

Abstract
Microbial abundance and community composition in marine sediments have been widely explored. However, high‐resolution vertical changes of benthic microbial diversity and co‐occurrence patterns are poorly described. The ecological contributions of abundant and rare species in sediments also remain largely unknown. Here, by analysing microbial populations at 14 depth layers of 10 subseafloor sediment cores (water depth 1,250–3,530 m) obtained in the South China Sea, we provided the vertical profiles of microbial β‐diversity and co‐occurrence influenced by subcommunities of different abundance. These 134 sediment samples were clustered into four groups according to sediment depth (1–2, 6–10, 30–90 and 190–790 cm) with obvious shifts in microbial community compositions. The vertical succession of microorganisms was consistent with redox zonation and influenced by terrestrial inputs. Partitioning of vertical β‐diversity showed extremely high species replacement between deep layers and the surface layer, indicating selection‐induced loss of rare species and dispersal of dormant cells and spores. By contrast, for horizontal β‐diversity, richness of rare species became increasingly significant in deep sediments. Accompanying this β‐diversity profile were clear changes in the association pattern, with microorganisms being less connected in deeper sediment layers, probably reflecting reduced syntrophic interactions. Rare species accounted for an indispensable proportion in the co‐occurrence network, and tended to form complex “small worlds.” The rare subcommunity also responded differently to various environmental factors compared with the abundant subcommunity. Our findings expand current knowledge on vertical changes of marine benthic microbial diversity and their association patterns, emphasizing the potential roles of rare species.
Keywords: co‐occurrence network, deep‐sea sediment, microbial community, rare biosphere, β‐diversity
1. INTRODUCTION
Marine sediments are the largest reservoir of organic carbon (OC) (Hedges & Keil, 1995) and are populated by a diverse microbial community (Parkes et al., 2000; Zinger et al., 2011). Bacteria and archaea in sediments play key roles in organic matter (OM) remineralization, nutrient cycling and energy transfer (Mason et al., 2009). Their metabolic activities profoundly impact the biogeochemical cycles of carbon, nitrogen and sulphur on a global scale (Berner, 1982; Nealson, 1997; Orcutt et al., 2011). As sediments accumulate over geological time, buried microbial life persists at decreasing growth rates due to the depletion of respiratory electron acceptors and the lesser reactivity of substrates with depth (D’Hondt et al., 2004, 2019; Jorgensen & Marshall, 2016), resulting in the assembly of the deep biosphere. It has been proposed that microbial communities in this deep biosphere represent descendants of surface communities that were buried in the past (Lever et al., 2015; Starnawski et al., 2017; Walsh et al., 2016). Selection is thus considered the predominant force driving vertical community succession in the sediment column, while other processes, including diversification, dispersal and drift, might impart minor changes mainly within the surface environment (Nemergut et al., 2013; Petro et al., 2017). In‐depth knowledge of microbial biodiversity and interaction patterns is important for understanding these biogeographical processes, which is particularly true for the large and complex microbial populations in marine sediments (Hoshino et al., 2020). Previous studies have discussed vertical benthic microbial diversity in varied depth ranges (Inagaki et al., 2003, 2015; Nunoura et al., 2016, 2018; Schauer et al., 2010; Starnawski et al., 2017; Vuillemin, Vargas, et al., 2020; Vuillemin et al., 2019; Walsh et al., 2016), and highly resolved vertical profiles of microbial communities in sediments will help to better depict the stratified community structure and continuous variations in microbial diversity. However, so far, the majority of these studies have often focused on specific types of sedimentary environment (Nunoura et al., 2018; Yang et al., 2020) or comparison of microbial community over large spatial or temporal scales (Böer et al., 2009; Hoshino et al., 2020; Hoshino & Inagaki, 2019). Parallel comparison of site‐to‐site microbial diversity at different depths in a less varied sediment region is scarce.
β‐Diversity of microbial communities, a long‐standing interest in the field of marine microbial ecology, indicates how microbial assemblages vary in space and along environmental gradients. It also provides insights into the mechanism driving biodiversity changes and their consequences for multiple ecosystem functions (Mori et al., 2018). β‐Diversity reflects two different phenomena: species replacement (also called turnover) and richness difference or nestedness (Baselga, 2010). By partitioning β‐diversity into replacement and richness components, we are able to interpret the underlying ecosystem processes. In marine sediments, species replacement implies the simultaneous gain and loss of species due to processes such as selection during burial or along environmental gradients, genetic diversification and historical events (Macalady et al., 2013; Martiny et al., 2006; Nemergut et al., 2013), while richness difference may reflect shifts in the relative abundance of taxa that are always present, typified by the persistence of certain surface communities in deep sediments (Legendre, 2014). Many studies have shown that although both components contribute to total β‐diversity, their relative contributions may vary across different biological groups (Baselga, 2010; Hu et al., 2019; Si et al., 2015), emphasizing the need for β‐diversity partitioning in order to discern the antithetic biological implications underlying overall community changes. Partitioning of β‐diversity gives more insights into the factors driving spatial variability in biotic communities compared with a consideration of total β‐diversity alone (Soininen et al., 2018). For example, β‐partitioning patterns of microbes in upper lake sediment clearly distinguished taxonomic changes caused by either microbial activity or those related to sediment burial (Wurzbacher et al., 2017). A nested community selected from the surface has also been proposed for the deep biosphere in lacustrine sediment based on partitioning of β‐diversity (Vuillemin et al., 2018). However, no studies have examined how replacement and richness components of microbial β‐diversity change with depth in marine sediments and their biogeographical implications.
Patterns of microbial dynamics are associated with interspecies interactions (Gorter et al., 2020). Thus, knowledge of alterations in microbial interactions underlying community changes will help to create a better understanding of microbial ecology. Co‐occurrence networks provide system‐level insights into the complex microbial interactions (Lv et al., 2019), although there is still debate over whether this method provides true and valid interactions (Liu et al., 2019). Reconstruction of microbial networks can advance our knowledge of the complex behaviours in microbial communities and predict the effects of perturbations on community dynamics (Röttjers & Faust, 2018). Previous studies have used network analysis to explore interactions between marine benthic microbes and microbe–environment relationships (Buongiorno et al., 2019; Wang et al., 2020; Zhang, Li, et al., 2020), but most of these studies focused only on shallow sediments and none of them revealed the vertical shift of microbial networks in the subseafloor. In addition, network analysis can be a powerful tool for inferring the keystone taxa in a microbial community which can drive community composition and function irrespective of their abundance (Banerjee et al., 2018). While keystone taxa underline the importance of numerically inconspicuous taxa in microbiome functioning (Banerjee et al., 2018), they are seldom discussed for marine sediments.
Microorganisms often present a skewed abundance distribution in a local community, with a few relatively abundant species coexisting alongside a large number of rare species referred to as the “rare biosphere” (Logares et al., 2014; Lynch & Neufeld, 2015; Pedrós‐Alió, 2012; Sogin et al., 2006). Abundant and rare taxa play distinct roles in an ecosystem. The abundant taxa are important for the fluxes of OM and biomass production due to their huge quantities (Pedrós‐Alió, 2012), whereas recent studies increasingly emphasize that the rare biosphere not only serves as reservoirs of genetic and functional diversity, but also has substantial functional roles (Lynch & Neufeld, 2015; Shade & Gilbert, 2015; Shade et al., 2014). Moreover, the rare subcommunities can serve as components of microbial seed banks, which may maintain a dormant state and recruit to higher density upon changes in environmental conditions, and the dispersal of microbial seed banks appears to primarily shape subsurface community compositions across oceans (Lennon & Jones, 2011). Rare microbes have also been reported to achieve essential functions in nutrient cycling, and may enhance the functionality of the abundant microbes (Bodelier et al., 2013; Pedrós‐Alió, 2007; Philippot et al., 2013), suggesting that there could be comprehensive interactions between abundant and rare species (Jousset et al., 2017). Therefore, rare microbes could be overlooked keystone species regulating the functions of diverse ecosystems. The distribution patterns of rare taxa have been studied in different environments (Jiao & Lu, 2020; Mo et al., 2018; Wu et al., 2017), with the results indicating that microbial community assembly in the rare biosphere can be driven by distinct processes and dependent on distinct environmental variables as compared with the abundant taxa. Nevertheless, the distribution of rare microbial taxa in deep marine sediments remains largely unexplored. Additionally, the moderate subcommunity has been increasingly discussed in recent studies (Bay et al., 2020; Sinkko et al., 2019; Xue et al., 2018; Zhang, Hou, et al., 2020), which reflects an intermediate state between the abundant and rare counterparts in several important aspects, including diversity and ecological processes that structure microbial community compositions. Whether the abundant, moderate and rare subcommunities in sediments show similar vertical changes or whether they respond differentially to environmental heterogeneity is yet to be answered.
The South China Sea (SCS) is one of the largest marginal seas in the world. The Shenhu Area, located on the northern slope between the Xisha Trough and Dongsha Islands of the SCS, is an important region for methane hydrate exploration (Wu et al., 2011). Organic‐rich sediments have accumulated since the late Mesozoic. The thickness of sediment is 1,000–7,000 m with 0.46%–1.9% OM (Wang et al., 2000). A previous study revealed that hydrate‐free and hydrate‐containing surface sediments in the Shenhu Area possess distinct microbial community compositions (Jiao et al., 2015). However, a comprehensive study of vertical microbial ecology in sediments near the Shenhu Area is lacking. Here, we collected 10 sediment cores ranging from 6 to 8 m and provided a highly resolved vertical profile of microbial abundance and distribution in this area, mainly aiming to evaluate (i) how microbial β‐diversity and its two distinct components change along sediment depth, (ii) what are the vertical patterns of microbial interaction networks, and (iii) the importance of the abundant and rare subcommunities regarding microbial diversity and interactions in marine sediments.
2. MATERIAL AND METHODS
2.1. Sample collection and environmental characterization
Ten sediment gravity cores were collected on the northern slope of the SCS (18.44–19.26°N, 114.25–115.30°E), ranging from 6.64 to 8.36 m. Water depths at these sites were 1,250–3,530 m. The geographical information of the 10 sampling sites is summarized in Figure 1a and Table S1. The sediment samples were collected by a gravity corer during cruise HYSH201901 organized by Guangzhou Marine Geological Survey from February 26 to March 6, 2019. The cores were sliced with a stainless‐steel cutter for subsampling. To reduce contamination, the outer 1 cm of sediment was sliced off with a red‐hot, sterile spatula. An unused, sterile spatula was used to carefully sample the uncontaminated centre of the remaining core sample at each depth. The sediments were transferred to sterilized plastic tubes and stored at −80°C before OM measurement and DNA extraction. We obtained 134 samples in total at 1, 2, 6, 10, 30, 50, 90, 190, 290, 390, 490, 590, 690 and 790 cm of these cores. Sediment porewater was extracted using a Rhizon sampler attached to a vacuum tube under N2 atmosphere (Seeberg‐Elverfeldt et al., 2005; Yao et al., 2014). After collection, porewater samples were further split into acidified or unacidified subsamples, and stored at 4°C for subsequent analysis of dissolved inorganic carbon (DIC), nutrients and other solutes (Zhao, Yao, et al., 2017). DIC was measured using the nondispersive infrared method with a precision of 0.1% and a detection limit of 0.2 µM (AS‐C3, Apollo SciTech) (Cai & Wang, 1998). Dissolved inorganic nutrients (,,, and ) in porewaters were determined with the standard colorimetric methods using a continuous flow analyser (Auto‐Analyzer 3; Seal Analytical) (Catalano, 1987; Worsfold et al., 2016). Major cations (Ca2+ and Mg2+) and dissolved Mn (assumed to be present as Mn2+) were determined using inductively coupled plasma optical emission spectrometry (ICP‐OES; iCAP6300, Thermo Fisher Scientific) (Zhao, Yao, et al., 2017). Major anions ( and Cl−) in porewater were analysed using ion chromatography (ICS‐3000, Dionex) on 1:800 diluted aliquots in Milli‐Q water (Yao et al., 2014). Total OC (TOC), total nitrogen (TN) and stable carbon isotope composition (δ13CTOC) in sediments were analysed after removing carbonates with HCl (6 m) on an elemental analyser (vario MICRO cube, Elementar) interfaced to a continuous flow isotope ratio mass spectrometer (Isoprime100, Isoprime Co.) as described by Yao et al. (2014).
FIGURE 1.
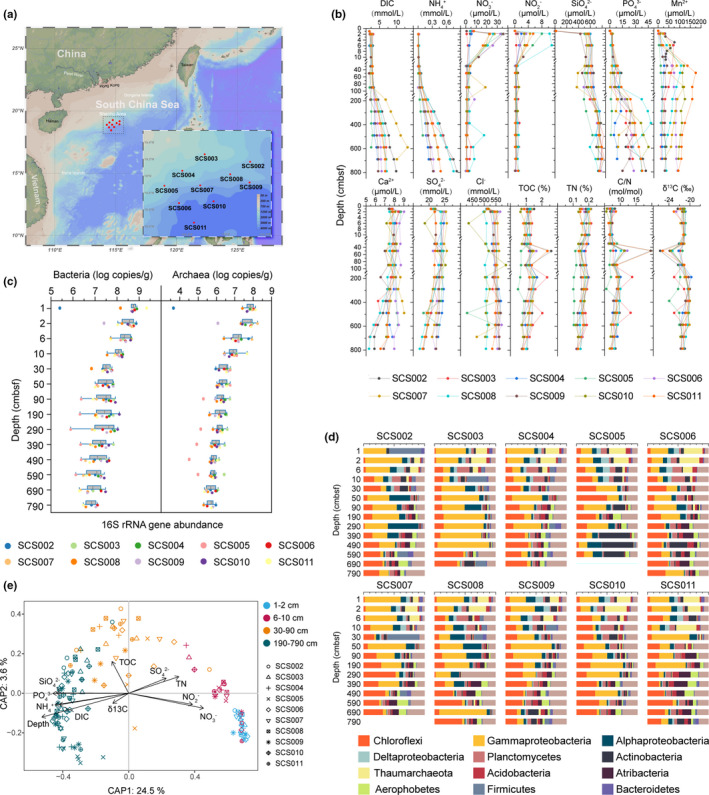
Site locations, environmental parameters, microbial abundance and communities in SCS sediments. (a) Sampling map of the northern slope of the SCS. (b) Depth profiles of key chemical characteristics. (c) Vertical changes of bacterial and archaeal 16S rRNA gene abundance. (d) Microbial community compositions at the phylum level and at class level of Proteobacteria. (e) db‐RDA illustrating the relationship between the microbial community at the OTU level and significantly influential environmental variables
2.2. DNA extraction, sequencing and reads processing
Genomic DNA was extracted from 0.25 g of sediment (wet weight) using the Power Soil DNA Isolation Kit (MoBio Laboratories) and a FastPrep‐24 cell disrupter (MP Biomedicals) according to the manufacturers’ instructions. The quality and quantity of the extracted DNA were measured by a Nanodrop ND‐2000 spectrophotometer (Thermo Fisher Scientific). The primer set of 515FmodF (5′‐GTGYCAGCMGCCGCGGTAA‐3′) and 806RmodR (5′‐GGACTACNVGGGTWTCTAAT‐3′) was used to amplify the V4 hypervariable region of the 16S rRNA gene from bacteria and archaea (Walters et al., 2016). These primers were modified according to Parada et al. (2016) and Apprill et al. (2015), which improves detection of Thaumarchaeota and clade SAR11 without degrading performance on taxa already amplified effectively by the original primer set. Polymerase chain reactions (PCRs) were conducted as follows: 3 min for denaturation at 95°C; 29 cycles of 30 s at 95°C, 30 s for annealing at 55°C, 45 s for elongation at 72°C, and 10 min for a final extension at 72°C. PCRs were conducted in triplicate 20 µl mixture comprising 0.4 µl of FastPfu Polymerase, 0.8 µl of each primer (5 μm), 2 µl of 2.5 mm deoxyribonucleoside triphosphates (dNTPs), 4 µl of 5× FastPfu Buffer, and 10 ng of template DNA. The PCR products were purified from 2% agarose gels using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences) and further quantified using QuantiFluor‐ST (Promega) following the manufacturers’ protocol. Purified amplicons were pooled in equimolar ratio and paired‐end sequenced (2 × 300) on an Illumina Miseq PE300 platform (Illumina) at Majorbio Bio‐Pharm Technology.
Raw reads were quality‐filtered by trimmomatic (Bolger et al., 2014) and merged by Fast Length Adjustment of Short reads (flash 1.2.11, https://ccb.jhu.edu/software/FLASH/) with the following criteria: (i) the reads were truncated at any site receiving an average quality score <20 over a 50‐bp sliding window; (ii) sequences whose overlap was greater than 10 bp were merged if there were no more than two nucleotide mismatches; (iii) sequences of each sample were separated by barcoding (exact matches) and primers (allowing two nucleotide mismatches); and (iv) reads containing ambiguous bases were deleted. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using uparse (Edgar, 2013) (version 7.0.1090, http://drive5.com/uparse/). Singleton OTUs that may represent sequencing errors were removed before downstream analyses. The taxonomy of each 16S rRNA gene sequence was analysed via the Ribosomal Database Project (RDP) classifier algorithm (version 2.11, http://rdp.cme.msu.edu/) (Wang et al., 2007) against the silva database (Release132, http://www.arb‐silva.de). To equalize sequencing depth, each sample was rarefied to 33,686 reads (the lowest sequence number across all samples) for further analysis. The raw data were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (accession no.: PRJNA606622).
2.3. Quantitative PCR
Quantitative PCR was performed to quantify the abundance of total bacteria and archaea using primer sets specific for the bacterial 16S rRNA gene (Eub338F/Eub518R) (Yin et al., 2013) and archaeal 16S rRNA gene (967F/1060R), respectively (Cadillo‐Quiroz et al., 2006). A 20‐µl mixture contained 10 µl of SYBR Premix ExTaq II (2×), 0.4 µl of ROX Reference Dye II (50×) (TaKaRa), 0.2 µl of primers for each gene (10 µm), and 2 µl of template. The thermal cycling steps for the bacterial 16S rRNA gene consisted of an initial denaturation at 95°C for 5 min, 40 cycles of 95°C for 30 s, 53°C for 1 min and 72°C for 15 s, and a final extension at 72°C for 10 min. The thermal cycling steps for the archaeal 16S rRNA gene consisted of an initial denaturation at 95°C for 30 s, 40 cycles of 95°C for 5 s, 50°C for 30 s and 72°C for 30 s, and a final extension at 72°C for 5 min. All assays were conducted in triplicate, with negative controls, using an ABI 7500 Real‐Time PCR System (Applied Biosystems). Standard curves were constructed by PCR amplifying a 10‐fold serial dilution of plasmids containing target gene fragments. The amplification curves showed good linear relationships (R 2 > .999) and the amplification efficiencies were 0.90 and 0.95 for the bacterial and archaeal 16S rRNA gene, respectively.
2.4. Data analysis and statistics
The alpha diversity indices, including Good's coverage, Chao 1, Shannon, Shannon‐even, and phylogenetic diversity (PD), were calculated using mothur (version 1.30.1) (Schloss et al., 2009). Analyses of similarities (ANOSIM) was performed with the “vegan” package in R. A linear discriminant analysis effect size (LEfSe) was applied at the genus level (all‐against‐all, LDA > 3.5; http://huttenhower.sph.harvard.edu/galaxy/) to identify the discriminant bacterial and archaeal clades at different depths.
We defined “abundant” OTUs as those having relative abundances above 0.1% of the total sequences, “moderate” OTUs as those having relative abundances between 0.01% and 0.1%, and “rare” OTUs as those having relative abundances below 0.01% (singletons, which may be caused by sequencing errors, were discarded) (Jiao & Lu, 2020). Nonmetric multidimensional scaling (NMDS) was performed based on Bray–Curtis distances with the “vegan” package in r. Tests of homogeneity of dispersions (“betadisper” in the “vegan” package) followed by ANOVAs were performed to compare the multivariate dispersions. Differences of within‐group dissimilarity were determined using the Kruskal–Wallis test followed by a post‐hoc analysis in originpro 2020 (OriginLab). Total β‐diversity and its partitioning into replacement and richness components (β repl and β rich) were calculated based on the Jaccard dissimilarity via the BAT package in r (Cardoso et al., 2015). We defined vertical β‐diversity as β‐diversity between the sum of OTUs in the surface layer (1 cm) and each deeper layer, and horizontal β‐diversity as β‐diversity calculated among samples from the same depth layer.
Co‐occurrence networks were constructed for all samples and four depth groups, respectively, using the “igraph” and “Hmisc” libraries in r. To reduce complexity, only OTUs occurring in more than 25% of samples were retained. The pairwise Spearman's correlations between OTUs were calculated, with a correlation coefficient > |.7| and a p value <.01 (Benjamini and Hochberg‐adjusted) being considered as a valid relationship. The network‐level (mean node degree, clustering coefficient, average path length, modularity, density, diameter, betweenness centralization and degree centralization) and node‐level (degree, transitivity, betweenness centrality and closeness centrality) topological features of a network were calculated. gephi (version 0.8.2 beta, http://gephi.org) was used for network visualization. Keystone species in each co‐occurrence network were defined as those with a high degree of ranking in the top 20% among all nodes, and low betweenness centrality, ranking in the bottom 20% among nodes with high degree (Berry & Widder, 2014).
We implemented the Mantel test using the “vegan” package in R to identify significant environmental factors correlated with the variations of microbial communities. The relative importance of factors in explaining community variation was investigated by variation partitioning based on distance‐based redundancy analysis (db‐RDA) of Hellinger‐transformed OTU abundance data using the “vegan” package in R.
3. RESULTS
3.1. General environmental characterization
Detailed sediment and porewater environmental parameters are shown in Figure 1b. In general, DIC, and showed a similar tendency to increase with depth, especially below 90 cm. and Mn2+ were also higher in deeper sediments than in surface layers (p < .001 in Mann–Whitney U tests). Conversely, higher NO3 − and NO2 − concentrations were detected in the top 10 cm (p < .001 in Mann–Whitney U tests). Ca2+ and exhibited a decreasing trend with depth. TOC and TN contents were constant with depth, but abrupt increases of TOC and C/N were observed at 50, 190 and 490 cm in several sites. δ13C was lower at 50 cm in most sites, in concert with the increases of TOC and C/N at this depth, indicating that there might be distinct sources of OC input compared with other depths.
3.2. General microbial abundance and α‐diversity in SCS sediment
Quantitative PCR showed that the 16S rRNA gene abundances of both bacteria and archaea were highest in surface sediments (1–2 cm; 108–109 copies g–1 for bacteria, 107–108 copies g–1 for archaea), and then declined gradually with depth by 1.5–3 orders of magnitude (Figure 1c). Differences were also observed among the studied sites but were less distinguishable than those among depths (ANOSIM global R: .0831, p = .001). Across all samples, the abundance of the bacterial 16S rRNA gene was higher (0.2–2.0 orders of magnitude) than that of archaea.
A total of 4,513,924 reads were obtained after quality control, discard of singletons and rarefaction, and were clustered into 29,520 OTUs at a 97% similarity level (25,896 bacterial OTUs and 3,539 archaeal OTUs). We further divided the OTUs into abundant, moderate and rare OTUs depending on their relative abundance. In the whole data set, 147 OTUs and 878 OTUs, representing 57.0% and 24.1% of all sequences, belonged to abundant and moderate OTUs, respectively; 28,495 OTUs contributing 18.9% of all sequences were classified as rare OTUs.
Good's coverage values ranged between 95.8% and 99.9% across all samples, indicating that sequences generated from these samples could represent most of the microbial community in the studied sites. All α‐diversity indices, including Chao 1, Shannon, Shannon evenness and PD, fluctuated with a similar trend with depth (Figure S1). Chao 1 and PD indices exhibited an evident median increase in the top 10 cm and a subsequent decrease to the deeper layers (290–490 cm). Shannon and Shannon evennness indices were slightly lower at 50–490 cm compared with those in the surface and deepest sediments.
3.3. Vertical and horizontal β‐diversity patterns
NMDS analysis based on the Bray–Curtis distance revealed that the sediment samples clustered more strongly by depth than by site. In general, samples were divided into four groups (Figure 2a) at the OTU level: 1–2, 6–10, 30–90 and 190–790 cm, for the whole data set (p = .001, R 2 = .36 in PERMANOVA) as well as the abundant (p = .001, R 2 = .38 in PERMANOVA), moderate (p = .001, R 2 = .40 in PERMANOVA), and rare subcommunities (p =.001, R 2 = .22 in PERMANOVA). However, the dispersion of these groups also differed significantly (“betadisper,” ANOVA, F = 16.0, p < .001). Rare OTUs best resembled the segregation of samples of the entire community. In general, the dissimilarity between samples shown by the rare subcommunity was highest compared with the abundant and moderate subcommunities at all depths, with least variation within each group (Figure 2b). Comparison of community variance at each depth group showed depth‐related horizontal heterogeneity (Figure 2b). For the entire community, within‐depth distances increased from 1–2, 6–10 to 30–90 cm, and then decreased at 190–790 cm (p < .001, χ2 = 444.1 in Kruskal–Wallis test). Similar patterns were observed for abundant (p < .001, χ2 = 274.4 in Kruskal–Wallis test) and moderate subcommunities (p < .001, χ2 = 571.9 in Kruskal–Wallis test), whereas the within‐depth distances increased with depth and were highest at 190–790 cm for rare OTUs (p < .001, χ2 = 761.5 in Kruskal–Wallis test).
FIGURE 2.
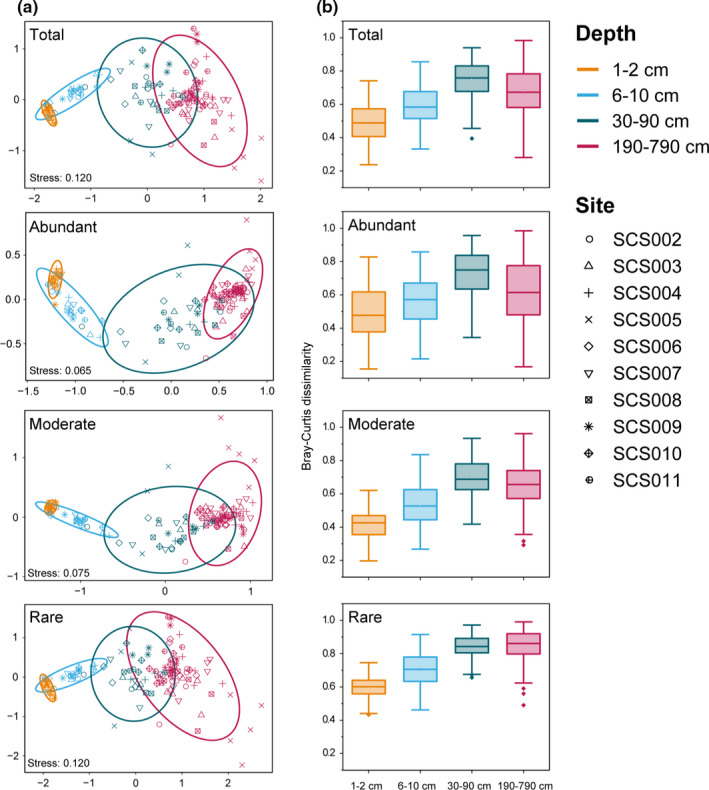
Comparison of microbial communities at the OTU level in SCS sediments. (a) Nonmetric multidimensional scaling ordination of total, abundant, moderate and rare subcommunities based on the Bray–Curtis dissimilarity. (b) Comparison of Bray–Curtis dissimilarity of microbial OTUs within different depth groups for each subcommunity
To explore the extent of community turnover with depth, we compared the deeper layers with the surface layer (1 cm). The β‐diversity based on the entire community increased with depth and approached 1 (i.e., no shared taxa) in the deepest layer (790 cm) (Figure 3a). Upon partitioning the overall β‐diversity into taxonomic richness (β rich) and replacement effects (β repl), taxonomic replacement increased in the top 90 cm and was consistently high at 190–790 cm (Figure 3a). In contrast, the effect of richness was high at 6–30 cm and then decreased with depth, accounting for only minor proportions at 190–790 cm. For abundant and moderate subcommunities, replacement between surface and deeper layers significantly decreased compared with that of the entire community, while the increased and decreased trends for replacement and richness effects, respectively, were similar to the entire community. In addition, vertical changes in replacement and richness effects for the rare subcommunity exactly reflected that of the entire community.
FIGURE 3.
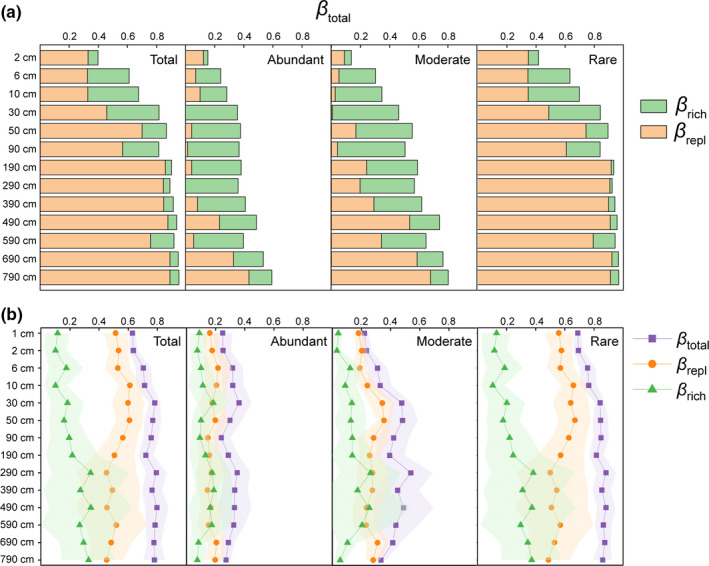
β‐Diversity and β‐diversity partitioning patterns for the entire community and subcommunities. (a) Vertical β‐diversity between the surface (1 cm) and each deeper layer. (b) Horizontal β‐diversity among samples from the same depth layer
We further analysed horizontal β‐diversity within samples from the same sediment depth, and the results showed that total horizontal β‐diversity increased in the top 30 cm and remained at a similar level in deeper layers (Figure 3b). Replacement effects were higher than richness effects in each layer above 190 cm, and showed a decreasing trend with sediment depth. Richness effects gradually increased from the surface to 190 cm (Figure 3b). Although there was no significant change of total horizontal β‐diversity at 290–790 cm, large variations of replacement and richness effects were observed between different sites. The patterns exhibited by the entire community exactly matched those of the rare subcommunity, with slightly higher horizontal distance among rare OTUs. The horizontal distances in abundant and moderate subcommunities were lower compared with that of the rare subcommunity, and fluctuated in different layers.
3.4. Microbial community compositions at different depths
In total, 80 phyla were observed in the SCS sediment samples. The most dominant phylum was Proteobacteria accounting for 47.25% of all sequences, followed by Chloroflexi (25.12%), Planctomycetes (8.58%) and Actinobacteria (6.62%) (Figure 1d). Dominant archaeal phyla included Thaumarchaeota (6.17%), Crenarchaeota (1.99%), Nanoarchaeaeota (1.62%), Hadesarchaeaeota (0.95%) and Euryarchaeota (0.83%). The relative abundance of different microbial groups changed significantly with depth (Figure 1d). Using LEfSe analysis, the surface sediments (1–2 cm) were found to be characterized by a high abundance of Nitrososphaeria, Dadabacteriia, Nitrospinia (within the phylum Nitrospirae), and several other groups (Figure S2). Lokiarchaeia, within the phylum Asgardaeota, was a distinct archaeal group enriched at 30–90 cm, while Hadesarchaeaeota was enriched in deeper layers (190–790 cm). Bacterial groups which were significantly more abundant at 190–790 cm contained several orders from Dehalococcoidia and Aminicenantia, as well as other groups within the Chloroflexi and Deltaproteobacteria.
The abundant OTUs were primarily affiliated with Proteobacteria (38.10%), Chloroflexi (22.45%) and Thaumarchaeota (8.84%), whereas the proportions of Proteobacteria was 23.01% and 17.09% in moderate and rare OTUs, respectively. Chloroflexi (24.06%) and Planctomycetes (10.93%) were two other primary taxa of moderate OTUs. Many more taxonomic groups were identified in rare OTUs, with Proteobacteria (17.09%), Planctomycetes (11.61%) and Chloroflexi (9.77%) being the major groups, as well as a high proportion of Nanoarchaeaeota (9.08%) and Patescibacteria (7.60%). The rare OTUs also contained considerably more unclassified taxa (7.45%) compared with those in abundant and moderate OTUs (0.68% and 2.05%, respectively). The proportions of subcommunities were similar in different depth groups: abundant OTUs, 0.46%–0.59%; moderate OTUs, 2.44%–3.27%; rare OTUs, 96.14%–96.98%.
3.5. Microbial co‐occurrence networks in SCS sediment
A co‐occurrence network was built based on correlation relationships across all samples from different depths (Figure 4a). The resulting network consisted of 1,468 nodes linked by 158,298 edges (Table S2). The entire network was clearly parsed into five major modules, of which modules 1, 2 and 3, respectively, accounted for 42.9%, 38.8% and 13.2% of the whole network. Most of the OTUs from module 1, the largest module, and module 3 had higher relative abundances in surface and shallow sediments, whereas the majority of the OTUs from module 2 had higher relative abundances in deep sediments (Figure 4b). The OTUs from Proteobacteria, Chloroflexi and Planctomycetes displayed wide correlations with others (Figure 4a). A much higher number of strong positive correlations (92.4%) were observed than negative ones (7.6%), with negative correlations mainly distributed between OTUs in module 2.
FIGURE 4.
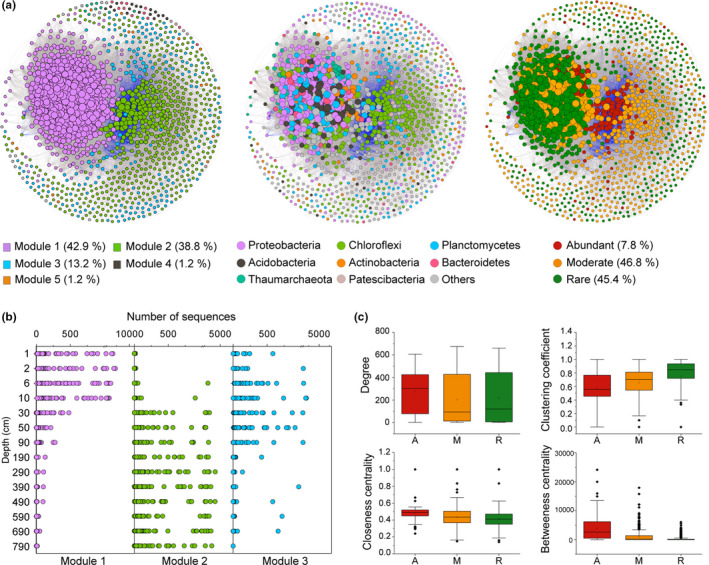
Co‐occurrence networks of the microbial community in all sediment samples and its properties. (a) Co‐occurrence networks with OTUs coloured by modularity, taxonomy and subcommunity, successively. Each connection shown has a correlation coefficient >|.7| and a p value <.01. The size of each node is proportional to the number of connections. (b) Numbers of sequences at a certain depth for each OTU in different modules. (c) Comparison of node‐level topological features among subcommunities. A, abundant; M, moderate; R, rare
By partitioning nodes into subcommunities, rare OTUs accounted for nearly the same proportions as moderate OTUs in the network (Figure 4a). We further compared node‐level topological features of abundant, moderate and rare subcommunities (Figure 4c). Abundant OTUs showed the highest degree level among the three subcommunities (p < .05, χ2 = 10.6 in Kruskal–Wallis test). Clustering coefficients increased significantly from abundant and moderate OTUs to rare OTUs (p < .001, χ2 = 186.5 in Kruskal–Wallis test), while the opposite pattern was observed for closeness (p < .001, χ2 = 57.4 in Kruskal–Wallis test) and betweenness centrality (p < .001, χ2 = 205.2 in Kruskal–Wallis test). No significant differences in eigenvector centrality values among the three subcommunities were found (data not shown).
We identified 59 keystone OTUs in total with high degree (>477) and low betweenness centrality (<311), including 41 rare OTUs, 16 moderate OTUs, and only two abundant OTUs, which may play key roles in maintaining the structure and function of microbial communities. The majority of keystone OTUs belong to Proteobacteria (25), Planctomycetes (13) and Acidobacteria (9). Other keystone OTUs belong to Bacteroidetes, Chloroflexi, Nitrospirae, Acidobacteria, Patescibacteria and Dadabacteria (Table S3).
3.6. Vertical changes of co‐occurrence networks
To study the shift of microbial co‐occurrence patterns along depth, we built separate co‐occurrence networks for samples from 1–2, 6–10, 30–90 and 190–790 cm (Figure 5). The complexity of the microbial network at 190–790 cm decreased significantly (Figure 5; Table S2). Both the number of OTUs involved in the network and the number of correlations decreased, as did the diameter, average degree and average path length. The power law distributions of networks in the shallow sediments (1–2 and 6–10 cm) conformed to scale‐free network topology, with a power‐law exponent γ approaching 2 (1.840 and 1.955, respectively), while networks of deep sediments (30–90 and 190–790 cm) less resembled the scale‐free networks (γ = 1.381 and 1.316, respectively) (Figure S3). However, although there were fewer nodes in the network at 30–90 cm compared with 6–10 cm, the edge number increased drastically at 30–90 cm compared with other depths. Nodes with a high degree in the 30–90 cm network were mainly Chloroflexi, with 4,474 edges in total connected to this phylum. Rare OTUs accounted for the majority of nodes (>60%) in the networks of the 1–2, 6–10 and 30–90 cm networks, and decreased to 28% in the 190–790 cm network (Figure S4), whereas the proportions of rare OTUs as keystone nodes were higher in layers other than 1–2 cm. Keystone OTUs identified at 1–2, 6–10 and 30–90 cm mainly belonged to Proteobacteria, Chloroflexi, and Planctomycetes, while keystone OTUs in the 190–790 cm layer were primarily Bacteroidetes and Firmicutes.
FIGURE 5.
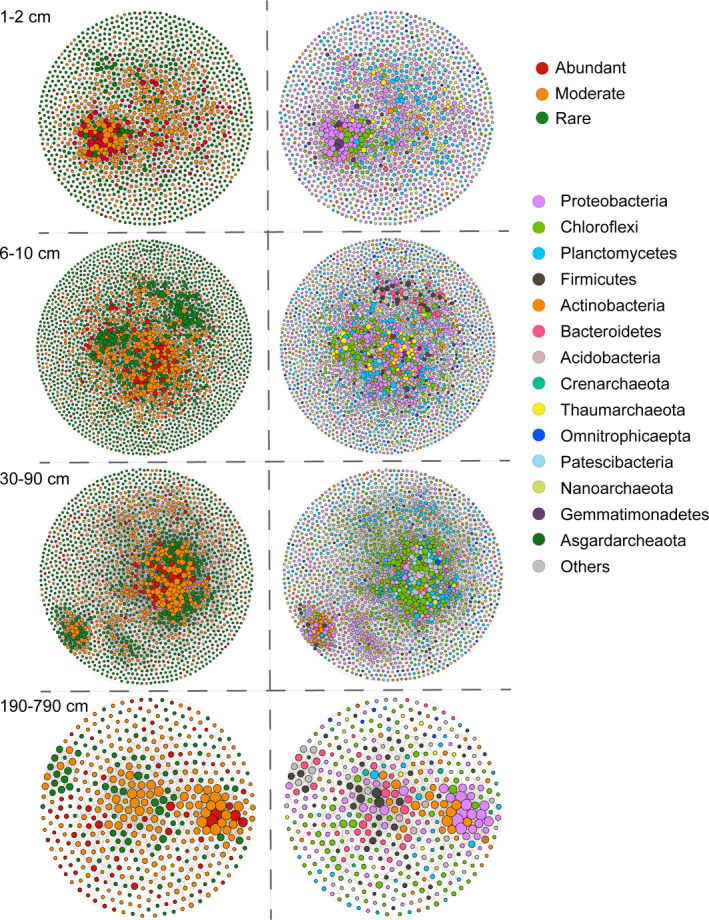
Co‐occurrence networks of the microbial community in each depth group. Each connection shown has a correlation coefficient > |.7| and a p value <.01. The size of each node is proportional to the number of connections. The OTUs were coloured by subcommunity and taxonomy, respectively
3.7. The influence of environmental factor on community variation
Environmental factors with significant influence on sediment communities as revealed by db‐RDA (p < .05) are shown in Figure 1e. DIC, NH4 +, PO4 3− and SiO4 2− significantly correlated with microbial community structure at 190–790 cm, while and were correlated with samples in the top 10 cm. Samples from 30–90 cm appeared to be separated from other samples by TOC. The Mantel test results indicated that changes of total, abundant, moderate and rare communities were correlated with DIC, , , , , , , Cl−, TN and C/N (Table S4). In addition, DIC, , and TN showed stronger correlations with rare OTUs than abundant OTUs, while and conversely showed stronger correlations with abundant OTUs than rare OTUs (Table S4). The α‐diversity of the entire community was significantly influenced by DIC, , , , , Ca2+, TOC, TN, C/N, and δ13C.
4. DISCUSSION
Knowledge of microbial community composition, diversity and association patterns is important for a better understanding of how environmental variables and biogeographical processes influence subsurface biospheres in marine sediments. We found that microbes in deeper sediment layers exhibited a distinct β‐diversity partitioning and interaction pattern compared with those in shallower sediments. Selection was the major process determining the community assembly in sediments, while other processes, such as diversification and dispersal from terrestrial inputs, may exert impacts on the rare subcommunity. In addition, we provided evidence that the rare subcommunity played a non‐negligible role in shaping microbial diversity and interactions in marine sediments.
4.1. Microbial communities shaped by geochemical zonation and palaeoenvironmental factors in the SCS sediments
SCS is one of the largest marginal seas in the world and is abundant in biotic resources. The microbial cell abundance in surface sediment of the northern slope of the SCS has been reported to vary between 106 and 109 cells g–1 sediment (Cui et al., 2019; Jiang et al., 2007; Jiao et al., 2015). The bacterial and archaeal 16S rRNA gene abundance in surface sediments of this study was 107–109 and 106–108 copies g–1, respectively, which was one to two orders of magnitude lower compared with those in sites located closer to the Pearl River Estuary (Dang et al., 2013) with high OM from terrestrial inputs. Due to fierce selection processes in the subseafloor sediment, bacterial and archaeal abundance decreased logarithmically with depth. The α‐diversity also showed a decreasing trend from 10 to 690 cm, in accordance with previously reported distribution patterns of microbial abundance and diversity (D’Hondt et al., 2019; Kallmeyer et al., 2012).
Successive community shifts were observed with increasing sediment depth, consistent with redox zones characterized by aerobic respiration, denitrification, reduction of oxidized manganese and sulphate (Figure 1b). was rapidly depleted by abundant ammonia‐oxidizing Thaumarchaeota in the top 10 cm (Vuillemin et al., 2019), and re‐accumulated in deeper layers. The ammonia‐oxidizing process led to high concentrations of and in shallow sediments, which might support denitrification in a wide diversity of bacteria (especially Proteobacteria) (Devol, 2015). The increased Mn2+ at 30 cm indicated the activity of Mn‐oxidizing communities, possibly accompanied by Fe oxidizers. Concentrations of started to decrease below 190 cm, due to increased sulphate‐reducing bacteria including Desulfuromonadales, Desulfovibrionales, Desulfobacterales and Desulfarculales in Deltaproteobacteria. However, these sulphate‐reducing bacteria only accounted for a small proportion in Deltaproteobacteria, outcompeted by fermentative members of Chloroflexi (Dehalococcoides), Atribacteria and Aerophobetes (Kerrigan et al., 2020; Vuillemin, Vargas, et al., 2020; Wang & Li, 2016). Fermentation of organic acid was also achieved by several archaeal groups enriched in deep sediments, including Lokiarchaeota (Orsi et al., 2020) and Bathyarchaeota (He et al., 2016). The oxidation of OM through sulphate reduction and fermentation was considered the main mechanism contributing to DIC production in deep sediments (Valdemarsen & Kristensen, 2010). Methanotrophic archaea (ANME) were not found in these samples, indicating that the sulphate–methane transition zone (SMTZ) and methanogenic zone in SCS sediments were much deeper (Graw et al., 2018).
Although the vertical changes of microbial communities corresponded with the succession of predominant terminal electron‐accepting pathways with sediment depth, terrestrial inputs also play a role in shaping microbial communities, as evidenced by TOC, C/N and δ13C. Surrounded by the Asian continent and Taiwan to the north (Figure 1a), 80% of the total SCS surface sediments were composed of river‐borne terrigenous sediments, especially in the northern slope (Liu et al., 2008). The sedimentation rate in the study area reached 0.77–8.12 cm ka–1 as reported by Schönfeld and Kudrass (1993) and 2–12 cm ka–1 by Shao et al. (2001), forming organic‐rich sediment with TOC remaining at a relatively high level (around 1% except for 50 cm) with depth. There was a sudden increase in TOC and change in δ13C percentage at 50 cm in most sites, reflecting terrestrial inputs affected by historical climate oscillations such as the glacial and interglacial cycles (Lai et al., 2020; Zhao, Liu, et al., 2017). Distinct TOC, C/N and δ13C profiles of SCS005 were observed at 50–490 cm and this may explain the peculiar microbial community compositions in deep sediments of SCS005 featured by large proportions of Actinobacteria, which were considered to be dispersed from different sources of terrestrial input compared with other sites.
Therefore, the microbial community in SCS sediment has resulted from a combination of palaeoenvironmental factors and substrate depletion with depth (Figure 1e). However, few studies have differentiated abundant and rare taxa from the total microbial community when assessing its response to environmental factors. We found that although abundant, moderate and rare subcommunities were all significantly correlated with the measured factors (Table S4), the correlation coefficients varied between certain factors and the subcommunities. These results indicated that the abundant, moderate and rare subcommunities may be differentially influenced by environmental factors.
4.2. β‐Diversity patterns indicate distinct processes shaping abundant and rare subcommunities in sediments
Microbial communities in sediments are considered to derive from selection from within the surface communities, as well as dispersal of microorganisms in seawater and the terrestrial environment. To explore the detailed succession of abundant and rare microorganisms with sediment depth and the processes behind these changes, we partitioned the β‐diversity into richness and replacement components. This can help to distinguish taxonomic changes that result from loss of surface species during burial processes and microbial diversification, and that from selection of surface populations. In a simplified, competing structural model proposed by Wurzbacher et al. (2017), the richness effect represents a community exclusively consisting of species filtered from the surface taxa, resulting in the transition from a complex and rich community at the surface to an increasingly depauperate community in deeper sediments. The replacement effect instead resembles a structured community of niche specialists at various layers that are well adapted to the specific environmental conditions. Our results showed clear changes of these distinct kinds of effects in marine sediments. For the abundant subcommunity, the richness effect consistently accounted for a high proportion of β‐diversity, indicating that the abundant species in deep sediment are a subset of surface populations primarily resulted from the process of selection. This was consistent with previous reports that selection is the major process determining the assembly of abundant communities (Starnawski et al., 2017). However, the observation of a decreasing trend in the richness effect in deepest layers (690 and 790 cm) for both abundant and moderate subcommunities indicated that the filtering effect might eventually be weakened as sediment depth increased. For the rare subcommunity, by contrast, the replacement effect showed overwhelming dominance below 190 cm, indicating loss of surface taxa and existence of novel species compared with those in the surface. Rare microorganisms are susceptible to local elimination through environmental or biological processes that select for the survival of abundant organisms (Pedrós‐Alió, 2012). During burial, many rare species tended to be filtered out due to their low abundance, resulting in the high replacement effect when compared with the surface. Meanwhile, distinct rare communities derived from terrestrial input and persisting in deep sediments for a long time as inactive seed banks might also be a possible explanation for the elevated replacement effect. Other processes, such as diversification, could occasionally occur in deep sediments, but may not be the dominant determinant for species replacement.
The β‐partitioning patterns above 30 cm in SCS sediments were highly similar to those reported by Wurzbacher et al. (2017) in lake sediment, featuring a consistently high replacement effect and increased richness effect. However, in contrast to their assumption, our results from deeper sediments revealed that the richness component did not show a further increase in a linear manner. In fact, the richness effect was abated below 30 cm and replacement took over in deeper layers, and such changes were predominantly determined by the rare subcommunity. Given the different environmental conditions such as salinity and sulphate concentrations, as well as sedimentation process in marine and freshwater sediments, whether this changing pattern of β‐diversity components can be applied to distinct sediment types is yet to be proven.
Although these SCS samples were collected on a small regional scale, site‐to‐site community variations were observed. As indicated by the increased horizontal β‐diversity, microbial communities at the same depth exhibited greater differences in deep sediments than in shallow sediments, in line with the results shown between two coastal habitats of the Mediterranean Sea (Luna et al., 2013). Despite this moderate increase in total horizontal β‐diversity with depth, the underlying changes in the two β‐diversity components were more pronounced and showed opposing trends, especially for the rare subcommunity. The replacement effect was stronger than the richness effect especially in the shallow sediments, reflecting species loss and gain by stochastic processes such as dispersal from seawater columns and distinct terrestrial inputs. However, during the burial processes over time, these communities have been confined to specific populations that can survive in the deep seafloor, resulting in an increasing contribution of the richness effect to horizontal β‐diversity. These results are the first to reveal the detailed contributions of spatial turnover (replacement) and nestedness (richness) underlying the total amount of β‐diversity at certain depths of marine sediments.
4.3. Co‐occurrence patterns indicated reduced microbial interactions in the deep seafloor and the importance of the rare subcommunity
Co‐occurrence networks can potentially provide novel and unique perspectives on microbial interactions and ecological assembly rules beyond those of diversity and composition. The correlation‐based network was divided into three major modules with species occupying either shallower (oxic) or deeper (anoxic) sediments, which clearly reflected niche differentiation related to sedimentary redox environment. Each module comprised a group of diverse microbial taxa, with large numbers of Proteobacteria, Chloroflexi and Planctomycetes in Modules 1, 2 and 3, respectively. A similar network pattern has been reported for the South Yellow Sea (SYS) and East China Sea (ECS) sediment samples from the top 30 cm (Qiao et al., 2018), in which a separation of microbes from shallower and deeper sediments into two modules was observed. Here, we created a co‐occurrence network involving deeper subseafloor samples and more OTUs (especially rare OTUs), which may represent a general model for microbial co‐occurrence patterns in marine sediments.
The topological features of the network revealed that the abundant OTUs had more connections with other OTUs and were more frequently located in central positions of the network compared with rare OTUs located relatively distantly (Ma et al., 2016), emphasizing the vital role of abundant OTUs in microbial networks (Zhang, Hou, et al., 2020). However, rare OTUs accounted for a considerable proportion of nodes (45.4%) and keystone OTUs (69.5%), indicating that rare OTUs constituted an indispensable part in the microbial network. Previous network analysis using selected OTUs based on relative abundance or read number to reduce complexity might have neglected the impacts of these rare species (Buongiorno et al., 2019; Liu et al., 2014, 2020). A higher clustering coefficient of rare OTUs indicated that they were more likely to present in an interconnected “small world,” emphasizing that rare OTUs contributed to the complexity of networks (Masuda et al., 2018). Rare OTUs with high degree were primarily found in Module 1, suggesting that rare OTUs might be more important in supporting community function and stability in shallow sediments than in deep sediments. Among them, OTUs belonging to Acidobacteria and Phycisphaerae in Planctomycetes are involved in the decomposition of various biopolymers (Hoshino et al., 2020; Quaiser et al., 2008), and some of these OTUs were identified as keystone OTUs (Table S3). These rare keystone taxa were considered to exert a pronounced influence by participating in specific single steps of broader processes such as OM decomposition (Banerjee et al., 2018). However, the function and metabolic features of most of these keystone OTUs remain largely unknown, such as the uncultured NB1‐j in Deltaproteobacteria. Their potential roles in sediment microbial networks have yet to be revealed. It was noticeable that the roles of rare OTUs should be interpreted with caution, as they might represent dead or dormant cells and extracellular DNA (cysts, spores and inactive seed banks) (Vuillemin et al., 2017), especially those belonging to Bacteroidetes, Firmicutes, Actinobacteria and Planctomycetes. They might have no functional roles in the network but only coexist with other species (Lennon & Jones, 2011), and their co‐occurrence reflected distal inflows from the Pearl River down to the continental slope. Despite this, the importance of rare taxa has been emphasized in the networks of various ecosystems including soil, rhizosphere and reservoir systems (Lupatini et al., 2014; Shi et al., 2016; Xue et al., 2018), and here we provide evidence suggesting a high proportion of rare species in sediment microbial networks and some of them may be involved in OM decomposition.
While the network of samples from all depths largely reflects shared niche preferences due to geochemical zonation in sediment, networks of separate depth groups may better represent microbial interactions at specific depths. Networks with reduced complexity in deep sediment implied that microbes in the deep subseafloor may interact less with each other, and this might be due to (i) the reduction of microbial abundance and the loss of many species compared with the surface environment; and (ii) the slowed growth rate and metabolism of these microorganisms with some of them entering the dormancy state to maintain survival (Jorgensen & Marshall, 2016). For instance, although Dehalococcoidia were highly enriched in deep sediment, the correlations involving Dehalococcoidia in deep sediments were less than those in the surface (189 and 238 edges, respectively). In the surface, Dehalococcoidia had more correlations with other microbial clades, such as members of the Gammaproteobacteria, Gemmatimonadetes and Phycisphaerae, and such corrections across different clades decreased in deep sediments. However, associations between Dehalococcoidia and sulfate‐reducing bacteria in Deltaproteobacteria were identified in deep sediments, suggesting that sulphate‐reducing activity may enhance dechlorination by Dehalococcoidia (May et al., 2008). Other studies have suggested that carbon inputs and shifts in carbon resource increased network complexity (Farrer et al., 2019; Shi et al., 2016), which could possibly explain the drastic increase of interactions in the network of 30–90 cm, as increased TOC and distinct δ13C was detected at ~50 cm. These results indicated that terrestrial inputs may also influence the structure of microbial co‐occurrence networks.
5. CONCLUSIONS
Through a comprehensive investigation of the microbial communities in surface and subseafloor SCS sediments, we showed distinct patterns of β‐diversity partitioning and co‐occurrence along sediment depth down to ~8 m. Generally, selection was the dominant processes determining the β‐diversity partitioning patterns. The high replacement effect between deep sediment and the surface layers may suggest loss of rare species during burial as well as dispersal of species with terrestrial inputs. Co‐occurrence network analysis further showed that microbes in the deep subseafloor tended to survive independently with less syntrophic relationships, due to the reduced abundance and metabolic activity. However, rare species accounted for a large proportion in the network, with several keystone taxa that may involve in OM decomposition. Our results expand current knowledge on the ecological significance of rare species related to microbial diversity and interactions in marine sediments.
AUTHOR CONTRIBUTIONS
X.‐H.Z., J.L. and Y.Z. conceived the study. Y.Z. and J.L. analysed the data and wrote the manuscript. P.Y. analysed the environmental factors. C.S. performed DNA extraction and qPCR experiments. X.S. performed sampling and S.L. provided the opportunity for sampling. All authors edited and approved the final manuscript.
ETHICAL APPROVAL
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Fundamental Research Funds for the Central Universities (202172002, 202072003), the Programs of China Geological Survey (DD20160221), the National Natural Science Foundation of China (91751202, 41730530, 41906099, 92051115 and 41976101) and China Postdoctoral Science Foundation (2019M652469). We thank all the scientists and crews for their assistance with sampling during the cruise organized by Guangzhou Marine Geological Survey. Sincere thanks go to Jianbin Yang, Xiao Dong and Bin Zhao for their technical assistance. We also thank Delei Song and Xiaoyu Zhu for help with sampling and Shuxian Yu with data analysis.
DATA AVAILABILITY STATEMENT
The raw sequencing data of 16S rDNA genes were deposited in the Sequence Read Archive database at NCBI under accession nos.: SRR11088488–SRR11088621 (PRJNA606622). The OTU table and representative sequences, as well as the R scripts used are provided in the Supporting information.
REFERENCES
- Apprill, A. , Mcnally, S. , Parsons, R. , & Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microbial Ecology, 75(2), 129–137. 10.3354/ame01753 [DOI] [Google Scholar]
- Banerjee, S. , Schlaeppi, K. , & van der Heijden, M. G. A. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nature Reviews Microbiology, 16(9), 567–576. 10.1038/s41579-018-0024-1 [DOI] [PubMed] [Google Scholar]
- Baselga, A. (2010). Partitioning the turnover and nestedness components of beta diversity. Global Ecology and Biogeography, 19(1), 134–143. 10.1111/j.1466-8238.2009.00490.x [DOI] [Google Scholar]
- Bay, S. K. , McGeoch, M. A. , Gillor, O. , Wieler, N. , Palmer, D. J. , Baker, D. J. , Chown, S. L. , & Greening, C. (2020). Soil bacterial communities exhibit strong biogeographic patterns at fine taxonomic resolution. mSystems, 5(4), 1–16. 10.1128/msystems.00540-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner, R. A. (1982). Burial of organic carbon and pyrite sulfur in the modern ocean. American Journal of Science, 282, 451–473. 10.2475/ajs.282.4.451 [DOI] [Google Scholar]
- Berry, D. , & Widder, S. (2014). Deciphering microbial interactions and detecting keystone species with co‐occurrence networks. Frontiers in Microbiology, 5, 1–14. 10.3389/fmicb.2014.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelier, P. L. E. , Meima‐Franke, M. , Hordijk, C. A. , Steenbergh, A. K. , Hefting, M. M. , Bodrossy, L. , von Bergen, M. , & Seifert, J. (2013). Microbial minorities modulate methane consumption through niche partitioning. ISME Journal, 7(11), 2214–2228. 10.1038/ismej.2013.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böer, S. I. , Hedtkamp, S. I. C. , Van Beusekom, J. E. E. , Fuhrman, J. A. , Boetius, A. , & Ramette, A. (2009). Time‐ and sediment depth‐related variations in bacterial diversity and community structure in subtidal sands. ISME Journal, 3(7), 780–791. 10.1038/ismej.2009.29 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buongiorno, J. , Herbert, L. C. , Wehrmann, L. M. , Michaud, A. B. , Laufer, K. , Røy, H. , Jørgensen, B. B. , Szynkiewicz, A. , Faiia, A. , Yeager, K. M. , Schindler, K. , & Lloyd, K. G. (2019). Complex microbial communities drive iron and sulfur cycling in Arctic Fjord sediments. Applied and Environmental Microbiology, 85(14), 1–16. 10.1128/AEM.00949-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadillo‐Quiroz, H. , Bräuer, S. , Yashiro, E. , Sun, C. , Yavitt, J. , & Zinder, S. (2006). Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environmental Microbiology, 8(8), 1428–1440. 10.1111/j.1462-2920.2006.01036.x [DOI] [PubMed] [Google Scholar]
- Cai, W. J. , & Wang, Y. (1998). The chemistry, fluxes, and sources of carbon dioxide in the estuarine waters of the Satilla and Altamaha Rivers. Georgia. Limnology and Oceanography, 43(4), 657–668. 10.4319/lo.1998.43.4.0657 [DOI] [Google Scholar]
- Cardoso, P. , Rigal, F. , & Carvalho, J. C. (2015). BAT ‐ Biodiversity Assessment Tools, an R package for the measurement and estimation of alpha and beta taxon, phylogenetic and functional diversity. Methods in Ecology and Evolution, 6(2), 232–236. 10.1111/2041-210X.12310 [DOI] [Google Scholar]
- Catalano, G. (1987). An improved method for the determination of ammonia in seawater. Marine Chemistry, 20(3), 289–295. 10.1016/0304-4203(87)90079-X [DOI] [Google Scholar]
- Cui, H. , Su, X. , Chen, F. , Holland, M. , Yang, S. , Liang, J. , Su, P. , Dong, H. , & Hou, W. (2019). Microbial diversity of two cold seep systems in gas hydrate‐bearing sediments in the South China Sea. Marine Environmental Research, 144, 230–239. 10.1016/j.marenvres.2019.01.009 [DOI] [PubMed] [Google Scholar]
- D’Hondt, S. , Jørgensen, B. B. , Miller, D. J. , Batzke, A. , Blake, R. , Cragg, B. A. , & Acosta, J. L. S. (2004). Distributions of microbial activities in deep subseafloor sediments. Science, 306(5705), 2216–2221. 10.1126/science.1101155 [DOI] [PubMed] [Google Scholar]
- D’Hondt, S. , Pockalny, R. , Fulfer, V. M. , & Spivack, A. J. (2019). Subseafloor life and its biogeochemical impacts. Nature Communications, 10(1), 1–13. 10.1038/s41467-019-11450-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, H. , Yang, J. , Li, J. , Luan, X. , Zhang, Y. , Gu, G. , Xue, R. , Zong, M. & Klotz, M. G. (2013). Environment‐dependent distribution of the sediment nifH‐harboring microbiota in the northern South China Sea. Applied and Environmental Microbiology, 79(1), 121–132. 10.1128/AEM.01889-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devol, A. H. (2015). Denitrification, anammox, and N2 production in marine sediments. Annual Review of Marine Science, 7, 403–423. 10.1146/annurev-marine-010213-135040 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10, 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Farrer, E. C. , Porazinska, D. L. , Spasojevic, M. J. , King, A. J. , Bueno de Mesquita, C. P. , Sartwell, S. A. , Smith, J. G. , White, C. T. , Schmidt, S. K. , & Suding, K. N. (2019). Soil microbial networks shift across a high‐elevation successional gradient. Frontiers in Microbiology, 10, 1–13. 10.3389/fmicb.2019.02887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter, F. A. , Manhart, M. , & Ackermann, M. (2020). Understanding the evolution of interspecies interactions in microbial communities. Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1798), 20190256. 10.1098/rstb.2019.0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graw, M. F. , D'Angelo, G. , Borchers, M. , Thurber, A. R. , Johnson, J. E. , Zhang, C. , Liu, H. , & Colwell, F. S. (2018). Energy gradients structure microbial communities across sediment horizons in deep marine sediments of the South China Sea. Frontiers in Microbiology, 9, 1–12. 10.3389/fmicb.2018.00729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y. , Li, M. , Perumal, V. , Feng, X. , Fang, J. , Xie, J. , Sievert, S. M. , & Wang, F. (2016). Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nature Microbiology, 1, 16035. 10.1038/nmicrobiol.2016.35 [DOI] [PubMed] [Google Scholar]
- Hedges, J. I. , & Keil, R. G. (1995). Sedimentary organic matter preservation: an assessment and speculative synthesis. Marine Chemistry, 49(2–3), 81–115. 10.1016/0304-4203(95)00008-F [DOI] [Google Scholar]
- Hoshino, T. , Doi, H. , Uramoto, G.‐I. , Wörmer, L. , Adhikari, R. R. , Xiao, N. , Morono, Y. , D’Hondt, S. , Hinrichs, K.‐U. , & Inagaki, F. (2020). Global diversity of microbial communities in marine sediment. Proceedings of the National Academy of Sciences of the United States of America, 117(44), 27587–27597. 10.1073/pnas.1919139117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, T. , & Inagaki, F. (2019). Abundance and distribution of Archaea in the subseafloor sedimentary biosphere. ISME Journal, 13, 227–231. 10.1038/s41396-018-0253-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G. , Wilson, M. C. , Wu, J. , Yu, J. , & Yu, M. (2019). Decoupling species richness variation and spatial turnover in beta diversity across a fragmented landscape. PeerJ, 7, e6714. 10.7717/peerj.6714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki, F. , Hinrichs, K. U. , Kubo, Y. , Bowles, M. W. , Heuer, V. B. , Hong, W. L. , Hoshino, T. , Ijiri, A. , Imachi, H. , Ito, M. , Kaneko, M. , Lever, M. A. , Lin, Y. S. , Methé, B. A. , Morita, S. , Morono, Y. , Tanikawa, W. , Bihan, M. , Bowden, S. A. … Yamada, Y. (2015). Exploring deep microbial life in coal‐bearing sediment down to ∼2.5 km below the ocean floor. Science, 349(6246), 420–424. 10.1126/science.aaa6882 [DOI] [PubMed] [Google Scholar]
- Inagaki, F. , Suzuki, M. , Takai, K. , Oida, H. , Sakamoto, T. , Aoki, K. , & Horikoshi, K. (2003). Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Applied and Environmental Microbiology, 69(12), 7224–7235. 10.1128/AEM.69.12.7224-7235.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. , Dong, H. , Ji, S. , Ye, Y. , & Wu, N. (2007). Microbila diversity in the deep marine sediment from the Qiongdongnan Basin in China Sea. Geomicrobiology Journal, 24(6), 505–517. 10.1080/01490450701572473 [DOI] [Google Scholar]
- Jiao, L. , Su, X. , Wang, Y. , Jiang, H. , Zhang, Y. , & Chen, F. (2015). Microbial diversity in the hydrate‐containing and ‐free surface sediments in the Shenhu area. South China Sea. Geoscience Frontiers, 6(4), 627–633. 10.1016/j.gsf.2014.04.007 [DOI] [Google Scholar]
- Jiao, S. , & Lu, Y. (2020). Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environmental Microbiology, 22(3), 1052–1065. 10.1111/1462-2920.14815 [DOI] [PubMed] [Google Scholar]
- Jorgensen, B. B. , & Marshall, I. P. G. (2016). Slow microbial life in the seabed. Annual Review of Marine Science, 8, 311–332. 10.1146/annurev-marine-010814-015535 [DOI] [PubMed] [Google Scholar]
- Jousset, A. , Bienhold, C. , Chatzinotas, A. , Gallien, L. , Gobet, A. , Kurm, V. , Küsel, K. , Rillig, M. C. , Rivett, D. W. , Salles, J. F. , van der Heijden, M. G. A. , Youssef, N. H. , Zhang, X. , Wei, Z. , & Hol, W. H. G. (2017). Where less may be more: How the rare biosphere pulls ecosystems strings. ISME Journal, 11(4), 853–862. 10.1038/ismej.2016.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallmeyer, J. , Pockalny, R. , Adhikari, R. R. , Smith, D. C. , & D’Hondt, S. (2012). Global distribution of microbial abundance and biomass in subseafloor sediment. Proceedings of the National Academy of Sciences of the United States of America, 109, 16213–16216. 10.1073/pnas.1203849109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, D. , Hedlund, B. P. , Xie, W. , Liu, J. , Phelps, T. J. , Friedrich, M. W. , & Webster, G. (2020). Impact of terrestrial input on seep‐sea benthic archaeal community structure in South China Sea sediments. Frontiers in Microbiology, 11, 572017. 10.3389/fmicb.2020.572017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre, P. (2014). Interpreting the replacement and richness difference components of beta diversity. Global Ecology and Biogeography, 23(11), 1324–1334. 10.1111/geb.12207 [DOI] [Google Scholar]
- Lennon, J. T. , & Jones, S. E. (2011). Microbial seed banks: The ecological and evolutionary implications of dormancy. Nature Reviews Microbiology, 9(2), 119–130. 10.1038/nrmicro2504 [DOI] [PubMed] [Google Scholar]
- Lever, M. A. , Rogers, K. L. , Lloyd, K. G. , Overmann, J. , Schink, B. , Thauer, R. K. , Hoehler, T. M. , & Jørgensen, B. B. (2015). Life under extreme energy limitation: A synthesis of laboratory‐ and field‐based investigations. FEMS Microbiology Reviews, 39(5), 688–728. 10.1093/femsre/fuv020 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Meng, Z. , Liu, X. , & Zhang, X.‐H. (2019). Microbial assembly, interaction, functioning, activity and diversification: a review derived from community compositional data. Marine Life Science & Technology, 1(1), 112–128. 10.1007/s42995-019-00004-3 [DOI] [Google Scholar]
- Liu, J. , Yang, H. , Zhao, M. , & Zhang, X. H. (2014). Spatial distribution patterns of benthic microbial communities along the Pearl Estuary. China. Systematic and Applied Microbiology, 37(8), 578–589. 10.1016/j.syapm.2014.10.005 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Zhu, S. , Liu, X. , Yao, P. , Ge, T. , & Zhang, X.‐H. (2020). Spatiotemporal dynamics of the archaeal community in coastal sediments: assembly process and co‐occurrence relationship. The ISME Journal, 14(6), 1463–1478. 10.1038/s41396-020-0621-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Tuo, S. , Colin, C. , Liu, J. T. , Huang, C.‐Y. , Selvaraj, K. , Chen, C.‐T. , Zhao, Y. , Siringan, F. P. , Boulay, S. , & Chen, Z. (2008). Detrital fine‐grained sediment contribution from Taiwan to the northern South China Sea and its relation to regional ocean circulation. Marine Geology, 255(3–4), 149–155. 10.1016/j.margeo.2008.08.003 [DOI] [Google Scholar]
- Logares, R. , Audic, S. , Bass, D. , Bittner, L. , Boutte, C. , Christen, R. , Claverie, J.‐M. , Decelle, J. , Dolan, J. R. , Dunthorn, M. , Edvardsen, B. , Gobet, A. , Kooistra, W. H. C. F. , Mahé, F. , Not, F. , Ogata, H. , Pawlowski, J. , Pernice, M. C. , Romac, S. , … Massana, R. (2014). Patterns of rare and abundant marine microbial eukaryotes. Current Biology, 24(8), 813–821. 10.1016/j.cub.2014.02.050 [DOI] [PubMed] [Google Scholar]
- Luna, G. M. , Corinaldesi, C. , Rastelli, E. , & Danovaro, R. (2013). Patterns and drivers of bacterial α‐ and β‐diversity across vertical profiles from surface to subsurface sediments. Environmental Microbiology Reports, 5(5), 731–739. 10.1111/1758-2229.12075 [DOI] [PubMed] [Google Scholar]
- Lupatini, M. , Suleiman, A. K. A. , Jacques, R. J. S. , Antoniolli, Z. I. , de Siqueira Ferreira, A. , Kuramae, E. E. , & Roesch, L. F. W. (2014). Network topology reveals high connectance levels and few key microbial genera within soils. Frontiers in Environmental Science, 2, 1–11. 10.3389/fenvs.2014.00010 [DOI] [Google Scholar]
- Lv, X. , Zhao, K. , Xue, R. , Liu, Y. , Xu, J. , & Ma, B. (2019). Strengthening insights in microbial ecological networks from theory to applications. Msystems, 4(3), 4–8. 10.1128/msystems.00124-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. D. J. , & Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nature Reviews Microbiology, 13(4), 217–229. 10.1038/nrmicro3400 [DOI] [PubMed] [Google Scholar]
- Ma, B. , Wang, H. , Dsouza, M. , Lou, J. , He, Y. , Dai, Z. , Brookes, P. C. , Xu, J. , & Gilbert, J. A. (2016). Geographic patterns of co‐occurrence network topological features for soil microbiota at continental scale in eastern China. ISME Journal, 10(8), 1891–1901. 10.1038/ismej.2015.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macalady, J. L. , Hamilton, T. L. , Grettenberger, C. L. , Jones, D. S. , Tsao, L. E. , & Burgos, W. D. (2013). Energy, ecology and the distribution of microbial life. Philosophical Transactions of the Royal Society B: Biological Sciences, 368(1622), 20120383. 10.1098/rstb.2012.0383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny, J. B. H. , Bohannan, B. J. M. , Brown, J. H. , Colwell, R. K. , Fuhrman, J. A. , Green, J. L. , Horner‐Devine, M. C. , Kane, M. , Krumins, J. A. , Kuske, C. R. , Morin, P. J. , Naeem, S. , Øvreås, L. , Reysenbach, A.‐L. , Smith, V. H. , & Staley, J. T. (2006). Microbial biogeography: Putting microorganisms on the map. Nature Reviews Microbiology, 4(2), 102–112. 10.1038/nrmicro1341 [DOI] [PubMed] [Google Scholar]
- Mason, O. U. , Di Meo‐Savoie, C. A. , Van Nostrand, J. D. , Zhou, J. , Fisk, M. R. , & Giovannoni, S. J. (2009). Prokaryotic diversity, distribution, and insights into their role in biogeochemical cycling in marine basalts. ISME Journal, 3(2), 231–242. 10.1038/ismej.2008.92 [DOI] [PubMed] [Google Scholar]
- Masuda, N. , Sakaki, M. , Ezaki, T. , & Watanabe, T. (2018). Clustering coefficients for correlation networks. Frontiers in Neuroinformatics, 12, 7. 10.3389/fninf.2018.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, H. D. , Miller, G. S. , Kjellerup, B. V. , & Sowers, K. R. (2008). Dehalorespiration with polychlorinated biphenyls by an anaerobic ultramicrobacterium. Applied and Environmental Microbiology, 74(7), 2089–2094. 10.1128/AEM.01450-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo, Y. , Zhang, W. , Yang, J. , Lin, Y. , Yu, Z. , & Lin, S. (2018). Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME Journal, 12, 2198–2210. 10.1038/s41396-018-0153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, A. S. , Isbell, F. , & Seidl, R. (2018). β‐Diversity, community assembly, and ecosystem functioning. Trends in Ecology and Evolution, 33(7), 549–564. 10.1016/j.tree.2018.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealson, K. H. (1997). Sediment bacteria: Who’s there, what are they doing, and what’s new? Annual Review of Earth and Planetary Sciences, 25, 403–434. 10.1146/annurev.earth.25.1.403 [DOI] [PubMed] [Google Scholar]
- Nemergut, D. R. , Schmidt, S. K. , Fukami, T. , O'Neill, S. P. , Bilinski, T. M. , Stanish, L. F. , Knelman, J. E. , Darcy, J. L. , Lynch, R. C. , Wickey, P. , & Ferrenberg, S. (2013). Patterns and processes of microbial community assembly. Microbiology and Molecular Biology Reviews, 77(3), 342–356. 10.1128/mmbr.00051-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoura, T. , Nishizawa, M. , Hirai, M. , Shimamura, S. , Harnvoravongchai, P. , Koide, O. , Morono, Y. , Fukui, T. , Inagaki, F. , Miyazaki, J. , Takaki, Y. , & Takai, K. (2018). Microbial diversity in sediments from the bottom of the challenger deep, the mariana trench. Microbes and Environments, 33(2), 186–194. 10.1264/jsme2.ME17194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoura, T. , Takaki, Y. , Shimamura, S. , Kakuta, J. , Kazama, H. , Hirai, M. , Masui, N. , Tomaru, H. , Morono, Y. , Imachi, H. , Inagaki, F. , & Takai, K. (2016). Variance and potential niche separation of microbial communities in subseafloor sediments off Shimokita Peninsula. Japan. Environmental Microbiology, 18(6), 1889–1906. 10.1111/1462-2920.13096 [DOI] [PubMed] [Google Scholar]
- Orcutt, B. N. , Sylvan, J. B. , Knab, N. J. , & Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiology and Molecular Biology Reviews, 75, 361–422. 10.1128/mmbr.00039-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi, W. D. , Vuillemin, A. , Rodriguez, P. , Coskun, Ö. K. , Gomez‐Saez, G. V. , Lavik, G. , Mohrholz, V. , & Ferdelman, T. G. (2020). Metabolic activity analyses demonstrate that Lokiarchaeon exhibits homoacetogenesis in sulfidic marine sediments. Nature Microbiology, 5, 248–255. 10.1038/s41564-019-0630-3 [DOI] [PubMed] [Google Scholar]
- Parada, A. E. , Needham, D. M. , & Fuhrman, J. A. (2016). Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environmental Microbiology, 18(5), 1403–1414. 10.1111/1462-2920.13023 [DOI] [PubMed] [Google Scholar]
- Parkes, R. J. , Cragg, B. A. , & Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: A review. Hydrogeology Journal, 8(1), 11–28. 10.1007/pl00010971 [DOI] [Google Scholar]
- Pedrós‐Alió, C. (2007). Dipping into the rare biosphere. Science, 315(5809), 192–193. 10.1126/science.1135933 [DOI] [PubMed] [Google Scholar]
- Pedrós‐Alió, C. (2012). The rare bacterial biosphere. Annual Review of Marine Science, 4(1), 449–466. 10.1146/annurev-marine-120710-100948 [DOI] [PubMed] [Google Scholar]
- Petro, C. , Starnawski, P. , Schramm, A. , & Kjeldsen, K. U. (2017). Microbial community assembly in marine sediments. Aquatic Microbial Ecology, 79(3), 177–195. 10.3354/ame01826 [DOI] [Google Scholar]
- Philippot, L. , Spor, A. , Hénault, C. , Bru, D. , Bizouard, F. , Jones, C. M. , Sarr, A. , & Maron, P.‐A. (2013). Loss in microbial diversity affects nitrogen cycling in soil. ISME Journal, 7(8), 1609–1619. 10.1038/ismej.2013.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, Y. , Liu, J. , Zhao, M. , & Zhang, X. H. (2018). Sediment depth‐dependent spatial variations of bacterial communities in mud deposits of the Eastern China marginal seas. Frontiers in Microbiology, 9, 1–12. 10.3389/fmicb.2018.01128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaiser, A. , López‐garcía, P. , Zivanovic, Y. , Henn, M. R. , Rodriguez‐valera, F. , & Moreira, D. (2008). Comparative analysis of genome fragments of Acidobacteria from deep Mediterranean plankton. Environmental Microbiology, 10, 2704–2717. 10.1111/j.1462-2920.2008.01691.x [DOI] [PubMed] [Google Scholar]
- Röttjers, L. , & Faust, K. (2018). From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiology Reviews, 42(6), 761–780. 10.1093/femsre/fuy030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer, R. , Bienhold, C. , Ramette, A. , & Harder, J. (2010). Bacterial diversity and biogeography in deep‐sea surface sediments of the South Atlantic Ocean. ISME Journal, 4(2), 159–170. 10.1038/ismej.2009.106 [DOI] [PubMed] [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , Lesniewski, R. A. , Oakley, B. B. , Parks, D. H. , Robinson, C. J. , Sahl, J. W. , Stres, B. , Thallinger, G. G. , Van Horn, D. J. , & Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld, J. , & Kudrass, H. R. (1993). Hemipelagic sediment accumulation rates in the South China sea related to late quaternary sea‐level changes. Quaternary Research, 40(3), 368–379. 10.1006/qres.1993.1090 [DOI] [Google Scholar]
- Seeberg‐Elverfeldt, J. , Schlüter, M. , Feseker, T. , & Kölling, M. (2005). Rhizon sampling of porewaters near the sediment‐water interface of aquatic systems. Limnology and Oceanography: Methods, 3, 361–371. 10.4319/lom.2005.3.361 [DOI] [Google Scholar]
- Shade, A. , & Gilbert, J. A. (2015). Temporal patterns of rarity provide a more complete view of microbial diversity. Trends in Microbiology, 23(6), 335–340. 10.1016/j.tim.2015.01.007 [DOI] [PubMed] [Google Scholar]
- Shade, A. , Jones, S. E. , Gregory Caporaso, J. , Handelsman, J. , Knight, R. , Fierer, N. , & Gilbert, J. A. (2014). Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. Mbio, 5(4), 1371–1385. 10.1128/mBio.01371-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, L. , Li, X. , Wei, G. , Liu, Y. , & Fang, D. (2001). Provenance of a prominent sediment drift on the Northern slope of the South China Sea. Science in China, Series D: Earth Sciences, 44(10), 919–925. 10.1007/BF02907084 [DOI] [Google Scholar]
- Shi, S. , Nuccio, E. E. , Shi, Z. J. , He, Z. , Zhou, J. , & Firestone, M. K. (2016). The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecology Letters, 19(8), 926–936. 10.1111/ele.12630 [DOI] [PubMed] [Google Scholar]
- Si, X. , Baselga, A. , & Ding, P. (2015). Revealing beta‐diversity patterns of breeding bird and lizard communities on inundated land‐bridge islands by separating the turnover and nestedness components. PLoS One, 10, e0127692. 10.1371/journal.pone.0127692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkko, H. , Hepolehto, I. , Lyra, C. , Rinta‐Kanto, J. M. , Villnäs, A. , Norkko, J. , Norkko, A. , & Timonen, S. (2019). Increasing oxygen deficiency changes rare and moderately abundant bacterial communities in coastal soft sediments. Scientific Reports, 9(1), 1–14. 10.1038/s41598-019-51432-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin, M. L. , Morrison, H. G. , Huber, J. A. , Welch, D. M. , Huse, S. M. , Neal, P. R. , Arrieta, J. M. , & Herndl, G. J. (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proceedings of the National Academy of Sciences, 103(32), 12115–12120. 10.1073/pnas.0605127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soininen, J. , Heino, J. , & Wang, J. (2018). A meta‐analysis of nestedness and turnover components of beta diversity across organisms and ecosystems. Global Ecology and Biogeography, 27(1), 96–109. 10.1111/geb.12660 [DOI] [Google Scholar]
- Starnawski, P. , Bataillon, T. , Ettema, T. J. G. , Jochum, L. M. , Schreiber, L. , Chen, X. , Lever, M. A. , Polz, M. F. , Jørgensen, B. B. , Schramm, A. , & Kjeldsen, K. U. (2017). Microbial community assembly and evolution in subseafloor sediment. Proceedings of the National Academy of Sciences of the United States of America, 114(11), 2940–2945. 10.1073/pnas.1614190114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdemarsen, T. , & Kristensen, E. (2010). Degradation of dissolved organic monomers and short‐chain fatty acids in sandy marine sediment by fermentation and sulfate reduction. Geochimica Et Cosmochimica Acta, 74(5), 1593–1605. 10.1016/j.gca.2009.12.009 [DOI] [Google Scholar]
- Vuillemin, A. , Ariztegui, D. , Horn, F. , Kallmeyer, J. , Orsi, W. D. , Anselmetti, F. , Corbella, H. , Francus, P. , Lücke, A. , Maidana, N. I. , Ohlendorf, C. , Zolitschka, B. , Schabitz, F. & Wastegård, S. (2018). Microbial community composition along a 50 000‐year lacustrine sediment sequence. FEMS Microbiology Ecology, 94(4), 1–14. 10.1093/femsec/fiy029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Horn, F. , Alawi, M. , Henny, C. , Wagner, D. , Crowe, S. A. , & Kallmeyer, J. (2017). Preservation and significance of extracellular DNA in ferruginous sediments from Lake Towuti, Indonesia. Frontiers in Microbiology, 8, 1440. 10.3389/fmicb.2017.01440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Kerrigan, Z. , D'Hondt, S. , & Orsi, W. D. (2020). Exploring the abundance, metabolic potential and gene expression of subseafloor Chloroflexi in million‐year‐old oxic and anoxic abyssal clay. FEMS Microbiology Ecology, 96(12), fiaa223. 10.1093/femsec/fiaa223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Vargas, S. , Coskun, Ö. K. , Pockalny, R. , Murray, R. W. , Smith, D. C. , D'Hondt, S. & Orsi, W. D. (2020). Atribacteria reproducing over millions of years in the Atlantic abyssal subseafloor. Mbio, 11(5), e01937–e2020. 10.1101/2020.07.10.198200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Wankel, S. D. , Coskun, Ö. K. , Magritsch, T. , Vargas, S. , Estes, E. R. , Spivack, A. J. , Smith, D. C. , Pockalny, R. , Murray, R. W. , D’Hondt, S. , & Orsi, W. D. (2019). Archaea dominate oxic subseafloor communities over multimillion‐year time scales. Science Advances, 5(6), 1–12. 10.1126/sciadv.aaw4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, E. A. , Kirkpatrick, J. B. , Rutherford, S. D. , Smith, D. C. , Sogin, M. , & D’Hondt, S. (2016). Bacterial diversity and community composition from seasurface to subseafloor. ISME Journal, 10(4), 979–989. 10.1038/ismej.2015.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters, W. , Hyde, E. R. , Berg‐Lyons, D. , Ackermann, G. , Humphrey, G. , Parada, A. , Gilbert, J. A. , Jansson, J. K. , Caporaso, J. G. , Fuhrman, J. A. , Apprill, A. , & Knight, R. (2016). Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems, 1(1), 9–15. 10.1128/msystems.00009-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Prell, W. L. , & Blum, P. (2000). Leg 184 summary: exploring the Asian monsoon through drilling in the South China Sea. In Proc. ODP, Sci. Results. Ocean Drilling Program. doi: 10.2973/odp.proc.ir.184.2000 [DOI] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 13, 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Tao, J. , Liu, H. , Li, P. , Chen, S. , Wang, P. , & Zhang, C. (2020). Contrasting bacterial and archaeal distributions reflecting different geochemical processes in a sediment core from the Pearl River Estuary. AMB Express, 10(1), 16. 10.1186/s13568-020-0950-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Gao, Z.‐M. , Li, J.‐T. , Bougouffa, S. , Tian, R. M. , Bajic, V. B. , & Qian, P.‐Y. (2016). Draft genome of an Aerophobetes bacterium reveals a facultative lifestyle in deep‐sea anaerobic sediments. Science Bulletin, 61(15), 1176–1186. 10.1007/s11434-016-1135-6 [DOI] [Google Scholar]
- Worsfold, P. , McKelvie, I. , & Monbet, P. (2016). Determination of phosphorus in natural waters: A historical review. Analytica Chimica Acta, 918, 8–20. 10.1016/j.aca.2016.02.047 [DOI] [PubMed] [Google Scholar]
- Wu, N. , Zhang, H. , Yang, S. , Zhang, G. , Liang, J. , Lu, J. , Su, X. , Schultheiss, P. , Holland, M. , & Zhu, Y. (2011). Gas hydrate system of Shenhu Area, Northern South China Sea: Geochemical results. Journal of Geological Research, 2011, 1–10. 10.1155/2011/370298 [DOI] [Google Scholar]
- Wu, W. , Logares, R. , Huang, B. , & Hsieh, C. H. (2017). Abundant and rare picoeukaryotic sub‐communities present contrasting patterns in the epipelagic waters of marginal seas in the northwestern Pacific Ocean. Environmental Microbiology, 19, 287–300. 10.1111/1462-2920.13606 [DOI] [PubMed] [Google Scholar]
- Wurzbacher, C. , Fuchs, A. , Attermeyer, K. , Frindte, K. , Grossart, H.‐P. , Hupfer, M. , Casper, P. , & Monaghan, M. T. (2017). Shifts among eukaryota, bacteria, and archaea define the vertical organization of a lake sediment. Microbiome, 5, 41. 10.1186/S40168-017-0255-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Y. , Chen, H. , Yang, J. R. , Liu, M. , Huang, B. , & Yang, J. (2018). Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME Journal, 12(9), 2263–2277. 10.1038/s41396-018-0159-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Xiao, X. , & Zhang, Y. (2020). Microbial diversity of sediments from an inactive hydrothermal vent field, Southwest Indian Ridge. Marine Life Science & Technology, 2(1), 73–86. 10.1007/s42995-019-00007-0 [DOI] [Google Scholar]
- Yao, P. , Zhao, B. , Bianchi, T. S. , Guo, Z. , Zhao, M. , Li, D. , Pan, H. , Wang, J. , Zhang, T. , & Yu, Z. (2014). Remineralization of sedimentary organic carbon in mud deposits of the Changjiang Estuary and adjacent shelf: Implications for carbon preservation and authigenic mineral formation. Continental Shelf Research, 91, 1–11. 10.1016/j.csr.2014.08.010 [DOI] [Google Scholar]
- Yin, Q. , Fu, B. , Li, B. , Shi, X. , Inagaki, F. , & Zhang, X. H. (2013). Spatial variations in microbial community composition in surface seawater from the ultra‐oligotrophic center to Rim of the South Pacific Gyre. PLoS One, 8, e55148. 10.1371/journal.pone.0055148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Li, Y. , Xiang, S.‐Z. , Yan, Y. U. , Yang, R. , Lin, M.‐P. , Wang, X.‐M. , Xue, Y.‐L. , & Guan, X.‐Y. (2020). Sediment microbial communities and their potential role as environmental pollution indicators in Xuande Atoll, South China Sea. Frontiers in Microbiology, 11, 1–13. 10.3389/fmicb.2020.01011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Hou, F. , Xie, W. , Wang, K. , Zhou, X. , Zhang, D. , & Zhu, X. (2020). Interaction and assembly processes of abundant and rare microbial communities during a diatom bloom process. Environmental Microbiology, 22(5), 1707–1719. 10.1111/1462-2920.14820 [DOI] [PubMed] [Google Scholar]
- Zhao, B. , Yao, P. , Bianchi, T. S. , Xu, Y. , Liu, H. , Mi, T. , Zhang, X.‐H. , Liu, J. , & Yu, Z. (2017a). Early diagenesis and authigenic mineral formation in mobile muds of the Changjiang Estuary and adjacent shelf. Journal of Marine Systems, 172, 64–74. 10.1016/j.jmarsys.2017.03.001 [DOI] [Google Scholar]
- Zhao, S. H. , Liu, Z. F. , Chen, Q. , Wang, X. X. , Shi, J. N. , Jin, H. Y. , Liu, J. J. , & Jian, Z. M. (2017b). Spatiotemporal variations of deep‐sea sediment components and their fluxes since the last glaciation in the northern South China Sea. Science China Earth Sciences, 60(7), 1368–1381. 10.1007/s11430-016-9058-6 [DOI] [Google Scholar]
- Zinger, L. , Amaral‐Zettler, L. A. , Fuhrman, J. A. , Horner‐Devine, M. C. , Huse, S. M. , Welch, D. B. M. , Martiny, J. B. H. , Sogin, M. , Boetius, A. , & Ramette, A. (2011). Global patterns of bacterial beta‐diversity in seafloor and seawater ecosystems. PLoS One, 6(9), 1–11. 10.1371/journal.pone.0024570 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
The raw sequencing data of 16S rDNA genes were deposited in the Sequence Read Archive database at NCBI under accession nos.: SRR11088488–SRR11088621 (PRJNA606622). The OTU table and representative sequences, as well as the R scripts used are provided in the Supporting information.