

Abstract
The demand for iron is high in pregnancy to meet the increased requirements for erythropoiesis. Even pregnant females with initially iron‐replete stores develop iron‐deficiency anemia, due to inadequate iron absorption. In anemic females, the maternal iron supply is dedicated to maintaining iron metabolism in the fetus and placenta. Here, using a mouse model of iron deficiency in pregnancy, we show that iron recycled from senescent erythrocytes becomes a predominant source of this microelement that can be transferred to the placenta in females with depleted iron stores. Ferroportin is a key protein in the molecular machinery of cellular iron egress. We demonstrate that under iron deficiency in pregnancy, levels of ferroportin are greatly reduced in the duodenum, placenta and fetal liver, but not in maternal liver macrophages and in the spleen. Although low expression of both maternal and fetal hepcidin predicted ferroportin up‐regulation in examined locations, its final expression level was very likely correlated with tissue iron status. Our results argue that iron released into the circulation of anemic females is taken up by the placenta, as evidenced by high expression of iron importers on syncytiotrophoblasts. Then, a substantial decrease in levels of ferroportin on the basolateral side of syncytiotrophoblasts, may be responsible for the reduced transfer of iron to the fetus. As attested by the lowest decrease in iron content among analyzed tissues, some part is retained in the placenta. These findings confirm the key role played by ferroportin in tuning iron turnover in iron‐deficient pregnant mouse females and their fetuses.
1. INTRODUCTION
Pregnancy is one of the phases of the mammalian life cycle most frequently characterized by the risk of iron deficiency. Consistent with the role of iron in many biological processes occurring in every cell of a growing organism, this microelement is indispensable for normal fetal development. Gestational iron deficiency anemia (IDA), a pathology resulting from the lack of iron, has been associated with pre‐term delivery, low birth weight and adverse pregnancy outcomes. 1 The causes of IDA in pregnancy are the especially high physiological demand for iron (iron requirements are increased nearly 10‐fold during pregnancy 2 ) and insufficient iron supply due to both low maternal reserves and inadequate dietary intake. The additional iron needs in pregnancy result mainly from the high rate of maternal erythropoiesis and red blood cell (RBC) expansion, as well as rapid embryonic/fetal growth and the formation of the placenta. 3 The use of iron supplements is strongly recommended during pregnancy, but this supposedly simple therapy is complicated by the need to consider dosing schedules that depend on the amount of supplemental iron, gestational age and the severity of anemia. 4 To address these issues, a better understanding of the handling of molecular iron in pregnancy is required. Moreover, in contrast to the well‐studied regulation of iron balance in adults or even in neonates, 5 the molecular mechanisms controlling iron movement between the maternal and fetal compartments at different stages of pregnancy have received little attention until quite recently. 6 For many years it was thought that iron metabolism during pregnancy is adjusted to meet the essential iron requirements of the fetus at the expense of the mother. 7 , 8 This means that much of the iron present in the maternal blood plasma in complex with transferrin is transferred across the placenta to the fetal circulation and delivered to sites of utilization in the fetus. However, it was recently proposed that iron entering the placenta is retained in this tissue during iron deficiency to be then transferred to the fetus. 6 The maternal blood plasma iron pool is replenished from three main sources: hepatocytes releasing iron stored in ferritin, reticuloendothelial macrophages recycling iron derived from senescent erythrocytes, and enterocytes of the proximal duodenum absorbing iron contained in the diet. A key participant in all these processes is ferroportin (Fpn), a transmembrane protein that transfers iron to plasma apo‐transferrin and the only iron exporter known in mammalian cells. 9 , 10 Furthermore, Fpn located on the basolateral membrane of syncytiotrophoblasts has been identified as the protein responsible for iron transport from the placenta to the fetal circulation. 11 , 12 The importance of Fpn in this process has been clearly demonstrated in mice by global and selective inactivation of the ferroportin (Slc40a1) gene. 11 A complete knockout resulted in no Fpn expression in the extraembryonic visceral endoderm (which is responsible for nutrient transfer from mother to fetus between days 5 and 10 of gestation) and early (E7.5) embryonic lethality. On the other hand, disruption of the Slc40a1 gene in all tissues except the extraembryonic visceral endoderm and placenta did not result in embryonic death. 11 Multiple regulatory mechanisms are involved in the control of Fpn expression. 10 , 13 Interestingly, regulation in response to inorganic iron involves competing control mechanisms, as exemplified by the post‐transcriptional induction of Fpn synthesis through the IRP/IRE system 14 and inhibition of its expression at the post‐translational level, depending on the interaction with hepcidin. 15 Such bidirectional Fpn regulation highlights its crucial role in fine tuning systemic iron bioavailability.
In the present study, we exposed pregnant mouse females and their E18.5 fetuses to severe nutritional iron deficiency and analyzed Fpn expression and its localization in sites critical for materno‐fetal iron movements. Under iron‐deficiency conditions, the level of Fpn protein was uniformly down‐regulated by about 50% compared to normal iron conditions in maternal duodenal enterocytes, the placenta and fetal liver. However, only a slight decrease occurred in maternal hepatic and splenic expression of Fpn, mainly localized in Browicz‐Kupffer cells and splenic macrophages. In the absence of negative regulation, resulting from drastically lowered maternal and fetal hepcidin, the Fpn level in the examined tissues was very likely dependent on intrinsic iron content.
In the light of this finding, we postulate that iron retrieved from phagocytosed senescent erythrocytes maintains Fpn expression in macrophages, and is then efficiently recycled to the bloodstream and preferentially delivered to the placenta at the expense of the mother. A certain part of this iron is retained in the placenta as evidenced by the relatively small drop in the placental iron content, while the rest is likely transferred to the fetal circulation by the remaining Fpn in the basolateral membrane of syncytiotrophoblasts.
2. METHODS
2.1. Experimental animals and tissue collection
Experiments were performed using 5‐month‐old pregnant female mice (strain B6; 129S7) bred in the Department of Molecular Biology, Institute of Genetics and Animal Biotechnology of the Polish Academy of Sciences. All mice were housed at constant temperature (22°C) under artificial light (12‐hour photoperiod). Two weeks prior to mating with males of the same strain, females were fed control or iron‐deficient diets (Harlan Laboratories, Madison, WI, USA) containing 48 and 4 ppm iron, respectively. The female mice were maintained on the same diets throughout pregnancy. For timed matings, the morning plug was identified and was considered E0.5. Plugged females were sacrificed by isoflurane overdose at E18.5. In a few experiments, non‐pregnant females exposed to the same dietary iron regimes were used. Blood samples were taken from the inferior vena cava of females using heparin‐coated syringes. The blood was collected in heparin‐coated tubes and centrifuged at 800 g/4°C/10 min. Plasma samples were then frozen in liquid nitrogen and stored at −80°C for later analysis. Fetuses were dissected from the uterus, sacrificed by decapitation and their livers were collected and frozen. Placentas from fetuses, chosen from each mother at random, were rapidly dissected and frozen in liquid nitrogen, before being stored at −80°C. Tissue samples (maternal and fetal livers, maternal duodenum, and placenta) for immunofluorescence analysis were fixed in 4% paraformaldehyde in PBS. Bone marrow cells for total cellular RNA extraction were flushed out from the femur of females with ice‐cold Hanks balanced salt solution. All procedures were approved by the 2nd Local Ethical Commission (permission number WAW2/53/2016).
2.2. Measurement of red blood cell indices, blood plasma iron parameters and non‐heme iron content in tissues
The red blood cell (RBC) count, hemoglobin concentration (HGB), hematocrit (HCT) and mean cell volume (MCV) were determined using an automated hematology analyser (Abacus Junior Vet 5; Diatron, Budapest, Hungary). The plasma iron concentration and total iron‐binding capacity (TIBC) were evaluated using an assay based on the colorimetric measurement of an iron‐chromazurol complex (absorbance at 630 nm) according to the manufacturers's protocol (Alpha Diagnostic, Poland). The percentage transferrin saturation (TSAT) was then calculated. The non‐heme iron content of liver and placenta fragments (100 mg) and of spleen fragments (40 mg) was determined by acid digestion of the samples at 100°C for 10 min., followed by colorimetric measurement of an iron‐ferrozine complex (absorbance at 560 nm) as described previously. 16
2.3. Real‐time quantitative PCR (RT‐qPCR)
Total cellular RNA was extracted from liver, bone marrow, kidney and placental tissue (20 mg) using Trizol reagent (Invitrogen) according to the manufacturer's protocol. Two micrograms of DNase‐treated total RNA were reverse transcribed using a Transcriptor First‐Strand cDNA Synthesis Kit (Roche, Switzerland). This cDNA was used as the template for real‐time quantitative PCR analysis with gene‐specific primer pairs (Table S1), performed in a Light Cycler U96 (Roche Diagnostics, Mannheim, Germany). The amplified products were detected using SYBR Green I (Roche Diagnostics). To confirm amplification specificity, the PCR products were subjected to melting curve analysis and agarose gel electrophoresis. Light Cycler U96 Software was used for data analysis. Transcript levels were normalized relative to the control reference genes selected using NormFinder software v0.953 (https://moma.dk/normfinder-software).
2.4. Western blot analysis
For the detection of ferroportin (Fpn) and L‐ferritin (L‐Ft) protein levels, membrane and cytosolic extracts, from maternal liver, spleen, duodenum, fetal liver, and from the placenta, were prepared as previously described 17 and separated by electrophoresis on 10% and 14% SDS‐PAGE gels, respectively. Electroblotting of the resolved proteins (100 μg) onto polyvinylidene difluoride membrane (Millipore), blocking and incubation with primary antibodies were performed as previously described. 18 The following primary antibodies were used: a rabbit antiserum raised against recombinant mouse L‐Ft, (kindly provided by Dr. P. Santambrogio, Department of BioTechnology, San Raffaele Scientific Institute, Milan, Italy), and a rabbit polyclonal anti‐Fpn antibody (Alpha Diagnostic), affinity purified rabbit polyclonal anti‐mouse Fpn antibody kindly provided by Dr. François Canonne‐Hergaux (INSERM UMR 1043, Centre de Physiopathologie de Toulouse Purpan, Toulouse, France), transferrin receptor 1 monoclonal antibody (Thermo Fisher), anti‐human Natural Resistance‐Associated Protein 2 (DMT1/NRAMP2), (Alpha Diagnostic Intl. Inc). Membranes were then washed and incubated with peroxidase‐conjugated anti‐rabbit secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature (20°C). Immunoreactive bands were detected using the ECL (enhanced chemiluminescence) Plus Western blotting detection system (Amersham Life Sciences). Band intensities were quantified by densitometric analysis using Quantity One software, v.4.6.6 (Bio‐Rad).
2.5. Immunofluorescence (IF) analysis and confocal microscopy of liver, placenta and duodenum sections
After laparotomy, livers and placentas from pregnant mothers (E18.5) were immediately dissected and fixed in 4% paraformaldehyde (Sigma) in phosphate‐buffered saline (PBS) at 4°C for 24 h. Following two 30‐minutes washes in PBS, the tissues were successively soaked in 12.5% and 25% sucrose (Bioshop) at 4°C for 2 h and 7 days, respectively. Hepatic and placental samples were then embedded in Cryomatrix medium (Thermo Scientific), frozen in liquid nitrogen and sectioned in 20‐μm slices using a cryomicrotome (Shandon). The sections were washed in PBS for 10 min and permeabilized by bathing in PBS/0.1% Triton X‐100 (Sigma) for 20 mins. Non‐specific antibody binding was blocked by incubating the tissue sections in PBS/3% BSA (Bioshop) at room temperature (RT) for 1.5 h. For protein detection, sections were incubated overnight at RT with primary antibodies diluted in PBS/3% BSA. As a negative control, some sections were incubated without primary antibody. The primary and fluorochrome‐conjugated secondary antibodies used in IF analysis are described in Table S2. Next, the sections were washed for 5 × 6 min with PBS/0.1% Triton X‐100 and incubated for 1.5 h at RT with secondary antibody diluted in PBS/3% BSA. Finally, sections were washed for 10 min in PBS and mounted in Vectashield medium with DAPI (Vector Labs). The presence of Fpn in Browicz‐Kupffer cells (liver macrophages) was determined by double immunofluorescence staining of the investigated protein and the macrophage marker F4/80. In this case, liver sections were first incubated with anti‐Fpn followed by anti‐F4/80 primary antibody, and then they were incubated with a mixture of secondary antibodies conjugated with different fluorochromes: Cy3 for Fpn and Alexa488 for F4/80. Note, IF was analysed with a Zeiss LSM 710 confocal microscope (Carl Zeiss) using a × 40 objective and Zeiss ZEN software.
2.6. ImageJ analysis of immunofluorescence images
ImageJ analysis was performed to characterize IF produced using the ferroportin antibody. Samples from each group of animals were prepared for analysis at the same time. Liver and placenta sections from three different females per experimental group were examined by confocal microscopy at 40 × magnification. For quantitative comparisons between experimental groups, the same acquisition parameters were applied to capture images within the same experimental set. Subsequently, ImageJ software (NIH, Bethesda) was used to measure mean fluorescence in each tubule section. The signal intensity was manually quantified to generate a Mean Gray Value: the sum of grey values in the selected area divided by the number of pixels within that area. For each experimental group, 30 measurements of livers and placentas were made, that is, 10 measurements per female.
2.7. Measurement of blood plasma erythroferrone (ERFE) level
An indirect ERFE ELISA kit designed for quantification of mouse erythroferrone in serum was purchased from Intrinsic Lifesciences The BioIron Company (catalog no. SKU# ERF‐200). Serum samples, standards and controls mixed with sample diluent (100 μL/well) were incubated for 1 h with immobilized polyclonal antibody in the pre‐coated plate. The plate was than washed and unbound mouse ERFE was removed. A second anti‐mouse Erfe antibody conjugated with biotin was added for 1 h, washed and followed with HRP‐SA conjugated which binds to biotinylated anti‐mouse erythroferrone for the next 30 min. After washing the enzyme‐TMB reaction formed a blue colored complex within 15 min. The reaction was terminated by the addition of a stop solution, which turned the reaction mixture yellow. Colorimetric measurement (absorbance at 450 nm) was performed using a microplate reader (ELx800 BIO‐TEK INSTRUMENTS. Inc, Winoski, VT, USA). The intensity of the yellow color was proportional to the Erfe concentration in the samples. A standard curve was prepared by plotting the log concentration of standard curve vs its corresponding OD (450).
2.8. Statistical analysis
Statistical analysis was performed using ANOVA and Kruskal‐Wallis ANOVA tests (for parametric and non‐parametric data distributions, respectively) combined with proper post‐hoc tests (Tukey and Dunn tests, respectively) with p ≤ .05 (*), p ≤ .01 (**) and p ≤ .001 (***) being considered statistically significant and highly significant, respectively.
3. RESULTS
3.1. Iron deficiency anemia (IDA) in pregnant females fed a low‐iron diet
Exposure of pregnant rodent females to an iron‐poor diet is a common procedure used to induce iron deficiency and iron‐deficiency anemia in pregnancy. 6 , 19 In this study we found that pregnant female mice subjected to dietary iron restriction had considerably decreased iron parameters in the blood plasma, such as iron concentration and transferrin saturation, compared with those fed a control diet (Figure S1). Consequently, pregnant iron‐deficient females manifested symptoms of microcytic anemia characterized by significantly diminished RBC count, hemoglobin concentration, hematocrit and MCV values (Table S3). Pregnancy in females fed the control diet, and iron deficiency in non‐pregnant females led to deterioration of RBC status, although to a lesser degree than in iron‐deficient mothers (Table S3).
3.2. Decreased hepatic iron stores in iron‐deficient pregnant females and their fetuses
Depletion of iron stores in the bone marrow and the liver remains the most definitive test for differentiating iron‐deficiency anemia from other microcytic anemias. 20 Measurement of the hepatic non‐heme iron content in anemic pregnant females revealed a five‐fold decrease compared with iron‐replete animals (Figure S2(A)). Unsurprisingly, the level of iron in the livers of fetuses from anemic mothers was also lowered; however, this decrease was only three‐fold. Ferritin is a major iron‐storage protein and its expression is highly positively correlated with cellular and tissue iron status. 21 Hepatic ferritin in mammals is mostly comprised of L‐chains (ca. 80%) and therefore the L‐ferritin level in the liver reflects hepatic iron abundance. The assessment of L‐ferritin protein levels in maternal and fetal livers by Western blotting largely confirmed the results of direct measurement of iron content. Iron‐deficient mothers and their fetuses both showed drastic reductions in the level of L‐ferritin, but again, this decrease was much greater in the former than the latter (Figure S2(B)).
3.3. Hepatic hepcidin and BMP6 mRNA levels in iron‐deficient females and fetuses
Hepcidin is a small peptide hormone produced by hepatocytes that orchestrates body iron fluxes (including iron transfer across the placenta) by adjusting iron supply to body iron requirements. Hepcidin binds to Fpn to induce its degradation, thus inhibiting iron release from exporting cells. 22 Its expression is controlled by iron levels (circulating and liver iron content), erythropoietic activity and inflammatory cues. 23 In pregnancy, maternal and fetal hepcidins are regulated by both maternal and fetal iron conditions. 24 To assess the functioning of the hepcidin‐ferroportin regulatory axis under iron deficiency in pregnancy, we examined hepcidin mRNA abundance in maternal and fetal livers, comparing the obtained results with hepcidin transcript levels in the livers of iron‐deficient non‐pregnant females (Figure 1(A)). In all examined iron‐deficient subjects, hepcidin mRNA expression was down‐regulated compared to the respective controls. Importantly, even in iron‐replete fetuses the hepcidin transcript was barely detectable. Comparison of hepcidin expression in pregnant and non‐pregnant females receiving the standard iron diet demonstrated that pregnancy triggers a nearly three‐fold drop in hepcidin mRNA abundance. Hepatic hepcidin expression in hepatocytes is activated predominantly at the transcriptional level by a member of the TGFβ superfamily, bone morphogenetic protein 6 (BMP6), 25 the expression of which is induced by iron in non‐parenchymal cells of the liver. 26 To establish the relationship between the expression of this activator and that of its target gene hepcidin, we examined BMP6 mRNA levels in the liver samples. In both iron‐deficient non‐pregnant and pregnant females, hepatic BMP6 expression was substantially reduced to 24% and 55% of the levels found in the respective iron‐replete controls (Figure 1(B)), and corresponded to the similar decreases seen in hepcidin mRNA expression. As in the case of hepcidin, pregnancy caused a three‐fold decrease in BMP6 transcript levels in animals receiving the standard iron diet. We observed no differences in hepatic BMP6 mRNA level between fetuses according to their iron status, which was consistent with their very low levels of hepcidin mRNA.
FIGURE 1.

Decrease in hepatic hepcidin and BMP6 mRNA levels in iron‐deficient non‐pregnant females, pregnant females and their E18 fetuses. RT‐qPCR analysis of hepcidin (A) and BMP6 (B) transcript levels in livers. The histograms display the relative hepcidin and BMP6 mRNA levels in arbitrary units (means ± S.D.). Liver samples were obtained from eight pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. For fetuses, values were calculated for three sets of pooled fetal liver samples obtained from three pregnant females of each group. The histogram displays the relative hepcidin and BMP6 mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (*p < .05, **p < .01, ***p < .001)
3.4. Increased expression of erythroid inhibitors of hepcidin in iron‐deficient pregnant females
Erythropoietic regulation of hepcidin is one of the major signaling pathways controlling hepatic hepcidin expression. 27 Enhancement of the erythropoiesis induced by increased erythropoietin (Epo) produced in kidney, causes suppression of hepcidin to increase iron availability for hemoglobin synthesis in erythroblasts. Assessment of Epo mRNA expression in the kidneys of anemic mothers by Real‐Time PCR revealed a substantial increase compared to control ones, but also to anemic non‐pregnant females (Figure S3(A)). Among the erythroid regulators that reduce hepcidin expression during expanded erythropoiesis, erythroferrone (ERFE) has been identified as a soluble factor produced and released by erythroblasts after bleeding or erythropoietin treatment. 28 To investigate the possible contribution of ERFE to the negative regulation of hepcidin in pregnant females fed a low‐iron diet, we measured its expression at the mRNA level in bone marrow as well as its concentration in blood plasma. We found that both indexes of ERFE expression were dramatically elevated compared to pregnant females receiving the standard iron diet and non‐pregnant iron‐deficient females (Figure S3(B) and (C)). Apart from ERFE, it has been proposed that other erythroid factors, including GDF15, 29 suppress hepcidin during erythropoiesis. Although this regulatory function of GDF15 has been questioned, 30 we found that GDF15 mRNA expression in bone marrow was significantly increased in iron‐deficient pregnant females (Figure S3(D)), suggesting that it may play a role in the negative regulation of hepcidin expression.
3.5. A slight decrease in hepatic and splenic ferroportin in iron‐deficient mothers vs a prominent decrease in other tissues
Ferroportin (Fpn), the sole exporter of non‐heme iron known to date, transfers cellular iron to apo‐transferrin, which then transports this microelement via the blood to various tissues in the body, including the bone marrow. 10 Ferroportin is particularly abundant in hepatic and splenic macrophages, where it exports iron recycled from senescent erythrocytes, a process of critical importance for sustaining physiological erythropoiesis. 31 Assessment of Fpn levels in the livers of anemic mothers by Western blotting revealed a relatively small decrease (14%) compared to control females (Figures 2(A) and (B)). A much greater decrease in hepatic Fpn protein abundance (>50%, Figure 2(B)) was detected in iron‐deficient E18.5 fetuses compared to the controls.
FIGURE 2.
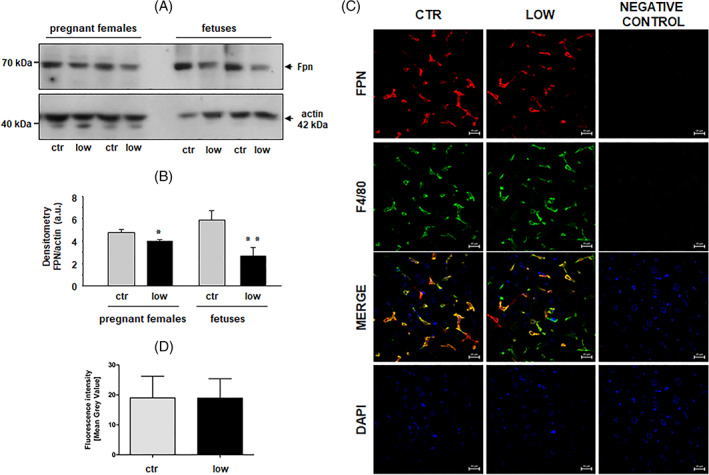
Differential decrease in hepatic ferroportin (Fpn) expression in pregnant females fed a low‐iron diet and in their E18 fetuses. (A) Western blot analysis of Fpn protein levels in liver membrane fractions prepared from pregnant females (individuals) of the iron‐replete (ctr) and iron‐deficient (low) groups, and their fetuses (pooled samples). A representative immunoblot is shown (B) Immunoreactive Fpn bands from separate blots performed on hepatic membrane extracts obtained from four females and three sets of pooled fetal liver samples obtained from three pregnant females of each group were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant differences are indicated (*p < .05, **p < .01). (C) Immunofluorescent staining of Fpn (red channel; top panel) and macrophage marker F4/80 (green channel; second panel from the top) in livers obtained from pregnant females of each group analyzed by confocal microscopy. Co‐localization of Fpn and F4/80 is shown in the merged image (second panel from the bottom) and the location of nuclei is disclosed by counterstaining with DAPI (bottom panel). To confirm the specificity of Fpn and F4/80 detection, liver sections of pregnant females were incubated with only the respective secondary antibodies. No staining was detected in these negative controls (third column). (D) Quantitative analysis of the Fpn fluorescent signal in female livers. Immunofluorescence in 30 liver sections from pregnant females of each group was quantified manually by ImageJ analysis and Fpn signal intensities as Mean Gray Values (means ± S.D.) are plotted in arbitrary units (a.u.). Scale bars correspond to 20 μm [Color figure can be viewed at wileyonlinelibrary.com]
Immunofluorescent (IF) co‐localization studies of Fpn and F4/80, a well‐known macrophage marker, 32 demonstrated that Fpn was mainly located in Browicz‐Kupffer cells in the livers of both control and iron‐deficient pregnant females (Figure 2(C)). Comparative quantitative analysis of the Fpn IF signal using ImageJ revealed no significant difference in the expression of Fpn between livers of control and iron‐deficient pregnant females (Figure 2(D)). Considering that spleen (ie, splenic macrophages) largely participates in recycling of iron from senescent RBC, we also determined Fpn protein level by Western blotting in the spleen of anemic mothers. Despite a strong decrease in splenic iron content (Figure S4(A)) we found no significant changes in Fpn level compared neither to pregnant females fed control diet nor to females from other experimental groups (Figure S4(B) and (C)). Taken together, the results of both Western blotting and IF suggested that the drop in hepatic and splenic Fpn level in anemic mothers is at best marginal. To check the role of duodenal Fpn in iron delivery to the circulation in pregnant females receiving the low‐iron diet, we assessed its localization on absorptive enterocytes in the duodenum and then quantified the intensity of the IF signal. In the duodenum of females from both experimental groups, Fpn was located along the basal and lateral membranes of absorptive enterocytes (Figure 3(A) and (B)). However, the intensity of the Fpn IF signal was much lower (46%) in the duodenum of iron‐deficient pregnant females compared to those fed the standard iron diet (Figure 3(C)). Western blot analysis of Fpn protein level in the duodenum of iron‐deficient, anemic mothers revealed even greater decrease (52%) compared to mothers fed control iron diet (Figure 3(D) and (E)).
FIGURE 3.

Substantial decrease in duodenal ferroportin (Fpn) protein level in pregnant females fed a low‐iron diet. (A) Immunofluorescent staining of Fpn in the duodenum of pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups analyzed by confocal microscopy. To confirm the specificity of Fpn detection, the duodenum sections were incubated with only the secondary antibody. No Fpn staining was detected in these negative controls. Counterstaining of nuclei was performed with DAPI. Scale bars correspond to 20 μm. (B) High magnification image of duodenal enterocytes showing clear basolateral localization of Fpn. (C) Quantitative analysis of the Fpn fluorescent signal in duodenum. Immunofluorescence in 30 duodenum sections from pregnant females of each group was quantified manually by ImageJ analysis and Fpn signal intensities as Mean Gray Values (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (***p < .001). D, Western blot analysis of Fpn protein levels in duodenum membrane fractions prepared from pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. Fe‐NTA BMDM, ferric nitrilotriacetate (100 μM) treated mouse bone marrow‐derived macrophages E, Immunoreactive Fpn bands from the blot shown in D were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant differences are indicated (*p < .05, ***p < .001). gray bars – non‐pregnant females; black bars – pregnant females [Color figure can be viewed at wileyonlinelibrary.com]
The placenta serves as the interface between mother and fetus, and it mediates nutrient transport to the latter, including the transport of iron. The uni‐directional transfer of iron taken up by the placenta to the fetal circulation is effected by Fpn located on the basolateral membrane of syncytiotrophoblasts. 12 In the placentas of iron‐deficient females, the Fpn level determined by Western blotting (Figure 4(A)) was decreased by about 42% compared to iron‐replete females (Figure 4(B)). Strong down‐regulation of Fpn was also clearly shown by IF analysis of placental sections from females receiving the iron‐deficient diet (Figure 4(C)). Microscopic analysis of IF staining indicated typical localization of Fpn in the basal (fetal facing) membrane of placental syncytiotrophoblasts (Figure 4(C)). Interestingly, the fall in iron content in the placentas of iron‐deficient females was the lowest (only two‐fold) among the three tissues examined in our study, that is, maternal and fetal liver, and placenta (Figure 4(D)).
FIGURE 4.
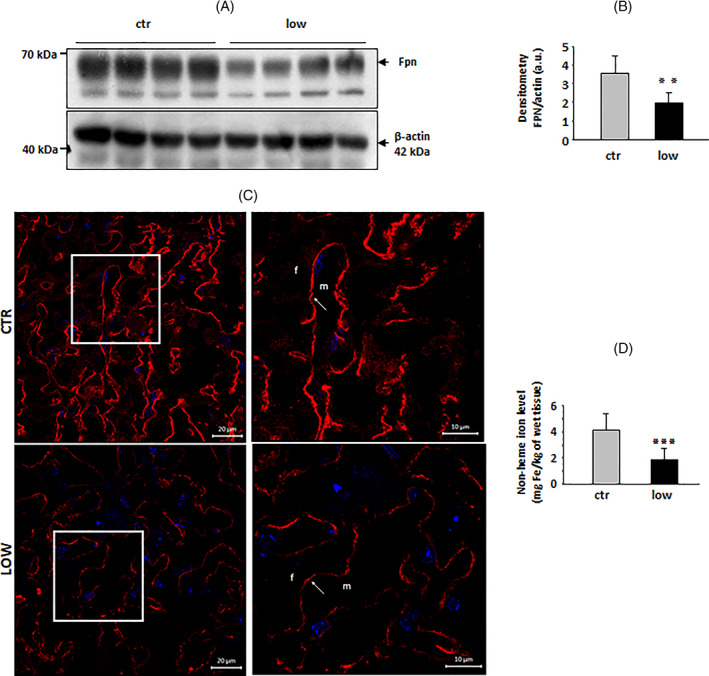
Decreased expression of ferroportin (Fpn) in the placenta of females fed a low‐iron diet. (A) Western blot analysis of Fpn protein levels in placenta membrane fractions prepared from pregnant females (individuals) of the iron‐replete (ctr) and iron‐deficient (low) groups. A representative immunoblot is shown. (B) Immunoreactive Fpn bands from separate blots performed on placental membrane extracts obtained from four females of each group were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (**p < .01). C Immunofluorescent staining of Fpn in the placenta of pregnant females of each group analyzed by confocal microscopy. Cell nuclei were counterstained with DAPI (blue). Scale bars correspond to 20 μm. The right‐hand panels show high magnification images of stained placenta (areas boxed in the left‐hand panels), confirming the basolateral localization of Fpn in syncytiotrophoblasts. Scale bars correspond to 10 μm. m, maternal compartment. f, fetal compartment; D, Non‐heme iron content in placenta samples obtained from six pregnant females of each group (means ± S.D.). Significant difference is indicated (***p < .001) [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Increased expression of iron importers in the placenta of iron‐deficient females
While Fpn mediates unidirectional transport of iron accumulated in syncytiotrophoblasts to the fetal circulation, transferrin receptor 1 (TfR1) and divalent metal transporter 1 (DMT1) are responsible for iron loading to these cells from the maternal circulation. 12 In the placenta of iron‐deficient females the expression of these two iron importers was strongly up‐regulated at the mRNA (Figures S5(A) and S6(A)) and protein (Figures S5(C),(D) and S6(C),(D)) levels, consistent with the paradigm of preferential iron transport across the placenta to satisfy fetal iron requirements 7 , 8 or placenta requirements as recently assumed. 6 Immunofluorescent detection of TfR1 and DMT1 in placental sections (Figures S5(B) and S6(B), respectively) showed clearly enhanced fluorescent signals for these proteins localized on the apical side of syncytiotrophoblasts facing the maternal circulation. Interestingly, while TfR1 displayed exclusively linear localization on the apical membrane (Figure S5(B)), DMT1 was also detected in intracellular vesicles in the form of discrete dots (Figure S6(B)). This localization pattern suggests that, according to a well‐characterized mechanism, DMT1 participates in the transport of iron liberated from transferrin within the endosome to the cytoplasm. 33
4. DISCUSSION
Pregnancy is a physiological condition frequently associated with iron deficiency anemia. 1 During pregnancy, iron needs are greatly increased because of the expansion of maternal red blood cell (RBC) mass, and growth of the placenta and the fetus. Efficient mobilization of iron from maternal hepatic stores is required to satisfy the iron requirements of both the mother and fetus. Relatively little is known about the regulation of iron fluxes between the iron‐deficient mother, placenta and fetus. In particular, there is a dearth of knowledge concerning gestational control of the expression of Fpn, the sole cellular iron exporter, operating not only in maternal and fetal compartments, but also at the maternal‐fetal interface, that is, the placenta. 10 , 12 Therefore, we performed a comprehensive evaluation of Fpn expression and distribution in the maternal duodenum, liver, spleen and in the placenta: tissues with a high potential to release iron. We also determined the expression of Fpn in the fetal liver. This analysis was performed on subjects under conditions of dietary iron restriction, which in pregnant females led to drastic decreases in both plasma iron concentration and transferrin saturation, and consequently a decline in RBC indices. Severe iron deficiency in pregnant mothers was also confirmed by substantial decreases in hepatic and splenic iron content and hepatic L‐ferritin protein level. Unsurprisingly, maternal iron deficiency caused a strong reduction in hepcidin, a liver peptide that negatively regulates dietary iron absorption and iron release from macrophages and hepatocytes by binding and degrading Fpn. 15 Suppression of hepcidin has been observed in normal and iron‐deficient pregnancies, 24 and is perceived as a means of regulating iron mobilization from stores to meet requirements for the expansion of maternal red cell mass, and placental and fetal growth. A possible mechanism underlying this modulation may involve down‐regulation of bone morphogenetic protein 6 (BMP6), a main activator of hepcidin, 25 expressed predominantly in hepatic endothelial sinusoidal cells and modulated by iron fluctuations. 26 Interestingly, in comparison with iron‐replete non‐pregnant females, hepatic BMP6 mRNA levels declined to the same extent in both non‐pregnant iron‐deficient females and in pregnant females fed the standard iron diet. The restriction of dietary iron in pregnancy did not induce any further reduction in maternal hepatic BMP6 expression. These data indicate that even with an adequate dietary iron supply during pregnancy, BMP6‐dependent signaling pathways regulating hepcidin are strongly attenuated to increase overall iron availability. It has been reported that apart from iron scarcity, hepcidin expression can also be suppressed by secreted erythroid factors, such as erythroferrone (ERFE) 28 and GDF15 29 thereby increasing iron availability for hemoglobin synthesis. Kautz and co‐workers described how ERFE synthesis is promoted by both erythropoietin (EPO) injection and expanded erythropoiesis caused by bleeding. 28 An increase in erythropoietic activity in the later stages of pregnancy appears to depend upon elevated plasma EPO levels and increases ERFE expression by erythroblasts. 34 Indeed, in iron‐deficient pregnant females, we observed a concerted regulations: increased EPO mRNA expression in the kidney and both dramatic rise in ERFE transcript level in bone marrow accompanied by highly elevated blood plasma concentrations of ERFE, which can lead to hepcidin suppression. Similarly, in the bone marrow cells of these animals we detected increased expression of GDF15, another inhibitor of hepcidin expression, originally reported to be secreted at high levels during erythroblast maturation. 29 Thus, our results suggest that the inhibition of maternal hepcidin in pregnancy is mediated by both iron deficiency and erythroid factors. The post‐translational regulation of Fpn by hepcidin means that in the absence of this peptide hormone or where levels are low, the concentration of Fpn on the cell surface is high. 15 In the case of absorptive enterocytes the lack of this negative regulation results in enhanced absorption of dietary iron, a phenomenon that is central to the pathophysiology of hereditary hemochromatosis. 35 However, it is important to bear in mind that apart from hepcidin‐mediated control, Fpn expression also depends on intracellular downstream regulatory circuits mediated by heme and iron. 36 , 37 These intrinsic adjustments determine a basal level of Fpn on the cell membrane, which is then susceptible to regulation by Hepc. In iron‐deficient pregnant mothers, Fpn expression was strongly down‐regulated in the duodenum and only slightly reduced or unchanged in the liver and spleen, respectively. We hypothesize that despite hepcidin deficiency, Fpn levels in the basolateral membrane of enterocytes are highly depressed as a result of post‐transcriptional IRP‐dependent regulation in response to low intra‐enterocytic iron levels caused by an iron‐deficient diet. 38 Where intra‐enterocytic iron concentrations are low, the iron absorption genes including Fpn are transcriptionally induced by stabilized hypoxia‐inducible factor 2α (HIF2α). 39 On the other hand, under these conditions, de novo synthesis of HIF2α is subject to IRP‐mediated translational inhibition due to the presence of an IRE in the 5’UTR of the HIF2α mRNA. 40 With prolonged iron deficiency this IRP‐dependent regulation can prevail and contributes to the decrease in Fpn. It should be underlined that Fpn transcript containing IRE sequence (and thus amenable to regulation by IRPs) accounts only for 25% of total Fpn mRNA in duodenum. 41 With regard to maternal hepatic Fpn, our IF colocalization and Western blot analyses clearly demonstrated that it is mainly expressed in liver macrophages (Browicz‐Kupffer cells), and this expression is only slightly decreased by an iron‐deficient diet. This may be due to the specificity of iron metabolism in Browicz‐Kupffer cells, which play an essential role in iron recycling through the phagocytosis of senescent RBCs, and thus continuously acquire iron in large amounts. 37 , 42 Although overall iron content in the liver was strongly decreased, it seems that this more accurately reflects the iron status in hepatocytes. In macrophages, Fpn is sequentially up‐regulated by heme‐dependent transcriptional induction followed by post‐transcriptional IRP/IRE system regulation mediated by iron continuously released by heme catabolism, as described previously. 37 Importantly, under absolute iron‐deficiency conditions, hypochromic and microcytic erythrocytes have a shortened lifespan coupled with accelerated reticuloendothelial cell sequestration once released into the circulation. 43 We therefore postulate that during severe maternal iron deficiency, iron recycling into the plasma through Fpn by erythrophagocytosing hepatic and splenic macrophages is the only efficient pathway of iron delivery for the mother and her fetuses.
Animal model studies indicate that a hierarchy of iron delivery during pregnancy is established in order to maintain fetal iron levels at the expense of the mother. 7 , 8 Despite this priority, a sufficient supply of iron to the fetus is difficult to maintain and iron deficiency can, lead to neonatal morbidity with an increased risk of prematurity, low birth weight and deleterious effects on brain development. 44 , 45 Recently, it was also demonstrated that under conditions of iron deficiency in pregnancy, this microelement is preferentially retained in the placenta at the expense of the fetus in order to maintain iron‐dependent placental processes that are indispensable for the overall function of this organ. 6 Our results indicate the strong potential of the placenta to take up iron from the maternal circulation as visualized by the high level expression of TfR1 and DMT1, proteins involved in iron uptake, localized to the apical membrane of the placental syncytiotrophoblasts. 12 Similar results have been reported for the placenta of iron‐deficient female rats. 46 In contrast, the level of Fpn expressed at the basal membrane 12 was only half that observed in control, iron‐replete pregnant mice. This expression pattern of iron transport proteins in pregnant females under severe iron deficiency only causes a moderate reduction in placental iron content, which is much less drastic than the decrease observed in both maternal and fetal livers and maternal spleens. This observation largely confirms recent results showing mildly decreased placental iron level at E18.5 in iron‐deficient pregnant females compared to iron replete ones. 6 Molecular mechanisms controlling iron transporter protein expression in the placenta seem to involve the concerted action of the post‐transcriptional intracellular IRP/IRE system and post‐translational regulation by fetal hepcidin. The importance of IRPs (especially IRP147) in handling placental iron has been clearly demonstrated in human 47 , 48 , 49 and animal 6 , 50 studies. Using an almost identical model of dietary iron deficiency in pregnant mouse females, Sanghke and co‐workers observed a 50% reduction in placental Fpn expression that was very likely mediated by IRP1. 6 It is tempting to speculate that under conditions of iron deficiency, this regulatory mechanism provides a compromise solution for maintaining the balance between iron delivery to developing fetuses and protection of the placenta from excessive iron depletion. In the light of our results, it is unlikely that hepcidin, which is barely detectable in the fetal liver, can modulate the Fpn protein level at the basolateral membrane of syncytiotrophoblasts and thus Fpn‐dependent export from the placenta to the fetal circulation under normal physiological conditions. Therefore, the findings of the present study and those of others 6 , 51 demonstrate that the contribution of fetal hepcidin to the regulation of placental Fpn in both normal and iron‐deficient conditions is of minor importance. Furthermore, the similar low‐level expression of BMP6 in the fetal liver under normal and iron‐deficient conditions, (despite a three‐fold difference in hepatic iron content), suggests that fetal BMP6 may be unresponsive to iron signaling and thus a BMP6‐dependent pathway of hepcidin regulation may be impaired.
In summary, our study emphasizes the complex role of Fpn, which is differentially expressed in maternal and fetal tissues, and in the placenta, in the distribution of iron between the mother, the fetus and the placenta under conditions of iron deficiency. First, the unaltered ferroportin‐dependent iron transport from liver and spleen macrophages seems to be the only efficient pathway of iron delivery to the maternal circulation. Second, the great decrease in placental ferroportin is responsible for the reduced iron export into the fetal circulation and for the maintenance of iron levels in the placenta. Finally, the reduction in fetal hepatic Fpn may secure greater iron availability for fetal erythropoiesis, which takes place in this organ.51‐53
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
Table S1 Primers used in Real Time PCR analysis
Table S2. Antibodies used in immunofluorescence analysis
Table S3. Decrease in red blood cell (RBC) indices of pregnant females fed a low‐iron diet. RBC indices were determined for 5–7 pregnant and non‐pregnant females of both the iron‐replete (ctr) and iron‐deficient (low) groups (means ± S.D.). RBC, red blood cell count; HCT, hematocrit; MCV, mean cell volume, HGB, hemoglobin level, RET, reticulocytes. ns –not significant.
Figure S1 Decrease in blood plasma iron parameters of pregnant females fed a low‐iron diet. Iron plasma level and transferrin saturation were determined for 6 females of both the iron‐replete (ctr) and iron‐deficient (low) groups (means ± S.D.). Significant differences are indicated (***p < 0.001).
Figure S2 Decreased hepatic iron status of pregnant females fed a low‐iron diet and their E18.5 fetuses. (A) Non‐heme iron content in the livers of pregnant females and fetuses (means ± S.D.). Liver samples were obtained from 6 pregnant females of both the iron‐replete (ctr) and iron‐deficient (low) groups. For hepatic iron measurements in the prenatal period, liver samples from 5 E18.5 fetuses of each group were pooled. Values were calculated for 3 sets of pooled fetal liver samples obtained from 3 pregnant females of each group. Significant differences are indicated (*p < 0.05, ***p < 0.001). (B) L‐ferritin (L‐Ft) levels in liver cytosolic extracts (100 μg/lane) were assessed by Western blotting. The analyses were performed in quadruplicate using extracts obtained from individual females of the iron‐replete (ctr) and iron‐deficient (low) groups and their fetuses, and representative results are shown. Recombinant mouse L‐Ft (rL‐Ft), a generous gift from Dr. P. Santambrogio (Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy), was used as a positive control.
Figure S3 Increase in erythropoietin (Epo), erythroferrone (Erfe) and GDF15 expression in pregnant females fed a low‐iron diet. (A) RT‐qPCR analysis of Epo transcript levels in the kideny of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative Epo mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (*p < 0.05, **p < 0.01). (B) RT‐qPCR analysis of Epo transcript levels in the bone marrow of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative Erfe mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (***p < 0.001). (C) Erfe levels were measured in the blood plasma of 5‐9 pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups using a competitive ELISA (means ± S.D.). In (A), (B), and (C) gray bars – non‐pregnant females; black bars pregnant females. (D) RT‐qPCR analysis of GDF15 transcript levels in the bone marrow of 8 pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative GDF15 mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (*p < 0.05).
Figure S4 No reduction in ferroportin (Fpn) protein level despite substantially decreased splenic iron content in pregnant females fed a low‐iron diet. (A) Non‐heme iron content in the spleens of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) (means ± S.D.). Significant differences are indicated (***p < 0.001). (B) Western blot analysis of Fpn protein levels in spleen membrane fractions prepared from pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. Fe‐NTA BMDM, ferric nitrilotriacetate (100 μM) treated mouse bone marrow‐derived macrophages (C) Immunoreactive Fpn bands from the blot shown in (b) were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (**p < 0.01). gray bars – non‐pregnant females; black bars – pregnant females
Figure S5 Increased expression of transferrin receptor 1 (TfR1) in the placenta of females fed a low‐iron diet. (A) RT‐qPCR analysis of TfR1 transcript levels in the placentas of 6 pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative TfR1 mRNA levels in arbitrary units (means ± S.D.). Significant difference is indicated (**p < 0.01). (B) Immunofluorescent staining of TfR1 (red channel) in placentas obtained from pregnant females of each group analyzed by confocal microscopy. Counterstaining of nuclei was performed with DAPI. Scale bars correspond to 20 μm. The right‐hand panels show high magnification images of stained placenta (areas boxed in the left‐hand panels). Scale bars correspond to 10 μm. m, maternal compartment; f, fetal compartment. (C) Western blot analysis of transferrin receptor 1 (TfR1) protein levels in placenta membrane fractions prepared from females of the iron‐replete (ctr) and iron‐deficient (low) groups. (D) Immunoreactive TfR1 bands from the blot shown in (C) were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant differences are indicated (**p < 0.01)
Figure S6 Increased expression of divalent metal transporter 1 (DMT1) in the placenta of females fed a lowiron diet. (A) RT‐qPCR analysis of DMT1 transcript levels in the placentas of 6 pregnant females of the ironreplete (ctr) and iron‐deficient (low) groups. The histogram displays the relative DMT1 mRNA levels in arbitrary units (means ± S.D.). (B) Immunofluorescent staining of DMT1 (red channel) in placentas obtained from pregnant females of each group analyzed by confocal microscopy. Counterstaining of nuclei was performed with DAPI. Scale bars correspond to 20 μm. The right‐hand panels show high magnification images of stained placenta (areas boxed in the left‐hand panels). Scale bars correspond to 10 μm. m, maternal compartment. f, fetal compartment. (C) Western blot analysis of divalent metal transporter 1 (DMT1) protein levels in placenta membrane fractions prepared from females of the iron‐replete (ctr) and iron‐deficient (low) groups. (D) Immunoreactive DMT1 bands from the blot shown in (C) were quantified by densitometry using a Molecular Imager, and DMT1 protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (***p < 0.001)
ACKNOWLEDGMENTS
We are very grateful to Dr. François Canonne‐Hergaux for helpful comments. We thank John Gittins for a critical reading and English editing of the paper prior to submission. This work was realized in the frame of India‐Polish Intergovernmental Science and Technology Co‐operation Program and by The National Science Centre, Poland, grants no. 2017/25/B/NZ9/01707 and no. 2019/33/B/NZ9/02566.
Mazgaj R, Lipiński P, Edison ES, et al. Marginally reduced maternal hepatic and splenic ferroportin under severe nutritional iron deficiency in pregnancy maintains systemic iron supply. Am J Hematol. 2021;96:659–670. 10.1002/ajh.26152
Funding information Narodowe Centrum Nauki, Grant/Award Numbers: 2017/25/B/NZ9/01707, 2019/33/B/NZ9/02566
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article and the data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Breymann C. Iron deficiency anemia in pregnancy. Semin Hematol. 2015;52:339‐347. [DOI] [PubMed] [Google Scholar]
- 2. Bothwell TH. Iron requirements in pregnancy and strategies to meet them. Am J Clin Nutr. 2000;72:257S‐264S. [DOI] [PubMed] [Google Scholar]
- 3. Fisher AL, Nemeth E. Iron homeostasis during pregnancy. Am J Clin Nutr. 2017;106:1567S‐1574S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brannon PM, Taylor CL. Iron Supplementation during pregnancy and infancy: uncertainties and implications for research and policy. Nutrients. 2017;9:1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lipiński P, Styś A, Starzyński RR. Molecular insights into the regulation of iron metabolism during the prenatal and early postnatal periods. Cell Mol Life Sci. 2013;70:23‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sangkhae V, Fisher AL, Wong S, et al. Effects of maternal iron status on placental and fetal iron homeostasis. J Clin Invest. 2020;130:625‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McArdle HJ, Lang C, Hayes H, Gambling L. Role of the placenta in regulation of fetal iron status. Nutr Rev. 2011;69:S17‐S22. [DOI] [PubMed] [Google Scholar]
- 8. Gambling L, Lang C, McArdle HJ. Fetal regulation of iron transport during pregnancy. Am J Clin Nutr. 2011;94:1903S‐1907S. [DOI] [PubMed] [Google Scholar]
- 9. Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776‐781. [DOI] [PubMed] [Google Scholar]
- 10. Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab. 2015;22(5):777‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191‐200. [DOI] [PubMed] [Google Scholar]
- 12. Bastin J, Drakesmith H, Rees M, et al. Localisation of proteins of iron metabolism in the human placenta and liver. Br J Haematol. 2006;134:532‐543. [DOI] [PubMed] [Google Scholar]
- 13. Beaumont C. Multiple regulatory mechanisms act in concert to control ferroportin expression and heme iron recycling by macrophages. Haematologica. 2010;95:1233‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lymboussaki A, Pignatti E, Montosi G, et al. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39:710‐715. [DOI] [PubMed] [Google Scholar]
- 15. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090‐2093. [DOI] [PubMed] [Google Scholar]
- 16. Torrance JD, Bothwell TH. Iron stores. Methods Hematol. 1980;1:90‐115. [Google Scholar]
- 17. Canonne‐Hergaux F, Gruenheid S, Ponka P, et al. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood. 1999;93:4406‐4417. [PubMed] [Google Scholar]
- 18. Starzyński RR, Canonne‐Hergaux F, Willemetz A, et al. Haemolytic anaemia and alterations in hepatic iron metabolism in aged mice lacking Cu,Zn‐superoxide dismutase. Biochem J. 2009;420:383‐390. [DOI] [PubMed] [Google Scholar]
- 19. Gambling L, Danzeisen R, Gair S, et al. Effect of iron deficiency on placental transfer of iron and expression of iron transport proteins in vivo and in vitro. Biochem J. 2001;356:883‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang M. Iron deficiency and other types of anemia in Infants and children. Am Fam Physician. 2016;93:270‐278. [PubMed] [Google Scholar]
- 21. Arosio P, Elia L, Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69:414‐422. [DOI] [PubMed] [Google Scholar]
- 22. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434‐1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017;8:126‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koenig MD, Tussing‐Humphreys L, Day J, et al. Hepcidin and iron homeostasis during pregnancy. Nutrients. 2014;6:3062‐3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41:478‐481. [DOI] [PubMed] [Google Scholar]
- 26. Rausa M, Pagani A, Nai A, et al. Bmp6 expression in murine liver non parenchymal cells: a mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS One. 2015;10:e0122696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ganz T. Erythropoietic regulators of iron metabolism. Free Radic Biol Med. 2019;133:69‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096‐1101. [DOI] [PubMed] [Google Scholar]
- 30. Casanovas G, Vujić Spasic M, Casu C, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knutson MD, Oukka M, Koss LM, et al. Iron release from macrophages after erythrophagocytosis is up‐regulated by ferroportin 1 overexpression and down‐regulated by hepcidin. Proc Natl Acad Sci U S A. 2005;102:1324‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirsch S, Austyn JM, Gordon S. Expression of the macrophage‐specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J Exp Med. 1981;154:713‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55‐63. [DOI] [PubMed] [Google Scholar]
- 34. Sangkhae V, Nemeth E. Placental iron transport: The mechanism and regulatory circuits. Free Radic Biol Med. 2019;133:254‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mastrogiannaki M, Matak P, Peyssonnaux C. The gut in iron homeostasis: role of HIF‐2 under normal and pathological conditions. Blood. 2013;122:885‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393‐408. [DOI] [PubMed] [Google Scholar]
- 37. Marro S, Chiabrando D, Messana E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica. 2010;95:1261‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delaby C, Pilard N, Puy H, Canonne‐Hergaux F. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron‐dependent protein expression. Biochem J. 2008;411:123‐131. [DOI] [PubMed] [Google Scholar]
- 39. Schümann K, Moret R, Künzle H, Kühn LC. Iron regulatory protein as an endogenous sensor of iron in rat intestinal mucosa. Possible implications for the regulation of iron absorption. Eur J Biochem. 1999;260:362‐372. [DOI] [PubMed] [Google Scholar]
- 40. Sanchez M, Galy B, Muckenthaler MU, et al. Iron‐regulatory proteins limit hypoxia‐inducible factor‐2alpha expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420‐426. [DOI] [PubMed] [Google Scholar]
- 41. Zhang L‐D, Hughes RM, Ollivierre‐Wilson H, et al. A ferroportin transcript that lacks an iron‐responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9:461‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beaumont C, Canonne‐Hergaux F. Erythrophagocytosis and recycling of heme iron in normal and pathological conditions; regulation by hepcidin. Transfus Clin Biol. 2005;12:123‐130. [DOI] [PubMed] [Google Scholar]
- 43. Allen LH. Anemia and iron deficiency: effects on pregnancy outcome. Am J Clin Nutr. 2000;71:1280S‐1284S. [DOI] [PubMed] [Google Scholar]
- 44. Lozoff B, Georgieff MK. Iron deficiency and brain development. Semin Pediatr Neurol. 2006;13:158‐165. [DOI] [PubMed] [Google Scholar]
- 45. Gambling L, Czopek A, Andersen HS, et al. Fetal iron status regulates maternal iron metabolism during pregnancy in the rat. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1063‐R1070. [DOI] [PubMed] [Google Scholar]
- 46. Georgieff MK, Berry SA, Wobken JD, et al. Increased placental iron regulatory protein‐1 expression in diabetic pregnancies complicated by fetal iron deficiency. Placenta. 1999;20:87‐93. [DOI] [PubMed] [Google Scholar]
- 47. Bradley J, Leibold EA, Harris ZL, et al. Influence of gestational age and fetal iron status on IRP activity and iron transporter protein expression in third‐trimester human placenta. Am J Physiol Regul Integr Comp Physiol. 2004;287:R894‐R901. [DOI] [PubMed] [Google Scholar]
- 48. Zaugg J, Melhem H, Huang X. Gestational diabetes mellitus affects placental iron homeostasis: mechanism and clinical implications. FASEB J. 2020;34:7311‐7329. 10.1096/fj.201903054R. [DOI] [PubMed] [Google Scholar]
- 49. Martin ME, Nicolas G, Hetet G, et al. Transferrin receptor 1 mRNA is downregulated in placenta of hepcidin transgenic embryos. FEBS Lett. 2004;574:187‐191. [DOI] [PubMed] [Google Scholar]
- 50. Kämmerer L, Mohammad G, Wolna M, et al. Fetal liver hepcidin secures iron stores in utero. Blood. 2020;136:1549‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sasaki K, Iwatsuki H. Origin and fate of the central macrophages of erythroblastic islands in the fetal and neonatal mouse liver. Microsc Res Tech. 1997;39:398‐405. [DOI] [PubMed] [Google Scholar]
- 52. Bednarz A, Lipiński P, Starzyński RR, et al. Role of the kidneys in the redistribution of heme‐derived iron during neonatal hemolysis in mice. Sci Rep. 2019;9:11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anderson C, Aronson I, Jacobs P. Erythropoiesis: erythrocyte deformability is reduced and fragility increased by iron deficiency. Hematology. 2000;4:457‐460. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used in Real Time PCR analysis
Table S2. Antibodies used in immunofluorescence analysis
Table S3. Decrease in red blood cell (RBC) indices of pregnant females fed a low‐iron diet. RBC indices were determined for 5–7 pregnant and non‐pregnant females of both the iron‐replete (ctr) and iron‐deficient (low) groups (means ± S.D.). RBC, red blood cell count; HCT, hematocrit; MCV, mean cell volume, HGB, hemoglobin level, RET, reticulocytes. ns –not significant.
Figure S1 Decrease in blood plasma iron parameters of pregnant females fed a low‐iron diet. Iron plasma level and transferrin saturation were determined for 6 females of both the iron‐replete (ctr) and iron‐deficient (low) groups (means ± S.D.). Significant differences are indicated (***p < 0.001).
Figure S2 Decreased hepatic iron status of pregnant females fed a low‐iron diet and their E18.5 fetuses. (A) Non‐heme iron content in the livers of pregnant females and fetuses (means ± S.D.). Liver samples were obtained from 6 pregnant females of both the iron‐replete (ctr) and iron‐deficient (low) groups. For hepatic iron measurements in the prenatal period, liver samples from 5 E18.5 fetuses of each group were pooled. Values were calculated for 3 sets of pooled fetal liver samples obtained from 3 pregnant females of each group. Significant differences are indicated (*p < 0.05, ***p < 0.001). (B) L‐ferritin (L‐Ft) levels in liver cytosolic extracts (100 μg/lane) were assessed by Western blotting. The analyses were performed in quadruplicate using extracts obtained from individual females of the iron‐replete (ctr) and iron‐deficient (low) groups and their fetuses, and representative results are shown. Recombinant mouse L‐Ft (rL‐Ft), a generous gift from Dr. P. Santambrogio (Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy), was used as a positive control.
Figure S3 Increase in erythropoietin (Epo), erythroferrone (Erfe) and GDF15 expression in pregnant females fed a low‐iron diet. (A) RT‐qPCR analysis of Epo transcript levels in the kideny of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative Epo mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (*p < 0.05, **p < 0.01). (B) RT‐qPCR analysis of Epo transcript levels in the bone marrow of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative Erfe mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (***p < 0.001). (C) Erfe levels were measured in the blood plasma of 5‐9 pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups using a competitive ELISA (means ± S.D.). In (A), (B), and (C) gray bars – non‐pregnant females; black bars pregnant females. (D) RT‐qPCR analysis of GDF15 transcript levels in the bone marrow of 8 pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative GDF15 mRNA levels in arbitrary units (means ± S.D.). Significant differences are indicated (*p < 0.05).
Figure S4 No reduction in ferroportin (Fpn) protein level despite substantially decreased splenic iron content in pregnant females fed a low‐iron diet. (A) Non‐heme iron content in the spleens of 5‐9 pregnant and nonpregnant females of the iron‐replete (ctr) and iron‐deficient (low) (means ± S.D.). Significant differences are indicated (***p < 0.001). (B) Western blot analysis of Fpn protein levels in spleen membrane fractions prepared from pregnant and non‐pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. Fe‐NTA BMDM, ferric nitrilotriacetate (100 μM) treated mouse bone marrow‐derived macrophages (C) Immunoreactive Fpn bands from the blot shown in (b) were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (**p < 0.01). gray bars – non‐pregnant females; black bars – pregnant females
Figure S5 Increased expression of transferrin receptor 1 (TfR1) in the placenta of females fed a low‐iron diet. (A) RT‐qPCR analysis of TfR1 transcript levels in the placentas of 6 pregnant females of the iron‐replete (ctr) and iron‐deficient (low) groups. The histogram displays the relative TfR1 mRNA levels in arbitrary units (means ± S.D.). Significant difference is indicated (**p < 0.01). (B) Immunofluorescent staining of TfR1 (red channel) in placentas obtained from pregnant females of each group analyzed by confocal microscopy. Counterstaining of nuclei was performed with DAPI. Scale bars correspond to 20 μm. The right‐hand panels show high magnification images of stained placenta (areas boxed in the left‐hand panels). Scale bars correspond to 10 μm. m, maternal compartment; f, fetal compartment. (C) Western blot analysis of transferrin receptor 1 (TfR1) protein levels in placenta membrane fractions prepared from females of the iron‐replete (ctr) and iron‐deficient (low) groups. (D) Immunoreactive TfR1 bands from the blot shown in (C) were quantified by densitometry using a Molecular Imager, and Fpn protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant differences are indicated (**p < 0.01)
Figure S6 Increased expression of divalent metal transporter 1 (DMT1) in the placenta of females fed a lowiron diet. (A) RT‐qPCR analysis of DMT1 transcript levels in the placentas of 6 pregnant females of the ironreplete (ctr) and iron‐deficient (low) groups. The histogram displays the relative DMT1 mRNA levels in arbitrary units (means ± S.D.). (B) Immunofluorescent staining of DMT1 (red channel) in placentas obtained from pregnant females of each group analyzed by confocal microscopy. Counterstaining of nuclei was performed with DAPI. Scale bars correspond to 20 μm. The right‐hand panels show high magnification images of stained placenta (areas boxed in the left‐hand panels). Scale bars correspond to 10 μm. m, maternal compartment. f, fetal compartment. (C) Western blot analysis of divalent metal transporter 1 (DMT1) protein levels in placenta membrane fractions prepared from females of the iron‐replete (ctr) and iron‐deficient (low) groups. (D) Immunoreactive DMT1 bands from the blot shown in (C) were quantified by densitometry using a Molecular Imager, and DMT1 protein levels (means ± S.D.) are plotted in arbitrary units (a.u.). Significant difference is indicated (***p < 0.001)
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article and the data that support the findings of this study are available from the corresponding author upon reasonable request.