless the host is killed before the virus had time to reproduce. Extreme virulence, with maximum killing efficiency is not sustainable in the long term, for want of hosts. This essential requirement directs us to look into functions expected to emerge as an epidemic unfolds. Unfortunately, understanding the various paths of evolution has led to a large number of simplifying hypotheses, shaped by anthropocentric views with an economic or moral flavour: the behaviour of a biological entity has been seen as « altruistic » or « selfish ». Mutations are perceived as « advantageous », « deleterious » or « neutral ». In the case of a viral pathogen, this is despite the fact that a great many processes leading to an active viral progeny cannot have pre‐existing reasons to elude inconsistencies. For example, even in the absence of an identified functional consequence in a gene product, a nucleotide change in the genome sequence can affect metabolic organization of the host, fine tuning of the virus replicase, interference with innate immunity, temperature, modulation of functional tRNA availability, co‐infection with another pathogen, and so forth. This has consequences for the multiplication of the virus, with constraints at all types of levels similar to those just listed. None of this is really « neutral ». Assuming neutrality or similar soft aspects is simply a means to describe processes we do not understand and to hide our ignorance. This is misleading if we hope to be able to anticipate at least some of the future of an epidemic, notwithstanding the inevitable role of ‘black swans’ in the way biological entities evolve (Taleb, 2008).
Whether a change in the virus genome has an effect will be seen in the course of time, with different possible outcomes depending whether the virus spread is surveyed in the short, medium or long term. What is important to us is not the model of evolution we would like to apply, but, rather, to make out the panel of features that may or will emerge as the pandemic expands, reaches a peak and calms down. In this respect, the present epidemic was highly predictable, and, in fact, predicted in many studies (Moya et al., 2004; Turinici and Danchin, 2007; Horby et al., 2013). Rather than use models to try and make predictions, we prefer here to try and identify the functions that enter into play during a viral infection, to see how they could help us anticipate at least some of the future of the epidemic.
Functional analysis: from function to sequence, not the reverse
The concept of function is notoriously difficult to grasp (Allen et al., 1998). Let us here use design in industry as a metaphor. With the purpose of creating an entity of interest, the design of industrial devices and processes begins with the understanding of the functions they are associated with, functional analysis [Fantoni et al. in (Norell Bergendahl and Stanford University, 2009)]. A common way to proceed is bottom‐up, listing parts and combining them into progressively more complex entities until the final contraption is obtained. By contrast, a better way works top‐down, starting from the end‐user point of view. We first identify the master function of the device (e.g. printing, for a paper printer) and then progress toward identification of the helper functions required for the master function to operate (e.g. feeding paper, supplying ink, providing energy, programming printing time, etc.), to end up with the basic components making the instrument. Understanding the functions of a virus, in particular its master functions, is not trivial however. We propose that two widely different but intertwined master functions are necessary here: reproduction (making a progeny) and exploration (reaching a host). The ways these functions can be implemented are essentially limitless. Yet, this does not imply that they do not encounter constraints: they must be embodied in the material entities that are associated with life, specific building blocks and macromolecules. Fortunately, this limits the span of our quest. Because viruses existed long before animals emerged, animals have devised general functions meant to counteract invasion by viruses.
Exploration
Let us begin with an open‐ended list of viral functions and host responses associated with the master function « exploration ». For any entity associated with life, this function requires a specific helper function, « addressing », repeatedly used at different scales. From our point of view, the target of pathogenic viruses is a human being. The first contact of the virus is the surface of an individual person. This highlights specific routes of entry. Here are common examples:
Face/gut tropism is favoured by the fact that we use our hands not only consciously but also unconsciously (just look at the number of people touching their face when you look around you). This tropism can then be split into eyes, nose, then respiratory tract, and mouth then gut, with a possible feedback in the ubiquitous oro‐faecal contamination routes, which were indeed identified at the onset of the COVID‐19 epidemic (Jin et al., 2020). The idea that the gut route retains its possible significance is reflected by the presence of the virus in wastewater (Sharma et al., 2021). At this time, it seems that infection by SARS‐CoV‐2 via this pathway is limited (Goldman, 2020). However, in our anticipation, it is important to be aware that this route may suddenly acquire a dominant role, as argued below.
Direct respiratory tract tropism when the virus is aerosol borne. The fine details of this route should be monitored with care as an infection privileging the nose would have different consequences from an infection going deep in the lungs. This would also reveal a likely change in host cells' entry doors.
Vector‐borne viruses: skin and the blood stream are the usual door of entry [notwithstanding possible more intricate situations when a pathogen is itself the host of a parasite for example, a feature that – curiously – has not yet been explored despite the risk for novel ways of infection (Ng et al., 2007)].
The COVID‐19 pandemic is relevant to the first two routes, but we should note that, depending on human behaviour (relaxing rules of hand and face hygiene for example) new windows of evolution for the virus are prone to open up.
Addressing the human host
COVID‐19 has many features of a disease of human societies (Danchin, 2003). This is reflected in the reluctance of people to accept even temporary changes in their behaviour (Cherif et al., 2016). The role of aerosols, for example, has been and still is disputed for various socio‐political reasons, especially because wearing masks is mainly useful to protect others, not so much the wearer (Wei et al., 2021), and that perception of the importance of the common good greatly differs in different societies (https://interactives.lowyinstitute.org/features/covid‐performance/). Yet, aerosols – which can keep the virus under airborne conditions for a very long time, even outdoors (Zhang et al., 2020) – are likely to be a major transmission route in the case of SARS‐CoV‐2 (Dumont‐Leblond et al., 2020; Lieber et al., 2021). Controversy in the domain is very damaging as it resulted in a large number of inappropriate recommendations about the wearing of masks (Czypionka et al., 2020). Failing to appreciate the importance of this route is a major risk for the virus to perpetuate itself, creating dangerous variants in the long term. Many features of human behaviour, namely the human appeal for socializing in crowded environments, also contribute to the spread and perpetuation of the disease. Here are a few examples of contamination sources, which highlight a variety of possible entry points of an infection, its propagation or persistence.
Environmental variations
Respiratory diseases have a strong seasonal component (Audi et al., 2020). However, many confounding factors may hide the true causal relationships between a well‐identified factor and infection. Seasonality, pollution, urbanization, biodiversity or latitude have all been suggested to have a causal role in infectious diseases (Wood et al., 2017). Curiously, the fact that temperature or rain may change the indoor/outdoor pattern of human groups has rarely been taken into consideration (Bulfone et al., 2020). Since probability of infection relates to virus concentration in air, dilution of virus after expulsion from infected individuals is key to transmission, so the high density of a human population in crowded spaces – including outdoors – is doomed to result in a high level of contagion (Derjany et al., 2020). Moreover, air stagnation in wind‐protected pockets in urban settings is less favourable to virus dilution than open rural settings. Pollution by particles has also been associated with the severity of the disease (Brauer et al., 2021). In an outdoor environment, the distribution of UV light will vary enormously depending on latitude and altitude, for example (Karapiperis et al., 2020). It is, therefore, likely that the evolution of the virus will differ in different settings, because the selection pressure for infection and entry routes will differ. In this respect, understanding of the role of the virus envelope, not only its capsid and spike, is crucial.
Lung and gut tropism
When anticipating the future of the present pandemic, both the intestinal and respiratory routes are of key importance, as, in the case of other coronaviruses, there has been a back and forth shuttle between them. For example, transmissible gastroenteritis alphacoronavirus (TGEV) replicates in both the villous epithelial cells of the small intestine and the lung cells of new‐born piglets, causing a mortality of nearly 100% that devastated pig farms in the United States as early as 1946 (Doyle and Hutchings, 1946). A second disease, this time infecting the respiratory tract and also caused by an alphacoronavirus, porcine respiratory coronavirus (PRCV), was identified in 1984 (Wesley et al., 1990). It was then found that PRCV is a mutant of TGEV carrying a few deletions that change the viral tropism from intestinal to respiratory epithelia (Rasschaert et al., 1990). Remarkably, PRCV infection protected pigs against TGEV, providing a spontaneous natural vaccine (Bernard et al., 1989). Change in tropism was thus associated to attenuation of viral virulence. This observation suggests that monitoring events involving alteration of intestinal health should be implemented seriously. Independent of tropism, this hopeful evolutionary trend is however rather unlikely as it rests on a sequence of lucky accidents: Attenuation, followed by efficient spreading of the attenuated variant and then immune cross‐protection against the primary pathogen. Although attenuated variants may emerge in parallel with viral spreading, chances are low that these three events naturally co‐occur in the short term. We should remember however that, surprisingly, two amino acid changes at the N‐terminus of the pig TGEV spike protein were enough to result in loss of enteric tropism (Ballesteros et al., 1997). Attenuated forms should therefore be actively looked for. Unfortunately however, recombination with other RNA viruses, a common process affecting coronaviruses (Zhang et al., 2005; Chen et al., 2019), might prevent the promising spread of attenuated infections (see further discussion below).
Human polymorphism
Features of the genetic structure of the human population should also be considered. This begins to be understood (Williams et al., 2020) but, at this date (21 March), searching PubMed for ‘human polymorphism’ AND (‘Covid‐19’ OR ‘SARS‐CoV‐2’) retrieved no result. Removing the quotes lists 281 references, with only a handful of relevant ones [e.g. (Ovsyannikova et al., 2020)]. Interestingly, a genetic study exploring coronavirus‐dependent polymorphisms suggested that the ancestors of East‐Asian populations did already adapt to coronavirus infections that infected people for thousands of years so that their descendants are likely less naïve and hence vulnerable to the present epidemic (Souilmi et al., 2020).
Before reaching its cell's target, SARS‐CoV‐2 must attach and go through layers of mucin and other secreted compounds. It happens that the human population is split into several groups, depending on the way they modify secretions with the so‐called Lewis system (Lemieux et al., 1979; Nordgren and Svensson, 2019). This should add yet another layer to the contribution of the various blood groups to the profile of infection (Bloch et al., 2021; Le Pendu et al., 2021; Schetelig et al., 2021). The virus also has to pass the barrier of adaptive immune response at the humoral and cellular level, and human polymorphism in this domain is huge. However, spread of the disease in isolated populations might yield important observations allowing us to anticipate some of the future of the epidemic if the viral genome sequences can be collected in these populations. In terms of environmental cues impacting adaptive immunity, past infections may have already featured epitopes presented by the new SARS‐CoV‐2 virus (Ng et al., 2020). The human leucocyte antigens (HLA) which define major markers of human polymorphism are likely to have an important contribution at this level. This needs to be explored in depth as it may happen that a particular viral epitope tagged by the HLA overlaps with a host factor, triggering a serious autoimmune response [see discussion of the case of narcolepsy, linked to the influenza H1N1 virus in a particular HLA group (Schinkelshoek et al., 2019)]. In general, however, the contribution of the adaptive immune response is likely to result in a very large panel of phenotypes, linked to the considerable human polymorphism. It is only via collection of a large number of virus genome sequences that this contribution might be understood (besides the commonplace features associated to the process of inflammation), allowing proper anticipation associating the metabolic consequences of adaptive immunity with specific features of human polymorphism.
At this stage, the virus must enter cells (Tortorici and Veesler, 2019). To this aim, it uses as a receptor, ACE2, angiotensin‐converting enzyme 2, discussed in the next section. Encoded in the X chromosome, the ACE2 gene displays significant polymorphism. This noteworthy feature implies that sexual dimorphism impacts infection. Variation in females, linked to random X‐chromosome inactivation in every cell of their body, leads to a mosaic polymorphism which may result in a continuum of propensity to infection and possibly severity of the disease. This genetic feature should be taken into account when investigating spread, severity and evolution of the virus (Khayat et al., 2020; Hamet et al., 2021).
Addressing cell surface receptors
Viruses recruit a variety of receptors and host factors to bind to their targets (Baranowski et al., 2001). In coronaviruses, the spike protein is the major determinant of tropism type associated with cognate receptors (Hulswit et al., 2016). Similar to its predecessor SARS‐CoV‐1 and unlike MERS‐CoV which uses another receptor, the spike protein S of SARS‐CoV‐2 mediates viral attachment and entry into the host cell by binding to its primary target receptor ACE2, an essential carboxypeptidase of the renin‐angiotensin hormone system (Gross et al., 2020). ACE2 is expressed in the heart, kidney, testis and the gastrointestinal system. In the lung, it is expressed at a low level in some alveolar type 2 cells, and the expression seems to be person‐specific (Hikmet et al., 2020; Zou et al., 2020). In the present context, it may also be noticed that ACE2 expression is induced by interferon (Ziegler et al., 2020), suggesting a feedforward loop in the process of infection, which may explain the recruitment of ACE2 as a receptor during coronaviruses evolution.
As a further interesting feature, ACE2 is co‐expressed with the transmembrane serine protease 2 (TMPRSS2) within nasal goblet secretory cells, cornea, lung alveolar type 2 cells, ileal absorptive enterocytes, intestinal epithelial cells and gallbladder (Lukassen et al., 2020; Trypsteen et al., 2020). Involvement of a proteolytic function has been identified as critical for viral infection (Laporte and Naesens, 2017), and proteolytic cleavage of the coronavirus Spike (S) glycoproteins activates the glycoprotein for host cell entry [see below (Hoffmann et al., 2020b)]. In an exercise of anticipation, it may be revealing to spot differences between highly related viruses and diseases. In the case of SARS‐CoV‐1 infection, cleavage of the ACE2 receptor itself at arginine and lysine residues enhanced viral infectivity. These residues are essential for cleavage by TMPRSS2 and human airway trypsin‐like protease [HAT, (Bertram et al., 2011)]. In contrast, ACE2 cleavage was dispensable for activation of the viral S protein for SARS‐CoV‐1 (Heurich et al., 2014). Yet another protease may be involved as a receptor: dipeptidyl peptidase 4 (DPP‐4) has been suggested to be a co‐receptor for SARS‐CoV‐2, but this has not been further substantiated (Badawi and Ali, 2021). In any event, the role of proteolysis in the first steps of infection has to be carefully surveyed as the epidemic develops. It is not only involved in the initial binding of the virus to its host's cells, but also mediates membrane fusion, as discussed below.
Cell‐mediated membrane fusion entry of the virus
Infection by SARS‐CoV‐2 proceeds in two steps: Immediately after binding of the S protein to its ACE2 receptor, the viral envelope fuses with the host cell membrane, using cellular proteases priming the spike glycoprotein S for cell entry. Their role in post‐translational modifications, protein kinase activities or for various types of inflammatory cells is crucial to understand the spread of human coronaviruses (Tharappel et al., 2020). The S protein is a homotrimer, each monomer consisting of the two functional subunits S1 and S2, which have different roles. In coronaviruses, proteolysis, known as priming, separates the S1 subunit, which contains the receptor‐binding domain and drives binding to the cell receptor, from the S2 subunit, which triggers the initiation of membrane fusion (Krueger et al., 2001; Bosch et al., 2003). Effective fusion requires S2 activation through its further cleavage into an S2' fragment (Chambers et al., 2020).
Surprisingly – and this is the major question posed by the still enigmatic origin of the virus – SARS‐CoV‐2 has recruited within the S gene an insertion fragment coding for a multibasic site motif, Arg‐Arg‐Ala‐Arg (RRAR), at the S1 and S2 boundary. This makes it a typical target for the Golgi apparatus subtilisin‐like protease furin (Coutard et al., 2020). The presence of this site, absent in SARS‐CoV‐1 which instead contains a single Arg residue, may support the involvement of several proteases and directly impinge on virulence and host selectivity. It may also enhance cell–cell fusion without impacting viral entry (Andersen et al., 2020). Substantiating this view, functional analyses support a predominant role of the host transmembrane serine protease 2 (TMPRSS2) and furin, acting jointly to promote viral membrane fusion at the cell surface (Hoffmann et al., 2020a; Papa et al., 2021). Cleavage of the S2 subunit primes the S protein for TMPRSS2 processing at the S2' site, triggering membrane fusion with neighbouring cells, and forming syncytia (Buchrieser et al., 2020; Papa et al., 2021). This highlights a large network of interactions mediating SARS‐CoV viruses entry in cells, leaving much room for evolution. It is therefore particularly important to follow emergence of mutations in this region and to correlate them with clinical data. Mutations in the region can be anticipated as likely to generate viruses which will enhance, or conversely alleviate, their invasion capacity of human cells. In a different scenario, viral entry by endocytosis may be supported by other proteases, including members of the cathepsin and furin family. The process of membrane fusion, triggered by the S2 domain of the spike protein, triggers endocytosis of the virus via the cell's lysosomes (Ballout et al., 2020). Among other entry routes not reviewed here, this process generates endosomes that merge with lysosomes, subsequently triggering the release of the coated viral genome in the cell's cytoplasm (Zhao et al., 2021), where it will begin its replication cycle (discussed below in the Section Reproduction).
Interestingly, fusion of a number of cells into a syncytium is a widespread infectious process, for example observed in the case of the infectious bronchitis virus (Yamada and Liu, 2009). An intriguing biochemical activity has also been proposed to account for the formation of membrane‐less compartments in the cell (Ditlev, 2021). This process is mediated by so‐called ‘disordered’ regions in proteins that have been demonstrated to lead to phase separations, a likely origin of membrane‐less compartments (Shea et al., 2021). In coronaviruses, the process may involve the N protein, in particular during the uncoating step, generating a virus‐specific chamber that progressively increases in size as the virus multiplies, further recruiting the cell's machinery and its metabolites (Dang et al., 2021; Lu et al., 2021). Besides ribosomes, involved in translation of the viral genome, multiple proteins of the host interacting with viral proteins are involved in compartmentalization processes, which remain poorly documented.
Virus‐specific protection
Natural selection has provided viruses with the means to avoid being easily recognized and inactivated either by physicochemical or biological processes. Coronaviruses are enveloped, which provides them with an extra layer of protection but requires a specific interaction with the lipid metabolism of their host. The SARS‐CoV‐2 viral genome encodes four structural proteins present in the mature virion – the spike (S), envelope (E), membrane (M) proteins and the genome compacting nucleoprotein (N). We have discussed the role of proteins S and N. The other two proteins have a protective role, a regulatory role and a structural role, shaping the virion. Furthermore, creating an inside and an outside, an envelope requires the implementation of a new function that copes with osmotic pressure and/or electric potential. Besides evident interference with the host immune response, this accounts for the ion channel activity of the E protein and possibly membrane‐bound accessory proteins (DeDiego et al., 2014).
Proteins are sensitive to proteolytic attack and they age spontaneously (Truscott et al., 2016). Post‐translational modification contributes to overcome these vulnerabilities. Viral proteins used for cell entry are extensively glycosylated for several reasons: to assist in protein folding, provide stability and most importantly, shield the virus from immune recognition by its host [this is often described as a ‘glycan shield’ (Walls et al., 2016)]. The level of glycosylation is somewhat variable and a role of sialylation, so important in influenza infection (Östbye et al., 2020), has not yet been carefully examined in the case of SARS‐CoV‐2. There are 22 potential glycosylation sites per S protein monomer (D. Wang et al., 2020a). Most of these sites are documented N‐glycosylations, while the occupancy of putative O‐glycosylations sites is lower. The corresponding amino acid residues in the proteins have therefore an important shielding role. Non‐synonymous mutations are likely to affect the progeny of the mutant strains and changes involving asparagine residues should be monitored in priority.
Maturation and release of the virus in the environment
In the second exploration stage, the virus is liberated from the infected cell, the infected organ and the organism. This stage of infection has yet to be fully characterized. It may play a critical role in the transmissibility of some virus variants (Lemey et al., 2021). Death of the host cells liberates virions, but it could also trap them. An active release is therefore a more efficient way to propagate the virus descent. This has long been witnessed as virus maturation by budding (Garoff et al., 1998). The host factors involved in coronavirus budding still have many unknowns but the overall sequence of steps within the cytoplasm, involving both the endoplasmic reticulum and the Golgi apparatus has been outlined (Boson et al., 2020). As in other viruses (Ortego et al., 2007), the SARS‐CoV‐2 E protein is important to drive the maturation and release pathways. Once replicated, the genome associates with lipids of the endoplasmic reticulum, via the nucleoprotein N and the M protein, driving the process of phase separation necessary for virus packaging (Lu et al., 2021). Following assembly of the nucleocapsid N/genome complex, the envelope is put together with proteins E, M and S. The virus is subsequently transported to the cell's surface and released to the environment by unconventionally high‐jacking lysosomal exocytic function (Ghosh et al., 2020). Overall, we stress again the omnipresent role of proteases as an important features of the virus cycle and which has to be carefully monitored for anticipating future virus evolution.
Finally, viral proteins interact with host proteins, creating complexes of diverse stabilities. It is to be expected that, during maturation, the viral envelope will trap some viral and host proteins within the virion. Some of these may be trapped non‐specifically whereas others, because of their specific interactions with viral proteins, may be trapped more consistently. This step is generally overlooked, despite its likely role in preparing the virus for the next round of infection. This is all the more important because these proteins, in particular the viral ones, will be injected into the recipient host cell immediately after infection. This inevitable « contamination » has certainly been shaped by evolution as a way for the virus both to manipulate its host and to speed up initiation of viral multiplication. The CoV3D database of protein structures (Gowthaman et al., 2021) is an important resource to help us anticipate possible networks of interactions mediated by these viral proteins.
Immediately upon entering cells, the viral genome is translated into two large polypeptides that are rapidly split into active non‐structural proteins by viral proteases. Their inclusion within the virion will obviously represent a selective advantage (Haas et al., 2021). As a case in point, the SARS‐CoV‐2 papain‐like cysteine protease, a domain of non‐structural protein Nsp3, is essential for virus maturation, interference with host inflammation and antiviral immune responses. The protein complement present in virions has been studied for SARS‐CoV‐1 using mass spectrometry and protein kinase profiling (Neuman et al., 2008). The experiments revealed that, besides a protein complement likely to manipulate the protein kinase regulatory potential of the host (Siddell et al., 1981), two viral proteases, Nsp3 and Nsp5, were indeed present in purified virions. Finding the large, multiple‐membrane‐spanning Nsp3 protein was especially unexpected, asking us to understand how it is incorporated into the virion, while identifying an important feature submitted to natural selection. In addition, several other proteins, about which there is currently limited functional knowledge, such as Orf3a, Orf9b and Nsp2 have also been found in mature virions. A complete characterization of virion components will be essential to our gaining an understanding of the dynamics of early stages of infection.
Reproduction
Binding of the virus to its host cell is followed by internalization, with involvement of the S protein but, as just described, also of other viral proteins, in a scenario which is still incompletely deciphered. Once inside the cell, the virus must reach the machinery that allows it to make multiple copies of itself. Because SARS‐CoV‐2 is enveloped, it must first uncoat its genome – bound to several proteins, in particular nucleocapsid N – before it can be translated into the enzymes required for genome replication and formation of a new envelope.
Translation
The RNA genome has reached a correct location in the endoplasmic reticulum during the process of entry into specific cell types. It must immediately engage the translation machinery. This asks for formation of active initiation complexes by engaged ribosomes via a long 5′UTR (Tidu et al., 2020). To this aim, the viral genome mimics standard cellular mRNAs. In particular, its 5′ end is capped. This allows it to be translated immediately upon entry (Yan et al., 2021). The subsequent sequence of events (Hartenian et al., 2020) and the number of functional proteins generated by the SARS‐CoV‐2 virus is known (Kim et al., 2020). However, translation comprises many steps that correspond to yet unknown functions, some of which are likely to be important for the future evolution of the virus (Neches et al., 2021). Besides ribosomes, multiple proteins of the host interacting with viral proteins are involved in the process, which also remains poorly documented.
The coding capacity of SARS‐CoV‐2 is substantial, directing synthesis of 26 proteins (Finkel et al., 2021). Remarkably, translation of the viral RNA is organized into an asymmetric program: Immediately after uncoating, the first two thirds of the genome is translated from a large coding region into two polypeptides, Orf1a and Orf1ab produced in uneven quantity from the same RNA sequence. Orf1ab has its carboxy‐terminal region translated in a process involving a pseudo knot and a −1 frameshift. This omnipresent feature of coronavirus translation, causing asymmetrical translation of Nsp proteins, creates a specific environment to either control the precise ratio of Orf1a and Orf1ab proteins or delay the production of Orf1ab products (which comprise RNA‐dependent RNA replicase, RdRp, Nsp12) until the products of Orf1a (Nsp 1–11) have created a suitable environment for RNA replication (Fehr and Perlman, 2015). These polypeptides are subsequently split into 16 non‐structural proteins, Nsp1‐16 (the end of Orf1a; Nsp11 may not have an authentic function), some of which are present, as we have seen, in the free virion after it has exited from the host. The distal part of the genome is transcribed as discussed below and translated into individual proteins, including the major proteins of the virion E, M, N and S.
Scrutiny of a considerable number of genome sequences of the virus allows identification of mutations emerging in all of the proteins it encodes. Figure 1 displays the highest allele frequency of all called mutations in each of the viral proteins calculated as the ratio between the highest residue mutation rate and lowest mutation rate. Table S1 provides the number of mutations in each protein. Besides the S protein, expected to be highly variable, the fact that RdRP (Nsp12) is also very variable is already telling us that finding antivirals targeting replication is likely to be a difficult enterprise. The same is true with protease Nsp3, although being multidomain, this may be less of a problem because it offers multiple targets. The function of Nsp2 remains poorly understood, but its high variation extent makes it of particular interest. Variations in Orf3 are particularly interesting to investigate, because the protein is likely to control the stability of the viral envelope via maintaining a correct response to osmotic pressure.
Fig 1.
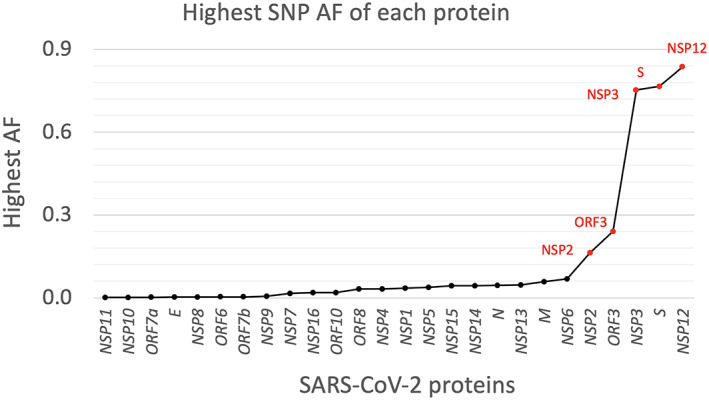
Distribution of the highest allele frequency (AF) of all called mutations (SNPs and deletions) in each protein from 193,687 strains. The label of the proteins with a percentage larger than 10% are shown and marked in red. AF = allele frequency. The y‐axis shows the highest mutation rate (i.e., the highest allele frequency) of all observed mutations from all 193 k isolates. [Color figure can be viewed at wileyonlinelibrary.com]
Control of translation is critical for virus development. This process is directly related to the availability of a precise tRNA complement. Remarkably, the human genome does not code for complete tRNA molecules. Precursor tRNAs must be matured first in the nucleus, and then a CCA complement must be added at their 3'end (Augustin et al., 2003). Together with control of many tRNA nucleotide modifications required to make tRNAs adapt to the codon usage bias of the genes of interest, this creates a bottleneck in the virus evolution that is likely to be reflected in virus‐encoded proteins. Nothing is known at this point about the expected functions, but monitoring mutations resulting in significant changes in the codon usage bias of viral protein genes would help us anticipate important deviations in the development of the epidemic.
Proteolysis
As just emphasized, proteolysis of virus‐encoded polyproteins is a key function of many viruses (Yost and Marcotrigiano, 2013). Immediately upon synthesis, the papain‐like protease domain of Nsp3 recognizes LXGG amino acid sequences and cleaves Orf1a and Orf1ab (Barretto et al., 2005; Gao et al., 2021). The 3‐D structure of the protein is known (Gao et al., 2021). Remarkably, this consensus cleavage sequence is also a sequence recognized by cellular deubiquitinating enzymes, allowing Nsp3 to interfere with its host cell regulatory functions. This protease also cleaves specifically and selectively IRF3, interferon regulatory factor 3. This cleavage might account for the weak Type‐I IFN response seen during SARS‐CoV‐2 infections (Moustaqil et al., 2021). Because it is acting on multiple important functions with a common recognition site possible scenarios of evolution of this protease are extremely limited. A second virus‐encoded protease, Nsp5, recognizing the [AVPT][VTKRM][LF]Q[ASN] sequence, cleaves Nsp4 at the end of Nsp3 and frees itself after cleavage at the end of Nsp4. Subsequently, Nsp5 cleaves off all Nsps from Nsp6 to Nsp16. As in the case of Nsp3, Nsp5 mediates cleavage of host proteins, in particular NLRP12, a potent mitigator of inflammation (Normand et al., 2018), and TAB1, TGFbeta activated kinase 1 binding protein 1, a component of the inflammatory response (Xu and Lei, 2020), pointing to a molecular mechanism for enhanced production of cytokines and inflammatory response (Moustaqil et al., 2021). Here, again, the potential for evolutionary events that impact disease severity, in this case, in particular the development of ‘long covid’ remains limited. In contrast, however, this highlights these proteases as good drug targets.
Transcription
Using the Nsp proteins translated and generated from the Orf1a and Orf1ab regions, in particular Nsp12 and Nsp13 (Arya et al., 2021), the distal third of the virus is subsequently expressed as transcripts coding individual proteins that contribute to inactivation of host defences while driving virus multiplication. The 3′ terminal end of the virus is transcribed into multiple transcripts that are translated into individual proteins, in particular the structural proteins E, M, N and S that have already been discussed. Oligomerization of protein N is necessary both for replication and for transcription (Ahamad et al., 2020). Transcripts, as is the complete viral genome, are capped. The mRNA capping complex comprises proteins Nsp14, Nsp16 and Nsp10. Nsp16 associated with Nsp10 to methylate cap‐0 at position 2′, completing the functional cap‐1 structure at the correct 5′‐ends of the virus so that they are recognized as authentic host functional RNAs and thus are not attacked by the antiviral response (Perveen et al., 2021). These proteins appear to have other functions, as is repeatedly observed with virus proteins, further restricting their evolutionary capacity. Nsp9 is also an RNA binding protein, but its function remains poorly characterized (see Section Quality Control, below). Besides capping, transcription initiation requires sequences folded into 3D RNA structures that are virus‐specific (Madhugiri et al., 2016) and do not, when mutated, allow for formation of productive viral genomes (mutation C241U examined below is an obvious exception). Importantly, the very fact that virus morphogenesis involves several essential transcription steps reveals that the sequence evolution in the transcription control regions is considerably constrained. In contrast to the lack of tolerance for mutations in the viral replication/transcription initiation sequences, protein coding sequences accumulate mutations that do permit synthesis of productive viral genomes. This, however, is protein‐dependent, some being more prone to accept mutations while others are less prone (Nagy et al., 2021).
Replication
Replication of the viral genome is central to virus multiplication. In the case of coronaviruses, which are positive‐sense single‐stranded RNA genome viruses, this process must be highly asymmetrical, with the complimentary copy of the virus genome synthesized in considerably lower amounts than the genome itself. The way this asymmetry is implemented is poorly understood. It is likely to be related to the presence of many factors associated with the RNA‐dependent RNA replication machinery, which is itself associated with the endoplasmic reticulum. As previously noted in the section discussing translation, the expression of proteins derived from Orf1a and Orf1ab is highly asymmetrical creating a suitable environment for RNA replication (Fehr and Perlman, 2015). Initiation of SARS‐CoV‐2 replication needs proteins Nsp7 and Nsp8 for priming at the structured 3′ end of the virus. Subsequently, Nsp12, the main subunit of RdRp, proceeds to replicate the genome, again with the help of Nsp7 and Nsp8 (Gao et al., 2020), while Nsp13 is involved in RNA unwinding. RNA‐dependent RNA polymerase is of unique importance as it dictates the efficiency and accuracy of the replication process. It has therefore been chosen as the target of many antiviral molecules. However, as shown in Fig. 1, many mutations have been retained in Nsp12, suggesting that the evolution of the virus offers a large panel of solutions to evade inhibition by drugs. This indicates that Nsp12 may not be a good target for anti‐virals. Another subunit, Nsp15, is a U‐specific endonuclease highly conserved in coronaviruses (Pillon et al., 2021). Its function is not well established but it appears to be involved in evading host antiviral responses (Zhao et al., 2020). Its evolution is, therefore, of interest to anticipate future trends in evolution of the virulence of the virus.
Quality control
Replication and transcription lack sufficient accuracy to limit errors in a genome as large as the 30,000 nucleotide long genome of SARS‐CoV‐2 (Bradwell et al., 2013). A proof‐reading complex that prevents accumulation of too many errors at each replication cycle has emerged as a consequence of this selection pressure. This is important because a large error rate leads to formation of defective viruses that rapidly become extinct (Pauly and Lauring, 2015). Reducing the accuracy in RNA virus replication is indeed the underlying idea driving drug design of some nucleotide analogs, such as favipiravir, so mutagenic that no descent of a virus can survive in its presence (Baranovich et al., 2013). Nsp14 is both a guanine‐N 7‐methyltransferase that produces the cap‐0 structure, and a proofreading 3′ to 5′ exonuclease removing mismatches that arise during genome replication (Ogando et al., 2020). Many mutations in this protein elevate the mutation load of the virus (Eskier et al., 2020). Scrutiny of mutations in this bifunctional enzyme has already revealed formation of ‘blooms’ of novel viral lineages, some of which are likely to result in attenuation (Cluzel et al., 2020).
Quality control of the proteolytic activities of the virus is also likely to have a significant role in its long term survival. It may thus be noted that dimeric Nsp9 binds in vitro to peptide LEVEL, which has similarity with the Nsp5 protease cleavage site (Littler et al., 2020). However, the main function of this protein is likely to involve modulation of activity of molecular chaperones. These factors are critical quality control elements, especially in stressful conditions such as during viral infection. It is, therefore, expected that SARS‐CoV‐2 codes for functions that fulfil and regulate the role of chaperones possibly via post‐translational modifications. Consistent with this, Nsp9 is modified by nucleotidylation of a glutamate residue (with a slight preference for UTP in vitro) by a manganese‐dependent activity associated with the NiRAN domain of Nsp12 (RNA‐dependent RNA polymerase, discussed previously). This residue belongs to a conserved N‐terminal NNE tripeptide. It is the only invariant residue in Nsp9 homologues in Coronaviridae (Slanina et al., 2021). Interestingly, the SelO(YdiU) counterpart of Nsp9 in Salmonella enterica Typhimurium, is a protein conserved in all three domains of life. It is a selenoprotein in mitochondria, substantiating its role in managing redox stresses. It also modifies molecular chaperones by uridylylation in conditions of ATP limitation (Y. Yang et al., 2020c). This role of this nucleotidylation should be explored with priority and placed in the perspective of the C > U trend, the origin of which is discussed in the following section.
Anticipating the future
It is trite to point out that predicting the future is difficult (Sullivan et al., 2013), especially as human behaviour is extremely erratic and varies from place to place. Yet, in many situations, we must make educated guesses. Even laypersons can propose interesting scenarios, as seen, for example, in the novel The End of October by Andrew Wright, who published early in 2020 a plausible development of what was just emerging as the COVID‐19 pandemic, thus demonstrating an authentic power of anticipation. Managing an epidemic to make it as short and innocuous as possible can save millions of lives and alleviate its tremendous economic burden. Using some of the enormous amount of knowledge that accumulates at a fast pace, after shifting out misinformation, we have highlighted here some of the more and less constrained evolutionary space that may be explored by natural selection in the course of SARS‐CoV‐2 evolution.
General pattern of evolution during the first year of the pandemic
While there has been a huge number of discussions in papers and on‐line publications, the human response to SARS‐CoV‐2 has been extremely biased and limited, leaving aside many points that should have been considered with urgency. Public discussions kept focusing on the most obvious feature of the virus, such as the protein sticking out of its envelope, the infamous spike protein (some of its roles are further discussed below), as well as pseudo‐treatments based on ex vivo experiments using uninformative cellular models. This is reflected by a simple Google search ‘SARS‐CoV‐2’ ‘spike’, which registered 6,000,000 pages on 3rd March (we note a decreasing trend, witnessing the power of fads: it was 9,000,000 pages on 15th February), compared with ‘SARS‐CoV‐2’ ‘Orf8’ which collected only 50,400 pages for example. Yet most, if not all, of the virus' proteins continue to evolve (Jaroszewski et al., 2020). As displayed in Fig. 2 and Table S1, mutations in the virus genes encompass much more than its spike protein, and there is a considerable variation between proteins. As discussed in the first part of this article, the selection pressure that stabilizes these changes is due to a great many causes, some of which are amenable to human intervention. Mutations that affected a large number of isolates (last columns in the histogram upper panel) are likely to be the most important for virus propagation.
Fig 2.
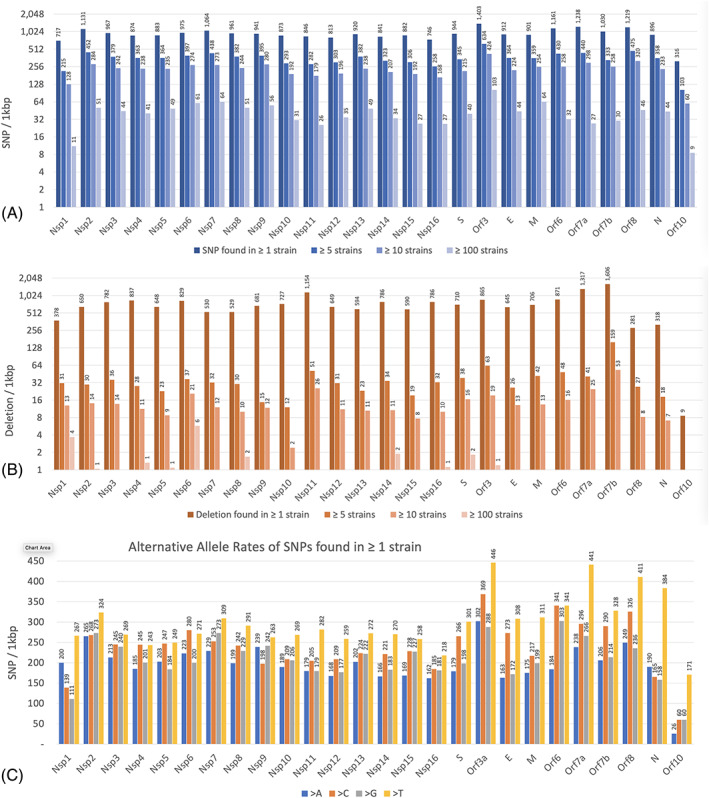
The mutation rate of each gene summarized from 193,687 SARS‐CoV‐2 strains.
A. SNP rate.
B. Deletion rate.
C. the alternative allele rates of SNPs found in ≥ 1 strain. ‘> A’ means transition of a SNP from non‐‘A’ to ‘A’. Using the length of each gene, the y‐axis is normalized to the number of mutations per 1 kbp. In (A) and (b), the y‐axis uses a base‐2 logarithmic scale, and the mutation rate of those found in ≥ 1 strain, ≥ 5 strains, ≥ 10 strains and ≥ 100 strains are shown. Information on how these mutation rates of each gene were generated is shown in the Supplementary Note. [Color figure can be viewed at wileyonlinelibrary.com]
Here is a specific example that may help us to anticipate future changes in the virus evolution. Protein Nsp1 is the first viral protein split out of the polyproteins Orf1a and Orf1ab. Surprisingly, the majority of viral variants in this protein retain a lower number of mutations than in the other Nsps of approximately the same size, suggesting strong selection pressure for long‐term evolution. Nsp1 has a critical role in translation initiation of the virus genome. Involved in discriminating the viral mRNA‐like genome from the bulk of cellular mRNAs, its translation features are distinct from those of the other proteins of the virus (Ou et al., 2020). Because it is submitted to such a considerable pressure for avoiding variation, the few mutations that have been retained in its sequence should be analysed with priority and their immediate consequences in terms of virulence reported.
Finally, as discussed previously, the selection pressure that stabilizes these changes is due to a great many causes, some of which may be amenable to human intervention. This calls for the urgent need to collect relevant metadata to build up the most out of the genomic information.
Environmental factors
We have stressed previously the questions asked by the many confounding factors that impact our understanding of respiratory diseases. It seems, however, well established that crowded environments, especially but not only indoors, provide the most significant contribution to person‐to‐person contamination. The structure of aeration systems is likely to have a strong impact, and this should prompt research in the way buildings are aerated, including using air filtration (Turgeon et al., 2014), while enforcing social distancing even in outdoor environments. To anticipate future respiratory diseases epidemics the construction of habitation, restaurants and office buildings as well as public transport should be submitted to compulsory regulation imposing specific rules on the control of air flow, such as those in surgery theatres (Timmis, 2020; Newsom et al., 2021).
Climate may have a contribution to the virus evolution. Studies such as that of (B. Chen et al., 2020a) do not contribute much insight simply because there are a considerable number of confounding factors to account for the development of the epidemic. Weather influences human behaviour: we tend to stay indoors when it is cold. Again, the contribution of indoors vs outdoors infection transmission is the best‐documented parameter, though most often only by inference. Indeed, this transmission parameter is linked to outside temperature (with a possible negative role of air‐conditioning when temperature/humidity is high). As a consequence, ventilation in closed spaces (Pease et al., 2021) might critically influence the evolution of the virus. This suggests several features that ought to be specifically monitored. In order to help anticipation, a thorough survey of metadata identifying infections in closed spaces should be associated with investigation (via genome sequencing) of the structure of the envelope and associated proteins, their binding to lipids as well as evolution of small hydrophobic proteins and links to ion salvage and transport. Another parameter of the environment, UV irradiation, should possibly be explored as well (Karapiperis et al., 2020). While killing pathogens, UV light is also mutagenic in a very specific way (Wurtmann and Wolin, 2009). Among more standard mutagenic nucleotide modifications, it generates uracil cyclobutane dimers. If significant, this would influence viral evolution in a highly biased way, meriting investigation, possibly via analysis of biases in the mutation patterns of the virus variants isolated from regions with high UV scores. However, we restrict here our discussion to well‐characterized features of the virus that are likely to contribute most to its future evolution and are amenable to experimental approaches.
In general, as noted in the vast literature dealing with the epidemic, emphasis was mainly placed on regulation of the host antiviral response. This perspective is helpful to allow us to link human habits with infection. For example, because smokers have more respiratory tract diseases, there may be some level of protection due to cross reactions with pathogens of previous infections (Saurabh et al., 2021), accounting for the initially paradoxical observation of a possible protective effect of smoking (Landoni et al., 2020), now established as a severity risk factor (Elliott et al., 2021). Multiple co‐infections may also be a source of recombination between viruses, a feature discussed below that is likely to have important consequences. This particular behaviour should be considered when anticipating the virus evolution – which, again, must be monitored by the sequence of its entire genome – in a fraction of the human population via connection with previous histories of respiratory and gut infections. In general, it is certainly critical to input, in the metadata linked to the genome sequence, patient clinical data that may reveal a tendency of virus tropism to evolve, in particular data relevant to both the respiratory and digestive functions.
Limitations in the viral host‐dependent development
Linking the virus and its host's metabolism is essential to account both for its short‐term and long‐term evolution. A virus, when it multiplies, must divert the host metabolism towards its own reproduction, i.e., viruses manipulate their host metabolism. Viruses thus have to accommodate the metabolic constraints of their host. In this respect, SARS‐CoV‐2 is a very interesting biological entity. Its multiplication has highlighted fascinating universal properties of cellular metabolism, based on an essentially overlooked property of growth. When cells grow, the bulk of metabolism takes place in the cytoplasm, where all the basic elements necessary for cellular growth are generated. This raises a neglected issue, well identified by economists, namely that of ‘non‐homothetic’ growth. The cell grows in three dimensions whereas the cell membrane is a surface that grows in two. Even more importantly, the genome is a linear polymer that grows in one dimension. This implies that there is an enormous metabolic pressure to make ‘too much’ of the membrane, and even greater pressure to make ‘too much’ of the genome. These features can, in principle, significantly benefit virus multiplication, not only for non‐enveloped viruses but also especially for enveloped viruses. The way these physical discordancies have evolved into cellular harmony needs to be unravelled.
In fact, this hurdle has been solved by natural selection via passing a whole section of metabolism through the synthesis of a single molecule, cytidine triphosphate [which gives the ‘C’ in the genome sequence (Danchin and Marlière, 2020; Ou et al., 2020)]. An unexpected feature of pyrimidine metabolism substantiates this observation by highlighting the ubiquitous absence of an anticipated enzyme activity. While phosphoribosyltransferases are omnipresent, scavenging purines, pyrimidines and other heterocyclic bases into mononucleotides, cytosine phophoribosyltransferase has not been identified in any living organism discovered to date (Ou et al., 2020). This is especially significant because closely related enzymes are ubiquitous. The missing enzyme should consistently emerge as a result of random mutations in existing counterparts, so its absence must reflect strong natural counter‐selection against outright salvage of cytosine nucleotides. A related consequence of this decreasing trend in cytosine in the SARS‐CoV‐2 genome is a parallel progressive loss of guanine residues, because G complements C during replication accounting for the somewhat parallel decrease in the G content of the virus. This will have interesting consequences, discussed below, as guanine is the target of specific defence reactions that involve reactive oxygen species (ROS).
Unsurprisingly, then, evolution has retained metabolic functions in hosts that interfere with the virus development via manipulating CTP synthesis. The synthesis of an antiviral analog of CTP – a fascinating natural analogy with the way human chemists create antiviral nucleotides – 3′‐deoxy‐3′,4′‐didehydro‐CTP (ddhCTP) by proteins of the viperin (Virus inhibitory protein, endoplasmic reticulum‐associated, interferon‐inducible) descent, is omnipresent in the innate antiviral arsenal in mammals (Kang et al., 2020) but also in oysters (Green et al., 2015) and even in Bacteria and Archaea (Bernheim et al., 2020). This inhibitor hampers simultaneously the four key functions of CTP: RNA synthesis, CCA addition to the 3′ end of tRNAs, cytosine nucleotide‐dependent synthesis of membrane lipids, and last but not least, synthesis of the universal carrier of protein glycosylation substrates, dolichyl phosphate (Ou et al., 2020). The consequence of this metabolic bottleneck is that SARS‐CoV‐2 evolves while shedding some of its cytosine content, unless a highly specific metabolic set up of the host has alleviated this constraint (see below). Cytosine residues that remain unchanged may therefore be evidence (whether at the RNA or encoded aminoacid level) for their importance in the perpetuation of the viral infection. This would point out important targets for COVID‐19 antivirals. It becomes therefore critical to monitor, during its short‐term evolution, how different lineages of the virus coped with this limitation. This includes pointing out possible reversion of the tendency to shed cytosine, as reversion would imply a change in the control of CTP synthetase and inhibition by viperin, while considerably opening up the virus evolution landscape. Again, this advocates for collecting as many as possible complete sequences of the virus genome.
The urgent need to gain a deeper understanding of the relationships between immunity and metabolism will by aided by the current massive increase in comparative phylogenetic studies of the virus in various species. Comparison with different metabolic setups, in particular with bat metabolism, should be extremely informative in this respect and help us anticipate some of the constraints of the evolutionary landscape of the virus. It is intriguing that these animals keep shedding viruses, generally maintained as non‐virulent commensals, while fast metabolism was selected as bats evolved flight (Shen et al., 2010). Flying is a high energy‐consuming activity that generates a large amount of ROS. Interestingly, however, ROS have a considerable positive role in defence against pathogens via the respiratory/oxidative burst by macrophages and neutrophils (Piacenza et al., 2019). While seldom explored, this positive outcome of fast metabolic turnover may contribute to the apparent harmlessness of viral infections in bats. Nevertheless, failure to restore redox homeostasis subsequent to antiviral responses triggered by infection may lead to unregulated release of ROS, pro‐oxidant cytokines and pathology from excessive inflammation. Regulation of this process is particularly relevant to multiple progressive events in lung infections, a common feature of severe COVID‐19 (and of course, ‘long covid’). In parallel, excess ROS will have a considerable impact on the viral genome, resulting in formation of 8‐oxoguanine, which triggers G > U transversion mutation events during replication. It is therefore expected that coronaviruses have evolved defences that alleviate the burden of this chemical assault. Monitoring consistent changes in the number of transversions in specific lineages of the virus is, therefore, of great importance (Cluzel et al., 2020).
Short‐term evolution of the virus: increased transmissibility
In the short term, a virus newly acquired from a different species must adapt the rates of its multiplication potential as well as its ability to infect its new hosts. This favours accumulation of mutations in all entities affecting these processes. The most likely early development in an epidemic is that the virus will increase its speed of propagation. This development stems from two general functions. The most likely one is an increase in transmission – many social parameters are relevant, including crowded environments, stable and prolonged production of infective aerosols by a subset of patients, increased latency phase, and so forth. A second critical feature would see an increase in viral replication success, with the reassuring consequence that a fast multiplication rate is usually associated with an increase in the mutation burden.
An average trend of approximately 22 mutations per genome per year has been observed during the first period of invasion of the human population by SARS‐CoV‐2. Consistent with the scenario outlined above, a general decrease in C nucleotides has been observed, starting from an as yet unexplained higher C content in most bat viral genomes (Matyášek and Kovařík, 2020; Ou et al., 2020; Simmonds, 2020). A website (http://www.bio8.cs.hku.hk/sarscov2) keeps track of the percentage of C in the new strains (Luo et al., 2020). The continuous emergence of mutations is also allowed by the expected functional versatility and resilience of protein sequences. Evolution of the proteome was monitored during the first 6 months of the pandemic (Lubin et al., 2020). A great many of the mutations lead to amino acid changes in viral proteins, suggesting that positive selection is operating at a significant level (Cluzel et al., 2020). While adaptation of the virus to a new host does not imply that it will cause a severe disease, some of the changes must have consequences in terms of severity of COVID‐19. We should note in particular that shedding C residues will make the ddhCTP interference less efficient, so that this might promote an increased virulence. Figure 3 displays the pattern of changes in all the proteins of the virus as a function of time. The average number of mutations per strain increased at a rate of ~1 mutation per month in the first 8 months of the pandemic (from December 2019 to July 2020), but then increased at a relatively lower rate at ~0.5 mutation per month (from August 2020 to January 2021).
Fig 3.
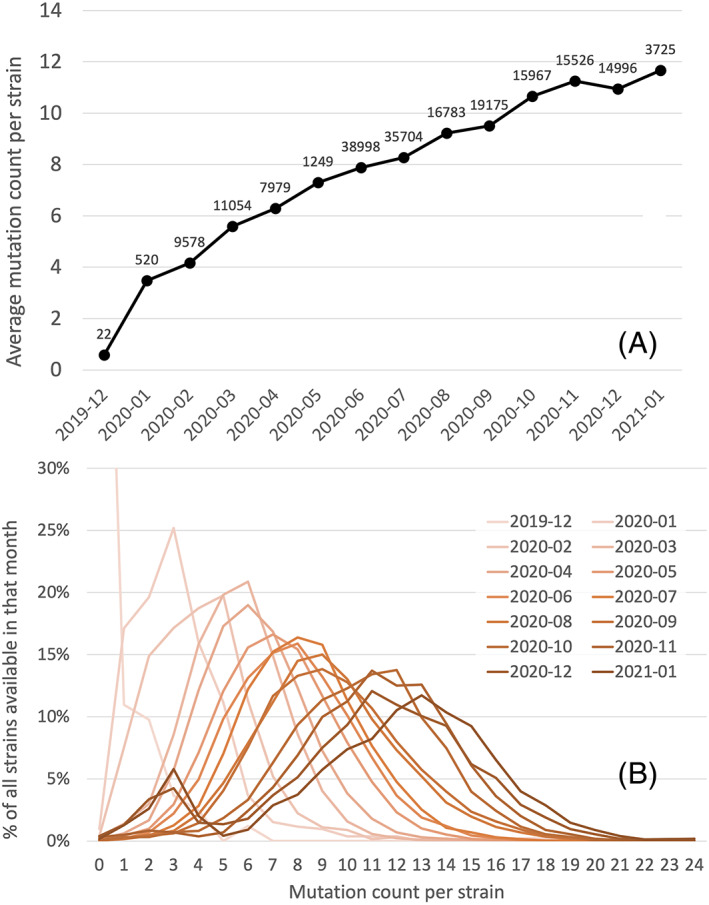
A. Average mutation count per strain in each month from December 2019 to January 2021.
B. The distribution of mutation counts per strain in each month from December 2019 to January 2021. In (A) the data label shows the number of available strains in that month in our analysis. [Color figure can be viewed at wileyonlinelibrary.com]
It is unlikely that the spontaneous mutation rate is very different along the genome sequence, yet some loci evolve rapidly, while other regions appear to be extremely constrained. In rapidly evolving regions, the selection pressure is relaxed with respect to the overall multiplication of the virus, so that it may rapidly adapt to the host's antiviral response. This observation should prompt monitoring whether evolution of specific functions correlates with environments with higher or lower transmission rates, in parallel with changes in the severity of the disease. Mutations that were often associated with mild disease affected proteins Orf8, Nsp6, Orf3a, Nsp4 and the nucleocapsid phosphoprotein N. In contrast, specific mutations located in the spike glycoprotein, in the RNA dependent RNA polymerase, and sometimes in Orf3a, Nsp3, Orf6 and N were associated with a serious outcome. Finally, mutations associated with a severe outcome have been located in Orf3a, again, and Nsp7. Unfortunately, the role of these proteins is rarely discussed in the context of the spread of the disease. Also, besides the pervasive role of S in triggering an immune response, among the 22 mutations that were associated with significant changes in the clinical outcome of the disease (either in the direction of mitigation or severity), four (three correlating with a severe disease, one with a mild disease) mapped onto a 10 amino acid long phosphorylated stretch of nucleocapsid N. This point to a highly relevant site in the viral genome (Nagy et al., 2021).
While some in silico and ex vivo analyses attempted to investigate whether mutations were deleterious for the virus, they tended to be structure‐oriented, rather than function‐oriented, which prejudices inferences that can be concluded. Whether a mutation is actually deleterious can only been understood if the related lineage does not persist for long. After a time when it was unclear whether rapid spread of some variants was due to a founder effect linked to superspreading events, it seems now established that some mutations are indeed affecting the spread of the disease (Borges et al., 2020). This view is essentially descriptive, after the fact. Also, it seldom takes into account the fact that a virus is an entity with highly integrated functions. To anticipate its evolution we must evaluate the functional epistasis between the various entities that form the virus, i.e. how the coupling of different mutations contributes to the viral fitness. In the present case, this type of analysis identified interactions between loci in SARS‐CoV‐2 genes Orf3a and Nsp2, Nsp12 and Nsp6, between Orf8 and Nsp4, and between loci in genes Nsp2, Nsp13 and Nsp14 (Zeng et al., 2020). This is revealing, as some of these interactions have not yet been linked to the severity of the disease (see above). It is, therefore, of considerable interest to further monitor how the sequence of proteins that were not already correlated to severity are evolving. As an important consequence of such a survey, identification of lineages associated with mild or severe cases of the disease, should enable more precise adaptation of the political management of disease transmission‐relevant behaviour (through regulation of social distancing, mask wearing, travel, etc.), to the evolution of key viral functions identified in the process.
The burden of the various steps of the virus cycle changes when the virus stays in a population for a long time. Coronaviruses co‐evolved with human populations, and there is some indication that a network of human genes may partially protect people from East Asia (Souilmi et al., 2020). Interestingly, some of the most essential proteins of the virus did not vary. For example, amino acid sequences of protease Nsp5 are highly conserved, well beyond SARS‐CoV‐2 variants and across all known coronaviruses as well. Non‐/weakly varying proteins conserved across virus families are potential targets for broad‐spectrum anti‐virals. SARS‐CoV‐2 Nsp5 is 95% identical in amino acid sequence to that of SARS‐CoV‐1. Its three‐dimensional structure could be used for designing inhibitors, an approach that was successful in the case of HIV (Lubin et al., 2020). Another key feature that must be linked with relevant metadata is the correlation between variation in lethality and specific mutations. Among 692 SARS‐CoV‐2 genome sequences, a statistically significant association with geographic origin and COVID‐19 case severity was observed. In particular, geographic variation in itself was associated with both case severity and allelic variation especially in strains from Indian origin (Goyal et al., 2021). This observation is fundamental and should prompt systematic sequencing of whole SARS‐CoV‐2 genome sequences while following their geographical evolution. Trends in virulence should be monitored carefully and local variations should trigger differentiated policies of containment.
A caveat in the management of our anticipations: fast spreading variants
As briefly outlined, the most likely early development of an epidemic is when the virus increases its speed of propagation. This may result from two general functions, an increase in transmission – here many parameters are relevant, including crowded environments, stable and prolonged production of infective aerosols by a subset of patients, and so forth – and an increase in viral replication success. The role of these processes is visible with several variant families that are spreading rapidly and superseding pre‐existing strains of the virus (https://nextstrain.org/ncov/global). The first documented example of this development was the D614G mutation in the spike protein, which enhances cleavage at the S1/S2 junction (Gobeil et al., 2021). Since then, at least eight major clades, as defined by the Global Initiative on Sharing All Influenza Data (GISAID): S, O, L, V, G, GH, GR and GV, have at the time of writing been found to span the planet (https://www.gisaid.org/phylodynamics/global/nextstrain/) with a specific pattern in Asia involving variants G, GH, GR, L, S, O (Sengupta et al., 2021), and in Europe where the virus is now evolving rapidly into multiple new lineages (Hodcroft et al., 2021b).
Unfortunately, the variant nomenclature is somewhat unstable and confusing: Nexstrain, an open source project to harness the scientific and public health potential of pathogen genome data (https://nextstrain.org) proposed five variant clades 19A, 19B, 20A, 20B and 20C. Qingtian Guan and coworkers also proposed that the lineages be distributed into five clades, but with different names: G614, S84, V251, I378 and D392 (Guan et al., 2020), which are somewhat related to the A, B, B.1, B.1.1 and B.1.177 clades proposed by Andrew Rambaut and coworkers, based on the epidemic in the UK (https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563).
Overlapping these classifications is yet another series of six clades and their underlying signature SNPs, which was validated by early availability of variant sequences. For example, a type VI clade was characterized by the four signature SNPs C241U (5′UTR), C3037U (Nsp3 F924F), C14408U (nsp12 P4715L) and A23403G (Spike D614G), with strong allelic associations. This variant became a dominant type very early on (H.‐C. Yang et al., 2020a) and is dominated by C to U evolution. As an illustration of the changes in clade dominance, in the Indian state of Telangana, the original (Wuhan) clade 19A was rapidly replaced by clade 20A with the omnipresent C241U mutation in the 5′UTR of the virus and the D614G spike mutation, followed by 20B, which is now dominant (Gupta et al., 2021). Subsequently, many other changes in the spike protein were found to propagate rapidly (Vilar and Isom, 2021), showing that the bulk of the selection pressure on this protein comes from adaptation to the host. We can therefore anticipate that this protein, and to a lesser extent the nucleocapsid protein, will evolve most rapidly under the selection pressure of vaccination. This is reflected in Fig. 4 and Fig. S1 which show how mutations propagated in the world. However, owing to extremely uneven data collection, this is only a gross overview of the situation, emphasizing the need for comprehensive programs of viral genome sequencing where the density of mutations is displayed as the virus develops in different countries. While the global mutation density of the spike protein increases gradually, countries such as Mexico and Brazil have shown a sharp increase in those since September 2020.
Fig 4.
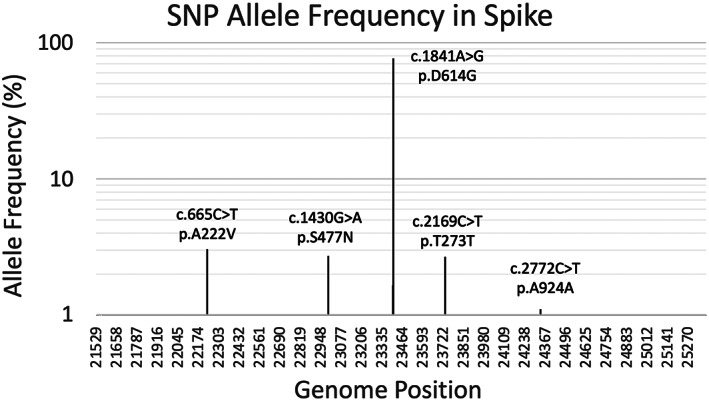
SNP allele frequency (AF) of the mutations in the spike protein in 193,687 SARS‐CoV‐2 full genomes. The y‐axis is on a base‐10 logarithmic scale. Only mutations with AF ≥ 1% are shown.
After publication of these studies, the appearance of other important variants was reported. At the time of writing, several variants with a number of mutations are circulating widely (https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html), and UK variant, B.1.1.7, South African variant, B.1.351, and Brazilian variant, B.1.248 (Firestone et al., 2021) are spreading worldwide. Many mutations vary simultaneously (https://covariants.org). This may result from convergent evolution, which would imply epistatic interactions between certain mutations. Identifying such mutations is likely to point out functional features of the virus that should enable anticipation of its further evolution, and possibly a tendency for it to attenuate its virulence. Among the features of these variants is, besides a flurry of mutations, the presence of a small deletion in the UK variant. In general, it is important to note that, during evolution, insertion/deletions are commonplace (see Fig. 2). As a matter of fact, an insertion in a bat virus genome is at the origin of SARS‐CoV‐2 infecting human beings. Covariation is illustrated in lineage B.1.525, an interesting new variant identified in 13 countries with spike protein mutations E484K, Q677H, F888L and a suite of deletions identical to those found in B.1.1.7 (https://cov-lineages.org/global_report_B.1.525.html). However, if we are to anticipate the future of the epidemic, we must remain aware of the potential importance of mutations other than those affecting the spike protein.
At this point, it is unclear whether or not the D614G mutation affects the severity of the disease. However, of the three major variants (Zhou et al., 2021) which have recently been identified, increased severity has been explicitly demonstrated in the B.1.1.7 variant (Davies et al., 2021). Many other variants are progressively replacing earlier strains of the virus, among which B.1.525 (Denmark, UK, Nigeria) is of unknown severity but increasing transmission potential.
Some examples of important variations outside of the spike protein
Most observations are simply descriptive, yet it is critical to identify alterations of viral functions, especially outside of expected spike protein variants, that impact its evolution. For example, we can expect that alteration of the replication process will have two widely contrasting consequences. On the one hand, it will prejudice variant propagation and lead to the demise of the virus in the long term, but, on the other hand, it may expand its evolutionary prospects in the short term. In this context, observing the tendency of the virus to create evolutionary « blooms » with an explosion of lineages should be connected, for example, with a possible reversal of the natural tendency of the virus to shed cytosine (Cluzel et al., 2020). Indeed, this process, valuable to anticipate the consequences of the viral evolution, generated several clades during the first 6 months of the epidemic (Koyama et al., 2020).
Among notable variations, the omnipresent C241U mutation in the 5′UTR of the viral genome, has not been linked to any specific phenotype. Commonly interpreted as neutral, it allows the virus to achieve a minor level of adaption to the metabolism of its host, with improved resistance to the action of viperin. Furthermore, it is located in the 5′UTR leader of the viral genome which is particularly important for its translational control and host specificity (Tidu et al., 2020). Following that change, there are many examples of blooms. For example, an interesting succession of mutations began with an early mutation, G11083U (protein Nsp6, L37F) now widely distributed worldwide and associated with widely different clades in India, for example (Banerjee et al., 2020). Yet another mutation, G1440A (G392D, protein Nsp2), followed by G2891A (A876T, ubiquitin‐like domain of protein Nsp3) was found in multiple countries (Liu et al., 2020), subsequently highlighting a conflict between translation of Orf7a and Orf7b. This showed that there is a cost / benefit dilemma for the expression of either one of these proteins. The descent of the virus at this locus is worth following up as it may result in interesting attenuated forms (Cluzel et al., 2020). In the same way, the Orf8 region of SARS‐related coronaviruses is hypervariable. It keeps changing during the course of epidemics, showing that it is subject to on‐going selection pressure, sometimes producing two peptides Orf8a and Orf8b (S. Chen et al., 2020b). During the first part of the epidemic Orf8 mutations displayed a branching that appeared in four different countries and in seven samples, spanning 6 weeks between the first and the last mutation (Cluzel et al., 2020). The Orf8 proteins are expressed at the end of the infection cycle. It will be important to monitor the way they contribute to the evolution of virulence of the virus (Neches et al., 2021).
Finally, it is of evident interest to monitor the evolution of the virus replicase, Nsp12. Early in the epidemic, yet another succession of mutations beginning with the widespread 5′end C241U mutation was followed by mutation C14408U (P314L) at the end of a zinc finger in Nsp12. This mutation appeared in many branches of the viral evolutionary tree. It altered the activity of the replicase in a noteworthy way, as this mutation was followed by « blooms » of novel lineages of the virus, suggesting that an altered replication process was mutagenic (Cluzel et al., 2020). One example, which should be carefully monitored, the very interesting succession: mutation of the spike protein A23403G (D614G), C3037U (synonymous), mutation G25563U (Q57H) in Orf3a forming potassium channels and supposed to negatively interfere with the function of the protein (Issa et al., 2020), C1059U (T265I) in protein Nsp2 and the triplet G4181A (A1305T) in the SUD‐N domain of protease Nsp3, then mutations G4285U (E1340D), and G28209U resulted in an end of translation at E106 of protein Orf8 appearing again as an important marker of the virus evolution (Neches et al., 2021). Now that 1 year has passed since the onset of the pandemic, new interpretations should be used to explore the mutations which continue to appear, especially in a context when vaccination is gathering speed.
Vaccination and its expected consequences
Vaccination has been the method of choice to control and even eradicate infectious diseases. However, while it is fairly straightforward in many cases, it has been difficult to create efficacious vaccines against several viruses, such as HIV (Oyston and Robinson, 2012). Many types of vaccines have been designed over the years (Smith, 2012), after the initial success due to injection of attenuated viruses (Theiler and Smith, 1937). In the case of coronaviruses, vaccines have been successfully used in animal diseases (Cruz et al., 2010; Singh et al., 2019). It is therefore expected that, at least during the early development of the disease, vaccination will be successful. However, as we have seen, the virus evolves very fast, so that there is a continuing risk that the virus will mutate in ways that render initial COVID‐19 vaccines less effective. Multiple strains are common evolutionary features, with likely impact on vaccination (Zeng et al., 2017). This seems to be the case with the B.1.351 variant first identified in South Africa, which may render initial vaccines less effective or even ineffective (Diamond et al., 2021). Furthermore, if a vaccination campaign is too slow, it will provide the virus with time to evolve variants that may totally escape vaccine‐induced immunity. In this respect, a vaccine based on a single protein of the virus or, even worse, on a domain of a viral protein (J. Yang et al., 2020b), will drive the evolutionary trajectory of the virus in such a way that variants carrying mutations in these proteins or protein domains will rapidly tend to accumulate.
Infections due to other causes may also have an influence on the evolution of the virus. For SARS‐CoV‐1 infection, the fact that certain populations appeared to be spared by the infection suggested that previous infections might have induced cross‐protection (Ng et al., 2003). We have noted above the apparently paradoxical correlation between smoking habits and milder infections. This implies that a link might appear between influenza, influenza vaccination and COVID‐19 disease progression. Infection by an influenza virus seems to enhance SARS‐CoV‐2 infectivity (Bai et al., 2021). However, retrospective studies did not find negative interactions between vaccination against influenza and COVID‐19, but rather the opposite (Green et al., 2020). Taken together all relevant observations call for implementing a fast vaccination program, while monitoring likely changes in the evolution of the virus genome as it continues to propagate in the partially vaccinated population. A consequence of these observations is that anticipation of the future of the disease must consider vaccinated and unvaccinated populations separately.
Long‐term evolution
The 1918–1919 flu epidemics may help us anticipate what may happen with the COVID‐19 epidemic. While it remains somewhat difficult to reconstruct an explicit scenario of what happened at the time, it is established that the epidemic developed in three phases. The first phase was similar to a serious flu epidemic; subsequently it developed into a very severe disease; finally, it adapted to the human host, eventually evolving in its still current form. The disease also passed from man to pig where it is still present (Shope, 1936). The apparent cause of the severe form was due to a reassortment event – the influenza virus is made of independent segments that may reassort upon co‐infection with a similar virus – which introduced unique variants of proteins that help the virus to multiply. Interestingly, these highly virulent variants did not involve the hemagglutinin or neuraminidase proteins that are required for the virus to infect and be released from host cells (Reid et al., 2004). This underscores again that it is crucial to consider virus features beyond proteins that are readily recognized by the adaptive immune system.
SARS‐CoV‐2 does not reassort because it is made of a single RNA element, but it is prone to recombine with available RNA. As a consequence, a most worrying evolutionary feature of the virus in the short term is that coronaviruses may undergo extensive recombination. RNA genomes usually recombine, but there is an inverse correlation between genome length and recombination rate because the longer genomes code for proofreading factors (Zhang et al., 2005; Goldstein et al., 2021). In SARS‐CoV‐2, the control of this process depends on the activity of protein Nsp14 (Gribble et al., 2021). Recombination can naturally stem from co‐infection with different strains of the same virus – and in this respect, this could reset Muller's ratchet as does sexual reproduction and more generally horizontal gene transfer, a process that may be amplified as travel restrictions are relaxed – but it can also use RNA from other viruses or even artificial constructs. In this respect, the replacement of uridine by pseudouridine in recent mRNA‐similar vaccines might have been a positive innovation that alleviated the pressure for accidental recombination events that would expand the evolutionary potential of the virus. However, this beneficial feature does not take into account temporary variants of the Nsp14 proofreading exonuclease. The descent of such mutants should be monitored carefully as they are likely to open up the evolutionary resources of the virus, generating hotspots in its spike, nucleocapsid and Orf8 proteins. Coronaviruses display a global pattern of recombination, particularly widespread in positive single stranded RNA viruses (Zhang et al., 2005; Patiño‐Galindo et al., 2021). Besides going backwards, recombination events may put together mutations that have appeared in widely different contexts. Since virus recombination occurs during co‐infections with different variants, it is favoured by conditions that create densely crowded environments, especially closed (indoor) environments, environments in which air breathed by many individuals is recirculated, and environments where vocal activity and hence virus expulsion is high. For this reason, it is essential to minimize exposure in such environments, at least until vaccination coverage is nearly complete or efficient antiviral drugs become available.
To anticipate the future of the epidemic we must carefully analyse how the tropism of the virus evolves. At this time, a respiratory tropism is dominant, but we know in other coronaviruses that this can evolve extremely rapidly. For example, three amino acid changes in the avian coronavirus spike protein allowed the virus to bind to kidney cells (Bouwman et al., 2020b). In mice, coronaviruses may display neurotropism (Pasick et al., 1994). Natural selection on cell entry and fusion is strongly related to the dynamic structure of the spike protein. We have seen that insertion of a furin cleavage site in a bat‐coronavirus enabled it to change its host and adapt to the human receptor ACE2 (Coutard et al., 2020). And now the virus has already found another receptor for entry, namely via interaction with protein S, CD147, a protein expressed in a variety of cells, including epithelial and neuronal cells, at least in models in vitro (K. Wang et al., 2020c). This is significant because, using multiple receptors allow shifting from one cell type to another and hence from one portal of entry organ to another. On the other hand, the MERS‐CoV receptor, dipeptidyl peptidase DPP4, is subject to polymorphism that negatively impacts virus entry (Kleine‐Weber et al., 2020). Other coronaviruses exploit yet other receptors: porcine delta coronavirus makes use of aminopeptidase N as an entry receptor and interacts with APN via domain S2 of its spike protein (Li et al., 2018). Mouse hepatitis coronavirus (MHV) is the only known coronavirus that uses the N‐terminal domain of its spike to recognize yet another protein receptor, CEACAM1a (Shang et al., 2020).
One other viral feature of note is the protein glycosylation that protects virus against degradation, and that may be used by the virus as a way to enter target cells. As a case in point, chicken coronavirus infectious bronchitis virus (IBV) enters host cells by binding of the viral heavily N‐glycosylated attachment protein spike to the alpha‐2,3‐linked sialic acid receptor Neu5Ac (Bouwman et al., 2020a). Human coronavirus OC43 apparently emerged from a bovine coronavirus (BCoV) spillover. It attaches to 9‐O‐acetylated glycan‐based receptor usage sialoglycans via its protein S with hemagglutinin‐esterase acting as a receptor‐destroying enzyme (Lang et al., 2020). The receptor‐interacting site is conserved in all coronavirus S glycoproteins that interact with 9‐O‐acetyl‐sialogycans, with an architecture similar to those of the ligand‐binding pockets of coronavirus hemagglutinin esterases and influenza virus C/D hemagglutinin‐esterase fusion glycoproteins (Tortorici and Veesler, 2019). Monitoring the antibody response against glycoproteins of the virus in parallel with changes in glycosylated amino acid residues as a consequence of mutations should therefore be developed.
Perspectives
More than 1 year after the onset of the COVID‐19 pandemic, we are at a transition moment, when the virus has been submitted to a variety of selection pressures that now place it on track for long‐term survival. Three major factors driving evolution of the virus towards possibly dangerous outcomes are now in place, recombination between strains, fairly slow vaccination campaigns and extremely limited research in the quest for antivirals. In parallel, the number of infected persons is very high, so that co‐infection with different variants of the virus in crowded environments is no longer a rare event. It becomes critical to be able to follow, in real time, the evolution of the complete genome sequence, so that we can pinpoint target sites in the proteins of virus that are likely to lead it to attenuation, or, conversely to more severe disease.
We advocate extensive collection of complete genome sequences of the virus. However, this only makes sense if we associate them with relevant metadata. In addition, questions about previous diseases are important metadata. The more metadata, the better. Metadata collection must be properly standardized, however.
The actions to be taken, which are urgent, are the following and address the key principle of Know thy enemy.
Sequence as many entire genomes (not just spike protein gene) of the virus as possible, everywhere. This should be possible at a time when sequencing technologies continue to improve. For example, experiments using Nanopore® sequencing of 752 clinical samples readily identified three clades of the virus (Bhoyar et al., 2021).
Associate significant metadata with these sequences (everything we can tell about the infected persons and clinical data) and couple metadata to specific mutations, without limiting investigation to the spike protein
Establish lineages and their propagation, and link to diverse parameters, including standard data such as age, sex, ethnicity – when allowed, weight, human genetic polymorphism features – HLA, Lewis secretory type, nutritional habits and general behaviour, such as smoking habits, and so forth. Also, because transmission requires human contacts, the exact place of infection (country, city, building) should be identified, and associated with meteorological parameters
Focus on changes in the pattern of evolution: formation of blooms of lineages, modification of the evolution of the nucleotide pattern (inversion of the trend in loss of C or G, transversions, etc.) and try to link this evolution to mutations in specific proteins of the virus
Based on this knowledge, identify lines that are being attenuated, and allow them to propagate, monitoring possible change in tropism from lung to gut and vice versa
Locate severe strains, and impose strict local containment
Trace transmission upstream, not only downstream which has little effect, and implement a strict control of movement of infected persons and their contacts
Accelerate as much as possible vaccination, especially in populations with high case incidence rates and with multiple circulating variants. Develop with urgency second and third generation vaccines based on the emergence of variants less affected by 1st generation vaccine immunity
Invest massively in the development of new antivirals, both target‐led, based on accumulating sequences of non/weakly varying viral proteins, and empirical non‐target‐based screens.
Finally, it seems likely that we will have to live with the progeny of SARS‐CoV‐2. This implies that, to control its negative consequences we will have to follow carefully the evolution of its antigenic determinants. We may end up with a situation somewhat similar to that of seasonal flu, and need a different vaccine every year. Co‐evolution with other respiratory diseases, flu in particular, has to be taken very seriously, as omitting to maintain stable herd immunity for the latter could lead to dire consequences.
Supporting information
Table S1 Mutation count and rate of each gene summarized from SARS‐CoV‐2 193,687 strains.
Fig. S1 Temporal‐geographical mutation density of the Spike proteins at four different time points in 2020.
Appendix S1 Supporting Information.
References
- Ahamad, S. , Gupta, D. , and Kumar, V. (2020) Targeting SARS‐CoV‐2 nucleocapsid oligomerization: insights from molecular docking and molecular dynamics simulations. J Biomol Struct Dyn 3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, C. , Bekoff, M. , and Lauder, G. (1998) Nature's Purposes. Cambridge, MA: MIT Press. [Google Scholar]
- Andersen, K.G. , Rambaut, A. , Lipkin, W.I. , Holmes, E.C. , and Garry, R.F. (2020) The proximal origin of SARS‐CoV‐2. Nat Med 26: 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya, R. , Kumari, S. , Pandey, B. , Mistry, H. , Bihani, S.C. , Das, A. , et al. (2021) Structural insights into SARS‐CoV‐2 proteins. J Mol Biol 433: 166725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audi, A. , AlIbrahim, M. , Kaddoura, M. , Hijazi, G. , Yassine, H.M. , and Zaraket, H. (2020) Seasonality of respiratory viral infections: will COVID‐19 follow suit? Front Public Health 8: 567184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin, M.A. , Reichert, A.S. , Betat, H. , Huber, R. , Mörl, M. , and Steegborn, C. (2003) Crystal structure of the human CCA‐adding enzyme: insights into template‐independent polymerization. J Mol Biol 328: 985–994. [DOI] [PubMed] [Google Scholar]
- Badawi, S. , and Ali, B.R. (2021) ACE2 Nascence, trafficking, and SARS‐CoV‐2 pathogenesis: the saga continues. Hum Genomics 15: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, L. , Zhao, Y. , Dong, J. , Liang, S. , Guo, M. , Liu, X. , et al. (2021) Coinfection with influenza A virus enhances SARS‐CoV‐2 infectivity. Cell Res 18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros, M.L. , Sánchez, C.M. , and Enjuanes, L. (1997) Two amino acid changes at the N‐terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 227: 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballout, R.A. , Sviridov, D. , Bukrinsky, M.I. , and Remaley, A.T. (2020) The lysosome: a potential juncture between SARS‐CoV‐2 infectivity and Niemann‐Pick disease type C, with therapeutic implications. FASEB J 34: 7253–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, A. , Sarkar, R. , Mitra, S. , Lo, M. , Dutta, S. , and Chawla‐Sarkar, M. (2020) The novel coronavirus enigma: phylogeny and analyses of coevolving mutations among the SARS‐CoV‐2 viruses circulating in India. JMIR Bioinform Biotech 1: e20735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranovich, T. , Wong, S.‐S. , Armstrong, J. , Marjuki, H. , Webby, R.J. , Webster, R.G. , and Govorkova, E.A. (2013) T‐705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol 87: 3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranowski, E. , Ruiz‐Jarabo, C.M. , and Domingo, E. (2001) Evolution of cell recognition by viruses. Science 292: 1102–1105. [DOI] [PubMed] [Google Scholar]
- Barretto, N. , Jukneliene, D. , Ratia, K. , Chen, Z. , Mesecar, A.D. , and Baker, S.C. (2005) The papain‐like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 79: 15189–15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvin, M.P. , and Anderson, K.V. (1996) A conserved signaling pathway: the Drosophila toll‐dorsal pathway. Annu Rev Cell Dev Biol 12: 393–416. [DOI] [PubMed] [Google Scholar]
- Bernard, S. , Bottreau, E. , Aynaud, J.M. , Have, P. , and Szymansky, J. (1989) Natural infection with the porcine respiratory coronavirus induces protective lactogenic immunity against transmissible gastroenteritis. Vet Microbiol 21: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim, A. , Millman, A. , Ofir, G. , Meitav, G. , Avraham, C. , Shomar, H. , et al. (2020) Prokaryotic viperins produce diverse antiviral molecules. Nature 589(7840):120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram, S. , Glowacka, I. , Müller, M.A. , Lavender, H. , Gnirss, K. , Nehlmeier, I. , et al. (2011) Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin‐like protease. J Virol 85: 13363–13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoyar, R.C. , Jain, A. , Sehgal, P. , Divakar, M.K. , Sharma, D. , Imran, M. , et al. (2021) High throughput detection and genetic epidemiology of SARS‐CoV‐2 using COVIDSeq next‐generation sequencing. PLoS ONE 16: e0247115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch, E.M. , Patel, E.U. , Marshall, C. , Littlefield, K. , Goel, R. , Grossman, B.J. , et al. (2021) ABO blood group and SARS‐CoV‐2 antibody response in a convalescent donor population. Vox Sang. https://onlinelibrary.wiley.com/doi/10.1111/vox.13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, V. , Isidro, J. , Cortes‐Martins, H. , Duarte, S. , Vieira, L. , Leite, R. , et al. (2020) Massive dissemination of a SARS‐CoV‐2 spike Y839 variant in Portugal. Emerg Microbes Infect 9: 2488–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, B.J. , van der Zee, R. , de Haan, C.A.M. , and Rottier, P.J.M. (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77: 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boson, B. , Legros, V. , Zhou, B. , Siret, E. , Mathieu, C. , Cosset, F.‐L. , et al. (2020) The SARS‐CoV‐2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus‐like particles. J Biol Chem 100111. 10.1074/jbc.RA120.016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman, K.M. , Habraeken, N. , Laconi, A. , Berends, A.J. , Groenewoud, L. , Alders, M. , et al. (2020a) N‐glycosylation of infectious bronchitis virus M41 spike determines receptor specificity. J Gen Virol 101: 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman, K.M. , Parsons, L.M. , Berends, A.J. , de Vries, R.P. , Cipollo, J.F. , and Verheije, M.H. (2020b) Three amino acid changes in avian coronavirus spike protein allow binding to kidney tissue. J Virol 94(2):e01363–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradwell, K. , Combe, M. , Domingo‐Calap, P. , and Sanjuán, R. (2013) Correlation between mutation rate and genome size in riboviruses: mutation rate of bacteriophage Qβ. Genetics 195: 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer, M. , Casadei, B. , Harrington, R.A. , Kovacs, R. , Sliwa, K. , and WHF Air Pollution Expert Group . (2021) Taking a stand against air pollution‐the impact on cardiovascular disease: a joint opinion from the World Heart Federation, American College of Cardiology, American Heart Association, and the European Society of Cardiology. Circulation 77(13):1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser, J. , Dufloo, J. , Hubert, M. , Monel, B. , Planas, D. , Rajah, M.M. , et al. (2020) Syncytia formation by SARS‐CoV‐2‐infected cells. EMBO J 39: e106267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfone, T.C. , Malekinejad, M. , Rutherford, G.W. , and Razani, N. (2020) Outdoor transmission of SARS‐CoV‐2 and other respiratory viruses: a systematic review. J Infect Dis 223(4):550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, J.P. , Yu, J. , Valdes, J.J. , and Arulanandam, B.P. (2020) SARS‐CoV‐2, early entry events. J Pathog 2020: 9238696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B. , Liang, H. , Yuan, X. , Hu, Y. , Xu, M. , Zhao, Y. , et al. (2020a) Predicting the local COVID‐19 outbreak around the world with meteorological conditions: a model‐based qualitative study. BMJ Open 10: e041397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Knutson, T.P. , Rossow, S. , Saif, L.J. , and Marthaler, D.G. (2019) Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Sci Rep 9: 3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Ning, X. , and Jiang, Z. (2017) Caspases control antiviral innate immunity. Cell Mol Immunol 14: 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zheng, X. , Zhu, J. , Ding, R. , Jin, Y. , Zhang, W. , et al. (2020b) Extended ORF8 gene region is valuable in the epidemiological investigation of severe acute respiratory syndrome‐similar coronavirus. J Infect Dis 222: 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherif, A. , Barley, K. , and Hurtado, M. (2016) Homo‐psychologicus: reactionary behavioural aspects of epidemics. Epidemics 14: 45–53. [DOI] [PubMed] [Google Scholar]
- Cluzel, N. , Lambert, A. , Maday, Y. , Turinici, G. , and Danchin, A. (2020) Biochemical and statistical lessons from the evolution of the SARS‐CoV‐2 virus: paths for novel antiviral warfare. C R Biol 343: 177–209. [DOI] [PubMed] [Google Scholar]
- Coutard, B. , Valle, C. , de Lamballerie, X. , Canard, B. , Seidah, N.G. , and Decroly, E. (2020) The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res 176: 104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, J.L.G. , Zúñiga, S. , Bécares, M. , Sola, I. , Ceriani, J.E. , Juanola, S. , et al. (2010) Vectored vaccines to protect against PRRSV. Virus Res 154: 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czypionka, T. , Greenhalgh, T. , Bassler, D. , and Bryant, M.B. (2020) Masks and face coverings for the lay public: a narrative update. Ann Intern Med M20–6625. https://www.acpjournals.org/doi/10.7326/M20-6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. (2003) Infection of society. As diseases have evolved to exploit the holes in our defences, including weaknesses in society, we have to reconsider our way of life, otherwise they will continue to haunt us. EMBO Rep 4: 333–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. , and Marlière, P. (2020) Cytosine drives evolution of SARS‐CoV‐2. Environ Microbiol 22: 1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, M. , Li, Y. , and Song, J. (2021) ATP biphasically modulates LLPS of SARS‐CoV‐2 nucleocapsid protein and specifically binds its RNA‐binding domain. Biochem Biophys Res Commun 541: 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, N.G. , Abbott, S. , Barnard, R.C. , Jarvis, C.I. , Kucharski, A.J. , Munday, J.D. , et al. (2021) Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science. eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeDiego, M.L. , Nieto‐Torres, J.L. , Jimenez‐Guardeño, J.M. , Regla‐Nava, J.A. , Castaño‐Rodriguez, C. , Fernandez‐Delgado, R. , et al. (2014) Coronavirus virulence genes with main focus on SARS‐CoV envelope gene. Virus Res 194: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derjany, P. , Namilae, S. , Liu, D. , and Srinivasan, A. (2020) Multiscale model for the optimal design of pedestrian queues to mitigate infectious disease spread. PLoS One 15: e0235891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, M. , Chen, R. , Xie, X. , Case, J. , Zhang, X. , VanBlargan, L. , et al. (2021) SARS‐CoV‐2 variants show resistance to neutralization by many monoclonal and serum‐derived polyclonal antibodies. Res Sq. 10.21203/rs.3.rs-228079/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditlev, J.A. (2021) Membrane‐associated phase separation: organization and function emerge from a two‐dimensional milieu. J Mol Cell Biol. https://academic.oup.com/jmcb/advance-article/doi/10.1093/jmcb/mjab010/6126818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, L.P. , and Hutchings, L.M. (1946) A transmissible gastroenteritis in pigs. J Am Vet Med Assoc 108: 257–259. [PubMed] [Google Scholar]
- Dumont‐Leblond, N. , Veillette, M. , Mubareka, S. , Yip, L. , Longtin, Y. , Jouvet, P. , et al. (2020) Low incidence of airborne SARS‐CoV‐2 in acute care hospital rooms with optimized ventilation. Emerg Microbes Infect 9: 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, J. , Bodinier, B. , Whitaker, M. , Delpierre, C. , Vermeulen, R. , Tzoulaki, I. , et al. (2021) COVID‐19 mortality in the UK biobank cohort: revisiting and evaluating risk factors. Eur J Epidemiol. https://link.springer.com/article/10.1007/s10654-021-00722-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskier, D. , Suner, A. , Oktay, Y. , and Karakülah, G. (2020) Mutations of SARS‐CoV‐2 nsp14 exhibit strong association with increased genome‐wide mutation load. PeerJ 8: e10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, A.R. , and Perlman, S. (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, Y. , Mizrahi, O. , Nachshon, A. , Weingarten‐Gabbay, S. , Morgenstern, D. , Yahalom‐Ronen, Y. , et al. (2021) The coding capacity of SARS‐CoV‐2. Nature 589: 125–130. [DOI] [PubMed] [Google Scholar]
- Firestone, M.J. , Lorentz, A.J. , Wang, X. , Como‐Sabetti, K. , Vetter, S. , Smith, K. , et al. (2021) First identified cases of SARS‐CoV‐2 variant B.1.1.7 in Minnesota – December 2020–January 2021. MMWR Morb Mortal Wkly Rep 70: 278–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Qin, B. , Chen, P. , Zhu, K. , Hou, P. , Wojdyla, J.A. , et al. (2021) Crystal structure of SARS‐CoV‐2 papain‐like protease. Acta Pharm Sin B 11: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , Yan, L. , Huang, Y. , Liu, F. , Zhao, Y. , Cao, L. , et al. (2020) Structure of the RNA‐dependent RNA polymerase from COVID‐19 virus. Science 368: 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff, H. , Hewson, R. , and Opstelten, D.J. (1998) Virus maturation by budding. Microbiol Mol Biol Rev 62: 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, S. , Dellibovi‐Ragheb, T.A. , Kerviel, A. , Pak, E. , Qiu, Q. , Fisher, M. , et al. (2020) β‐Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183: 1520–1535.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil, S.M.‐C. , Janowska, K. , McDowell, S. , Mansouri, K. , Parks, R. , Manne, K. , et al. (2021) D614G mutation alters SARS‐CoV‐2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep 34: 108630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, E. (2020) Exaggerated risk of transmission of COVID‐19 by fomites. Lancet Infect Dis 20: 892–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, S.A. , Brown, J. , Pedersen, B.S. , Quinlan, A.R. , and Elde, N.C. (2021) Extensive recombination‐driven coronavirus diversification expands the pool of potential pandemic pathogens. bioRxiv (Evol Biol) https://www.biorxiv.org/content/10.1101/2021.02.03.429646v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowthaman, R. , Guest, J.D. , Yin, R. , Adolf‐Bryfogle, J. , Schief, W.R. , and Pierce, B.G. (2021) CoV3D: a database of high resolution coronavirus protein structures. Nucleic Acids Res 49: D282–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal, M. , De Bruyne, K. , Belkum, A. V. , & West, B. (2021). Different SARS‐CoV‐2 haplotypes associate with geographic origin and case fatality rates of COVID‐19 patients Infection. Genetics and Evolution 90:104730. 10.1016/j.meegid.2021.104730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, I. , Ashkenazi, S. , Merzon, E. , Vinker, S. , and Golan‐Cohen, A. (2020) The association of previous influenza vaccination and coronavirus disease‐2019. Hum Vaccin Immunother. https://www.tandfonline.com/doi/full/10.1080/21645515.2020.1852010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, T.J. , Speck, P. , Geng, L. , Raftos, D. , Beard, M.R. , and Helbig, K.J. (2015) Oyster viperin retains direct antiviral activity and its transcription occurs via a signalling pathway involving a heat‐stable haemolymph protein. J Gen Virol 96: 3587–3597. [DOI] [PubMed] [Google Scholar]
- Gribble, J. , Stevens, L.J. , Agostini, M.L. , Anderson‐Daniels, J. , Chappell, J.D. , Lu, X. , et al. (2021) The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog 17: e1009226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, L.Z.F. , Sacerdoti, M. , Piiper, A. , Zeuzem, S. , Leroux, A.E. , and Biondi, R.M. (2020) ACE2, the receptor that enables infection by SARS‐CoV‐2: biochemistry, structure, allostery and evaluation of the potential development of ACE2 modulators. ChemMedChem 15: 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, Q. , Sadykov, M. , Mfarrej, S. , Hala, S. , Naeem, R. , Nugmanova, R. , et al. (2020) A genetic barcode of SARS‐CoV‐2 for monitoring global distribution of different clades during the COVID‐19 pandemic. Int J Infect Dis 100: 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A. , Sabarinathan, R. , Bala, P. , Donipadi, V. , Vashisht, D. , Katika, M.R. , et al. (2021) A comprehensive profile of genomic variations in the SARS‐CoV‐2 isolates from the state of Telangana, India. J Gen Virol. https://www.microbiologyresearch.org/content/journal/jgv/10.1099/jgv.0.001562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, P. , Muralidharan, M. , Krogan, N.J. , Kaake, R.M. , and Hüttenhain, R. (2021) Proteomic approaches to study SARS‐CoV‐2 biology and COVID‐19 pathology. J Proteome Res 20: 1133–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamet, P. , Pausova, Z. , Attaoua, R. , Hishmih, C. , Haloui, M. , Shin, J. , et al. (2021) SARS‐CoV‐2 receptor ACE2 gene is associated with hypertension and severity of COVID 19: interaction with sex, obesity and smoking. Am J Hypertens. https://academic.oup.com/ajh/advance-article/doi/10.1093/ajh/hpaa223/6056793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian, E. , Nandakumar, D. , Lari, A. , Ly, M. , Tucker, J.M. , and Glaunsinger, B.A. (2020) The molecular virology of coronaviruses. J Biol Chem 295: 12910–12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich, A. , Hofmann‐Winkler, H. , Gierer, S. , Liepold, T. , Jahn, O. , and Pöhlmann, S. (2014) TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88: 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikmet, F. , Méar, L. , Edvinsson, Å. , Micke, P. , Uhlén, M. , and Lindskog, C. (2020) The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16: e9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft, E.B. , Domman, D.B. , Snyder, D.J. , Oguntuyo, K. , Van Diest, M. , Densmore, K.H. , et al. (2021) Emergence in late 2020 of multiple lineages of SARS‐CoV‐2 Spike protein variants affecting amino acid position 677. medRxiv . preprint. 10.1101/2021.02.12.21251658. [DOI]
- Hoffmann, M. , Kleine‐Weber, H. , and Pöhlmann, S. (2020a) A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell 78: 779–784.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , Schroeder, S. , Krüger, N. , Herrler, T. , Erichsen, S. , et al. (2020b) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby, P.W. , Pfeiffer, D. , and Oshitani, H. (2013) Prospects for emerging infections in east and Southeast Asia 10 years after severe acute respiratory syndrome. Emerg Infect Dis 19: 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulswit, R.J.G. , de Haan, C.a.M. , and Bosch, B.‐J. (2016) Coronavirus spike protein and tropism changes. Adv Virus Res 96: 29–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur, S. (2019) Double‐stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa, E. , Merhi, G. , Panossian, B. , Salloum, T. , and Tokajian, S. (2020) SARS‐CoV‐2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems 5: e00266‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski, L. , Iyer, M. , Alisoltani, A. , Sedova, M. , and Godzik, A. (2020) The interplay of SARS‐CoV‐2 evolution and constraints imposed by the structure and functionality of its proteins. bioRxiv . preprint. 10.1101/2020.08.10.244756. [DOI] [PMC free article] [PubMed]
- Ji, W. , Wang, W. , Zhao, X. , Zai, J. , and Li, X. (2020) Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J Med Virol 92: 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X. , Lian, J.‐S. , Hu, J.‐H. , Gao, J. , Zheng, L. , Zhang, Y.‐M. , et al. (2020) Epidemiological, clinical and virological characteristics of 74 cases of coronavirus‐infected disease 2019 (COVID‐19) with gastrointestinal symptoms. Gut 69: 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, D. , Gao, S. , Tian, Z. , Huang, D. , Guan, G. , Liu, G. , et al. (2020) Ovine viperin inhibits bluetongue virus replication. Mol Immunol 126: 87–94. [DOI] [PubMed] [Google Scholar]
- Karapiperis, C. , Kouklis, P. , Papastratos, S. , Chasapi, A. , Danchin, A. , and Ouzounis, C.A. (2020) Preliminary evidence for seasonality of Covid‐19 due to ultraviolet radiation. F1000Res 9: 658. [Google Scholar]
- Khayat, A.S. , de Assumpção, P.P. , Meireles Khayat, B.C. , Thomaz Araújo, T.M. , Batista‐Gomes, J.A. , Imbiriba, L.C. , et al. (2020) ACE2 polymorphisms as potential players in COVID‐19 outcome. PLoS One 15: e0243887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.‐K. , Knapp, J.J. , Kuang, D. , Chawla, A. , Cassonnet, P. , Lee, H. , et al. (2020) A comprehensive, flexible collection of SARS‐CoV‐2 coding regions. G3 (Bethesda, Md) 10: 3399–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine‐Weber, H. , Schroeder, S. , Krüger, N. , Prokscha, A. , Naim, H.Y. , Müller, M.A. , et al. (2020) Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg Microbes Infect 9: 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama, T. , Platt, D. , and Parida, L. (2020) Variant analysis of SARS‐CoV‐2 genomes. Bull World Health Organ 98: 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, D.K. , Kelly, S.M. , Lewicki, D.N. , Ruffolo, R. , and Gallagher, T.M. (2001) Variations in disparate regions of the murine coronavirus spike protein impact the initiation of membrane fusion. J Virol 75: 2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landoni, G. , Zangrillo, A. , Romero García, C.S. , Faustini, C. , Di Piazza, M. , Conte, F. , et al. (2020) Nations with high smoking rate have low SARS‐CoV‐2 infection and low COVID‐19 mortality rate. Acta Biomed 91: e2020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, Y. , Li, W. , Li, Z. , Koerhuis, D. , van den Burg, A.C.S. , Rozemuller, E. , et al. (2020) Coronavirus hemagglutinin‐esterase and spike proteins coevolve for functional balance and optimal virion avidity. Proc Natl Acad Sci U S A 117: 25759–25770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte, M. , and Naesens, L. (2017) Airway proteases: an emerging drug target for influenza and other respiratory virus infections. Curr Opin Virol 24: 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pendu, J. , Breiman, A. , Rocher, J. , Dion, M. , and Ruvoën‐Clouet, N. (2021) ABO blood types and COVID‐19: spurious, anecdotal, or truly important relationships? A reasoned review of available data. Viruses 13(2): 160. 10.3390/v13020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey, P. , Ruktanonchai, N. , Hong, S. , Colizza, V. , Poletto, C. , den Broeck, F.V. , et al. (2021) SARS‐CoV‐2 European resurgence foretold: interplay of introductions and persistence by leveraging genomic and mobility data. Res Sq. https://www.researchsquare.com/article/rs-208849/v1. [Google Scholar]
- Lemieux, R.U. , LePendu, J. , and Hindsgaul, O. (1979) The Lewis antigens and secretor status. Jpn J Antibiot 32: S21–S31. [PubMed] [Google Scholar]
- Li, W. , Hulswit, R.J.G. , Kenney, S.P. , Widjaja, I. , Jung, K. , Alhamo, M.A. , et al. (2018) Broad receptor engagement of an emerging global coronavirus may potentiate its diverse cross‐species transmissibility. Proc Natl Acad Sci U S A 115: E5135–E5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber, C. , Melekidis, S. , Koch, R. , and Bauer, H.‐J. (2021) Insights into the evaporation characteristics of saliva droplets and aerosols: levitation experiments and numerical modeling. J Aerosol Sci 154: 105760. 10.1016/j.jaerosci.2021.105760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler, D.R. , Gully, B.S. , Colson, R.N. , and Rossjohn, J. (2020) Crystal structure of the SARS‐CoV‐2 non‐structural protein 9, Nsp9. iScience 23: 101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Shen, J. , Yang, L. , Hu, C.‐D. , and Wan, J. (2020) Distinct genetic spectrums and evolution patterns of SARS‐CoV‐2. medRxiv(Health Informatics). 10.1101/2020.06.16.20132902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S. , Ye, Q. , Singh, D. , Cao, Y. , Diedrich, J.K. , Yates, J.R. , et al. (2021) The SARS‐CoV‐2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane‐associated M protein. Nat Commun 12: 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin, J.H. , Zardecki, C. , Dolan, E.M. , Lu, C. , Shen, Z. , Dutta, S. , et al. (2020) Evolution of the SARS‐CoV‐2 proteome in three dimensions (3D) during the first six months of the COVID‐19 pandemic. bioRxiv . preprint. 10.1101/2020.12.01.406637 [DOI] [PMC free article] [PubMed]
- Lukassen, S. , Chua, R.L. , Trefzer, T. , Kahn, N.C. , Schneider, M.A. , Muley, T. , et al. (2020) SARS‐CoV‐2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J 39: e105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Wong, Y.‐S. , and Lam, T.‐W. (2020) Tracking cytosine depletion in SARS‐CoV‐2. bioRxiv(Bioinformatics). 10.1101/2020.10.26.354787 [DOI] [Google Scholar]
- Madhugiri, R. , Fricke, M. , Marz, M. , and Ziebuhr, J. (2016) Coronavirus cis‐acting RNA elements. Adv Virus Res 96: 127–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenkov, V. , Mazoure, B. , Rabusseau, G. , and Legendre, P. (2021) Horizontal gene transfer and recombination analysis of SARS‐CoV‐2 genes helps discover its close relatives and shed light on its origin. BMC Ecol Evol 21: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyášek, R. , and Kovařík, A. (2020) Mutation patterns of human SARS‐CoV‐2 and bat RaTG13 coronavirus genomes are strongly biased towards C>U transitions, indicating rapid evolution in their hosts. Genes (Basel) 11(7): 761. 10.3390/genes11070761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustaqil, M. , Ollivier, E. , Chiu, H.‐P. , Van Tol, S. , Rudolffi‐Soto, P. , Stevens, C. , et al. (2021) SARS‐CoV‐2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect 10: 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya, A. , Holmes, E.C. , and González‐Candelas, F. (2004) The population genetics and evolutionary epidemiology of RNA viruses. Nat Rev Microbiol 2: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, Á. , Pongor, S. , and Győrffy, B. (2021) Different mutations in SARS‐CoV‐2 associate with severe and mild outcome. Int J Antimicrob Agents 57: 106272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan, Y. , Nan, G. , and Zhang, Y.‐J. (2014) Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 6: 4999–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neches, R.Y. , Kyrpides, N.C. , and Ouzounis, C.A. (2021) Atypical divergence of SARS‐CoV‐2 Orf8 from Orf7a within the coronavirus lineage suggests potential stealthy viral strategies in immune evasion. mBio 12(1): e03014‐20. 10.1128/mBio.03014-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman, B.W. , Joseph, J.S. , Saikatendu, K.S. , Serrano, P. , Chatterjee, A. , Johnson, M.A. , et al. (2008) Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J Virol 82: 5279–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsom, R.B. , Amara, A. , Hicks, A. , Quint, M. , Pattison, C. , Bzdek, B.R. , et al. (2021) Comparison of droplet spread in standard and laminar flow operating theatres: SPRAY study group. J Hosp Infect 110: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, K.W. , Faulkner, N. , Cornish, G.H. , Rosa, A. , Harvey, R. , Hussain, S. , et al. (2020) Preexisting and de novo humoral immunity to SARS‐CoV‐2 in humans. Science 370: 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T.‐W. , Turinici, G. , Ching, W.‐K. , Chung, S.‐K. , and Danchin, A. (2007) A parasite vector‐host epidemic model for TSE propagation. Med Sci Monit 13: BR59–BR66. [PubMed] [Google Scholar]
- Ng, T.W. , Turinici, G. , and Danchin, A. (2003) A double epidemic model for the SARS propagation. BMC Infect Dis 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgren, J. , and Svensson, L. (2019) Genetic susceptibility to human norovirus infection: an update. Viruses 11(3): 226. 10.3390/v11030226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norell Bergendahl, M. , and Stanford University (eds). (2009) Design Theory and Research Methodology. Glasgow: Design Society. [Google Scholar]
- Normand, S. , Waldschmitt, N. , Neerincx, A. , Martinez‐Torres, R.J. , Chauvin, C. , Couturier‐Maillard, A. , et al. (2018) Proteasomal degradation of NOD2 by NLRP12 in monocytes promotes bacterial tolerance and colonization by enteropathogens. Nat Commun 9: 5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando, N.S. , Zevenhoven‐Dobbe, J.C. , van der Meer, Y. , Bredenbeek, P.J. , Posthuma, C.C. , and Snijder, E.J. (2020) The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS‐CoV and SARS‐CoV‐2. J Virol 94(23): e01246–20. 10.1128/JVI.01246-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortego, J. , Ceriani, J.E. , Patiño, C. , Plana, J. , and Enjuanes, L. (2007) Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 368: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östbye, H. , Gao, J. , Martinez, M.R. , Wang, H. , de Gier, J.‐W. , and Daniels, R. (2020) N‐linked glycan sites on the influenza A virus neuraminidase head domain are required for efficient viral incorporation and replication. J Virol 94(19): e00874–20. 10.1128/JVI.00874-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, Z. , Ouzounis, C. , Wang, D. , Sun, W. , Li, J. , Chen, W. , et al. (2020) A path towards SARS‐CoV‐2 attenuation: metabolic pressure on CTP synthesis rules the virus evolution, Genome Biol Evol. 12(12): 2467–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulas, A. , Zanti, M. , Tomazou, M. , Zachariou, M. , Minadakis, G. , Bourdakou, M.M. , et al. (2021) Generalized linear models provide a measure of virulence for specific mutations in SARS‐CoV‐2 strains. PLoS One 16: e0238665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsyannikova, I.G. , Haralambieva, I.H. , Crooke, S.N. , Poland, G.A. , and Kennedy, R.B. (2020) The role of host genetics in the immune response to SARS‐CoV‐2 and COVID‐19 susceptibility and severity. Immunol Rev 296: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyston, P. , and Robinson, K. (2012) The current challenges for vaccine development. J Med Microbiol 61: 889–894. [DOI] [PubMed] [Google Scholar]
- Papa, G. , Mallery, D.L. , Albecka, A. , Welch, L.G. , Cattin‐Ortolá, J. , Luptak, J. , et al. (2021) Furin cleavage of SARS‐CoV‐2 spike promotes but is not essential for infection and cell‐cell fusion. PLoS Pathog 17: e1009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasick, J.M. , Kalicharran, K. , and Dales, S. (1994) Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL‐21 neuronal cell line. J Virol 68: 2915–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patiño‐Galindo, J.Á. , Filip, I. , and Rabadan, R. (2021) Global patterns of recombination across human viruses. Mol Biol Evol. https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msab046/6135088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly, M.D. , and Lauring, A.S. (2015) Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J Virol 89: 3584–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease, L.F. , Wang, N. , Salsbury, T.I. , Underhill, R.M. , Flaherty, J.E. , Vlachokostas, A. , et al. (2021) Investigation of potential aerosol transmission and infectivity of SARS‐CoV‐2 through central ventilation systems. Build Environ. https://www.sciencedirect.com/science/article/pii/S0360132321000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perveen, S. , Khalili Yazdi, A. , Devkota, K. , Li, F. , Ghiabi, P. , Hajian, T. , et al. (2021) A high‐throughput RNA displacement assay for screening SARS‐CoV‐2 nsp10‐nsp16 complex toward developing therapeutics for COVID‐19. SLAS Discov 2472555220985040. https://journals.sagepub.com/doi/10.1177/2472555220985040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacenza, L. , Trujillo, M. , and Radi, R. (2019) Reactive species and pathogen antioxidant networks during phagocytosis. J Exp Med 216: 501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillon, M.C. , Frazier, M.N. , Dillard, L.B. , Williams, J.G. , Kocaman, S. , Krahn, J.M. , et al. (2021) Cryo‐EM structures of the SARS‐CoV‐2 endoribonuclease Nsp15 reveal insight into nuclease specificity and dynamics. Nat Commun 12: 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasschaert, D. , Duarte, M. , and Laude, H. (1990) Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. J Gen Virol 71: 2599–2607. [DOI] [PubMed] [Google Scholar]
- Reid, A.H. , Fanning, T.G. , Janczewski, T.A. , Lourens, R.M. , and Taubenberger, J.K. (2004) Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J Virol 78: 12462–12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurabh, S. , Verma, M.K. , Gautam, V. , Kumar, N. , Jain, V. , Goel, A.D. , et al. (2021) Tobacco, alcohol use and other risk factors for developing symptomatic COVID‐19 vs asymptomatic SARS‐CoV‐2 infection: a case‐control study from western Rajasthan, India. Trans R Soc Trop Med Hyg. https://academic.oup.com/trstmh/advance-article/doi/10.1093/trstmh/traa172/6097955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetelig, J. , Baldauf, H. , Wendler, S. , Heidenreich, F. , Real, R. , Kolditz, M. , et al. (2021) Blood group A epitopes do not facilitate entry of SARS‐CoV‐2. J Intern Med. https://onlinelibrary.wiley.com/doi/10.1111/joim.13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkelshoek, M.S. , Fronczek, R. , Kooy‐Winkelaar, E.M.C. , Petersen, J. , Reid, H.H. , van der Heide, A. , et al. (2019) H1N1 hemagglutinin‐specific HLA‐DQ6‐restricted CD4+ T cells can be readily detected in narcolepsy type 1 patients and healthy controls. J Neuroimmunol 332: 167–175. [DOI] [PubMed] [Google Scholar]
- Sengupta, A. , Hassan, S.S. , and Choudhury, P.P. (2021) Clade GR and clade GH isolates of SARS‐CoV‐2 in Asia show highest amount of SNPs. Infect Genet Evol 89: 104724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Wan, Y. , Liu, C. , Yount, B. , Gully, K. , Yang, Y. , et al. (2020) Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Pathog 16: e1008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, V.K. , Jinadatha, C. , Lichtfouse, E. , Decroly, E. , van Helden, J. , Choi, H. , and Chatterjee, P. (2021) COVID‐19 epidemiologic surveillance using wastewater. Environ Chem Lett s10311‐021‐01188‐w. https://link.springer.com/article/10.1007%2Fs10311-021-01188-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea, J.‐E. , Best, R.B. , and Mittal, J. (2021) Physics‐based computational and theoretical approaches to intrinsically disordered proteins. Curr Opin Struct Biol 67: 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y.‐Y. , Liang, L. , Zhu, Z.‐H. , Zhou, W.‐P. , Irwin, D.M. , and Zhang, Y.‐P. (2010) Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc Natl Acad Sci 107: 8666–8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shope, R.E. (1936) The incidence of neutralizing antibodies for swine influenza virus in the sera of human beings of different ages. J Exp Med 63: 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell, S.G. , Barthel, A. , and ter Meulen, V. (1981) Coronavirus JHM: a virion‐associated protein kinase. J Gen Virol 52: 235–243. [DOI] [PubMed] [Google Scholar]
- Simmonds, P. (2020) Rampant C→U hypermutation in the genomes of SARS‐CoV‐2 and other coronaviruses: causes and consequences for their short‐ and long‐term evolutionary trajectories. mSphere 5: e00408‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, G. , Singh, P. , Pillatzki, A. , Nelson, E. , Webb, B. , Dillberger‐Lawson, S. , and Ramamoorthy, S. (2019) A minimally replicative vaccine protects vaccinated piglets against challenge with the porcine epidemic diarrhea virus. Front Vet Sci 6: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slanina, H. , Madhugiri, R. , Bylapudi, G. , Schultheiß, K. , Karl, N. , Gulyaeva, A. , et al. (2021) Coronavirus replication‐transcription complex: vital and selective NMPylation of a conserved site in nsp9 by the NiRAN‐RdRp subunit. Proc Natl Acad Sci U S A 118(6): e2022310118. 10.1073/pnas.2022310118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, K.A. (2012) Louis Pasteur, the father of immunology? Front Immunol 3: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souilmi, Y. , Lauterbur, M.E. , Tobler, R. , Huber, C.D. , Johar, A.S. , and Enard, D. (2020) An ancient viral epidemic involving host coronavirus interacting genes more than 20,000years ago in East Asia. bioRxiv(Evol Biol). 10.1101/2020.11.16.385401. [DOI] [Google Scholar]
- Sullivan, Gregory, F. , and Garson, O. (2013) It's difficult to make predictions, especially about the future. Quote Investig. https://quoteinvestigator.com/2013/10/20/no-predict/. [Google Scholar]
- Taleb, N. (2008) The Black Swan: The Impact of the Highly Improbable. New York: Random House. [Google Scholar]
- Tharappel, A.M. , Samrat, S.K. , Li, Z. , and Li, H. (2020) Targeting crucial host factors of SARS‐CoV‐2. ACS Infect Dis 6: 2844–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiler, M. , and Smith, H.H. (1937) The use of yellow fever virus modified by in vitro cultivation for human immunization. J Exp Med 65: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidu, A. , Janvier, A. , Schaeffer, L. , Sosnowski, P. , Kuhn, L. , Hammann, P. , et al. (2020) The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS‐CoV‐2 translation. RNA 27(3): 253–264. 10.1261/rna.078121.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis, K. (2020) covid ‐19 transmission: economy‐boosting investment should target innovation in pandemic containment strategies to minimize restrictions of civil liberties. Environ Microbiol 22: 4527–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici, M.A. , and Veesler, D. (2019) Structural insights into coronavirus entry. Adv Virus Res 105: 93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscott, R.J.W. , Schey, K.L. , and Friedrich, M.G. (2016) Old proteins in man: a field in its infancy. Trends Biochem Sci 41: 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trypsteen, W. , Van Cleemput, J. , van Snippenberg, W. , Gerlo, S. , and Vandekerckhove, L. (2020) On the whereabouts of SARS‐CoV‐2 in the human body: a systematic review. PLoS Pathog 16: e1009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon, N. , Toulouse, M.‐J. , Martel, B. , Moineau, S. , and Duchaine, C. (2014) Comparison of five bacteriophages as models for viral aerosol studies. Appl Environ Microbiol 80: 4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turinici, G. , and Danchin, A. (2007) The SARS case study: an alarm clock? In Encyclopedia of Infectious Diseases, Tibayrenc, M. (ed). Hoboken, NJ, USA: John Wiley & Sons, pp. 151–162. [Google Scholar]
- Vilar, S. , and Isom, D.G. (2021) One year of SARS‐CoV‐2: how much has the virus changed? Biology (Basel) 10(2): 91. 10.3390/biology10020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A.C. , Tortorici, M.A. , Frenz, B. , Snijder, J. , Li, W. , Rey, F.A. , et al. (2016) Glycan shield and epitope masking of a coronavirus spike protein observed by cryo‐electron microscopy. Nat Struct Mol Biol 23: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Baudys, J. , Bundy, J.L. , Solano, M. , Keppel, T. , and Barr, J.R. (2020a) Comprehensive analysis of the glycan complement of SARS‐CoV‐2 spike proteins using signature ions‐triggered electron‐transfer/higher‐energy collisional dissociation (EThcD) mass spectrometry. Anal Chem 92: 14730–14739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Li, X. , Li, T. , Zhang, S. , Wang, L. , Wu, X. , and Liu, J. (2020b) The genetic sequence, origin, and diagnosis of SARS‐CoV‐2. Eur J Clin Microbiol Infect Dis 39: 1629–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Chen, W. , Zhang, Z. , Deng, Y. , Lian, J.‐Q. , Du, P. , et al. (2020c) CD147‐spike protein is a novel route for SARS‐CoV‐2 infection to host cells. Signal Transduct Target Ther 5: 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, J. , Guo, S. , Long, E. , Zhang, L. , Shu, B. , and Guo, L. (2021) Why does the spread of COVID‐19 vary greatly in different countries? Revealing the efficacy of face masks in epidemic prevention. Epidemiol Infect 149: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley, R.D. , Woods, R.D. , Hill, H.T. , and Biwer, J.D. (1990) Evidence for a porcine respiratory coronavirus, antigenically similar to transmissible gastroenteritis virus, in the United States. J Vet Diagn Invest 2: 312–317. [DOI] [PubMed] [Google Scholar]
- Williams, F.M.K. , Freidin, M.B. , Mangino, M. , Couvreur, S. , Visconti, A. , Bowyer, R.C.E. , et al. (2020) Self‐reported symptoms of COVID‐19, including symptoms most predictive of SARS‐CoV‐2 infection, are heritable. Twin Res Hum Genet 23(6): 316–321. 10.1017/thg.2020.85. [DOI] [PubMed] [Google Scholar]
- Wood, C.L. , McInturff, A. , Young, H.S. , Kim, D. , and Lafferty, K.D. (2017) Human infectious disease burdens decrease with urbanization but not with biodiversity. Phil Trans R Soc B 372: 20160122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y.‐M. , Wang, W. , Song, Z.‐G. , et al. (2020) A new coronavirus associated with human respiratory disease in China. Nature 579: 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtmann, E.J. , and Wolin, S.L. (2009) RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol 44: 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y.‐R. , and Lei, C.‐Q. (2020) TAK1‐TABs complex: a central semiosome in inflammatory responses. Front Immunol 11: 608976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, Y. , and Liu, D.X. (2009) Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin‐dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 83: 8744–8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, L. , Ge, J. , Zheng, L. , Zhang, Y. , Gao, Y. , Wang, T. , et al. (2021) Cryo‐EM structure of an extended SARS‐CoV‐2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell 184: 184–193.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H.‐C. , Chen, C.‐H. , Wang, J.‐H. , Liao, H.‐C. , Yang, C.‐T. , Chen, C.‐W. , et al. (2020a) Analysis of genomic distributions of SARS‐CoV‐2 reveals a dominant strain type with strong allelic associations. Proc Natl Acad Sci U S A 117: 30679–30686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Wang, W. , Chen, Z. , Lu, S. , Yang, F. , Bi, Z. , et al. (2020b) A vaccine targeting the RBD of the S protein of SARS‐CoV‐2 induces protective immunity. Nature 586: 572–577. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Yue, Y. , Song, N. , Li, C. , Yuan, Z. , Wang, Y. , et al. (2020c) The YdiU domain modulates bacterial stress signaling through Mn2+‐dependent UMPylation. Cell Rep 32: 108161. [DOI] [PubMed] [Google Scholar]
- Yost, S.A. , and Marcotrigiano, J. (2013) Viral precursor polyproteins: keys of regulation from replication to maturation. Curr Opin Virol 3: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, H.‐L. , Dichio, V. , Rodríguez Horta, E. , Thorell, K. , and Aurell, E. (2020) Global analysis of more than 50,000 SARS‐CoV‐2 genomes reveals epistasis between eight viral genes. Proc Natl Acad Sci U S A 117: 31519–31526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z. , Li, T.‐T. , Jin, X. , Peng, F.‐H. , Song, N.‐H. , Peng, G.‐Q. , and Ge, X.‐Y. (2017) Coexistence of multiple genotypes of porcine epidemic diarrhea virus with novel mutant S genes in the Hubei Province of China in 2016. Virol Sin 32: 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. , Li, Y. , Zhang, A.L. , Wang, Y. , and Molina, M.J. (2020) Identifying airborne transmission as the dominant route for the spread of COVID‐19. Proc Natl Acad Sci U S A 117: 14857–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.W. , Yap, Y.L. , and Danchin, A. (2005) Testing the hypothesis of a recombinant origin of the SARS‐associated coronavirus. Arch Virol 150: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Sun, L. , Zhao, Y. , Feng, D. , Cheng, J. , and Zhang, G. (2020) Coronavirus endoribonuclease ensures efficient viral replication and prevents protein kinase R activation. J Virol 95. https://jvi.asm.org/content/95/7/e02103-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Qin, P. , and Huang, Y.‐W. (2021) Lysosomal ion channels involved in cellular entry and uncoating of enveloped viruses: implications for therapeutic strategies against SARS‐CoV‐2. Cell Calcium 94: 102360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D. , Dejnirattisai, W. , Supasa, P. , Liu, C. , Mentzer, A.J. , Ginn, H.M. , et al. (2021) Evidence of escape of SARS‐CoV‐2 variant B.1.351 from natural and vaccine induced sera. Cell S0092867421002269. https://www.cell.com/cell/fulltext/S0092-8674(21)00226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N. , Zhang, D. , Wang, W. , Li, X. , Yang, B. , Song, J. , et al. (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382: 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler, C.G.K. , Allon, S.J. , Nyquist, S.K. , Mbano, I.M. , Miao, V.N. , Tzouanas, C.N. , et al. (2020) SARS‐CoV‐2 receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181: 1016–1035.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, X. , Chen, K. , Zou, J. , Han, P. , Hao, J. , and Han, Z. (2020) Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med 14: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Mutation count and rate of each gene summarized from SARS‐CoV‐2 193,687 strains.
Fig. S1 Temporal‐geographical mutation density of the Spike proteins at four different time points in 2020.
Appendix S1 Supporting Information.