

Abstract
The term “privileged structure” refers to a single molecular substructure or scaffold that can serve as a starting point for high‐affinity ligands for more than one receptor type. In this report, a hitherto overlooked group of privileged substructures is addressed, namely aromatic oligoamides, for which there are natural models in the form of cystobactamids, albicidin, distamycin A, netropsin, and others. The aromatic and heteroaromatic core, together with a flexible selection of substituents, form conformationally well‐defined scaffolds capable of specifically binding to conformationally well‐defined regions of biomacromolecules such as helices in proteins or DNA often by acting as helices mimics themselves. As such, these aromatic oligoamides have already been employed to inhibit protein–protein and nucleic acid–protein interactions. This article is the first to bring together the scattered knowledge about aromatic oligoamides in connection with biomedical applications.
Keywords: aromatic oligoamides, cystobactamids, DNA-binding, dystamycin, foldamers, protein–protein interactions
Privileged! Aromatic oligoamides based on pyrroles, pyridines, and p‐aminobenzoic acids are presented as privileged structures that efficiently target nucleic acids and proteins as well as complexes derived therefrom. Their design can be related to natural products such distamycin A and the cystobactamids.
1. Introduction
Several thousand naturally occurring peptides are known, which show a wide range of biological properties. These include hormones, neurotransmitters, growth factors, ion channel ligands, anti‐infectives, anticancer agents, immunosuppressants, and others. [1] Whether they naturally are of ribosomal origin [2] or are biosynthesized by the non‐ribosomal peptide synthase (NRPS), [3] in both cases, they are usually based on α‐amino acids (Figure 1, top) and in selected cases also on β‐, γ‐, or ω‐amino acids.
Figure 1.
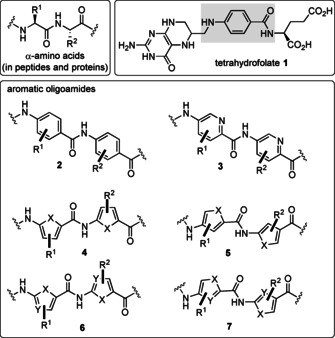
Amide backbone of peptides and proteins composed of natural α‐amino acids (top), p‐aminobenzoic acids (marked in gray) is part of tetrahydrofolic acid (1) and backbones of aromatic and heteroaromatic oligoamides 2–7 (bottom) (X=N, S).
Medicinal chemists have used the peptide motif to broaden the scope of these architectures by utilizing other types of aminocarboxylic acids, including arenes and hetarenes (Figure 1, bottom). For example, p‐aminobenzoic acid (PABA) is a constituent of folic acid (1) and is biosynthesized through the shikimate acid pathway. [4] p‐Aminobenzoic acid is also an important building block frequently found as a structural element in synthetic drugs. In 2002, it was analyzed that out of 12 111 commercial drugs, 1.5 % (184 drugs) contained the PABA moiety (see 2 and pyridine analog 3) with broad therapeutic potential including sun‐screening, antibacterial, antineoplastic, local anesthetic, anticonvulsant, antiarrhythmic, antiemetic, gastrokinetic, antipsychotic, neuroleptic, and migraine prophylactic properties. [5]
Likewise, five‐membered heteroarenes were also developed through structural insertions between an amino and a carboxyl group, which function as monomers of oligoamides 4–7. Key work on the design of such specific DNA topographies recognizing hetarene‐containing oligoamides has been published by the Dervan group over a period of several decades (see Section 3).
The present report is intended to provide an overview on oligoamides based on arenes and hetarenes covering the three major topics—natural products, medicinal chemistry, and supramolecular chemistry—commonly with a focus on drug development and chemical biology. We would like to draw the reader's attention to the fact that such oligoamides, despite the diversity of the fields in which they have been studied, are privileged in terms of control of their conformation and their ability to sequence specifically recognized biomacromolecules including proteins and nucleic acids. They are even able to intervene in protein–protein and protein–nucleic acid interactions and inhibit such complexes.
Our report starts with naturally occurring aromatic oligoamides, moves on to medicinal chemistry programs, especially in the recognition of double‐stranded (ds) DNA, and will then address structural aspects of these oligoamides as foldamers.
2. Natural Antibiotics Based on Oligo‐p‐Aminobenzoic Acids
2.1. Cystobactamids
In 2014, Müller and co‐workers reported on a new class of oligoamide natural products that are characterized by building blocks derived from p‐aminobenzoic acid. These were isolated from myxobacteria of the genius Cystobacter sp. Cbv34 and were later also found in strains of the genera Cystobacter, Myxococcus, and Corallococcus. [6] These new oligoamides were called cystobactamids 919‐1 (8) and 919‐2 (9; Figure 2). Two subclasses have been described, having either an iso‐β‐methoxyasparagine or a β‐methoxyasparagine unit linking the oligoarylamides. Later, more derivatives such as the cystobactamids 920‐1 (10), 920‐2 (11), and 861‐2 (12) were disclosed, which structurally differ in the E‐ring and the aspartate hinge region. [7]
Figure 2.
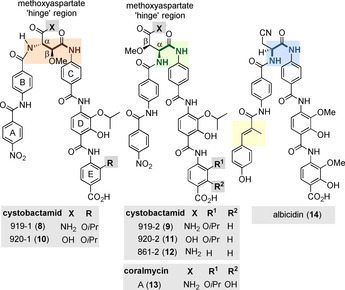
Structures of selected natural cystobactamids 919‐91 (8), 919‐2 (9), 920‐1 (10), 920‐2 (11), and 861‐2 (12) and coralmycin A (13) (rings are labeled A–E).
Cystobactamid 862‐1 (12) was found to be the most active natural member. It inhibits several clinically relevant Gram‐positive and Gram‐negative strains (Acinetobacter baumannii: MIC=0.5 μg mL−1, Citrobacter freundii: MIC=0.06 μg mL−1, carbapenem‐resistant E. coli WT‐III marRΔ74bp: MIC=0.5 μg mL−1, fluoroquinolone‐resistant P. aeruginosa CRE: MIC=1.0 μg mL−1, and Proteus vulgaris: MIC=0.25 μg mL−1) [7] by inhibiting bacterial type IIa topoisomerases. The broad‐spectrum antibacterial activity of the cystobactamids is associated with inhibition of topoisomerases type IIA, namely DNA gyrase and topoisomerase IV.
Later, Kim and co‐workers also isolated cystobactamid 919‐2 (9) along with two other derivatives, named coralmycins A (13) and B (β‐epi‐cystobactamid 920‐2 epi‐11). Here, cultures of Corallococcus coralloides myxobacteria served as the source of these natural oligoamides. [8] In contrast to the original stereochemical assignment, Kim proposed that the configuration of the methoxyasparagine hinge region of 12 should be revised to 2S,3R, which was later confirmed by the total synthesis of both diastereomers (2S,3S and 2S,3R) of cystobactamid 920‐2 (11). [9]
Several focused libraries were prepared by total synthesis to obtain chemically more stable derivatives (the amide bond between the C and D rings is easily hydrolyzed under chemical and enzymatic conditions), improved water solubility, and a broader antibacterial spectrum of activity against Gram‐negative and ‐positive bacteria. The structural changes included the replacement of the amide group by urea or by the 1,2,3‐triazole heterocycle, the latter being able to serve as a substitute for the amide group in cystobactamids. [10]
2.2. Albicidin
An even more remarkable story can be told about albicidin 14, which is structurally closely related to the cystobactamids (Figure 2). First isolated from the phytopathogenic bacterium Xanthomonas albilineans in the 1980s, it took more than 30 years before the structure could finally be elucidated. [11] The natural product showed bactericidal properties by inhibiting DNA replication. [12] Albicidin 14 efficiently inhibits DNA supercoiling catalyzed by E. coli DNA gyrase with an IC50 value of approximately 40 nm. Stabilization of the cleavage complex between gyrase and DNA, which is ATP‐dependent, also makes albicidin 14 an effective compound against Gram‐positive and Gram‐negative bacterial strains. [13] It must be stressed that cystobactamids are structurally related to albicidin 14 with similar antibacterial properties.[ 11 , 14 ] The pentapeptide backbone consists of four aromatic amino acids and the non‐canonical β‐l‐cyano‐alanine. At the N‐terminus, there is 4‐hydroxy‐coumaric acid with an additional methyl group in the α‐position. Besides two unsubstituted p‐aminobenzoic acids flanking the chiral amino acid, the molecule also consists of two C‐terminal 4‐amino‐3‐hydroxy‐2‐methoxybenzoic acids (PMBA). A clear disparity between albicidin 14 and the cystobactamids is the central amino acid. It has been shown that substitution by natural amino acids results in albicidin analogs with IC50 values comparable to those of the natural product. However, some of these derivatives failed in cell‐based assays because they showed reduced MIC values. The replacement of the cyano group by a 1 H‐1,2,3‐triazole provided access to an analog with improved properties, with regard to susceptibility to hydrolysis.[ 14b , 14c ] The N‐terminal functionality seems to have an important influence on the activity. The modification is limited to the substitution of the hydroxyl group (e.g., CF3 or F). [14d] The carbamoylation of this group proved to be beneficial for the antibacterial activity. [14e] Other substitution patterns or different molecule lengths led to weak inhibitors or even to the deletion of the activity. [14d] It is noteworthy that other albicidins were later isolated, showing structural features also known from cystobactamids. The albicidin analog with an l‐β‐OMe‐Asn instead of the β‐l‐cyanalanine shows only reduced antibacterial properties. Other structural combinations of both aromatic oligopeptide classes also only led to reduced activities. However, this combinatorial approach confirmed the crucial role of the N‐terminal aromatic moiety with respect to the substitution pattern and molecular length. Furthermore, the beneficial effect of larger alkoxy groups (OEt or OiPr) at the C‐terminal PABA units was shown.[ 14c , 14f ] In addition, albicidin 14 and related compounds are hydrolyzed by the enzyme AlbD. [15] By introducing peptide bioisosters between the chiral amino acid and the C‐terminus, the triazole unit proved to be suitable to avoid hydrolysis while maintaining activity, [14c] as was also reported for cystobactamids. [10]
Structurally, cystobactamids 8–13 and albicidin 14 are remarkable because they contain aromatic elements that have been independently studied in the field of supramolecular chemistry and the design of oligoaromatic foldamers. These can form helical structures by intramolecular hydrogen bonding. A number of aromatic elements I–VII of such helical oligoamides are summarized in Figure 3 and noteworthy rings E and F in the cystobactamids reflect elements II and IV. These structural units reveal different scenarios of intramolecular hydrogen bonding in which the amide bond and aromatic substituents or heteroatoms (pyridine) with hydrogen‐donor or ‐acceptor properties are involved. Once such elements are part of aromatic oligoamides, defined foldamers with specific binding properties for different biological receptors are generated (a detailed discussion is found in Section 4).
Figure 3.
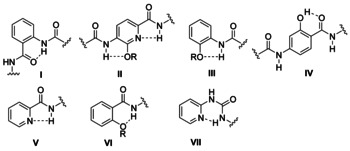
Structures of different classes of oligoaromatic backbone monomers I–VII and their hydrogen‐bonding patterns.
Bacterial type IIa topoisomerases are important targets for antibiotics, of which aminocoumarins that bind to the ATPase active site and the quinolones that bind to the enzyme are the most well‐known classes. In the case of the cystobactamids and albicidin, the exact biological target on type II bacterial topoisomerases is not yet fully known, the structural knowledge of such foldamers and the way they can intervene in protein–protein as well as protein–nucleic acid complexes will be helpful to shed light on the exact mode of action of these natural PABA‐containing oligoamides (see also Section 4).
3. Oligomers Based on Pyrrole Carboxamide Units
3.1. Natural oligopyrrole amides
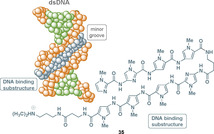
The link between foldamers and cystobactamids and albicidin has not yet been established, but another class of natural products served as a starting point for a medicinal chemistry program that centrally considered concepts of hydrogen bonds with the biological target structure DNA. These are based on oligopyrrole carboxamides. Indeed, DNA is a pharmacological target for various drugs that are currently in clinical application. [16] One way for small aromatic ligand molecules to interact with double‐stranded (ds) DNA is by non‐covalent binding to its minor groove. The aromatic oligoamides distamycin A (15) and netropsin (16) were the first known natural products to interact with the minor groove of dsDNA, followed by anthelvencin C (17) and kikumycin B (18) (Figure 4). These oligoamide antibiotics share cationic charges, hydrogen‐bond donors, a crescent shape, and an oligopyrrole carboxamide skeleton. Netropsin (16) was discovered in 1951 by Finlay et al. as a fermentation product of Streptomyces netropsis. [17] Distamycin A (15) was isolated from Streptomyces distallicus in 1958 by F. Arcamone and collaborators. [18] Both oligoamides show antibiotic (5 to 1000 μg mL−1) and antiviral (50 to 100 μm) activities owing to their reversible binding to DNA.[ 17 , 19 ] For distamycin A (15) and netropsin (16), it was shown that they interact with high selectivity with certain sequences within the minor groove of the double helical DNA (Figure 5).
Figure 4.

Structures of natural pyrrole bearing oligoamides distamycin A (15), netropsin (16), anthelvencin C (17), and kikumycin B (18).
Figure 5.
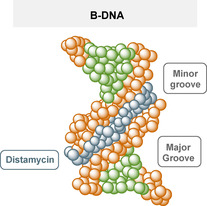
Simplified presentation of distamycin A 15 (blue) binding to the minor groove of B‐DNA (orange and green). [20]
In fact, four to five consecutive dAdT base pairs are affected (Figure 6). [21] This observation was generalized by Dervan et al. who found that a ligand with n amide groups allows a binding site size of n+1 base pairs. This conclusion was based on NMR and X‐ray crystallographic investigations of a netropsin‐oligonucleotide complex. [22] Two possible types of these complexes have been described, the first being a 1:1 peptide‐DNA complex (Figure 6). [23] The position‐specific binding of these cationic ligands to DNA results from hydrogen bonds, electrostatic attraction, and van der Waals interactions. [22]
Figure 6.
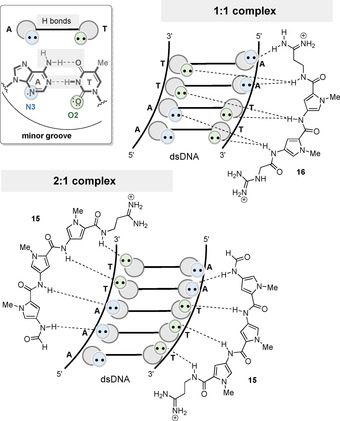
Hydrogen bonding of AT pairs and structural representations of 1:1 and 2:1 complexes of dsDNA with netropsin (16) and distamycin (15), respectively.
In addition, the width of the minor groove of these A,T‐rich sequences bound to the ligand is narrower than that of standard B‐DNA. [24] Two or three N‐methylpyrrolecarboxamides twist in a spiral to give the DNA good complementarity, whereas the NH groups are involved in bifurcated hydrogen bonds to adenine N3 and thymine O2 atoms of bases on both DNA strands. The second structural type is a 2:1 peptide‐DNA complex first reported by Wemmer and Pelton. They used higher ligand/DNA stoichiometries in which two distamycin molecules bind antiparallel to the same DNA sequence. [25] The minor groove width of the 2:1 complex is probably twice as large as that of the 1:1 complex, and each ligand only forms hydrogen bonds with bases on a single strand. [26]
Netropsin (16) and distamycin A (15) have binding affinities ranging from K a=4.0×108 to 5.0×108 m −1 to poly(dA), poly(dT), and poly(dAdT) sequences. [27] It is noteworthy that promoter regions contain many of these (dAdT)‐rich sequences, which then interact with distamycin (15) and netropsin (16), possibly inhibiting DNA‐dependent DNA (cell replication) or RNA (transcription) synthesis.[ 27 , 28 ] The reason why distamycin and netropsin bind AT‐rich sequences is related to the hydrogen‐donor character of their amide protons, which coordinate with the free electron pairs (hydrogen‐bond acceptors) of adenine N3 and thymine O2 in the DNA nucleotide sequence (Figure 6).
Furthermore, Beerman et al. showed that distamycin A (15) modulates the activity of mammalian topoisomerase I. At 0.5 and 1.0 μm, they observed that distamycin stimulates topoisomerase I relaxation of supercoiled DNA, whereas distamycin inhibits the enzyme at concentrations above 2.0 μm. [29] Unfortunately, both natural products show only limited cytotoxicity, which is why their clinical development was never pursued further.
The anthelvencins (specifically anthelvencin C (17)) were isolated in 1965 from cultures of Streptomyces venezuelae ATCC 14 583 and 14 585 and show moderate antibacterial, antifungal (MIC range=1.56–50.0 μg mL−1), and anthelmintic activities. [30] Lee et al. showed that anthelvencin A binds in the minor groove of DNA along the sequence 5’‐A4A5T6T7‐3’. [31] Finally, the kikumycins (kikumycin B (18)) were isolated from the culture filtrate of Streptomyces phaochromogenes R‐719 and they show antiviral properties in the 10 μg mL−1 range. [32]
These structural and binding studies of naturally occurring oligopyrrolamides set the stage for a remarkable academic medicinal chemistry program that clearly showed an evolutionary character since 1951 when netropsin (16) was first discovered. It is briefly summarized in the following subsections.
3.2. Functionalized distamycin derivatives
The results of the natural products suggest that oligopyrroleamides are well suited to recognize and target AT‐rich regions of DNA. With this in mind, Dervan's group prepared the bifunctional distamycin‐EDTA molecule (DE⋅FeII 19) in the early 1980s (Figure 7). It was known that iron(II)‐EDTA complexes are capable of inducing DNA strand breakage in the presence of oxygen. Therefore, the conjugation of EDTA‐FeII to a distamycin analog provided a sequence‐specific DNA cleavage agent. [33] In line with the DNA binding properties of the natural oligopyrroles described above, it was not unexpectedly observed that the cleavage site was in the immediate vicinity of sequences consisting of four to five base pairs dAdT.
Figure 7.
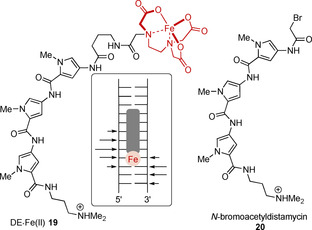
Structure of a bifunctional distamycin‐EDTA molecule (DE⋅FeII 19), its cleavage sites indicated by arrows, and structure of N‐bromoacetyldistamycin 20.
Some of these derivatives have been further developed to give them additional DNA‐alkylating capabilities. The first group to design such analogs was that of Arcamone. They combined small units known to have alkylating properties (nitrogen mustard, aziridine, oxirane, and haloacetyl) with distamycin analogs and created cytotoxic and antitumor active constructs (Figure 8). [34] The cytotoxicity (in vitro cytotoxicity: 0.01 to 29.5 ng mL−1) of such constructs increased with increasing number of up to five pyrrole units, which also led to improved in vivo potency and antileukemic activity (optimal non‐toxic dose: 0.20 to 0.78 mg kg−1). Subsequently, Dervan et al. confirmed the sequence‐specific cleavage of DNA when using N‐bromoacetyldistamycin 20 as alkylating agent. [35]
Figure 8.
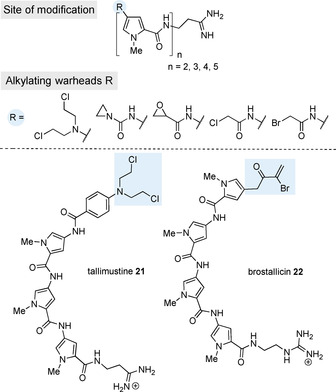
Top: Concept of oligopyrrolamides attached with functional groups able to alkylate DNA. Bottom: Structures of tallimustine 21 and brostallicin 22. Alkylating substituents are marked in light blue.
Since then, the literature has reported various other studies on distamycin derivatives, but these did not lead to new entities that showed improved efficacy compared with classical anticancer drugs such as doxorubicin or cisplatin. However, the combination of the in vivo antileukemic activity of the nitrogen mustard component with the cytotoxic activity of the anticancer drugs chlorambucil and melphalan produced a hybrid of nitrogen mustard distamycin and a para‐aminobenzoic acid component known as tallimustine (21) (Figure 8). Tallimustine (21) is one of the few distamycin analogs that made it through phase I and II clinical trials. Although the para‐nitrogen‐mustard‐benzoic acid moiety is a very mild alkylating agent, which may not exert unwanted non‐specific alkylations, the myelotoxicity of tallimustine (21) is too high compared with its antitumor effect (in vitro IC50=24.4 ng mL−1). [36] Another related compound that has made it into phase II clinical trials is brostallicin (22), but this could not be pushed into phase III clinical trials either. [37]
3.3. Chain‐extended derivatives of distamycin and netropsin
As distamycin A (15), netropsin (16), and their analogs bind AT‐rich DNA sequences, the question arose as to how the binding preference of DNA binding agents can be tailored to additional base sequences with the aim of developing new drugs for cancer therapies. Rational structural modifications of the natural products led to the development of lexitropsin 23, a member of a larger group of oligoamides called lexitropsins (Figure 9). As it was known how the oligopyrrole natural compounds interact with the nucleobases, it was recognized that the use of a suitable hydrogen‐bond acceptor instead of a pyrrole unit could lead to a change in the base site recognition from T‐A to G‐C. Certain heterocyclic units (e.g., imidazole, furan, pyrazole, thiazole, or triazole) incorporated into the initial oligopyrrole chain can accept hydrogen bonding from the exocyclic amino group of guanine (Figure 9). With this rationale, a large number of new derivatives could be generated and biologically validated. [38] However, it was noted that lexitropsins (exemplified by 23) often show a reduced DNA affinity compared with the oligopyrroles. [39]
Figure 9.
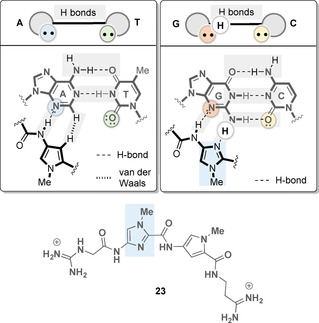
Rationale for the development of lexitropsins (imidazole ring marked in light blue).
In addition, Dervan et al. also designed molecules derived from distamycin, which bind sequences with both G,C and A,T base pairs (Figure 10). [40] These oligoamides carry a pyridine ring or alternatively an imidazole ring at the N‐terminus. Depending on the sequence, both oligomers can bind either as a 1:1 or 2:1 oligoamide‐DNA complex and, in contrast to distamycin, 2‐ImN binds the 5′‐TGACT‐3′ sequence as a 2:1 complex even at low ligand/DNA stoichiometries. [41]
Figure 10.
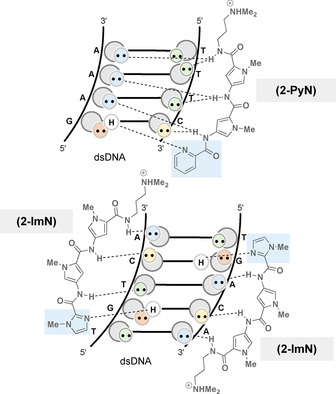
The 1:1 complex of 2‐PyN with 5’‐GAAA‐3’ and 2:1 complex of 2‐ImN with 5’‐TGTCA‐3’ (DNA symbolism see Figure 9; pyridine and imidazole rings marked in light blue).
A further step to improve sequence specificity was the design of a molecule that is able to bind DNA in a 2:1 ratio, resulting in specific contacts with each strand of the double helix. As a result, bis‐netropsin and distamycin derivatives were developed with flexible polymethylene tethers of different lengths, which are connected at the central pyrrole rings. [42] Despite the conformational freedom, these dimers (e.g., 24) can also only bind to five base pair long sequences. But they are able to establish specific contacts with each strand of the sequence 5′‐TGTCA‐3′, whereas 2‐PyN and 2‐ImN could only address this sequence as a 1:1 complex with binding affinities around 105 m −1.[ 42b , 43 ] The binding affinity of the tethered molecule 24 shown in Figure 11 is increased to K a=1.1×106 m −1.
Figure 11.
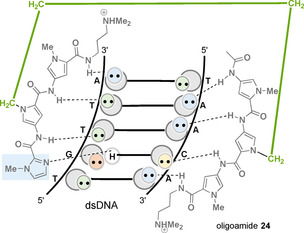
Complex formed between the covalent heterodimer 24 and the nucleotide sequence 5’‐TGTTA‐3’ (DNA symbolism see Figure 9; linker element marked in green, imidazole ring marked in light blue).
Lown et al. also used methylene linkers of different lengths and different numbers of N‐methylpyrrole units similar to bis‐netropsin analog 24, which were positioned between the central N‐methylpyrrole units (Figure 12). In their studies, they focused on the biological activity of netropsin and those bis‐netropsin analogs that show interference with the activity of DNA topoisomerases I and II. The compounds actually inhibited the catalytic activity of the isolated topoisomerase II and interfered with the stabilization of the cleavable complexes of topoisomerase I and II in the cell nucleus. A noteworthy positive aspect is that the dimers with linkers consisting of 1–4 and 6–9 methylene groups and no methylene group (see structures 26 and 27) have a far greater inhibitory effect against the isolated enzyme (IC90=7–150 μm) as well as in the nuclear system than netropsin (16; IC90=200 μm). Increasing the number of individual N‐methylpyrrole units to a total of up to six (see structure 25) also enhanced the inhibitory properties up to 5 μm. This study unraveled that the length of the linker and the number of pyrrole units determines the biological activity of the aromatic oligoamides that bind to the minor groove of the double helical DNA. [44] There may be a correlation between the sequence binding site of the aromatic ligand and the topoisomerase recognition site. For example, matrix‐associated regions (MAR) often contain several topoisomerase binding sites containing AT‐rich elements or are located in their vicinity. [45]
Figure 12.
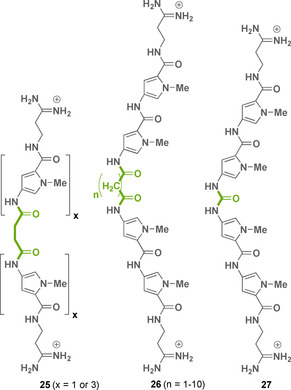
Structures of bis‐netropsin analogs 25–27 (linker elements marked in green).
3.4. Hairpin motifs
Knowing that 2:1 ligand/DNA complexes carry antiparallel side‐by‐side dimers, Dervan et al. established a structural motif in which these dimers are linked head‐to‐tail (Figure 13). These aromatic oligoamides were supposed to show increased sequence specificity and binding affinity to DNA. Different amino acid linkers were chosen and it was shown that 4‐aminobutyric acid is the optimal turn unit for hairpin binding of the six‐ring hairpin oligoamide 28. The binding affinity of the generated construct to the sequence 5′‐TGTTA‐3′ was determined to be K a=7.6×107 m −1. [46]
Figure 13.

Complex formed between oligoamide 28 and oligonucleotide 5’‐TGTTA‐3’ (DNA symbolism see Figure 9; linker element marked in green, imidazole ring marked in light blue).
As part of the evolutionary improvement, eight‐ring hairpin polyamides were synthesized that bind to DNA sequences along six base pairs with equilibrium association constants between 108 and 1010 m −1, similar to the affinity of natural DNA‐binding transcription factors that typically bind four to six base pairs of DNA. [47] However, for therapeutic reasons, it is not yet possible to target a unique sequence in the human genome because a sequence of four to six base pairs is too short for this purpose. The ligand must reach at least 15–17 base pairs, a number that would occur only once in about 3×109 base pairs on the human genome. [48] Isohelical analyses showed, however, that pyrrole carboxamides are about 20 % longer than required for a perfect adaptation to the base pair increase along the minor groove. [49] As an approach to circumvent this phase problem when binding four contiguous base pairs, β‐alanine/β‐alanine pairs were introduced instead of pyrrole/pyrrole pairs. This design allows a corresponding extension of the chain (polyamide 29, Figure 14). [50] Furthermore, β‐alanine/imidazole and imidazole/β‐alanine pairs could be used to target C‐G and G‐C base pairs. [51] Several research groups also have installed flexible β‐alanine fragments to reduce the molecular rigidity of polyamides.
Figure 14.
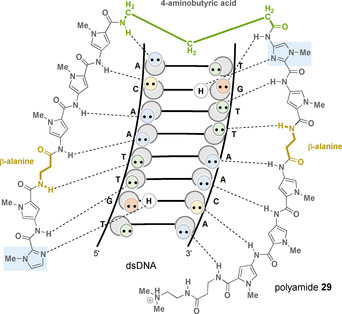
Complex of polyamide 29 with 5’‐TGTTAACA‐3’ (DNA symbolism see Figure 9; linker element marked in green, imidazole rings marked in light blue, and β‐alanine fragments marked in yellow).
With the realization that an imidazole/pyrrole or an imidazole/β‐alanine pair allows differentiation between the guanine/cytosine base pair from the cytosine/guanine couple, the challenge of differentiating between the adenine/thymine from thymine/adenine nucleobase pairs was pursued. It was found that the pyrrole/hydroxypyrrole pair provides a solution here. [52] The additional hydroxy functionality added to the pyrrole ring is able to form an additional hydrogen bond to thymine O2 and its two free electron pairs can interact with the hydroxypyrrole‐containing oligoamide 30 (Figure 15). In contrast, adenine N3 with only one free electron pair is not able to form that additional hydrogen bond with the hydroxy group. [53]
Figure 15.
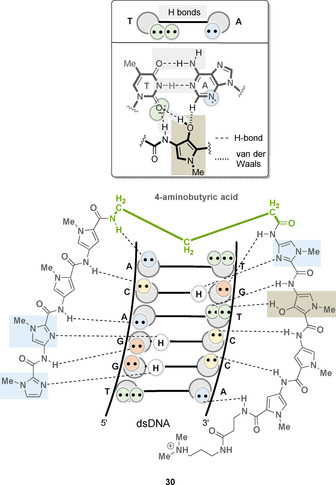
Complex of oligoamide 30 with 5′‐TGTCCA‐3′ (see Figure 9 for DNA symbolism; linker element highlighted in green, imidazole ring highlighted in light blue, hydroxypyrrole ring highlighted in bright brown) and rationale for the preferential interaction of hydroxypyrrole with thymine.
However, the hairpin polyamides that have been best investigated to date like 31 or 32 consist of eight five‐membered heterocycles with a chiral 2,4‐ or 3,4‐diaminobutyric acid linker [54] and an isophthalic acid residue at the C‐terminus, which allows favorable uptake into the cell nucleus (Figure 16). [55] These oligomers have been investigated in more detail in various biological and pharmaceutical applications. [59] It was found that these molecules block the elongation reaction catalyzed by RNA polymerase II (31: K i=50 nm) [56] and inhibit topoisomerase binding to DNA. [57] For example, by binding a DNA sequence near a TATA element (promoter region), the polyamides showed that they inhibit the DNA binding activity of TATA box binding proteins and transcription by RNA polymerase II. [58] In cell culture experiments, these oligomers also revealed interference with protein‐DNA complexes, which disrupt the transcription factor‐DNA interfaces, thus inhibiting oncogenic signal transduction in various disease models (prostate and breast cancer, inflammatory disorders, solid tumors). [59]
Figure 16.
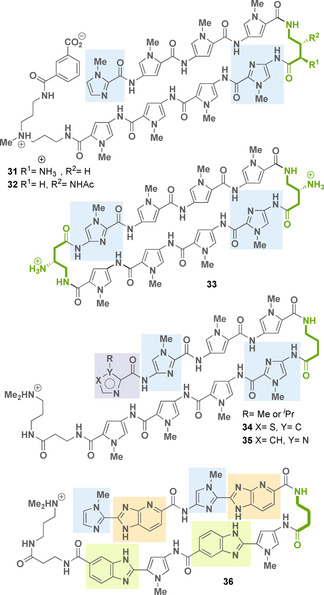
Structures of hairpin polyamides 31–36 (linker element marked in green, imidazole rings marked in light blue, thiazole or imidazole rings marked in light violet, benzimidazole rings marked in light green, and imidazopyridine rings marked in light orange).
Crystal structures of cyclic pyrrole‐imidazole polyamides like 33 (Figure 16), bound to duplex DNA oligonucleotides, showed a minor groove expansion of up to 4–5 Å, which led to compression of the major groove. The minor groove widening could be sufficient to alter the surface of the major groove in such a way that the binding of the transcription factor in the major groove is disturbed. [60] In this way, the expression of certain genes could be inhibited by altering the local DNA surface in certain DNA areas. [61] For this reason, this class of oligoamides could become potential therapeutic agents against cancer, which are triggered by excessive activity of transcription factors. [62]
Recently, Welte, Burley, and co‐workers disclosed a detailed structural and quantitative biophysical study of the DNA binding affinity, kinetics, and sequence selectivity of hairpin polyamide analogs that differ in the N‐terminal heterocycle (compounds 34–35). [63] This position influences DNA structural perturbations. It was shown that an increase of the steric bulk of an isopropyl group at the terminal imidazole unit does not impact dsDNA binding affinity but seems to have an impact on DNA major groove compression. Such hairpin polyamides also exhibit high affinity up to low nanomolar–picomolar values for their target dsDNA sequence.
Along this line, other hairpin oligomers such as 36 have been developed, which contain benzimidazole and imidazopyridine rings as monomeric units. These elements are regarded as mimicking amide groups. Some of these novel oligomers were found to target the guanine‐rich DNA sequence 5′‐WGGGGW‐3′ (36: K a=1.1–1.6×109 m −1), a core sequence in the DNA‐binding site of NF‐κB, a prolific transcription factor with high affinity. [64a]
3.5. Activation of gene expression
In contrast to the inhibition of cell function in cancer cells, pyrrole‐imidazole polyamides can also serve as activators of gene expression when fused with a peptide sequence. These bifunctional oligoamides act as DNA‐protein dimerizers because they consist of a DNA‐binding substructure and a second substructure that can bind to a natural transcription factor. In 2003, the Dervan group published such a protein DNA dimerizer 37, which binds both to the natural transcription factor Exd and sequence specifically in the minor groove of the target DNA via the oligoamide substructure (Figure 17). [64b]
Figure 17.
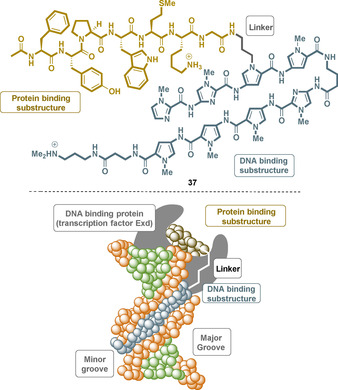
A protein‐DNA dimerizer 37 (top) based on a sequence‐specific polyamide and a short peptide (brown) that specifically binds to the transcription factor Exd and the minor groove of the DNA (bottom).
3.6. Binding to quadruplex DNA
Distamycin A (15) was found to bind not only to duplex DNA but also to G‐quadruplex DNA. Various distamycin derivatives were competitively tested for their binding affinity between duplex and G‐quadruplex DNA. The trimer 38 (Figure 18) proved to be the only oligoamide that revealed selectivity toward quadruplex DNA in this regard. However, these derivatives are structurally very similar to the minor groove binder distamycin A (15) and whereas distamycin A (15) is able to inhibit telomerase, trimer 38 shows no effect on telomerase activity. [65]
Figure 18.
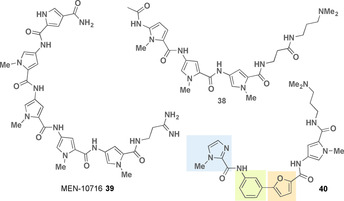
Structures of G‐quadruplex DNA binding oligoamides (imidazole ring marked in light blue, furan ring marked in light orange, benzene ring marked in light green).
The distamycin derivative MEN 10 716 39 (Figure 18) was able to inhibit telomerase in a cellular extract with an IC50 of 24 μm, and furthermore, cancer cells consistently showed >85 % reduced telomerase activity in the presence of 39. [66]
Given the competition between groove binding of duplex DNA and quadruplex DNA binding, a new class of aromatic oligoamides based on the distamycin structure was developed. By introducing a furan‐containing biaryl system, compound 40 (Figure 18) is bent such that the curvature forms a U‐shape that is in alignment with a terminal G‐quartet. Binding to a duplex DNA groove is thus unfavorably affected. The biaryloligoamides show cell growth inhibition against various cancer cell lines. [67]
Quinoline‐based oligoamides have also been discussed to selectively bind to quadruplex DNA versus duplex DNA. [68] Finally, a series of polyamides composed of pyrrole and imidazole motifs are reported that have been assigned potential as gene regulators and for cancer therapy. [69]
4. Synthetic PABA‐Based Oligoamide Foldamers
As shown above, several p‐aminobenzoic acid derivatives are present in the antibacterial natural products cystobactamids 8–13 and albicidin 14. Ether substituents or exchange of the benzene ring by pyridine can lead to conformational restrictions through intramolecular hydrogen bonding (Figure 3; substructures I–VII and Figure 19 A and B). Once incorporated with oligoamides, defined foldamers are generated that adopt a helical topography. These foldamers have emerged as architectures to mimic secondary structures of proteins or nucleic acids. Depending on additional substituents on the aromatic ring, foldamers based on benzamides are commonly stabilized by intramolecular hydrogen bonding. Such oligoamides display folding modes very different from peptidic foldamers. Several hydrogen‐bonding patterns have been designed and these can be based on either four‐membered, five‐membered, or six‐membered rings, some of which are depicted in Figure 19 A. In particular, the groups of Hamilton and Wilson have developed foldamers based on such linear aromatic oligobenzamides. [70] Several designs of oligoaromatic foldamers bear scaffold properties that match a helical topography. These can contain side chains, which are positioned in a way that mimics the spatial orientation of key recognition residues.[ 71 , 72 ] As already mentioned above, such foldamers are stabilized mainly by intramolecular hydrogen bonding, which create folding modes very different from peptides and peptidic foldamers I–IV. In part, these were confirmed by X‐ray analyses.[ 73 , 74 ] Supramolecular and biological properties of such foldamers have extensively been studied and are covered in the following chapter.
Figure 19.
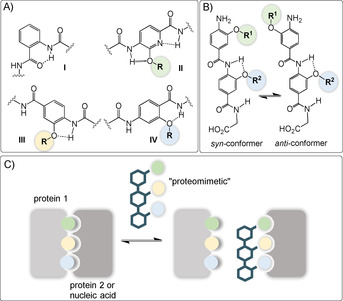
A) Structures of different classes I–IV of oligoaromatic backbone monomers with key intramolecular hydrogen bonding interaction. B) Possible syn‐/anti‐conformers discussed in ref. [74b] and C) aromatic oligoamides as potential inhibitors for α‐helix mediated protein–protein or protein–nucleic acid interactions (R can be flexibly varied for fine‐tuning properties of molecular recognition with the biological target. The color code of the side chain is not linked to a specific functional group).
4.1. Protein–protein interaction inhibitors
Sequence specifically designed synthetic aromatic oligoamides were shown to inhibit α‐helix mediated[ 75 , 76 , 77 ] protein–protein interactions (Figure 19 C).[ 78 , 79 ] These oligoamides serve as small molecule scaffolds occasionally termed “proteomimetics” [80] to orient functionality in a manner that reproduces the spatial and angular positioning of key side chains presented by the helix donor. [75]
4.1.1. Inhibitors of hDM2‐p53 interaction
A first‐generation foldamer of type 41 a acted as an α‐helix mimetic capable of inhibiting the p53‐hDM2 protein–protein interaction. For enhancing the solubility of such aromatic oligoamides, the architecture was complemented with an oligo‐ethyleneglycol tag, which had little impact on the binding affinity (Figure 20). [81]
Figure 20.
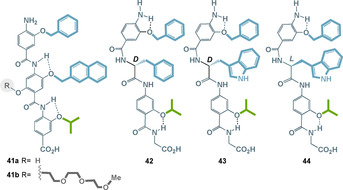
Aromatic oligoamides as inhibitors for α‐helix mediated protein–protein interactions between hDM2 and p53. (the color code of the side chain: green=aliphatic and blue=aromatic).
The design of such foldamers was later extended to chiral oligoaromatic amide foldamers 42–44 that recognize their target protein hDM2, thereby inhibiting protein–protein interaction with the tumor suppressor protein p53, and hence repressing p53 transcriptional activity. [82] The absolute configuration of the helix mimetic was critical dependent for the selectivity for p53/hDM2 inhibition versus Mcl‐1/NOXA‐B inhibition. For example, hDM2 displays a twofold preference for the l‐tryptophane hybrid 43 over 42.
In addition, it was found that hybrid architectures closely related to 42–44 replace a segment of the protein structure (the S‐peptide from RNase S) with a foldamer that itself mimics this α‐helix. Interestingly, such a non‐covalent complex with the S protein led to the restoration of catalytic function, thus showing that the foldamers can act as a component of a functional quaternary structure that performs RNA hydrolysis. [83]
In addition to the O‐alkylated foldamers discussed in detail here, there is one work on corresponding trimeric N‐alkylated oligoamides that effectively bind hDM2 by mimicking key residues of p53. In addition, these proteomimetics have also been shown to bind Mcl‐1/NOXA‐B in both biophysical assays and in a cellular context. [84]
4.1.2. Islet amyloid polypeptide (IAPP) antagonists
Islet amyloid polypeptide (IAPP, also called amylin) is a 37‐residue hormone peptide and associated with type 2 diabetes mellitus (DM2). This neuropancreatic peptide is co‐secreted with insulin from β‐cells and has a random coiled structure, as an α‐helix bound to the membrane or also as a β‐sheet structure. Each of these three conformations is associated with different functions. The aggregation/fibrillation of IAPP leads to oligomers that are assumed to exert cytotoxicity against their host β‐cells by interacting with the cell membrane. The aggregation of membrane‐bound α‐helices is critical for IAPPs dysfunction. Therefore, the stabilization of those helices is one approach to inhibit IAPP‐induced cytotoxicity. Helix–helix aggregation arises from interaction of certain sidechains on the helix surface with defined distances of i, i+3/i+4, i+7 (hydrogen bond between NH of amino acid I and carbonylamide of amino acid II after a helix turn leads to a spatial alignment of the side chains on the helix surface; the number refers to the position of the amino acid in the peptide backbone away from starting point I). Aromatic oligoamides were found to be suitable antagonists as they appear as foldamers with complementary binding surfaces. Optimization of the sidechain functionalities led to anionic foldamers that correspond with the cationic surface of IAPP α‐helices (Figure 21 A). However, the exclusive use of anionic sidechains is not sufficient. The aliphatic interactions at certain surface positions also affects the activity and the curvature. The latter can be tuned by backbone hydrogen bonding. Benzamide 45 and pyridylamide 46 both show activity against IAPP fibrillation. Still, hydrogen bonding in 46 results in a helical conformation superior to 45 and consequently to an improved antagonist activity. [85]
Figure 21.
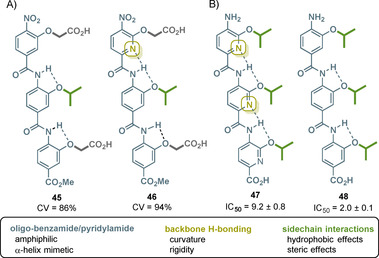
A) Structures of trimeric benzamide 45 and pyridylamide 46 with hydrogen‐bonding abilities and cell viability (CV) on IAPP‐mediated toxicity in INS cells at 20 μm. B) Structures of trimeric pyridylamide 47 and benzamide 48 with hydrogen‐bonding abilities and IC50 against cell proliferation of Bcl‐x L overexpressing cancer cell line DLD‐1 (the color code of the side chain: green=aliphatic and dark gray=carboxylate).
Sidechain optimization by the Hamilton and Miranker groups provided the next generation of oligoamides with two different backbones, which bear similar sidechain architectures, although only the carboxylate groups and not the aliphatic residues appear at the same position when overlaying both structures. However, tripyridylamide ADM‐3 49 and tetraquinoline amide ADM‐116 50 both inhibit IAPP fibrillation and are able to rescue INS‐1 cells from IAPP‐induced toxicity (Figure 22, top). [86]
Figure 22.
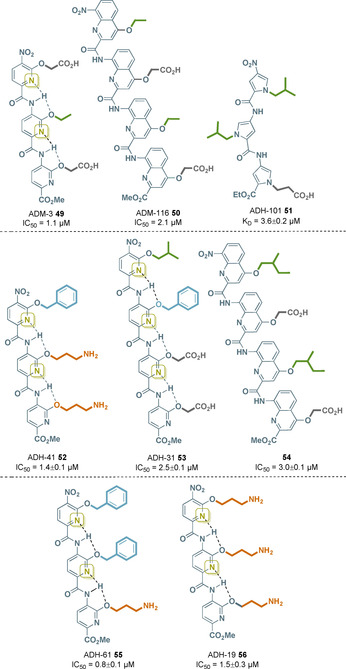
Structures of IAPP inhibitors 49–51 with different oligoaryl backbone units (IC50 values at 13 μm IAPP, K D at 20 nm IAPPF), Aβ inhibitors 52–54 (IC50 at 5 μm Aβ), PRC2 inhibitor 55 (IC50 values at 7.5 nm PRC2), and HIV‐1 TAR RNA inhibitor 56 (IC50 values at 0.2 μm TAR‐RNA; side chain color code: green=aliphatic, dark gray=carboxylate, blue=aromatic, and orange=amine). For further details on hydrogen bonds in quinoline‐bearing oligoamide backbones (as in 50 and 54) see Section 4.2.2 and Figure 23.
N‐Substituted oligopyrrolamides display an alternative foldamer backbone (see Section 3 and the natural products distamycin A (15) and netropsin (16)). These oligomers form secondary structures in which the N‐substituents are in close proximity with the sidechains in the oligopyridylamides. Modulation of the IAPP conformation was achieved by exposure to ADH‐101 51, a pyrrole‐based oligoamide (Figure 22, top). This foldamer inhibits IAPP aggregation and hence amyloidogenesis without observable cytotoxicity. [87]
4.1.3. Bcl‐xL inhibitors
Fletcher and his collaborators conducted a study of the structure–activity relationship of the backbone of a number of oligoamide‐foldamer‐based α‐helix mimetics. They were expected to resemble the Bak‐BH3 helix, which is able to interrupt the Bak‐Bcl‐x L interaction. They positioned three hydrophobic isopropoxy residues on the surface of the oligoamide.
By varying the benzene‐to‐pyridine ratio in the aromatic backbone, the most flexible and hydrophobic inhibitor 48 appeared to be superior to the more rigid pyridylamide 49 (Figure 21 B). [88] Interestingly, this trend is reversed as with the IAPP inhibitors 45 and 46. [85]
4.1.4. Aβ antagonists
Amyloid β peptide (Aβ) is formed by cleavage of the transmembrane protein β‐ and δ‐secretases. Isoforms of these species are prone to amyloidogenesis. The resulting oligomers (which then can form β‐sheet rich fibers) are associated with neurotoxicity and further to Alzheimer's disease (AD). Oligopyridylamides were shown to be suitable α‐helix mimetics to inhibit Aβ oligomerization, fibrillation, and cytotoxicity by bearing corresponding binding functionalities on their surface.
The inhibitory ability was proven in cellular milieu. Although mouse neurobastoma cells (N2a) treated with Aβ suffered from reduced viability, the latter could be almost completely maintained by applying ADH‐41 52 (Figure 22, middle).
Structure–activity relationship studies revealed that a decreasing hydrophobicity as well as the spatial composition of the sidechains led to diminished antagonist activity towards Aβ42. Protein selectivity was proven by performing the experiments with IAPP instead of Aβ, enzymes that share approximately 50 % sequence similarity, without any inhibitory effect. A second oligopyridylamide ADH‐31 53 was shown to also inhibit Aβ oligomerization (Figure 22, middle). In contrast, this α‐helix mimetic is anionic, resulting in less selectivity as it permits IAPP inhibition. More detailed studies showed ADH‐41 52 to be a more potent antagonist in the initial nucleation, whereas ADH‐31 53 is the superior antagonist in oligomerization. Consequently, both molecules bind to different subdomains of the protein. A combinatorial treatment with ADH‐31 53 and ADH‐41 52 is not beneficial. [89]
Oligoquinolines were also tested for their antagonistic activity against oligomerization of Aβ. Suppression of Aβ aggregation was achieved at an equimolar ratio with compound 54 by inducing an α‐helical conformation and stabilization of the helices (Figure 22, middle). Variation of the sidechain functionalities led to dramatically decreased inhibition indicating the specific activity of compound 54. N2a cell viability remained by 89 % after treatment with Aβ and compound 54 at an equimolar ratio, whereas no cytotoxic effects were observed when treating the cells with oligoquinoline 54 alone. Moreover, it was shown that 54 also disrupts already formed oligomers. Its comparable ability to inhibit Aβ oligomerization suggests its binding site to be the same as for ADH‐31 52. [90]
4.1.5. PRC2 inhibitors
Polycomb Repression Complex 2 (PRC2) catalyzes the methylation of histone H3‐lysine 27 (H3K27) to H3K27me2/3, which is associated with repressive chromatin. It is therefore a regulator of transcriptional activities within a cell and its dysregulation can cause disease or cancer. PRC2 has three subunits: EZH1/2, EED, and SUZ12, and it has been found that interference of the interaction between SRM and the SET‐I domain of EZH2 inhibits allosteric activation of PRC2. Owing to the design of the oligopyridylamide ADH‐61 55, which carries complementary side chains, the SRM/SET‐I interaction mediated by the α‐helix was effectively disrupted (Figure 22, bottom). [91]
4.2. Protein–nucleic acid interaction inhibitors
Such aromatic oligoamides with defined three‐dimensional conformation, achieved by the formation of internal hydrogen bonds, are not only capable of intervening in pharmaceutically relevant protein–protein interactions, but have also been shown to serve as substitutes in protein–nucleic acid interactions, similar to the oligopyrrolamides described above. Thus, both types of oligoamides are not only structurally related and significantly different from peptides based on α‐amino acids, but can also interact with complexes based on the same combinations of biomacromolecules. Two examples are briefly discussed below.
4.2.1. HIV‐1 TAR RNA inhibitor
The transactivation response (TAR) element, a 59‐nucleotide‐stem‐loop‐ncRNA at the 5′‐untranslated region (UTR) of the viral genome, was found to be a structurally and functionally important element of the HIV virus. This is because the transcription of the HIV virus is increased by complexing the TAR RNA with the TAT protein, a viral protein responsible for the recruitment of host transcriptional proteins. The TAR‐RNA also functions as micro‐RNA, which reduces viral apoptosis. Therefore, TAR‐RNA is a therapeutic target in HIV treatment. Oligopyridylamide foldamers have been found to target these protein–RNA interactions, and drug screening revealed that ADH‐19 56 is a promising antagonist (Figure 22, bottom). Its functional side chains are located in close proximity to the key residues of the TAT protein. ADH‐19 56 has been shown to selectively bind TAR‐RNA and therefore inhibit the TAR‐RNA‐TAT complex by mimicking the cationic protein surface. It was even reported that ADH‐19 56 is able to save TZM‐bI cells with an in vivo efficacy of IC50=25 μm. Another structurally similar oligoamide, ADH‐41 52 (Figure 22, middle), is also capable of inhibiting TAR‐RNA in vitro, but with reduced effectiveness. [92]
4.2.2. DNA mimetics
Huc and his team developed a series of aromatic oligoamides, the architecture of which is based on quinoline monomers. His group was able to show that these de novo engineered compounds are able to mimic the DNA surface by forming α‐helices. The anionic side chains on the outside of these single‐stranded foldamers resemble those on the surface of B‐DNA.
For example, the phosphonate‐modified oligoamide 57 efficiently inhibits human topoisomerase 1 (Top1) and human immunodeficiency virus 1 integrase (HIV‐1 IN) by recognizing their peripheral anionic character. However, it was found that these interactions are not sequence‐specific. Structure–activity relationship studies demonstrated the importance of the negatively charged functional group in conjunction with the target enzyme (Figure 23).
Figure 23.
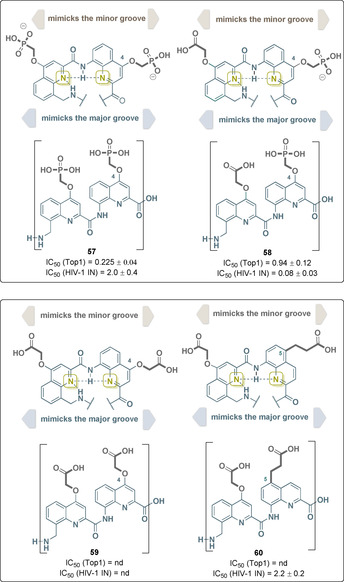
Structures of selected quinoline‐based oligoamide DNA mimetics with binding capabilities to Top1 and HIV‐1 IN (IC50 values are given) and dependence on the anionic functional groups and the location of the side chains on the quinoline backbone (the color code of the side chain: dark gray=carboxylate and phosphonate; dotted lines refer to hydrogen bonds).
Although Top1 is best inhibited by oligoamide 57, HIV‐1 IN inhibition increases by two orders of magnitude when phosphonate and carboxylate side chains are combined as found in foldamer 58. Other parameters such as side chain length and location on the quinoline backbone also proved to be crucial for activity. Shifting the anionic residue from position 4 in oligoamide 59 to position 5 as in oligoamide 60 by modifying the groove sizes restored activity against HIV‐1 IN, whereas Top1 is not inhibited. [93]
5. Miscellaneous
In contrast to oligobenzamides with PABA‐derived monomers, which have already been discussed here, the two broadly effective antiviral agents benzavir‐1 and ‐2 61 and 62 consist of ortho‐substituted aromatic rings similar to substructure I in Figures 3 and 18 (Figure 24). Their conformation has not yet been investigated in detail. Benzavir‐2 62 effectively inhibits DNA viruses, including both human adenovirus (HAdV) and herpes simplex virus type 1 and 2 (HSV‐1, HSV‐2). [94] In addition, its activity against the RNA virus Rift Valley Fever Virus (RVFV) has been reported. [95] Recently, benzavir‐2 62 has also been shown to inhibit the in vitro infection of various flaviviruses. [96] However, the cellular target of benzavir‐2 62 is not known. Lately, a few antimicrobial investigational drugs have been developed of which bricalidin 63 is a non‐peptide chemical mimic bearing an arylamide foldameric backbone designed to replicate amphiphilic properties of antimicrobial peptides. [97]
Figure 24.
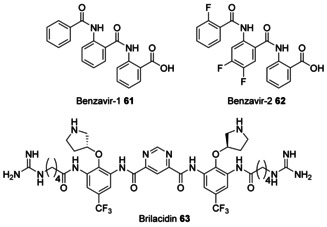
Structures of antiviral ortho‐connected oligoamides benzavir‐1 61, benzavir‐2 62, and of brilacidin 63.
Another side aspect of such aromatic polyamides are macrocyclic architectures. The combination of meta‐substituted diaminobenzenes with isophthalic acid derivatives can be used to produce macrocyclic oligomers. When phenols or ether substituents are positioned in the ortho‐positions, favorable three‐center intramolecular hydrogen bonds are formed, conformationally constraining the macrocyclic backbone structures. Such macrocycles create organic pore‐like structures 64, which aggregate and experience strong tubular π‐stacking via the aromatic systems (Figure 25). It has been found that the inner cavities can be adjusted to between 5 and 30 Å diameter. [98]
Figure 25.
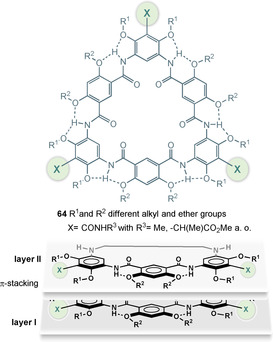
Structures of macrocyclic oligomers 64 based on substituted m‐diaminobenzenes and isophthalic acid and graphical presentation of π‐stacking of individual macrocycles.
Studies on these architectures have focused on their supramolecular and ion‐binding properties, whereas studies on their biological potential and pharmaceutical properties are still missing.
Related to these macrocyclic architectures are foldamers, which can form spiral hyperstructures (Figure 26). Huc and co‐workers developed a series of linear aromatic oligoamides that fold into capsules via metal bonding (e.g., with copper) and these can recognize carbohydrate guests in solution. [99] The authors created a “quasi alphabet” of monomers 65–73, which, when coupled by amide bonds, lead to oligomers that allow fine tuning of folding and carbohydrate recognition.
Figure 26.
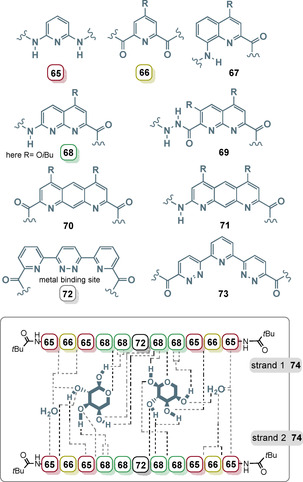
Heteroaromatic building blocks 65–73 developed for the design of oligoamides that form capsules in the presence of metals with R commonly OiBu. Model of double helix of oligoamide 74 with the capture of α‐d‐xylopyranose, β‐d‐arabinopyranose, and water. Hydrogen bonds in dotted lines as suggested in ref. [99a] (dotted lines end between two numbers: NH of the amide bond serves as the hydrogen‐bonding partner; dotted lines end at the frame of the number: Ns of the heterocycles serve as the hydrogen‐bonding partner and in the case of 68 both N atoms are involved).
An example is the sequence 74, which, after binding of metals, yields a double helix and as a consequence acquires the ability to absorb pentoses. For oligoamide 74, it was reported that it is able to sequester a heteromeric pair of pentoses, namely α‐d‐xylopyranose and one molecule of β‐d‐arabinopyranose, which become part of a double helix composed of two molecules of 74. [99a] Remarkably, this supramolecular structure forms without the need of metals.
In principal, these aromatic oligoamides act as molecular receptors for the recognition of complex target guests. These studies do not yet refer to biomedical or pharmaceutical applications. However, the relationship of these host molecules to the DNA mimetics discussed in Section 4.2.2 can be emphasized as their backbones are based in part on similar heteroaromatic monomers.
6. Conclusion
The term “privileged structure”, a single molecular substructure or backbone that can serve as a starting point for high‐affinity ligands for more than one receptor type, [100] was introduced as early as 1988. [101] As such, these motifs are often found in a variety of bioactive natural products. These privileged substructures function as evolutionary pre‐validated platforms and starting points for the design of medicinal chemistry programs. Natural products discussed in this context are terpenoids, polyketides, phenylpropanoids, and alkaloids. [102]
In this report, we dealt with a hitherto overlooked group of privileged substructures of aromatic oligoamides, for which there are natural models in the form of cystobactamids, albicidin, distamycin A, netropsin, and others. The aromatic and heteroaromatic core together with a flexible selection of substituents provide conformationally well‐defined scaffolds capable of binding to regions of biomacromolecules, especially proteins [103] and DNA, with well‐defined conformations such as α‐helices. As such, these aromatic oligoamides have already been used to inhibit DNA–protein, RNA–protein and protein–protein interactions as summarized in Figure 27.
Figure 27.
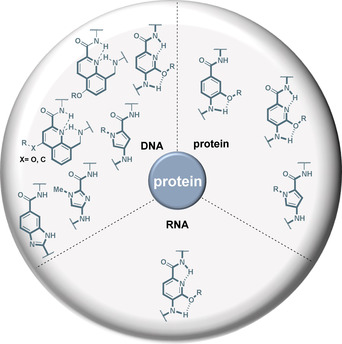
Common aromatic substructures in oligoamides and foldamers and their ability to affect protein–protein, protein–RNA, and protein–DNA complexes as discussed in this report.
The natural products distamycin A 15 and netropsin 16 served as the starting point for the development of heteroaromatic oligoamides that recognize sequence‐specifically double‐stranded DNA with binding to the minor groove. This enabled the blocking of protein binding, especially transcription factors. In the case of PABA and picolinic acid‐based oligoamides, the story can be told in reverse (Figure 28). Structural and conformational considerations served as a starting point for the development of (helical) foldamers, a concept which proved to be successful. Owing to their design, these foldamers are able to intervene in protein–protein and RNA–protein interactions that are based on defined, specifically helical secondary structures. Only later were cystobactamids and albicidin isolated from natural sources and structurally elucidated. Here, substituents and substitution patterns that reveal a strong relationship with oligobenzamides such as 44 are contained. It can therefore be assumed that similar hydrogen‐bonding patterns in the C–E rings are present in these natural oligoamides such as cystobactamid 919‐2 (9), which produce foldamer‐like substructures.
Figure 28.
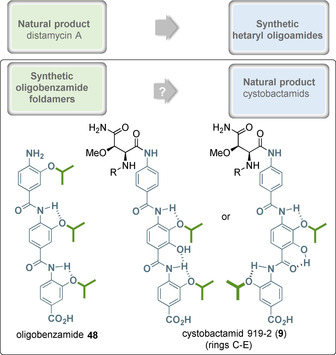
A summary: From natural products to privileged structures and vice versa (two possible hydrogen‐bond binding patterns in cystobactamid 9, as they could be derived from PABA‐foldamers like 48—these reveal significantly different conformations).
Structural data on the exact binding site of cystobactamids or albicidin, respectively, to gyrase or to DNA or to the gyrase‐DNA complex during passage of the DNA strand through the N‐gate are not yet available. [104] Initial investigations suggest that the oligobenzamidic part of the cystobactamids (rings C–E) could bind to the minor groove of the dsDNA. [10e] Therefore, the mode of action of these antibiotics could not yet be elucidated in full detail. But the knowledge gathered for the oligobenzamide foldamers with defined conformations could be helpful in deciphering the molecular basis for the antibacterial activity of these natural products.
We hope that this review will spark a broader and eventually deeper interest in these privileged aromatic oligoamides in the fields of medicinal chemistry and chemical biology.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Tim Seedorf has a B.Sc. in biochemistry. He received his master′s degree from Leibniz Universität Hannover, Germany, in Medicinal and Natural Products chemistry. During his master′s studies, he also worked in Prof. Berit Olofsson′s group at Stockholm University, Sweden, as part of the ERASMUS exchange program. He is currently a PhD student, supervised by Prof. Dr. Andreas Kirschning, working in the fields of medicinal chemistry and total synthesis.

Biographical Information
Andreas Kirschning studied chemistry at the University of Hamburg and at Southampton University (UK). He received his PhD in 1989 working in the field of organosilicon chemistry in the group of Prof. E. Schaumann. After a postdoctoral stay at the University of Washington (Seattle, USA) with Prof. Heinz G. Floss, he started his independent research at the Clausthal University of Technology in 1991, where he finished his habilitation in 1996. In 2000 he moved to the Leibniz University Hannover. His research interests cover structure elucidation as well as the semi‐, total synthesis of natural products. Recently, he initiated a research program on the role of coenzymes and cofactors being involved in the origin of life.

Biographical Information
Danny Solga completed his bachelor′s degree in biochemistry at Leibniz University Hannover and Hannover Medical School in 2016. He then completed a master′s degree in medicinal and natural products chemistry at Leibniz University and, sponsored by the German Academic Exchange Service (DAAD), at Stanford University (Stanford, USA) under the supervision of Prof. Dr. Paul Wender. He is currently a PhD student under the supervision of Prof. Dr. Andreas Kirschning at Leibniz University Hannover. His research involves oligopeptides in the field of synthetic organic and medicinal chemistry.

Acknowledgements
We are indebted to the BMBF (OpCyBac; grant number 16GW0219K) for funding our research on the cystobactamids. Open access funding enabled and organized by Projekt DEAL.
T. Seedorf, A. Kirschning, D. Solga, Chem. Eur. J. 2021, 27, 7321.
References
- 1. Fosgerau K., Hoffmann T., Drug Discovery Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- 2. McIntosh J. A., Donia M. S., Schmidt E. W., Nat. Prod. Rep. 2009, 26, 537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Süssmuth R. D., Mainz A., Angew. Chem. Int. Ed. 2017, 56, 3770–3821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3824–3878. [Google Scholar]
- 4. Gorelova V., Bastien O., De Clerk O., Lespinats S., Rébeillé F., van Der Straeten D., Sci. Rep. 2019, 9, 5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kluczyk A., Popek T., Kiyota T., de Macedo P., Stefanowicz P., Lazar C., Konishi Y., Curr. Med. Chem. 2002, 9, 1871–1992. [DOI] [PubMed] [Google Scholar]
- 6. Baumann S., Herrmann J., Raju R., Steinmetz H., Mohr K. I., Hüttel S., Harmrolfs K., Stadler M., Müller R., Angew. Chem. Int. Ed. 2014, 53, 14605–14609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14835–14839. [Google Scholar]
- 7. Hüttel S., Testolin G., Herrmann J., Planke T., Gille F., Moreno M., Stadler M., Brönstrup M., Kirschning A., Müller R., Angew. Chem. Int. Ed. 2017, 56, 12760 – 12764; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12934–12938. [Google Scholar]
- 8. Kim Y. J., Kim H. J., Kim G. W., Cho K., Takahashi S., Koshino H., Kim W. G., J. Nat. Prod. 2016, 79, 2223–2228. [DOI] [PubMed] [Google Scholar]
- 9. Planke T., Moreno M., Fohrer J., Gille F., Kanakis A., Norris M. D., Siebke M., Wang L. L., Hüttel S., Müller R., Kirschning A., Org. Lett. 2019, 21, 1359–1363. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Moreno M., Elgaher W. A. M., Herrmann J., Schläger N., Hamed M. M., Baumann S., Müller R., Hartmann R. W., Kirschning A., Synlett 2015, 26, 1175–1178; [Google Scholar]
- 10b. Moeller M., Norris M. D., Planke T., Cirnski K., Herrmann J., Müller R., Kirschning A., Org. Lett. 2019, 21, 8369–8372; [DOI] [PubMed] [Google Scholar]
- 10c. Planke T., Cirnski K., Herrmann J., Müller R., Kirschning A., Chem. Eur. J. 2020, 26, 4289–4296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Testolin G., Cirnski K., Rox K., Prochnow H., Fetz V., Grandclaudon C., Mollner T., Baiyoumy A., Ritter A., Leitner C., Krull J., van den H. A. Vassort J., Sordello S., Hamed M. M., Elgaher W. A. M., Herrmann J., Hartmann R. W., Müller R., Brönstrup M., Chem. Sci. 2020, 11, 1316–1334; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Elgaher W. A. M., Hamed M. M., Baumann S., Herrmann J., Siebenbürger L., Krull J., Cirnski K., Kirschning A., Brönstrup M., Müller R., Hartmann R. W., Chem. Eur. J. 2020, 26, 7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cociancich S., Pesic D., Petras D., Uhlmann S., Kretz J., Schubert V., Vieweg L., Duplan S., Marguerettaz M., Noell J., Pieretti I., Hügelland M., Kemper S., Mainz A., Rott P., Royer M., Süssmuth R. D., Nat. Chem. Biol. 2015, 11, 195–197. [DOI] [PubMed] [Google Scholar]
- 12. Birch R. G., Patil S. S., J. Gen. Microbiol. 1985, 131, 1069–1075. [DOI] [PubMed] [Google Scholar]
- 13. Hashimi S. M., Wall M. K., Smith A. B., Maxwell A., Birch R. G., Antimicrob. Agents Chemother. 2007, 51, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Kretz J., Kerwat D., Schubert V., Grätz S., Pesic A., Semsary S., Cociancich S., Royer M., Süssmuth R. D., Angew. Chem. Int. Ed. 2015, 54, 1969–1973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1992–1996; [Google Scholar]
- 14b. Grätz S., Kerwat D., Kretz J., von Eckardstein L., Semsary S., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1499–1502; [DOI] [PubMed] [Google Scholar]
- 14c. Behroz I., Durkin P., Grätz S., Seidel M., Rostock L., Spinczyk M., Weston J. B., Süssmuth R. D., Chem. Eur. J. 2019, 25, 16538–16543; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. Kerwat D., Grätz S., Kretz J., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1899–1903; [DOI] [PubMed] [Google Scholar]
- 14e. Petras D., Kerwat D., Pesic A., Hempel B.-F., von Eckardstein L., Semsary S., Arasté J., Marguerettaz M., Royer M., Cociancich S., Süssmuth R. D., ACS Chem. Biol. 2016, 11, 1198–1204; [DOI] [PubMed] [Google Scholar]
- 14f. von Eckardstein L., Petras D., Dang T., Cociancich S., Sabri S., Grätz S., Kerwat D., Seidel M., Pesic A., Dorrestein P. C., Royer M., Weston J. B., Süssmuth R. D., Chem. Eur. J. 2017, 23, 15316–15321. [DOI] [PubMed] [Google Scholar]
- 15. Vieweg L., Kretz J., Pesic A., Kerwat D., Grätz S., Royer M., Conciancich S., Mainz A., Süssmuth R. D., J. Am. Chem. Soc. 2015, 137, 24, 7608–7611. [DOI] [PubMed] [Google Scholar]
- 16. Aldossary S. A., Biomed. Pharmacol. J. 2019, 12, 7–15. [Google Scholar]
- 17. Finlay A. C., Hochstein F. A., Sobin B. A., Murphy F. X., J. Am. Chem. Soc. 1951, 73, 341–343. [Google Scholar]
- 18. Arcamone F., Penco S., Orezzi P., Nicolella V., Pirelli A., Nature 1964, 203, 1064–1065. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Becker Y., Monogr. Virol. 1976, 11, 1–130; [PubMed] [Google Scholar]
- 19b. Hoffmann C. E. in Annual Reports in Medicinal Chemistry, Vol. 11 (Ed.: Gordon M.), Academic Press, New York, 1976, pp. 128–137; [Google Scholar]
- 19c. Corcoran J. W., Hahn F. E., Snell J. F., Arora K. L., Mechanism of Action of Antimicrobial and Antitumor Agents, Springer, Berlin, 1975. [Google Scholar]
- 20. Marchini S., Broggini M., Sessa C., D′Incalci M., Expert Opin. Invest. Drugs 2001, 10, 1703–1714. [DOI] [PubMed] [Google Scholar]
- 21. Zimmer C. in Nucleic Acid Research and Molecular Biology, Vol. 15 (Ed.: Cohn W. E.), Academic Press, New York, 1975, pp. 285–318. [DOI] [PubMed] [Google Scholar]
- 22. Kopka M. L., Yoon C., Goodsell D., Pjura P., Dickerson R. E., Proc. Natl. Acad. Sci. USA 1985, 82, 1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coll M., Frederick C. A., Wang A. H., Rich A., Proc. Natl. Acad. Sci. USA 1987, 84, 8385–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoon C., Privé G. G., Goodsell D. S., Dickerson R. E., Proc. Natl. Acad. Sci. USA 1988, 85, 6332–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Pelton J. G., Wemmer D. E., Proc. Natl. Acad. Sci. USA 1989, 86, 5723–5727; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Pelton J. G., Wemmer D. E., J. Am. Chem. Soc. 1990, 112, 1393–1399. [Google Scholar]
- 26. Dwyer T. J., Geierstanger B. H., Bathini Y., Lown J. W., Wemmer D. E., J. Am. Chem. Soc. 1992, 114, 5911–5919. [Google Scholar]
- 27. Zimmer C., Wähnert U., Progr. Biophys. Mol. Biol. 1986, 47, 31–112. [DOI] [PubMed] [Google Scholar]
- 28. Hahn F. E., Pharmacol. Ther. Part A 1977, 1, 475–485. [Google Scholar]
- 29. McHugh M. M., Woynarowski J. M., Sigmund R. D., Beerman T. A., Biochem. Pharmacol. 1989, 38, 2323–2328. [DOI] [PubMed] [Google Scholar]
- 30. Probst G. W., Hoehn M. M., Woods B. L., Antimicrob. Agents Chemother. (1961–70) 1965, 5, 789–795. [PubMed] [Google Scholar]
- 31. Lee M., Shea R. G., Hartley J. A., Kissinger K., Pon R. T., Vesnaver G., Breslauer K. J., Dabrowiak J. C., Lown J. W., J. Am. Chem. Soc. 1989, 111, 345–354. [DOI] [PubMed] [Google Scholar]
- 32. Kikuchi M., Kumagai K., Ishida N., Ito Y., Yamaguchi T., Furumai T., Okuda T., J. Antibiot. 1965, 18, 243–250. [PubMed] [Google Scholar]
- 33.
- 33a. Shultz P. G., Taylor J. S., Dervan P. B., J. Am. Chem. Soc. 1982, 104, 6861–6863; [Google Scholar]
- 33b. Taylor J. S., Schultz P. G., Dervan P. B., Tetrahedron 1984, 40, 457–465. [Google Scholar]
- 34. Arcamone F., Mongelli N., Animati F., Distamycin Derivatives and Process for Their Preparation GB2178037A, 1985.
- 35. Baker B. F., Dervan P. B., J. Am. Chem. Soc. 1985, 107, 8266–8268. [Google Scholar]
- 36.
- 36a. Cozzi P., Il Farmaco 2000, 55, 168–173; [DOI] [PubMed] [Google Scholar]
- 36b. Cozzi P., Mongelli N., Curr. Pharm. Des. 1998, 4, 181–201. [PubMed] [Google Scholar]
- 37. Lee D. Y., Staddon A. P., Shabason J. E., Sebro R., Cancer Med. 2019, 8, 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Lown J. W., Krowicki K., Bhat U. G., Skorobogaty A., Ward B., Dabrowiak J. C., Biochemistry 1986, 25, 7408–7416; [DOI] [PubMed] [Google Scholar]
- 38b. Lown J. W., Antiviral Res. 1992, 17, 179–196. [DOI] [PubMed] [Google Scholar]
- 39. Lown J. W., J. Mol. Recognit. 1994, 7, 79–88. [DOI] [PubMed] [Google Scholar]
- 40. Wade W. S., Mrksich M., Dervan P. B., J. Am. Chem. Soc. 1992, 114, 8783–8794. [Google Scholar]
- 41. Mrksich M., Wade W. S., Dwyer T. J., Geierstanger B. H., Wemmer D. E., Dervan P. B., Proc. Natl. Acad. Sci. USA 1992, 89, 7586–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Mrksich M., Dervan P. B., J. Am. Chem. Soc. 1993, 115, 9892–9899; [Google Scholar]
- 42b. Mrksich M., Dervan P. B., J. Am. Chem. Soc. 1994, 116, 3663–3664. [Google Scholar]
- 43. Wade W. S., Mrksich M., Dervan P. B., Biochemistry 1993, 32, 11385–11389. [DOI] [PubMed] [Google Scholar]
- 44. Beerman T. A., Woynarowski J. M., Sigmund R. D., Gawron L. S., Rao K. E., Lown J. W., Biochim. Biophys. Acta, Gene Struct. Expression 1991, 1090, 52–60. [DOI] [PubMed] [Google Scholar]
- 45. Cockerill P. N., Garrard W. T., FEBS Lett. 1986, 204, 5–7. [DOI] [PubMed] [Google Scholar]
- 46. Mrksich M., Parks M. E., Dervan P. B., J. Am. Chem. Soc. 1994, 116, 7983–7988. [Google Scholar]
- 47. Trauger J. W., Baird E. E., Dervan P. B., Nature 1996, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- 48. Ghosh M. K., Cohen J. S. in Progress in Nucleic Acid Research and Molecular Biology, Elsevier, Amsterdam, 1992, pp. 79–126. [DOI] [PubMed] [Google Scholar]
- 49. Goodsell D. S., Dickerson R. E., Nucleic Acids Res. 1994, 22, 5497–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Trauger J. W., Baird E. E., Mrksich M., Dervan P. B., J. Am. Chem. Soc. 1996, 118, 6160–6166; [Google Scholar]
- 50b. Trauger J. W., Baird E. E., Dervan P. B., Chem. Biol. 1996, 3, 369–377. [DOI] [PubMed] [Google Scholar]
- 51. Turner J. M., Swalley S. E., Baird E. E., Dervan P. B., J. Am. Chem. Soc. 1998, 120, 6219–6226. [Google Scholar]
- 52. White S., Szewczyk J. W., Turner J. M., Baird E. E., Dervan P. B., Nature 1998, 391, 468–471. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Urbach A. R., Szewczyk J. W., White S., Turner J. M., Baird E. E., Dervan P. B., J. Am. Chem. Soc. 1999, 121, 11621–11629; [Google Scholar]
- 53b. Kielkopf C. L., White S., Szewczyk J. W., Turner J. M., Baird E. E., Dervan P. B., Rees D. C., Science 1998, 282, 111–115. [DOI] [PubMed] [Google Scholar]
- 54. Dose C., Farkas M. E., Chenoweth D. M., Dervan P. B., J. Am. Chem. Soc. 2008, 130, 6859–6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nickols N. G., Jacobs C. S., Farkas M. E., Dervan P. B., Nucleic Acids Res. 2007, 35, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu L., Wang W., Gotte D., Yang F., Hare A. A., Welch T. R., Li B. C., Shin J. H., Chong J., Strathern J. N., Dervan P. B., Wang D., Proc. Natl. Acad. Sci. USA 2016, 113, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hargrove A. E., Martinez T. F., Hare A. A., Kurmis A. A., Phillips J. W., Sud S., Pienta K. J., Dervan P. B., Agoulnik I. U., PLoS ONE 2015, 10, e0143161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ehley J. A., Melander C., Herman D., Baird E. E., Ferguson H. A., Goodrich J. A., Dervan P. B., Gottesfeld J. M., MCB 2002, 22, 1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dervan P. B., Kurmis A. A., Finn P. B. in DNA-Targeting Molecules as Therapeutic Agents (Ed.: Waring M. J.), RSC, London, 2018, pp. 298–325. [Google Scholar]
- 60.
- 60a. Chenoweth D. M., Dervan P. B., Proc. Natl. Acad. Sci. USA 2009, 106, 13175–13179; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60b. Chenoweth D. M., Dervan P. B., J. Am. Chem. Soc. 2010, 132, 14521–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang F., Nickols N. G., Li B. C., Marinov G. K., Said J. W., Dervan P. B., Proc. Natl. Acad. Sci. USA 2013, 110, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Darnell J. E., Nat. Rev. Cancer 2002, 2, 740–749. [DOI] [PubMed] [Google Scholar]
- 63. Aman K., Padroni G., Parkinson J. A., Welte T., Burley G. A., Chem. Eur. J. 2019, 25, 2757–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.
- 64a. Chenoweth D. M., Poposki J. A., Marques M. A., Dervan P. B., Bioorg. Med. Chem. 2007, 15, 759–770; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64b. Arndt H.-D., Hauschild K. E., Sullivan D. P., Lake K., Dervan P. B., Ansari A. Z., J. Am. Chem. Soc. 2003, 125, 13322–13323. [DOI] [PubMed] [Google Scholar]
- 65. Moore M. J. B., Cuenca F., Searcey M., Neidle S., Org. Biomol. Chem. 2006, 4, 3479–3488. [DOI] [PubMed] [Google Scholar]
- 66. Zaffaroni N., Lualdi S., Villa R., Bellarosa D., Cermele C., Felicetti P., Rossi C., Orlandi L., Daidone M. G., Eur. J. Cancer 2002, 38, 1792–1801. [DOI] [PubMed] [Google Scholar]
- 67. Rahman K. M., Reszka A. P., Gunaratnam M., Haider S. M., Howard P. W., Fox K. R., Neidle S., Thurston D. E., Chem. Commun. 2009, 4097–4099. [DOI] [PubMed] [Google Scholar]
- 68. Müller S., Laxmi-Reddy K., Jena P. V., Baptiste B., Dong Z., Godde F., Ha T., Rodriguez R., Balasubramanian S., Huc I., ChemBioChem 2014, 15, 2563–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.
- 69a. Yu Z., Pandian G. N., Hidaka T., Sugiyama H., Adv. Drug Delivery Rev. 2019, 147, 66–85; [DOI] [PubMed] [Google Scholar]
- 69b. Malinee M., Kumar A., Hidaka T., Horie M., Hasegawa K., Pandian G. N., Sugiyama H., Bioorg. Med. Chem. 2020, 28, 115248; [DOI] [PubMed] [Google Scholar]
- 69c. Zhang M., Liang J., Jiang S.-K., Xu L., Wu Y.-L., Awadasseid A., Zhao X.-Y., Xiong X.-Q., Sugiyama H., Eur. J. Med. Chem. 2020, 207, 112704. [DOI] [PubMed] [Google Scholar]
- 70.Review: Gopalakrishnan R., Frolov A. I., Knerr L., W. J. Drury III , Valeur E., J. Med. Chem. 2016, 59, 9599–9621. [DOI] [PubMed] [Google Scholar]
- 71. Azzarito V., Long K., Murphy N. S., Wilson A. J., Nat. Chem. 2013, 5, 161–173. [DOI] [PubMed] [Google Scholar]
- 72. Ripka A. S., Rich D. H., Curr. Opin. Chem. Biol. 1998, 2, 441–452. [DOI] [PubMed] [Google Scholar]
- 73. Hamuro Y., Geib S. J., Hamilton A. D., Angew. Chem. Int. Ed. Engl. 1994, 33, 446–448; [Google Scholar]; Angew. Chem. 1994, 106, 465–467. [Google Scholar]
- 74. Hamuro Y., Geib S. J., Hamilton A. D., J. Am. Chem. Soc. 1996, 118, 7529–7541. [Google Scholar]
- 75.
- 75a. Arrata I., Grison C. M., Coubrough H. M., Prabhakaran P., Little M. A., Tomlinson D. C., Webb M. E., Wilson A. J., Org. Biomol. Chem. 2019, 17, 3861–3867; [DOI] [PubMed] [Google Scholar]
- 75b. Campbell F., Plante J. P., Edwards T. A., Warriner S. L., Wilson A. J., Org. Biomol. Chem. 2010, 8, 2344–2351. [DOI] [PubMed] [Google Scholar]
- 76. Edwards T., Wilson A., Amino Acids 2011, 41, 743–754. [DOI] [PubMed] [Google Scholar]
- 77. Bullock B. N., Jochim A. L., Arora P. S., J. Am. Chem. Soc. 2011, 133, 14220–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wells J. A., McClendon C. L., Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- 79. Keskin O., Gursoy A., Ma B., Nussinov R., Chem. Rev. 2008, 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- 80. Orner B. P., Ernst J. T., Hamilton A. D., J. Am. Chem. Soc. 2001, 123, 5382–5383. [DOI] [PubMed] [Google Scholar]
- 81. Prabhakaran P., Barnard A., Murphy N. S., Kilner C. A., Edwards T. A., Wilson A. J., Eur. J. Org. Chem. 2013, 3504–3512. [Google Scholar]
- 82. Azzarito V., Miles J. A., Fisher J., Edwards T. A., Warriner S. L., Wilson A. J., Chem. Sci. 2015, 6, 2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hegedus Z., Grison C. M., Miles J. A., Rodriguez-Marin S., Warriner S. L., Webb M. E., Wilson A. J., Chem. Sci. 2019, 10, 3956–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barnard A., Long K., Martin H. L., Miles J. A., Edwards T. A., Tomlinson D. C., Macdonald A., Wilson A. J., Angew. Chem. Int. Ed. 2015, 54, 2960–2965; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.
- 85a. Kulikov O. V., Kumar S., Magzoub M., Knipe P. C., Saraogi I., Thompson S., Miranker A. D., Hamilton A. D., Tetrahedron Lett. 2015, 56, 3670–3673; [Google Scholar]
- 85b. Hebda J. A., Saraogi I., Magzoub M., Hamilton A. D., Miranker A. D., Chem. Biol. 2009, 16, 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.
- 86a. Kumar S., Schlamadinger D. E., Brown M. A., Dunn J. M., Mercado B., Hebda J. A., Saraogi I., Rhoades E., Hamilton A. D., Miranker A. D., Chem. Biol. 2015, 22, 369–378; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86b. Kumar S., Birola M., Miranker A. D., Chem. Commun. 2016, 52, 6391–6394; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86c. Kumar S., Birol M., Schlamadinger D. E., Wojcik S. P., Rhoades E., Miranker A. D., Nat. Commun. 2016, 7, 11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kumar S., Vogel M. C., Hamilton A. D., Org. Biomol. Chem. 2018, 16, 733–741. [DOI] [PubMed] [Google Scholar]
- 88. Yap J. L., Cao X., Vanommeslaeghe K., Jung K.-Y., Peddaboina C., Wilder P. T., Nan A., A. D. MacKerell, Jr. , Smythe W. R., Fletcher S., Org. Biomol. Chem. 2012, 10, 2928–2933. [DOI] [PubMed] [Google Scholar]
- 89.
- 89a. Kumar S., Hamilton A. D., J. Am. Chem. Soc. 2017, 139, 5744–5755; [DOI] [PubMed] [Google Scholar]
- 89b. Kumar S., Henning-Knechtel A. H., Magzoub M., Hamilton A. D., J. Am. Chem. Soc. 2018, 140, 6562–6574. [DOI] [PubMed] [Google Scholar]
- 90. Kumar S., Henning-Knechtel A., Chehade I., Magzoub M., Hamilton A. D., J. Am. Chem. Soc. 2017, 139, 17098–17108. [DOI] [PubMed] [Google Scholar]
- 91. Lee C.-H., Yu J.-R., Kumar S., Jin Y., LeRoy G., Bhanu N., Kaneko S., Garcia B. A., Hamilton A. D., Reinberg D., Mol. Cell 2018, 70, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Maity D., Kumar S., Curreli F., Debnath A. K., Hamilton A. D., Chem. Eur. J. 2019, 25, 7265–7269. [DOI] [PubMed] [Google Scholar]
- 93.
- 93a. Ziach K., Chollet C., Parissi V., Prabhakaran P., Marchivie M., Corvaglia V., Bose P. P., Laxmi-Reddy K., Godde F., Schmitter J.-M., Chaignepain S., Pourquier P., Huc I., Nat. Chem. 2018, 10, 511–518; [DOI] [PubMed] [Google Scholar]
- 93b. Corvaglia V., Carbajo D., Prabhakaran P., Ziach K., Mandal P. K., Dos Santos V., Legeay C., Vogel R., Parissi V., Pourquier P., Huc I., Nucleic Acids Res. 2019, 47, 5511–5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.
- 94a. Andersson E. K., Strand M., Edlund K., Lindman K., Enquist P.-A., Spjut S., Allard A., Elofsson M., Mei Y.-F., Wadell G., Antimicrob. Agents Chemother. 2010, 54, 9, 3871–3877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94b. Öberg C. T., Strand M., Andersson E. K., Edlund K., Tran N. P. N., Mei Y.-F., Wadell G., Elofsson M., J. Med. Chem. 2012, 55, 3170–3181; [DOI] [PubMed] [Google Scholar]
- 94c. Strand M., Islam K., Edlund K., Öberg C. T., Allard A., Bergström T., Mei Y.-F., Elofsson M., Wadell G., Antimicrob. Agents Chemother. 2012, 56, 5735–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Islam Md. K., Strand M., Saleeb M., Svensson R., Baranczewski P., Artursson P., Wadell G., Ahlm C., Elofsson M., Evander M., Sci. Rep. 2018, 8, 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gwon Y.-D., Strand M., Lindqvist R., Nilsson E., Saleeb M., Elofsson M., Överby A. K., Evander M., Viruses 2020, 12, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.
- 97a. Tew G. N., Scott R. W., Klein M. L., DeGrado W. F., Acc. Chem. Res. 2010, 43, 30–39; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97b. Mensa B., Howell G. L., Scott R., DeGrado W. F., Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.
- 98a. Wu X. X., Rui Liu R., Sathyamoorthy B., Yamato K., Liang G. X., Shen L., Ma S. F., Sukumaran D. K., Szyperski T., Fang W. H., He L., Chen X. B., Gong B., J. Am. Chem. Soc. 2015, 137, 5879–5882; [DOI] [PubMed] [Google Scholar]
- 98b. Kline M. A., Wei X. X., Horner I. J., Liu R., Chen S., Chen S., Yung K. Y., Yamato K., Cai Z. H., Bright F. V., Zeng X. C., Gong B., Chem. Sci. 2015, 6, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.
- 99a. Mateus P., Chandramouli N., Mackereth C. D., Kauffmann B., Ferrand Y., Huc I., Angew. Chem. Int. Ed. 2020, 59, 5797–5805; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5846–5854; [Google Scholar]
- 99b. Saha S., Kauffmann B., Ferrand Y., Huc I., Angew. Chem. Int. Ed. 2018, 57, 13542–13546; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13730–13734; [Google Scholar]
- 99c. Mateus P., Wicher B., Ferrand Y., Huc I., Chem. Commun. 2018, 54, 5078–5081. [DOI] [PubMed] [Google Scholar]
- 100. Kim J., Kim H., Park S. B., J. Am. Chem. Soc. 2014, 136, 14629–14638. [DOI] [PubMed] [Google Scholar]
- 101. Evans B. E., Rittle K. E., Bock M. G., DiPardo R. M., Freidinger R. M., Whitter W. L., Lundell G. F., Veber D. F., Anderson P. S., J. Med. Chem. 1988, 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- 102. Davison E. K., Brimble M. A., Curr. Opin. Chem. Biol. 2019, 52, 1–8. [DOI] [PubMed] [Google Scholar]
- 103.
- 103a. Alex J. M., Corvaglia V., Hu X., Engilberge S., Huc I., Crowley P. B., J. Am. Chem. Soc. 2017, 139, 2928–2931;28170240 [Google Scholar]
- 103b. Vallade M., Reddy P. S., Fischer L., Huc I., Eur. J. Org. Chem. 2018, 5489–5498; [Google Scholar]
- 103c. Alex J. M., Corvaglia V., Hu X., Engilberge S., Huc I., Crowley P. B., Chem. Commun. 2019, 55, 11087–11090; [DOI] [PubMed] [Google Scholar]
- 103d. Reddy P. S., d'Estaintot B. L., Granier T., Mackereth C. D., Fischer L., Huc I., Chem. Eur. J. 2019, 25, 11042–11047. [DOI] [PubMed] [Google Scholar]
- 104. Soczek K. M., Grant T., Rosenthal P. B., Mondragón A., eLife 2018, 7, e41215. [DOI] [PMC free article] [PubMed] [Google Scholar]