

ABSTRACT
Paget's disease of bone (PDB) is a late‐onset chronic progressive bone disease characterized by abnormal activation of osteoclasts that results in bone pain, deformities, and fractures. PDB is very rare in Asia. A subset of PDB patients have early onset and can develop malignant giant cell tumors (GCTs) of the bone (PDB/GCTs), which arise within Paget bone lesions; the result is a significantly higher mortality rate. SQSTM1, TNFRSF11A, OPG, VCP, and HNRNPA2B1 have been identified as pathogenic genes of PDB, and ZNF687 is the only confirmed gene to date known to cause PDB/GCT. However, the molecular mechanism underlying PDB/GCT has not been fully elucidated. Here, we investigate an extended Chinese pedigree with eight individuals affected by early‐onset and polyostotic PDB, two of whom developed GCTs. We identified a heterozygous 4‐bp deletion in the Profilin 1 (PFN1) gene (c.318_321delTGAC) by genetic linkage analysis and exome sequencing for the family. Sanger sequencing revealed another heterozygous 1‐bp deletion in PFN1 (c.324_324delG) in a sporadic early‐onset PDB/GCT patient, further proving its causative role. Interestingly, a heterozygous missense mutation of PFN1 (c.335 T > C) was identified in another PDB/GCT family, revealing that not only deletion but also missense mutations in PFN1 can cause PDB/GCT. Furthermore, we established a Pfn1‐mutated mouse model (C57BL/6J mice) and successfully obtained Pagetic phenotypes in heterozygous mice, verifying loss of function of PFN1 as the cause of PDB/GCT development. In conclusion, our findings reveal mutations in PFN1 as the pathological mechanism in PDB/GCT, and we successfully established Pfn1‐mutated mice as a suitable animal model for studying PDB‐associated pathological mechanisms. The identification of PFN1 mutations has great diagnostic value for identifying PDB individuals predisposed toward developing GCTs. © 2021 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: GENETIC ANIMAL MODEL, GIANT CELL TUMOR, MUTATIONS, PAGET'S DISEASE OF BONE, PFN1
Introduction
Paget's disease of bone (PDB [MIM: 167250]) is a chronic progressive metabolic bone disease characterized by abnormal activation of osteoclasts that results in abnormal bone resorption and compensatory osteogenic sclerosis.( 1 ) PDB can affect one (monostotic form) or more (polyostotic form) regions of the skeleton, with multiple constitutive symptoms, including bone pain, bone deformity, pathological fracture, tinnitus, hearing loss, vision loss, and osteoarthritis. In European countries, PDB is a relatively common chronic bone disorder, affecting 1% to 5% of those older than 50 years of age.( 2 ) In recent years, a decline in the prevalence of the disease has been reported, and it has been suggested that the clinical severity of the disease may also be attenuated. The disease has a distinct geographical distribution; it predominantly affects people of European descent and is rarely diagnosed in Asian populations.( 3 ) For example, the prevalence of PDB in Japan is only about 0.00028%.( 4 ) The deep differences in the prevalence and possible severity of the disease strongly suggest that genetic background and environmental factors are involved in its pathogenesis.
Neoplastic degeneration of Pagetic bone is the most severe complication of PDB, occurring in less than 1% of cases and resulting in osteosarcoma (OS; with an incidence of 0.3%) and, less frequently, fibrosarcoma (FS), chondrosarcoma (CS), and giant cell tumor (GCT).( 5 ) The severe subtype of early‐onset PDB that leads to the development of giant cell tumor of the bone (PDB/GCT) is extremely rare. Indeed, fewer than 150 cases have been reported in the literature to date, and little is known about its etiopathogenesis and management. Compared with PDB patients without GCTs, PDB‐GCT patients show a higher male/female ratio, more severe disease, and a higher mortality rate. Moreover, PDB‐GCT cases show elevated serum alkaline phosphatase (ALP) levels at neoplasm diagnosis and a higher prevalence of multifocal GCTs and have a positive familial history for PDB.( 6 )
Genetic factors are important in the pathogenesis of PDB. Mutations affecting the SQSTM1 gene (MIM: 601530), encoding the p62 protein, have been found in 35% and 10% of familial and sporadic cases of the uncomplicated form of PDB, respectively.( 7 , 8 , 9 ) In addition to SQSTM1, other genes associated with PDB‐related syndromes (TNFRSF11A [MIM: 603499],( 10 ) TNFRSF11B [MIM: 602643],( 11 , 12 , 13 ) VCP [MIM: 601023],( 14 ) HNRNPA2B1 [MIM: 600124]( 15 )) have been identified. Regarding the pathogenesis of PDB/GCT, ZNF687 (MIM: 610568) has been reported as the gene responsible for PDB/GCT in several patients from southern Italy.( 16 , 17 ) However, the molecular pathogenesis of PDB and PDB/GCT has not been fully elucidated; the known genes mentioned above can explain only a portion of cases, and it remains uncertain whether genes other than ZNF687 may be associated with PDB/GCT. Overall, potential pathogenetic genes and the molecular basis of tumorigenesis in GCTs developing in PDB need to be clarified.
Here, we report two families and one individual with sporadic disease of early‐onset and severe polyostotic Paget's disease of bone complicated with GCT caused by heterozygous deletion and missense mutations in the Profilin 1 (PFN1) gene, respectively. To confirm the pathogenicity of the mutation, we established a mouse model with the Pfn1 c.318_321delTGCC mutation and successfully obtained Pagetic phenotypes in heterozygous mice. The identification of PFN1 mutations has great diagnostic value for identifying PDB individuals predisposed toward developing GCTs. Meanwhile, our results expanded the pathogenic gene spectrum in PDB and undoubtedly made a significant contribution to reveal the pathogenesis of PDB/GCT.
Materials and Methods
Subjects
Two Chinese Han families with early‐onset severe PDB complicated with GCT were included in this study. A replication cohort of 30 unrelated PDB individuals negative via screening for mutations in SQSTM1, ZNF687, or other genes associated with PDB‐related syndromes was also included. Moreover, 570 healthy Han Chinese volunteers were included as controls. In addition, 113 ordinary GCT patients without PDB from our Pathology Department were included to further investigate somatic mutations in ordinary giant cell tumors.
This study was approved by the Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People's Hospital. Each subject signed informed consent before participation.
DNA isolation and genetic linkage analysis
Genomic DNA was extracted from the peripheral blood samples of all subjects using a Quick Gene DNA whole‐blood kit and Nucleic Acid Isolation System QuickGene‐610 L (Fujifilm, Tokyo, Japan) according to the manufacturer's protocol.
Genotyping and genomewide linkage analysis of 15 individuals in family 1 were carried out using an Illumina Human Omni ZhongHua‐8 BeadChip (Illumina, San Diego, CA, USA). Multipoint parametric linkage analysis was performed in Merlin by using pruned autosomal single‐nucleotide polymorphisms (SNPs) and assuming a dominant inheritance model for the disease phenotype.
Whole‐exome sequencing and validation
Whole‐exome sequencing was carried out for six PDB members of family 1. Exome capture was performed with SureSelect XT Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed via 150 base pair (bp) paired‐end sequencing using a HiSeq4000 instrument (Illumina). Reads were obtained and aligned to the human reference genomic sequence (GRCh37/hg19) with the Burrows‐Wheeler Aligner algorithm. We focused on functional variants (missense, nonsense, splicing site, and frameshift) and excluded those with a minor allele frequency higher than 1% in dbSNP131,1000 genomes and ExAC03. After filtering, we compared data for the six affected individuals to obtain shared variants and then compared them to the critical region previously identified by linkage analysis. We validated true positive variants through PCR followed by Sanger sequencing.
Sanger sequencing
Sanger sequencing of all three exons of the PFN1 gene and flanking intronic sequences was performed to search for mutations in family 2 and sporadic PDB cases from our biobank. The primer sequences were designed using Primer3 (v0.4.0). After amplification, the products were purified and sequenced with a DNA sequencing system (3730XL; ABI). All identified mutations were further verified in 570 healthy control volunteers using the same protocol. In addition, to identify somatic mutations, the genomic DNA was extracted from every tissue sample of 113 ordinary GCT patients without PDB and was subjected to sequence at the entire coding region of PFN1 gene.
Bioinformatics analysis
The online databases PolyPhen‐2 (Polymorphism Phenotyping v2), PROVEAN (Protein Variation Effect Analyzer), and Mutation Taster were used to assess the damaging effects of missense mutations in silico. To predict the effects on protein structure of the mutations compared with wild type (WT), molecular models of WT and mutant Profilin 1 were constructed with the SWISS‐MODEL server and Swiss‐Pdb Viewer, which retrieved the template structure 1AW1 from Protein Data Bank (PDB: 1AW1). The resulting models were evaluated on the basis of energy values calculated with the MODELER and SWISS model‐structure assessment tools. The mutant structure was also built with Swiss‐Pdb Viewer. Structural graphics and visualization were based on PyMol and Swiss‐Pdb Viewer.
Generation of Pfn1‐mutated mice
A mouse model of the Pfn1 c.318‐321delTGCC mutation was generated by using CRISPR‐Cas9 technology. In brief, Cas9 mRNA and guide RNA were obtained by in vitro transcription. A donor vector was constructed by in‐fusion cloning, involving a 3.0 kb 5′ homologous arm, a PM region, and a 3.0 kb 3′ homologous arm. The Cas9 mRNA, guide RNA, and donor vector were micro‐injected into the fertilized eggs of C57BL/6J mice to obtain F0‐generation mice. Three F0 mice with correct homologous recombination were obtained by PCR product sequencing. F0‐generation mice were mated with C57BL/6J mice to obtain positive F1‐generation mice. The colony was maintained by breeding heterozygotes, and the WT and heterozygous (Pfn1 +/mut) animals used in this study were littermates. All animal experiments were approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital, and all experiments conformed to the relevant regulatory standards.
Micro‐CT analysis
The hindlimbs of mice aged 8 weeks were fixed in 4% formalin‐buffered saline and stored in 80% ethanol. Micro‐CT analysis was performed using a Skyscan 1176 system (Bruker, Kontich, Belgium). For assessment of bone morphometry, the left tibia of 4‐month‐old animals was dissected free of most soft tissue and scanned at a resolution of 9 μm. The reconstruction and trabecular bone parameters, including the volumetric bone mineral density (vBMD), bone volume fraction (bone volume/total volume [BV/TV]), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), trabecular number (Tb.N), and cortical thickness (Ct.Th) of the bone were analyzed using the Skyscan, NRecon, Data Viewer, CTAn, CTvox, and Batman software in a stack of 200 slices immediately distal from the growth plate as described.
Radionuclide bone imaging
Mice aged 8 weeks were injected with 99 m Tc‐MDP via the tail vein. The postcontrast agent travels through the blood circulation to the bone for 4 hours; SPECT/CT tomography was then performed to observe the radioactivity.
Bone histology and immunohistochemistry staining
Mice were killed by cervical dislocation, and the hindlimbs were fixed for 24 hours in 4% formalin‐buffered saline and stored in 70% ethanol. Paraffin‐embedded decalcified sections were stained with hematoxylin and eosin or toluidine blue‐O (pH 4.5). To visualize osteoclasts, sections were stained with substrate for tartrate‐resistant acid phosphatase (TRAP) using TRAP Staining Kit (Sigma‐Aldrich, St. Louis, MO, USA).
For immunohistochemistry staining, sections were dewaxed in xylene, rehydrated in alcohol, and incubated in 3% hydrogen peroxide for 10 minutes to block endogenous peroxidase activity. Antigen retrieval was performed by microwave oven heating (10 minutes) in 0.01 M sodium citrate buffer (pH 6.0). Sections were incubated with 10% normal goat serum in PBS for 15 minutes at room temperature to block nonspecific binding. Then, the sections were incubated with rabbit anti‐Profilin 1 antibodies (1:500; EPR6304, Abcam, Cambridge, MA, USA) overnight at 4°C. The bound antibodies were visualized with a PV‐9000 2‐Step Plus Poly‐HRP Anti‐Rabbit IgG Detection System (ZSGB‐BIO, Beijing, China) and a Liquid DAB Substrate Kit (Invitrogen, Carlsbad, CA, USA). Positive reactions were defined as those showing brown signals in the cell cytoplasm.
Cell culture and differentiation
Mononuclear cells obtained from the peripheral blood of one healthy and one affected (III‐9 in family 1) individual were plated in 24‐well plates at approximately 2 × 105 cells/mL in alpha‐MEM supplemented with 10% FBS and penicillin/streptomycin in the presence of macrophage colony‐stimulating factor (M‐CSF; 30 ng/mL; Peprotech, Rocky Hill, NJ, USA) and RANKL (50 ng/mL; Peprotech). Osteoclastic differentiation was usually monitored for 5 days while periodically changing the medium and fixing and staining for TRAP activity.
Bone marrow cells obtained from WT and Pfn1 +/mut mouse femurs and tibias were plated in 25‐cm culture bottles at approximately 5 × 106 cells/mL in alpha‐MEM supplemented with 10% FBS and penicillin/streptomycin. After 24 hours of incubation at 37°C, 5% CO2, nonadherent bone marrow cells were replated on 24‐well plates at 5 × 104 cells/well. For the osteoclastogenesis assay, bone marrow cells were cultured for 72 hours in the presence of M‐CSF (30 ng/mL; Peprotech); the medium was then changed to medium containing both receptor activator of NF‐κB ligand (RANKL; 50 ng/mL; Peprotech) and M‐CSF. Osteoclastic differentiation was usually monitored for 5 days while periodically changing the medium. The cells were fixed and stained for TRAP activity.
TRAP staining
Bone marrow cells were plated and differentiated as described above. Cells were fixed with 2.5% glutaraldehyde, and TRAP was stained with a TRAP Staining Kit (Sigma‐Aldrich). TRAP‐positive cells with >3 nuclei were defined as mature osteoclasts. The number of mature osteoclasts was counted by Simple PCI software.
Resorption pit staining
A resorption pit forming assay was carried out using bone slices. Briefly, bone slices were adhered to 24‐well plates, and bone marrow cells were plated and differentiated as described above. On day 20, the slices were fixed with 2.5% glutaraldehyde and dehydrated using graded ethanol. The resorption pit of the bone slices was observed and photographed by scanning electron microscopy after gold spraying.
Serum bone turnover markers
Serum of WT and Pfn1 +/mut mice was collected and stored at −80°C. Expression levels of bone turnover markers (procollagen of type l collagen N‐prepeptide [P1NP] and tartrate‐resistant acid phosphatase 5b [TRACP‐5b]) were estimated using mouse P1NP ELISA Kit (Cusabio, Wuhan, China) and mouse TRACP‐5b ELISA Kit (Mlbio, Shanghai, China), respectively, according to the manufacturer's protocols.
Statistical analysis
The data are presented as median (interquartile range) for quantitative variables and number (percentage) for qualitative variables. All statistical analyses and graphical preparations were carried out using the SPSS 26.0 statistical software (IBM Corp., Armonk, NY, USA) and GraphPad Prism for Windows (La Jolla, CA, USA). Quantitative variables between the means of three or more independent unrelated groups were analyzed by one‐way ANOVA. Variables between two averages were analyzed by Student's t test. Differences were considered statistically significant at p < .05.
Results
Clinical history of the PDB/GCT pedigree
Two Chinese Han families with early‐onset severe PDB complicated with GCT were included in our study.
Family 1, with 24 members of three generations in total, among whom three members died, was from Anhui Province and visited our department in May 2017 (Fig. 1A ). According to clinical manifestations and auxiliary examination (measurement of bone turnover markers, skeletal X‐rays, and bone scintigraphy; Fig. 1B–D ), 10 family members were diagnosed with PDB in an autosomal dominant pattern. All affected subjects presented polyostotic PDB, with at least three compromised skeletal sites and early onset of the disease (Table 1). All affected subjects had skull involvement and presented with skull deformity, thickened and enlarged skull plate barrier, and sclerotic foci. Other lesion sites include ilium, femur, and vertebra, like deformity of femur, enlarged and disordered trabecular bone and osteoporosis, etc. In some affected subjects (II‐9, III‐4, III‐9), the lesion of femur presented as cortical thickening, sclerosis, and narrowed medullary cavity. Some affected subjects (II‐2, II‐4, II‐5, II‐9, and III9) with vertebral body involvement showed endplate sclerosis and vertebral compression. Interestingly, two PDB subjects in the pedigree (II‐4 and III‐4) developed GCTs, one each located in the left ilium and right nasal cavity. Specifically, subject III‐4 was first diagnosed with a nasal tumor after repeated epistaxis and nasal obstruction at age 34 years and was treated with surgery and radiotherapy. The pathology revealed GCT. However, the tumor recurred after two operations and 12 radiotherapies. Subsequently, he was diagnosed with severe polyostotic PDB, with areas of affected regions at the skull, spine, pelvis, humeri, ulnas, femurs and tibias at X‐ray examination after an accidental fracture of the spine and scapula. However, no osteolysis sites was found. The pathology of his lesion revealed GCT and the nasal tumor recurred after two operations and 12 times radiotherapy. Subsequently, he was diagnosed as severe polyostotic PDB with skull, spine, pelvis, humeri, ulna, femur, and tibia involvement by X‐ray examination after an accidental fracture of the spine and scapula. However, no osteolysis at any sites was found. Subject II‐4, the mother of III‐4, developed PDB at age 40 years. In her early 40s, she developed progressive deformity of the lower limbs and hearing loss. Afterward, she complained of increased pain and bone enlargement of the ilium. She was diagnosed with GCT of the ilium and was treated with radiotherapy in other medical institutions.
Fig 1.
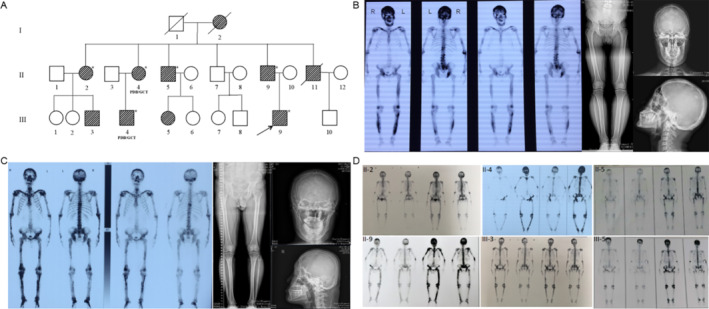
Pedigree and clinical features of family 1. (A) The pedigree of family 1 is shown. Paget's disease of bone (PDB) members are indicated with black slash symbols. Family members with PDB/giant cell tumor of bone (GCT) phenotype are labeled, and members marked with asterisk (*) indicate the subjects included in exome sequencing. The black arrow indicates the proband. (B) X‐ray examination and whole‐body bone scintigraphy of the proband (III‐9) show radionuclide uptake in the skull, the spine, the pelvis, the humeri, the ulnas, the femurs, and the tibias. The skull was deformity, and the skull plate barrier was thickened and enlarged with sclerotic foci. He had deformity of femur, large and disordered trabecular bone, and cortical thickening, sclerosis, and narrow medullary cavity. (C) X‐ray examination and whole‐body bone scintigraphy of subject III‐4 shows radionuclide uptake in the skull, the clavicles, the spine, the pelvis, the humeri, the ulnas, the femurs, and the tibias. The X‐ray findings of skull, femur, and tibia were similar to those of III‐9 patient. (D) Whole‐body bone scintigraphy of other PDB members all show radionuclide uptake in multiple bones, mainly in the skull, spine, pelvis, and limbs.
Table 1.
Clinical Characteristics of Subjects With Familial and Sporadic PDB and Positive for PFN1 Mutations
| Subject | Sex | PFN1 mutation | Age at visit (years) | Age of onset (years) | Symptoms | GCT | Serum ALP (U/L) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bone pain | Headache | Hearing loss | Bone deformity | Fracture | |||||||
| Family 1 | |||||||||||
| II‐2 | Female | c.318_321delTGAC | 66 | 40 | + | + | + | + | − | No | 199 |
| II‐4 (PDB/GCT) | Female | c.318_321delTGAC | 63 | 40 | + | + | + | + | − | Yes | 267 |
| II‐5 | Male | c.318_321delTGAC | 61 | NA | − | − | − | − | − | No | 275 |
| II‐9 | Male | c.318_321delTGAC | 53 | NA | − | − | − | − | + | No | 285 |
| III‐3 | Male | c.318_321delTGAC | 35 | 35 | − | + | + | − | − | No | 210 |
| III‐4 (PDB/GCT) | Male | c.318_321delTGAC | 38 | 34 | − | + | − | − | + | Yes | 235 |
| III‐5 | Female | c.318_321delTGAC | 33 | 33 | − | − | − | − | − | No | 378 |
| III‐9 | Male | c.318_321delTGAC | 26 | 26 | − | − | − | − | + | No | 642 |
| Family 2 | |||||||||||
| I‐2 | Female | c.335 T > C | 57 | NA | − | − | − | − | − | No | 62 |
| II‐2 (PDB/GCT) | Female | c.335 T > C | 32 | 20 | + | + | − | + | − | Yes | 2124 |
| Sporadic PDB/GCT | |||||||||||
| PDB‐2 (PDB/GCT) | Female | c.324_324delG | 21 | 18 | + | + | − | − | − | Yes | 434 |
PDB = Paget's disease of bone; GCT = giant cell tumor of bone; ALP = alkaline phosphatase; NA = data not available.
The normal range of serum ALP is 15 to 112 U/L.
The second family, with four members of two generations, was from Jiangsu Province and visited our department in November 2019 (Fig. 2A ). According to the clinical manifestations and auxiliary examinations, the proband and her mother were diagnosed with PDB in an autosomal dominant pattern. The proband (II‐2) developed deformity and bone pain of the limbs and skull in her early 20s. Subsequently, she developed multiple gradually enlarged bumps on the forehead at age 29 years (Fig. 2B ). The larger bump had a diameter of approximately 7 cm and was spherical, soft, purplish red, and painful when pressed; the smaller bump had a diameter of approximately 3 cm at her first visit to our department. She was diagnosed with severe polyostotic PDB. The bone scintigraphy showed increased tracer uptake in affected bones, including skull, clavicle, spine, humeri, ulna, pelvis, femur, and tibia. X‐ray examination showed that Paget lesions such as skull, femur, and vertebral body were similar to those of affected subjects of family 1 introduced above. However, the deformity of femurs and tibias was more obvious, and there were many osteolytic foci (Fig. 2D ). She was treated with an operation to remove tumors, and the pathology revealed GCT (Fig. 2C ).
Fig 2.
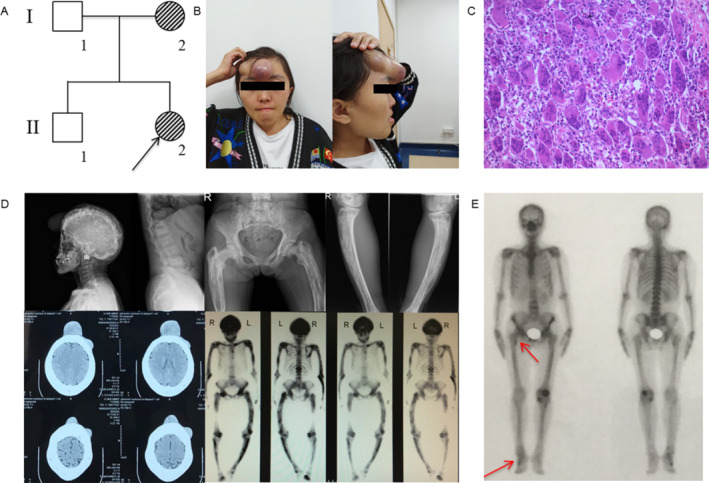
Pedigree and clinical features of family 2. (A) The pedigree of family 2 is shown. Paget's disease of bone (PDB) members are indicated with black slash symbols. Family members with PDB/giant cell tumor of bone (GCT) phenotype are labeled. The black arrow indicates the proband. (B) Bumps on the forehead of the proband (II‐2). The larger one has a diameter of about 7 cm and is spherical and purplish red; the smaller one has a diameter of about 3 cm. (C) Pathological image of tumors resected from the forehead of II‐2 (H&E staining 100×) shows numerous multinucleated giant cells. (D) X‐ray examination and whole‐body bone scintigraphy of subject II‐2 shows radionuclide uptake in the skull, the clavicles, the spine, the pelvis, the humeri, the ulnas, the femurs, and the tibias. The skull was deformed, and the skull plate barrier was thickened and enlarged with sclerotic foci. The deformity of femurs and tibias were more obvious with large and disordered trabecular bone, and osteoporosis with osteolysis sites. She had vertebral body involvement, showing endplate sclerosis and vertebral compression. (E) Whole‐body bone scintigraphy of subject I‐2 shows radionuclide uptake in the superior right femur, the pelvis, and the right foot.
This study also included 30 patients with sporadic PDB from our biobank, and none of the patients had any known genetic mutations associated with PDB (Table 2). One case was complicated with GCT of the nasal cavity and ilium. This subject was a 21‐year‐old female (PDB‐2) from Anhui Province who visited our department in February 2015 and complained of gradually increased bone pain of the right hip for 3 years and repeated epistaxis and nasal obstruction for 2 months. Her serum ALP level was 434 u/L (normal range: 56–112 U/L). She was diagnosed with severe polyostotic PDB, including skull, spine, pelvis, femur, and left tibia involvement. The affected long bone showed osteosclerosis and narrow marrow cavity, but no osteolysis was detected (Fig. 3A ). The combined GCT at the nasal cavity and the right ilium was operated subsequently (Fig. 3B , 3C ). Neither of her parents showed any similar signs of PDB or GCT. In addition, we recruited 570 healthy Han Chinese volunteers as controls to examine the frequency of mutations in the healthy population.
Table 2.
Clinical Characteristics of Patients With Sporadic PDB Patients and Non‐Pagetic GCT
| Characteristics | Sporadic PDB | Non‐Pagetic GCT |
|---|---|---|
| Age at visit (years) | 54 (48.5–67.5) | 36 (29–50) |
| Age of onset (years) | 51.5 (46–67.5) | 36 (29–50) |
| Sex, male/female | 19/11 | 70/43 |
| Symptoms | ||
| Bone pain | 27 (90%) | 108 (95.6%) |
| Headache | 2 (6.7%) | 0 |
| Hearing loss | 1 (3.3%) | 0 |
| Bone deformity | 8 (26.7%) | 28 (24.8%) |
| Fracture | 1 (3.3%) | 0 |
| No. of PDB sites | ||
| Monostotic | 16 (53.33%) | 113 (100%) |
| Polyostotic | 14 (46.67%) | 0 |
| GCT | 1 (3.33%) | 113 (100%) |
| Serum ALP (U/L) | 249 (149–440) | 108 (86–228) |
| Treatment | Zoledronate (5 mg) | Surgery (100%)/denosumab 120 mg (15, 13.3%) |
PDB = Paget's disease of bone; GCT = giant cell tumor of bone; ALP = alkaline phosphatase.
Data are the median (interquartile range) for quantitative variables and no. (%) of patients for qualitative variables. The normal range of serum ALP is 15 to 112 U/L.
Fig 3.
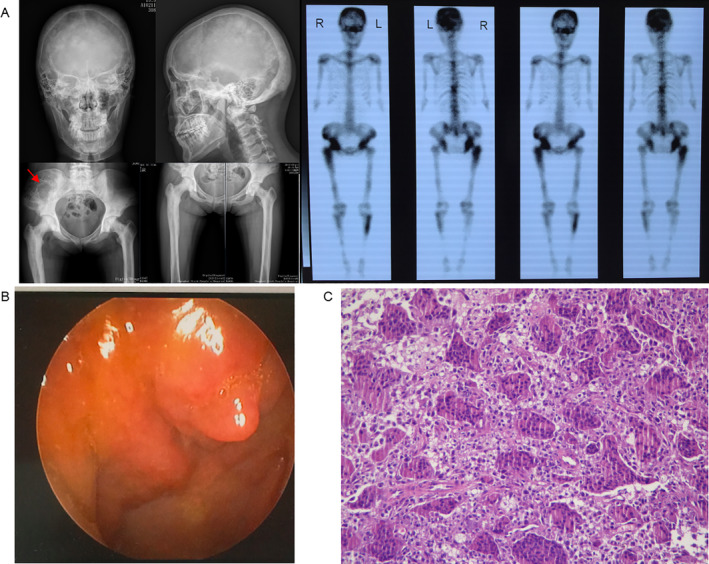
Clinical features of a patient with sporadic Paget's disease of bone/giant cell tumor of bone (PDB/GCT). (A) X‐ray examination and whole‐body bone scintigraphy of the subject shows radionuclide uptake in the skull, the clavicles, the spine, the pelvis, the humeri, the femurs, and the tibias. X‐ray findings of skull and vertebral body are similar to II‐2 of family 2. The affected long bone showed osteosclerosis and narrow marrow cavity, and large and disordered trabecular bone without osteolysis. The GCT was found at the right ilium. (B) Image of the nasal endoscope, showing tumors on the nasal cavity. (C) Pathological image of tumors resected from the nasal cavity (H&E staining 200×), showing numerous multinucleated giant cells.
All of the patients with PFN1 mutations mentioned above were treated immediately after diagnosis with zoledronate (Aclasta 5 mg, Sandoz, Holzkirchen, Germany) or denosumab (Xgeva 120 mg, Amgen, Thousand Oaks, CA, USA), with a follow‐up period up to 2 years (Table 3). Symptomatic relief was not significant in most of the patients, especially for those with GCTs. Serum ALP and C‐terminal telopeptide of type 1 collagen (β‐CTX) concentrations gradually decreased after treatment; however, we observed a poor response to treatment in those with PFN1 mutations and even a rise after 6 months in patient II‐2 from family 2, which reflected that persistent and long‐lasting active disease might be relevant for neoplastic degeneration rather than the mutation per se.
Table 3.
Treatment and Follow‐Up of Patients With PFN1 Mutations
| Subject | PFN1 mutation | Treatment | Serum ALP (U/L) | Serum β‐CTX (ng/L) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before therapy | 3 months after therapy | 6 months after therapy | 1 year after therapy | 2 years after therapy | Before therapy | 3 months after therapy | 6 months after therapy | 1 year after therapy | 2 years after therapy | |||
| Family 1 | ||||||||||||
| II‐2 | c.318_321delTGAC | Zoledronate(5 mg) | 199 | 107 | ||||||||
| II‐4 | c.318_321delTGAC | Zoledronate(5 mg) | 267 | 150 | 1951 | 1043 | ||||||
| II‐9 | c.318_321delTGAC | Zoledronate(5 mg) | 285 | 143 | 2887 | 1447 | ||||||
| III‐3 | c.318_321delTGAC | Zoledronate(5 mg) | 210 | 110 | ||||||||
| III‐4 | c.318_321delTGAC | Zoledronate(5 mg) | 235 | 120 | 113 | 1299 | 699 | 954 | ||||
| III‐5 | c.318_321delTGAC | Zoledronate(5 mg) | 378 | 257 | ||||||||
| III‐9 | c.318_321delTGAC | Zoledronate(5 mg) | 642 | 297 | 214 | 194 | 133 | 3574 | 2244 | 1538 | ||
| Family 2 | ||||||||||||
| II‐2 | c.335 T > C | Denosumab(120 mg) | 2124 | 1161 | 315 | 697 | 3040 | 2650 | 2374 | 3204 | ||
ALP = alkaline phosphatase.
The normal range of serum ALP is 15 to 112 U/L. The normal range of serum β‐CTX is 112 to 497 ng/L for females and 100 to 612 ng/L for males.
Linkage analysis and whole‐exome sequencing reveal a PFN1 truncating mutation as a cause of PDB/GCT in family 1
To identify the causative gene for this severe form of PDB complicated with GCT, we performed genetic linkage analysis for 15 members and whole‐exome sequencing for six affected members (II‐2, II‐4, II‐5, II‐9, III‐4, and III‐9) in family 1. Multipoint parametric linkage analysis achieved a maximal LOD score of 2.70 for chromosomes 17:8, 547–5, 657, and 423 (GRCh37) (Fig. 4A ). We identified 73 functional variations by WES, but only two were within the critical linkage regions. Only one showed precise cosegregation with the disease phenotype by Sanger sequencing: the c.318_321delTGAC heterozygous mutation (p. Asp107Argfs*3) in the PFN1 gene, coding Profilin 1, located on chromosome 17p13 (Fig. 4B ). The 4‐bp deletion leads to a frameshift with a premature stop codon, resulting in a putative protein of 108 amino acids instead of 140 residues.
Fig 4.
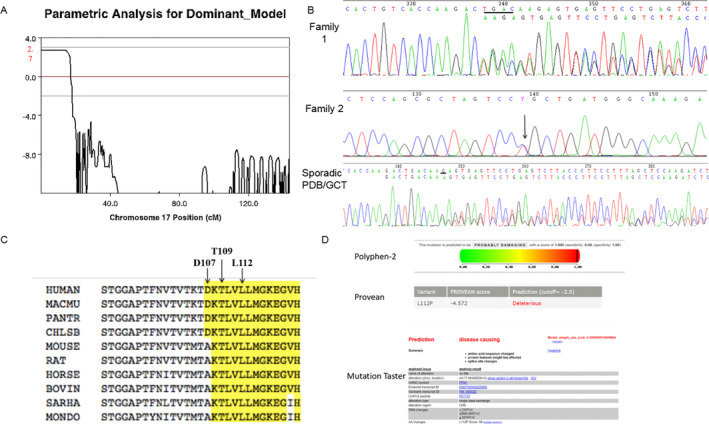
Mutation in the PFN1 gene underlies familial and sporadic Paget's disease of bone/giant cell tumor of bone (PDB/GCT). (A) Genetic linkage analysis achieved a maximal LOD score of 2.70 for chromosomes 17:8, 547–5, 657, 423 (GRCh37). (B) The DNA sequence of PFN1 in familial and sporadic PDB/GCT. Electropherogram shows heterozygous frameshift deletion in PFN1 c.318_321delTGAC in family 1 and c.324_324delG in sporadic PDB/GCT, heterozygous missense mutation c.335 T > C in family 2. (C) Evolutionary conservation of the mutation sites among different species. Arrows and highlights mark the mutated residues. (D) Mutational analysis of the c.335 T > C mutation in PFN1, showing the mutation predicted to be deleterious by PolyPhen‐2, Provean, and MutationTaster.
Sanger sequencing reveals novel truncating and missense mutations of PFN1 in family 2 and sporadic cases of PDB/GCT
To assess whether additional PDB individuals harbor PFN1 mutations, we sequenced the coding region of the PFN1 gene in family 2 and sporadic PDB cases from our biobank negative for mutations in any known genes related to PDB. Another deletion mutation at the adjacent location, the c.324_324delG heterozygous mutation (p.T109Rfs*2), in the PFN1 gene was identified in a patient with sporadic disease (PDB‐2) (Fig. 4B ). Interestingly, the subject carrying the deletion showed the most severe phenotype, and among all sporadic cases, this was the only one complicated with GCT. Moreover, the proband (II‐2) of family 2, who had a very severe form of PDB complicated with GCT, and her mother (I‐2) harbored a missense mutation at the adjacent location: the c.335 T > C heterozygous mutation (p.Leu112Pro) in the PFN1 gene (Fig. 4B ), suggesting that not only truncating mutations but also missense mutations in PFN1 are pathogenic mutations for PDB‐GCT. In general, early‐onset, polyostotic, severe, and complicated GCTs are clinical features shared among the affected subjects who harbored PFN1 mutations. Neither of the three mutations was detected in a cohort of 570 healthy individuals.
Bioinformatics analysis
All mutations were predicted to be deleterious by Provean, PolyPhen‐2, or MutationTaster, and all mutation sites are highly conserved according to the homology analysis among different animal species (Fig. 4C, D ), which supports the pathogenicity of the mutations.
To illustrate the effects on protein structure, we modeled the crystal structure of Profilin 1, which revealed that both the truncating mutation (c.318_321delTGAC) and missense mutation (c.335 T > C) affect the tertiary structure of the protein (Fig. 5) and the structure of Profilin 1 bound to actin (Supplemental Fig. S1).
Fig 5.
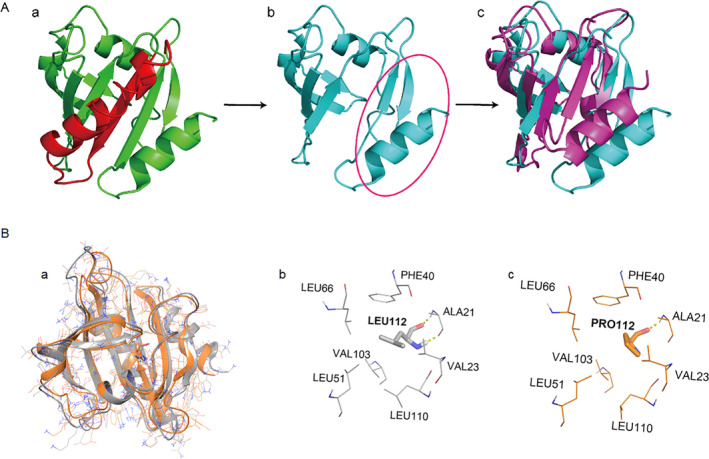
Protein modeling of the PFN1 truncating mutation and missense mutation. (A) Protein modeling of the truncating mutation. The wild‐type structure of Profilin 1 is indicated in (a); red represents the structure deleted by the truncated mutation. The mutant structure of Profilin 1 is indicated in (b); a comparison between the wild‐type structure (purple) and mutant structure (blue) is indicated in (c). (B) Protein modeling of the truncating mutation. A comparison between the wild‐type structure (gray) and mutant structure (orange) is indicated in (a). The local structure of wild type is indicated in (b), and the mutant structure is indicated in (c).
Somatic mutations of PFN1 in giant cell tumors
It is well known that somatic mutations are the most important tumorigenesis mechanisms involved in the development of tumors.
To further investigate whether somatic PFN1 mutations are related to ordinary giant cell tumors, we sequenced the coding region of the PFN1 gene using DNA samples extracted from the tumors of 113 ordinary GCT patients (clinical characteristics shown in Table 2) from our Pathology Department. No somatic mutations in the PFN1 gene were detected (data not shown), suggesting that the relationship between PFN1 and GCT may only exist in the germline and not in somatic tissue.
Osteoclast differentiation in patients with PFN1 mutation
To verify whether osteoclasts from patients show the typical features of Pagetic osteoclasts, we isolated mononuclear cells from the peripheral blood of one healthy and one affected (III‐9 in family 1) individual and differentiated them into osteoclasts. As expected, we observed a greater number of osteoclasts derived from the PFN1‐mutated PDB cells compared with control cells (Fig. 6).
Fig 6.
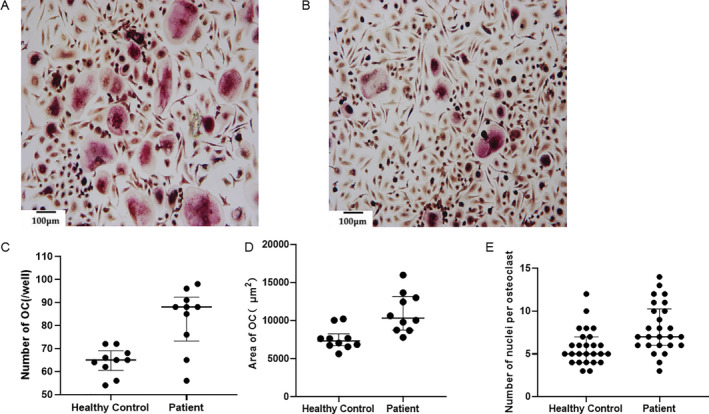
Osteoclast differentiation activity of peripheral blood mononuclear cells between patients with PFN1 mutations and healthy controls (100×). (A) TRAP staining of osteoclasts in patients with PFN1 mutations. (B) TRAP staining of osteoclasts in healthy controls. (C) Comparison of the numbers of TRAP‐positive osteoclasts, p = 0.0008. (D) Comparison of the area of TRAP‐positive osteoclasts, p = 0.0017. (E) Comparison of the number of nuclei per osteoclast, p = 0.0027. All data were analyzed by Student's t test.
Phenotypes of WT and Pfn1‐mutated mice
Growth failure and skeletal deformity arise postneonatally in Pfn1‐mutated mice
To investigate the roles of PFN1 mutation in PDB pathogenesis, Pfn1‐mutated mice were generated using CRISPR‐Cas9 technology in 2018. After repeated mating between the heterozygous males and females, homozygous mice could not be obtained, suggesting that homozygous mutation of the c.318–321 deletion in Pfn1 are fatal for mouse embryogenesis. Sanger sequencing of heterozygous Pfn1‐mutated mice confirmed the presence of a frameshift mutation (c.318‐321delTGCC), which is predicted to cause premature translation termination (p.Asp107Argfs*3).
The Pfn1 +/mut mice tended to be smaller in size from birth to adulthood, with shortened body and femur length, as well as deformed craniofacial bones and spines in appearance (Fig. 7A ), compared with WT mice. The wide arched calva, shortened maxilla, and curved spine contributed to the altered facial appearance, with a steep nose‐to‐head curvature and arched body on their profile.
Fig 7.
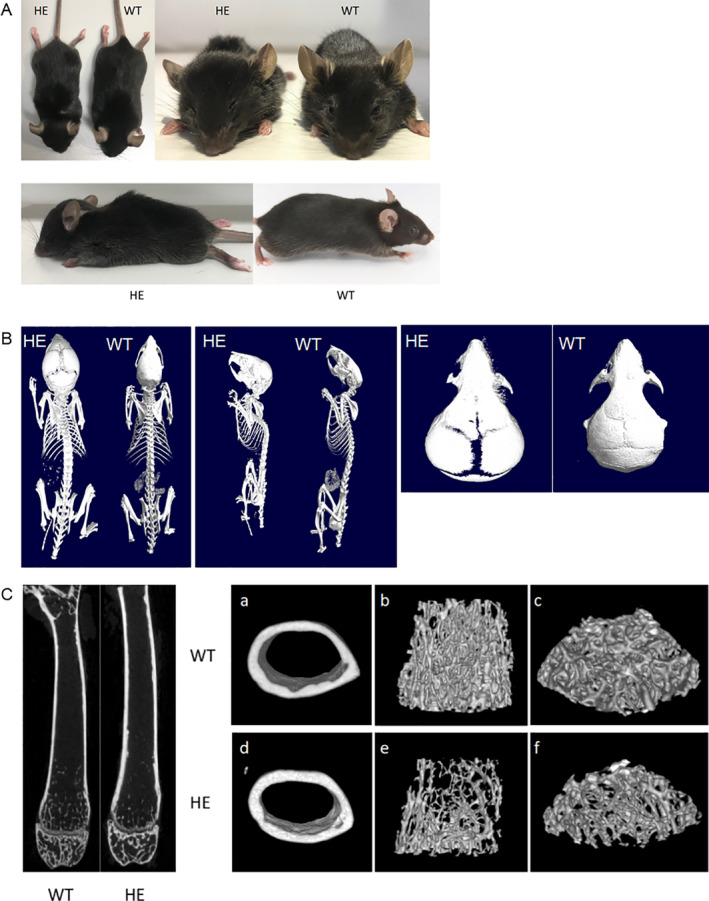
Comparison of appearance and 3D reconstruction between WT and Pfn1 +/mut mice. (A) Appearance of WT and Pfn1 +/mut mice. The wide arched calva, shortened maxilla, and curved spine contributed to the altered facial appearance with a steep nose‐to‐head curvature and arched body on their profile of Pfn1 +/mut mice compared with WT mice. (B) Images of 3D reconstruction of WT and Pfn1+/mut mice demonstrated wide cranial sutures and deformed craniofacial bones and spines in heterozygous mice compared with WT mice. (C) Images of 3D reconstruction of the femurs in WT and Pfn1 +/mut mice. All mice shown here were 8 weeks old and female.
Skeletal deformity and osteopenic nature of trabecular femurs in Pfn1‐mutated mice revealed by micro‐CT
To analyze skull deformities more intuitively and quantitatively, we utilized reconstructed 3D micro‐CT analysis of the whole skeleton and morphometric analysis of trabecular and cortical femurs of mice aged 8 weeks. The 3D reconstruction demonstrated a wide cranial suture and deformed craniofacial bones and spines in Pfn1 +/mut mice compared with WT mice (Fig. 7B ). Regarding the femurs, the trabecular bone parameters of Pfn1 +/mut mice (n = 8 for each group) indicated the osteopenic condition by lower BV/TV, Tb.Th, Tb.N, Tb.Sp, and vBMD. However, the cortical bone was thicker than that of WT mice, with a higher vBMD and Ct.Th (Supplemental Table S1), which may be due to active bone resorption in the trabecular bone but secondary sclerosis in the cortical bone with the Pagetic‐like phenotype in Pfn1 +/mut mice, as indicated by cross‐sectional images (Fig. 7C ).
Radioactivity concentration in Pfn1‐mutated mice by radionuclide bone imaging
To analyze differences in bone metabolism and the range of lesions, radionuclide bone imaging with 99 m Tc‐MDP was performed. The results revealed radioactivity concentration in the left femur of one Pfn1 +/mut mouse, which indicated the presence of active bone metabolism lesions (Supplemental Fig. S2). The concentration of radioactivity in the left femur was significantly higher than that of the contralateral side; other high spots showed normal concentrations in the joints and bladder.
Enhanced osteoclast differentiation and resorption in Pfn1‐mutated mice
To evaluate whether the Pfn1 mutation affects osteoclast differentiation, bone marrow macrophages (BMMs) were generated after isolation of total bone marrow cells from the hindlimbs of WT and Pfn1 +/mut mice for differentiation into osteoclasts. After 5 days of differentiation, the number of osteoclasts generated from Pfn1 +/mut mice was significantly greater than that generated from WT littermates (Fig. 8A, C ).
Fig 8.
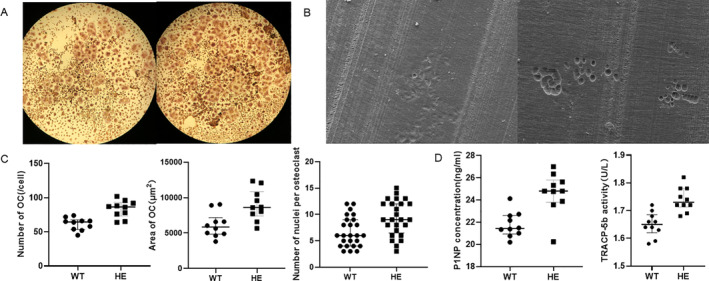
The Pfn1 truncating mutation results in accelerated osteoclast differentiation and increased bone resorption in mice. (A) TRAP staining of osteoclasts derived from WT and Pfn1 +/mut mice (40×). (B) Electron microscopy images of the resorption pits of WT and Pfn1 +/mut osteoclasts cultured on bone slices. (C) Comparison of the numbers of TRAP‐positive osteoclasts, p = .0003. Comparison of the area of TRAP‐positive osteoclasts, p = .0047. Comparison of the number of nuclei per osteoclast, p = .0041 (n = 6 for each group). (D) Comparison of the levels of serum bone turnover markers between WT and Pfn1 +/mut mice (n = 10 for each group), p = .0006 for P1NP and p = .0002 for TRACP‐5b. All data were analyzed by Student's t test.
In addition, the resorption pit formation assay revealed that osteoclasts derived from Pfn1 +/mut mice tended to resorb more mineralized matrix by larger and deeper pits (Fig. 8B ). This suggested that the mutation resulted in amplified matrix‐resorbing activity of osteoclasts, consistent with the increased bone resorption phenotype of PDB patients.
Increased serum bone turnover markers in Pfn1‐mutated mice
Bone turnover markers include enzymes and their decomposition products derived from bone cells and bone matrix components, reflecting the activity of bone formation and bone resorption. We determined expression levels of P1NP and TRACP‐5b by ELISA, and the results showed a significant increase in both P1NP and TRACP‐5b in Pfn1 +/mut mice compared with WT mice (Fig. 8D ), suggesting the active state of bone turnover in the former. This is also consistent with the increased bone resorption phenotype of PDB patients.
Pfn1‐mutated mice exhibit a reduced number of trabeculae by bone histology, an increased number of osteoclasts by TRAP staining, and decreased expression of Profilin 1 by immunohistochemistry staining
To investigate differences in bone histology, frontal sections of the distal femurs of mice aged 8 weeks were compared using hematoxylin and eosin staining. We noticed a reduced number and disorder of trabeculae in Pfn1 +/mut mice compared with WT mice (Fig. 9A ). We then examined whether osteoclastic bone resorption was indeed increased in the histological sections of Pfn1 +/mut mice. TRAP staining detected prominent numbers of multinucleated TRAP‐positive cells in Pfn1 +/mut mice at the endosteal regions of the metaphysis to the diaphysis, whereas osteoclast distribution was usually rare in WT mice (Fig. 9B ). We next evaluated expression of Profilin 1 in femur sections using immunohistochemistry staining. The results showed a significant decrease in expression in Pfn1 +/mut mice compared with WT mice (Fig. 9C ), consistent with the truncating mutation leading to loss of function of Profilin 1.
Fig 9.
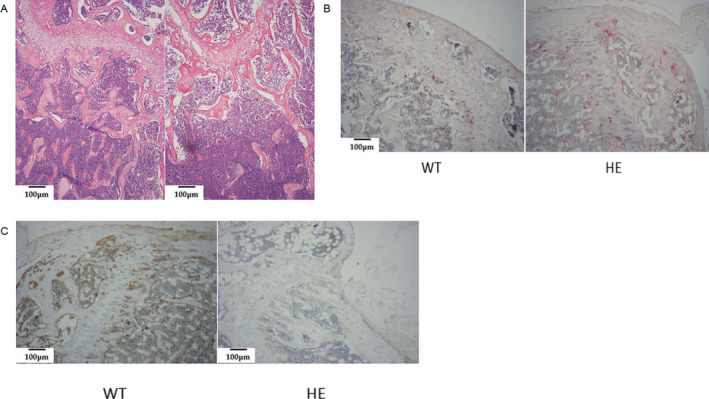
Bone histology, TRAP staining, and immunohistochemistry staining of distal femur sections from WT and Pfn1 +/mut mice. (A) H&E staining shows reduced and disordered trabeculae in Pfn1 +/mut mice compared with WT mice (100×). (B) TRAP staining indicates increased multinucleated osteoclasts at the trabecular surface of the Pfn1 +/mut femurs. (C) Immunohistochemistry staining showed a significant decrease in Profilin 1 expression in Pfn1 +/mut mice compared with WT mice.
To further investigate the potential pathogenesis, we detected expression of TDP‐43 in osteoclasts derived from bone marrow cells obtained from WT and Pfn1 +/mut mice. Unfortunately, we observed no difference between mutated and wild‐type cells (data not shown).
Discussion
The PFN1 gene is located at 17p13.3 (MIM: 176610) and contains 3 exons, encoding the small protein Profilin 1, which is highly evolutionarily conserved (15,054 Da) and contains 140 amino acids. Profilin 1 is a small actin monomer‐binding protein that is considered to be an essential control element for actin polymerization and cell migration.( 18 , 19 ) Profilin 1 is ubiquitously expressed in all cell types and in 231 organs and tissues throughout the body and interacts with a plethora of ligands, highlighting its involvement in controlling several cellular functions, including cytoskeletal organization, membrane trafficking, mRNA processing, and cell cycle regulation.( 18 , 20 , 21 , 22 , 23 , 24 ) In addition to these cellular processes, Profilin 1 has been implicated in tumorigenesis as a tumor suppressor, and its downregulation has been observed in multiple types of carcinoma (breast, pancreas, and bladder), correlating with shortened patient survival.( 25 , 26 , 27 )
Here, we report two Chinese families with 10 individuals affected by early‐onset polyostotic PDB, three of whom developed GCTs, and one patient with sporadic early‐onset PDB/GCT. Moreover, we identified three heterozygous mutations in the PFN1 gene as being responsible for the severe subtype of early‐onset Paget's disease, with progression to GCT, including two deletion truncating mutations and one missense mutation at the adjacent location. Regarding clinical characteristics, patients with PFN1 mutations exhibit persistent and long‐lasting active bone resorption, resulting in severe skeletal deformities, high levels of serum ALP and bone turnover markers, and giant cell tumors. Therefore, the bone radionuclide scans and the radiological images of these patients show more skeletal sites involved and more severe expansion and deformity in bones. All patients have skull involvement, which is a typical characteristic of the disease caused by PFN1 mutation. However, classic, late‐onset PDB showed milder imaging features. In addition, the patients in our study had relatively low response to the treatment of zoledronic acid or denosumab compared with the classic PDB. This is one of the key clinical features of PFN1 mutation, which is different from other pathogenic gene mutations.
To confirm the pathogenicity of the mutation, we established a mouse model with the Pfn1c.318_321delTGCC mutation. Although homozygous mice were not obtained (homozygous lethality was considered), heterozygous mice had obvious phenotypes, including abnormal growth and development, deformities of the craniofacial spine, accelerated osteoclast differentiation and increased bone resorption, differences in bone morphometry by micro‐CT, and higher bone turnover markers. These results demonstrate that mutation of Pfn1 can result in significant Pagetic phenotypes.
The increased osteoclast differentiation and bone deformity observed in our Pfn1‐mutated mouse model is also consistent with the skeletal phenotype recently described in osteoclast‐specific conditional Pfn1‐deficient mice (Cathepsin K‐Cre driver), which display dwarfism, craniofacial hypoplasia, and long bone deformities with osteolytic appearance.( 28 ) However, we have not observed neoplastic degeneration in the mutated mice, although the current maximum observation period is 14 months, which may reflect the differences in genetic function between mice and humans of PFN1, as well as the influence of environment on genetic phenotypes.
During our article preparation, Italian scientists published work with findings consistent with ours in January 2020( 29 ) and April 2020.( 30 ) They reported the same truncating PFN1 mutation c.318_321delTGAC, resulting in early‐onset severe PDB complicated with osteosarcoma and a germline heterozygous deletion of PFN1 in 4 cases of 218 PDB individuals based on copy number screening, which were not found in our study. Indeed, since the first description of PDB by Sir James Paget in 1877, it has been clear that the severe and polyostotic form of PDB has the potential for cancer progression in affected bones; the most frequent tumor is osteosarcoma, and the rarest is GCT. Our study confirmed these results, indicating that a truncating PFN1 mutation causes PDB. Furthermore, we identified two novel mutations in the PFN1 gene, including the missense mutation c.335 T > C (p.Leu112Pro), which expands the mutant spectrum of PFN1; thus, not only a truncating mutation but also a missense mutation can cause PDB/GCT. These two mutations are not found in any of the reference genomic databases derived from persons of European descent. Moreover, we first successfully established a Pfn1‐mutated mouse model, which is strong evidence for the pathogenesis of the PFN1 gene in PDB. This model is also a useful tool that can be utilized in further investigations of PFN1, PDB, and cancer progression in PDB.
Missense mutations of the PFN1 gene were previously identified to cause amyotrophic lateral sclerosis (ALS 18 [MIM: 614808]). ALS is a rare neurodegenerative disease that causes gradual loss of motor activity in the spinal cord, brain stem, and cerebral cortex, leading to muscular atrophy, difficulty in movement, and even death from respiratory failure at 3 to 5 years after diagnosis. Ten percent of cases are familial, usually being inherited in an autosomal dominant pattern. Studies have found that mutations in the PFN1 gene are one of the causes of family ALS. Currently, reported mutation sites are A20T, C71G, T109M, M114T, E117G, G118V, R136W, and Q139L, all of which are missense mutations( 31 , 32 ) (Fig. 10). Studies to date suggest a high correlation between the genetic background of ALS and PDB, which can both be included in multisystem proteinopathy (MSP).
Fig 10.
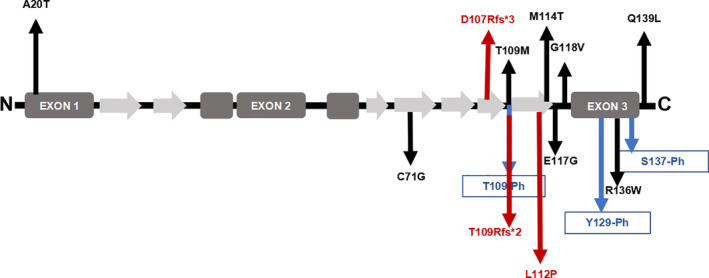
Profilin 1 protein structure and mutations linked to amyotrophic lateral sclerosis (ALS) or Paget's disease of bone (PDB). Schematic presentation of Profilin 1, including five α‐helices (dark gray) and seven β‐sheets (light gray). Eight of the ALS‐linked mutations are indicated by black arrows. Three amino acid residues with notable phosphorylation sites mutated in ALS are indicated by blue arrows. Three of the PDB‐linked mutations are indicated by red arrows.
MSP is an inherited pleiotropic degenerative disorder that can affect the muscle, bone, and nervous system.( 33 ) An operational definition of MSP is a combination of two or more of inclusion body myopathy (IBM), PDB, and ALS/frontotemporal dementia (FTD). Histopathologically, MSP‐affected tissues show ubiquitin‐positive inclusions that contain RNA‐binding proteins, such as TDP‐43, hnRNPA1, and hnRNPA2B1, but may also exhibit positive staining for proteins that mediate ubiquitin‐dependent autophagy, including p62/SQSTM1, VCP, optineurin, and ubiquilin‐2. At present, confirmed MSP pathogenic genes include SQSTM1, VCP, HNRNPA1, HNRNPA2B, and TDP43. As PFN1 mutations have been confirmed to be pathogenic for ALS, it is very likely that PFN1 mutations may also cause PDB. All known mutations of PFN1 in ALS are missense mutations. Mutations of the PFN1 gene causing PDB identified in our study include two truncating mutations and one missense mutation, and the L112P missense mutation has not been previously reported in ALS. None of the subjects in the present study displayed any ALS‐related symptoms. The effects of different mutation types on Profilin 1 function, as well as the mechanisms of different missense mutations resulting in different diseases, remain unclear. Further studies are needed to identify the underlying mechanisms.
In this study, we confirmed that a truncating mutation in PFN1 can accelerate osteoclast differentiation in animal models. However, the underlying molecular mechanism remains unclear. There are four hypotheses according to the mechanism of PFN1 mutations leading to enhanced osteoclast differentiation and bone resorption. First, studies have shown that mutated Profilin 1 induces accumulation of TDP‐43, which alters the activity of the NF‐κB signaling pathway as a subunit of NF‐κB/P65 coactivators in ALS.( 34 ) NF‐κB signaling activity has a direct effect on the differentiation and function of osteoclasts. However, we did not observe any difference in TDP‐43 expression between mutated and wild‐type osteoclasts in this study, which does not support the hypothesis of the accumulation of TDP‐43 in osteoclasts. Nevertheless, osteoclast precursor cells are of vital importance in the differentiation of osteoclasts, and accumulation of TDP‐43 in osteoclast precursor cells, but not osteoclasts, can alter NF‐κB pathway activity and lead to accelerated osteoclast differentiation. Further studies are needed to test this hypothesis. Second, the “actin ring” component Profilin 1 is an actin‐binding protein, and actin ring formation is a prerequisite for osteoclast bone resorption. The function of the actin ring in osteoclasts is to recombine its cytoskeleton to form a “sealing zone” structure located at the contact surface of osteoclasts and bone.( 35 ) The sealing zone serves to attach osteoclasts to bone and maintain the acidity of the resorbing lacunae. In the present study, protein structure modeling revealed that both a truncating mutation (c.318_321delTGAC) and a missense mutation (c.335 T > C) affect the structure of Profilin 1 bound to actin. Therefore, we hypothesize that PFN1 mutation might cause changes in the protein's binding to actin, affecting the formation of the actin ring and in turn impacting the process of osteoclast bone resorption. Third, regarding cell movement, recent studies have found that deletion of the PFN1 gene enhances osteoclast movement; although the number and volume of osteoclasts do not change significantly, the bone‐resorbing activity of osteoclasts does increase significantly.( 28 ) Profilin 1 normally controls cell movement by regulating the aggregation of actin. Therefore, we hypothesize that PFN1 mutation might enhance the movement of osteoclasts and thus increase bone‐resorbing activity. Last, Profilin 1 directly suppresses NF‐κB activity in the regulation of osteoclast differentiation and prevents degradation of the phosphatase PTEN, which is known to negatively regulate osteoclast differentiation.( 23 , 36 ) Therefore, PFN1 loss‐of‐function mutation may result in acceleration of osteoclastogenesis via simultaneous hyperactivation of NF‐κB signaling and elimination of the negative modulator PTEN.
In conclusion, we herein report three different mutations in the PFN1 gene, including two truncating mutations and one missense mutation, responsible for the early‐onset severe form of Paget's disease of bone complicated with GCT. A mouse model with a Pfn1‐truncating mutation was established and presented Paget's disease‐like phenotypes, thus confirming the loss of function of Profilin 1, which may activate the NF‐κB pathway as the cause of PDB/GCT development. In our study, we defined Pfn1 truncated mutant mice as a suitable model for studying PDB‐associated pathological mechanisms, which will tremendously help to unravel the etiology and pathomechanisms underlying early‐onset PDB in humans in the future. Meanwhile, our findings also expanded the pathogenic gene spectrum in PDB, and the identification of PFN1 mutation possesses great diagnostic value to identify PDB individuals predisposed to develop GCT. Further studies are needed to identify the clear mechanisms of PFN1 mutation in PDB/GCT.
Disclosures
All authors state that they have no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4275.
Supporting information
Appendix S1. Supplementary Information
Acknowledgments
We are grateful to the patients and their relatives for participating in the research. We thank the BGI‐Shenzhen company and the GENESKY company for whole‐exome sequencing and Sanger sequencing. This work was supported by the National Key R&D Program of China (2018YFA0800801), the National Natural Science Foundation of China (81974126 and 81770874), the Clinical Science and Technology Innovation Project of Shanghai Shenkang Hospital Development Center (SHDC12018120), and the Shanghai Key Clinical Center for Metabolic Disease, Shanghai Health Commission Grant (2017ZZ01013).
Authors' roles: Zhenlin Zhang and Hua Yue designed the study, and reviewed and approved the final manuscript. Zhe Wei investigated, analyzed data and wrote manuscript draft. Shanshan LI, Xiaohui Tao, Guoying Zhu, Zhenkui Sun, Zhangying Wei, Qiong Jiao, and Huizhen Zhang investigated and analyzed data. Zhenlin Zhang, Hua Yue, Baojie Li and Lin Chen take responsibility for the integrity of the data and the accuracy of the data analysis.
Author contributions: Zhenlin Zhang and Hua Yue: Designed the study. Zhe Wei: Conceptualization; data curation; formal analysis; investigation; methodology; validation; visualization; writing‐original draft. Shan‐shan Li: Methodology; software. Xiaohui Tao: Data curation; validation. Zhu Guoying: Methodology; resources; supervision. Zhenkui Sun: Methodology; resources. Zhanying Wei: Methodology; software. Qiong Jiao: Methodology; resources. Huizhen Zhang: Methodology; resources. Lin Chen: Formal analysis; funding acquisition; resources; supervision. Baojie Li: Formal analysis; methodology; resources; supervision; validation.
Contributor Information
Zhenlin Zhang, Email: zhangzl@sjtu.edu.cn.
Hua Yue, Email: yueyinglonghua@163.com.
References
- 1. Tuck SP. Adult Paget disease of bone: a tale of two guidelines. Rheumatology (Oxford). 2020;59:2197‐2198. [DOI] [PubMed] [Google Scholar]
- 2. Appelman‐Dijkstra NM, Papapoulos SE. Paget's disease of bone. Best Pract Res Clin Endocrinol Metab. 2018;32:657‐668. [DOI] [PubMed] [Google Scholar]
- 3. Chung PY, Van Hul W. Paget's disease of bone: evidence for complex pathogenetic interactions. Semin Arthritis Rheum. 2012;41:619‐641. [DOI] [PubMed] [Google Scholar]
- 4. Takata S, Hashimoto J, Nakatsuka K, et al. Guidelines for diagnosis and management of Paget's disease of bone in Japan. J Bone Miner Metab. 2006;24:359‐367. [DOI] [PubMed] [Google Scholar]
- 5. van Staa TP, Selby P, Leufkens HGM, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget's disease of bone in England and Wales. J Bone Miner Res. 2002;17:465‐471. [DOI] [PubMed] [Google Scholar]
- 6. Rendina D, De Filippo G, Ralston SH, et al. Clinical characteristics and evolution of giant cell tumor occurring in Paget's disease of bone. J Bone Miner Res. 2015;30:257‐263. [DOI] [PubMed] [Google Scholar]
- 7. Reddy SV. Etiology of Paget's disease and osteoclast abnormalities. J Cell Biochem. 2004;93:688‐696. [DOI] [PubMed] [Google Scholar]
- 8. Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582‐1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain‐specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum Mol Genet. 2002;11:2735‐2739. [DOI] [PubMed] [Google Scholar]
- 10. Nakatsuka K, Nishizawa Y, Ralston SH. Phenotypic characterization of early onset Paget's disease of bone caused by a 27‐bp duplication in the TNFRSF11A gene. J Bone Miner Res. 2003;18:1381‐1385. [DOI] [PubMed] [Google Scholar]
- 11. Whyte MP, Obrecht SE, Finnegan PM, et al. Osteoprotegerin deficiency and juvenile Paget's disease. N Engl J Med. 2002;347:175‐184. [DOI] [PubMed] [Google Scholar]
- 12. Naot D, Choi A, Musson DS, et al. Novel homozygous mutations in the osteoprotegerin gene TNFRSF11B in two unrelated patients with juvenile Paget's disease. Bone. 2014;68:6‐10. [DOI] [PubMed] [Google Scholar]
- 13. Saki F, Karamizadeh Z, Nasirabadi S, Mumm S, McAlister WH, Whyte MP. Juvenile paget's disease in an Iranian kindred with vitamin D deficiency and novel homozygous TNFRSF11B mutation. J Bone Miner Res. 2013;28(6):1501‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet. 2004;36:377‐381. [DOI] [PubMed] [Google Scholar]
- 15. Kim HJ, Kim NC, Wang YD, et al. Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Divisato G, Formicola D, Esposito T, et al. ZNF687 mutations in severe Paget disease of bone associated with giant cell tumor. Am J Hum Genet. 2016;98:275‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Divisato G, Scotto di Carlo F, Petrillo N, Esposito T, Gianfrancesco F. ZNF687 mutations are frequently found in Pagetic patients from South Italy: implication in the pathogenesis of Paget's disease of bone. Clin Genet. 2018;93:1240‐1244. [DOI] [PubMed] [Google Scholar]
- 18. Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14:461‐469. [DOI] [PubMed] [Google Scholar]
- 19. Ding Z, Bae YH, Roy P. Molecular insights on context‐specific role of profilin‐1 in cell migration. Cell Adh Migr. 2012;6:442‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Witke W, Podtelejnikov AV, Di Nardo A, et al. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 1998;17:967‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hein MY, Hubner NC, Poser I, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712‐723. [DOI] [PubMed] [Google Scholar]
- 22. Ertych N, Stolz A, Valerius O, Braus GH, Bastians H. CHK2‐BRCA1 tumor‐suppressor axis restrains oncogenic Aurora‐A kinase to ensure proper mitotic microtubule assembly. Proc Natl Acad Sci U S A. 2016;113:1817‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaidi AH, Manna SK. Profilin‐PTEN interaction suppresses NF‐kappaB activation via inhibition of IKK phosphorylation. Biochem J. 2016;473:859‐872. [DOI] [PubMed] [Google Scholar]
- 24. Lu H, Shamanna RA, de Freitas JK, et al. Cell cycle‐dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double‐strand break repair. Nat Commun. 2017;8:2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding Z, Joy M, Bhargava R, et al. Profilin‐1 downregulation has contrasting effects on early vs late steps of breast cancer metastasis. Oncogene. 2014;33:2065‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gronborg M, Kristiansen TZ, Iwahori A, et al. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics. 2006;5:157‐171. [DOI] [PubMed] [Google Scholar]
- 27. Zoidakis J, Makridakis M, Zerefos PG, et al. Profilin 1 is a potential biomarker for bladder cancer aggressiveness. Mol Cell Proteomics. 2012;11:M111.009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shirakawa J, Kajikawa S, Bottcher RT, et al. Profilin 1 negatively regulates osteoclast migration in postnatal skeletal growth, remodeling, and homeostasis in mice. JBMR Plus. 2019;3:e10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scotto di Carlo F, Pazzaglia L, Esposito T, Gianfrancesco F. The loss of profilin 1 causes early onset Paget's disease of bone. J Bone Miner Res. 2020;35:1387‐1398. [DOI] [PubMed] [Google Scholar]
- 30. Merlotti D, Materozzi M, Bianciardi S, et al. Mutation of PFN1 gene in an early onset, polyostotic Paget‐like disease. J Clin Endocrinol Metab. 2020;105:dgaa252. [DOI] [PubMed] [Google Scholar]
- 31. Wu CH, Fallini C, Ticozzi N, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alkam D, Feldman EZ, Singh A, Kiaei M. Profilin1 biology and its mutation, actin(g) in disease. Cell Mol Life Sci. 2017;74:967‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor JP. Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology. 2015;85:658‐660. [DOI] [PubMed] [Google Scholar]
- 34. Swarup V, Phaneuf D, Dupre N, et al. Deregulation of TDP‐43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB‐mediated pathogenic pathways. J Exp Med. 2011;208:2429‐2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson SR, Peters C, Saftig P, Bromme D. Cathepsin K activity‐dependent regulation of osteoclast actin ring formation and bone resorption. J Biol Chem. 2009;284:2584‐2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugatani T, Alvarez U, Hruska KA. PTEN regulates RANKL‐ and osteopontin‐stimulated signal transduction during osteoclast differentiation and cell motility. J Biol Chem. 2003;278:5001‐5008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary Information