

Abstract
Cortexolone 17α‐propionate, also known as clascoterone, is a potent androgen receptor inhibitor intended for the topical treatment of skin diseases associated with androgenic pathway alterations. In nonclinical studies, cortexolone 17α‐propionate was found to have a weak inhibitory effect on human Ether‐à‐go‐go‐Related Gene (hERG) potassium channels, which are vital for normal electrical activity in the heart. When used in a cream formulation, little cortexolone 17α‐propionate is absorbed. However, the solution formulation developed for the treatment of androgenetic alopecia leads to a measurable systemic concentration and accumulation of the antiandrogen. This phase 1 study assessed the effect of cortexolone 17α‐propionate on the QTc interval using concentration‐effect analysis and the effect of a meal on QTc to confirm assay sensitivity. Thirty‐two volunteers were randomly assigned to receive the active drug or a matching vehicle as placebo. Participants were dosed twice daily on days 1 to 3 (225 mg applied topically as a 7.5% solution 12 hours apart) and once on day 4. Pharmacokinetic and electrocardiogram assessments were performed after supratherapeutic doses. Assay sensitivity was successfully confirmed by using the food effect on the QTc interval. The results of this concentration‐QTc analysis demonstrate that cortexolone 17α‐propionate and its metabolite/degradation product had no effect on the QTc interval in the concentration range tested.
Keywords: androgen receptor inhibitor, androgenetic alopecia, antiandrogen, cardiac safety, cortexolone 17α‐propionate, QT interval
Cortexolone 17α‐propionate (Figure S1) is a topical androgen receptor inhibitor with weak glucocorticoid effects currently being developed by Cassiopea S.p.A (Lainate, Italy) for the treatment of androgenetic alopecia (AGA) and acne. It exerts local antiandrogenic activity by binding human androgen receptors to displace endogenous androgenic hormones 1 and is rapidly metabolized in plasma into cortexolone (also known as 11‐deoxycortisol)—a naturally occurring corticosteroid. 2
The use of available topical and oral antiandrogenic therapies for AGA and acne is currently limited because of undesirable systemic side effects, such as reduced libido, impairment of spermatogenesis, and feminization of male fetuses in pregnant women. 3 , 4 Cortexolone 17α‐propionate has been designed as a topical androgen receptor inhibitor with comparable efficacy to minoxidil for the treatment of AGA 5 and tretinoin for the treatment of acne, 6 , 7 but without the significant systemic activity seen with currently available oral antiandrogenic therapies.
Inhibition of human Ether‐à‐go‐go‐Related Gene (hERG) potassium channels causes QT interval prolongation and can lead to life‐threatening ventricular arrhythmias. 8 QT prolongation is a potential concern with any antiandrogenic drug, given that testosterone, a major endogenous androgen, appears to shorten the QTc interval. 9 , 10 At the same time, there is little research into the effects of antiandrogenic medications on the QT interval. One study in elderly male patients on antitestosterone treatment found little evidence of any QT‐prolonging effect. 11 Similarly, research by Saribayev et al (2014) suggested that corticosteroids have a QT‐shortening effect because of the activation of potassium channels through glucocorticoid signaling in cardiac myocytes. 12
In vitro assays investigating the effect of cortexolone 17α‐propionate on hERG potassium channels found the compound to inhibit hERG weakly in a dose‐dependent manner ranging from 2.6% to 40% at concentrations of 1 × 10–8 M (corresponding to a Cmax of 4 × 10–3 μg/mL) to 3 × 10–5 M.13 Subsequently, an in vivo safety pharmacology study was carried out in beagle dogs, which were attached to cardiac telemetry devices and administered subcutaneous doses of cortexolone 17α‐propionate. This study found no effects on arterial blood pressure, heart rate, body temperature, or QT interval at 10, 50, and 250 mg/kg (data available on request). However, the findings require further investigation to determine the effects of cortexolone 17α‐propionate on the QT interval in humans.
To date, clinical studies testing the 1% cream formulation have not resulted in significant systemic exposure, and it has been suggested that studies using a 5% solution (50 mg per application) would therefore provide sufficient exposures applicable to both the cream and solution formulation. In a 28‐day study testing multiple daily applications of a cortexolone 17α‐propionate 5% solution in healthy volunteers, maximum mean plasma concentrations of 3.63 ng/mL were observed that were similar to the lowest in vitro concentration that previously led to hERG inhibition. The authors did not detect any adverse cardiovascular signals. 1 The downstream metabolites, cortexolone 21‐propionate, and cortexolone are both 21‐hydroxysteroids. Cortexolone 21‐propionate has not been explored in terms of pharmacological activity, and cortexolone is an intermediate in the biosynthesis of cortisol. 13
However, none of these studies were specifically designed to evaluate the cardiac safety of cortexolone 17α‐propionate using high‐quality electrocardiograms (ECGs) and plasma concentration measurements for the analysis of concentration effects required to demonstrate the absence of an effect on the QT interval consistent with the International Council for Harmonization (ICH) E14 (R3) guidelines. 14 In addition, given the potential for patients to dose themselves in excess of doses recommended in clinical use, a cardiac safety study was deemed necessary to examine any QT interval effects of cortexolone 17α‐propionate and its metabolites/degradation products at supratherapeutic exposures.
Here, we describe the results of a phase 1 study investigating the effects of cortexolone 17α‐propionate on the QTc interval at supratherapeutic doses in healthy male and female volunteers, using a concentration‐effect analysis on the QTc interval validated by meal effects. In addition, we assessed the safety, tolerability, and pharmacokinetics (PK) of cortexolone 17α‐propionate.
Methods
Study Design and Procedures
This single‐center, randomized, double‐blind, placebo‐controlled study was conducted at Richmond Pharmacology (London, UK). The study aimed to assess the QTc interval in addition to the safety, tolerability, and pharmacokinetics of multiple doses of cortexolone 17α‐propionate in healthy adults. The study protocol (NCT03665194) was reviewed and approved by a National Health Service Research Ethics Committee (London Bridge, UK), and the Medicines and Healthcare Products Regulatory Authority (London, UK). The study was conducted according to the ethical principles enshrined in UK law, the Declaration of Helsinki, and Good Clinical Practice guidelines.
Study Volunteers
A total of 32 healthy adult volunteers (male and female, aged 18 to 40 years inclusive, body mass index between 18.0 and 25.0 kg/m2) were targeted for enrollment in this study. Volunteers had to be bald, have a shaved head, or be willing to have their heads shaved and be on acceptable forms of contraception to be enrolled in this study.
Written and signed informed consent was obtained from each participant before taking part in the study. Volunteers were excluded if they had (1) known structural cardiac abnormalities; (2) a family history of long QT syndrome; (3) cardiac syncope or recurrent, idiopathic syncope; (4) exercise‐related clinically significant cardiac events; or (5) any clinically significant abnormalities in rhythm, conduction, or morphology of resting ECG or clinically important abnormalities that might have interfered with the interpretation of QTc interval changes. These included but were not limited to sinus node dysfunction, clinically significant PR (PQ) interval prolongation, intermittent second‐ or third‐degree atrioventricular block, complete bundle branch block, abnormal T‐wave morphology, and a QT interval corrected using the Fridericia's formula (QTcF) > 450 milliseconds.
Eligible participants were randomly assigned to a treatment regimen according to a schedule generated by an independent statistician using PROC Plan. Volunteers were randomly assigned on day 1 in a 3:1 ratio to either cortexolone 17α‐propionate or matching vehicle placebo.
Procedures
The study outline is shown in Figure 1. The dosing regimen for 24 volunteers consisted of a morning and evening dose (12 hours apart) of 225 mg (3 mL) cortexolone 17α‐propionate applied topically to the scalp and both thighs as a 7.5% solution (75 mg in 1 mL), representing a total daily dose of 450 mg (6 mL) per participant. The cortexolone 17α‐propionate dose in this study was 4.5‐fold higher than the highest dose previously tested in the 28‐day multiple‐dose study (50 mg per application) and was chosen to achieve a minimum 2‐fold mean exposure relative to the previous study. In addition, 8 volunteers received a matching vehicle placebo. Twice‐daily dosing was planned on days 1 to 3 followed by a single dose on day 4, all under fed conditions. Matching standard meals were given on days 1 and 4, where breakfast was served 30 minutes prior to the morning dose; lunch was given 5 hours after the morning dose, and no other meals were served before completion of the cardiac assessments 2 to 4 hours postmeal. Dinner was served 10 hours after the morning dose.
Figure 1.

Study outline flowchart.
Pharmacokinetic Assessments
To achieve primary and secondary objectives, blood samples for plasma pharmacokinetics were collected on days 1 and 4 at −1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 12 hours, at −1 hour on days 2 and 3, and 24 and 48 hours following the last dose on day 4. Plasma samples to determine concentrations of cortexolone 17α‐propionate and its main metabolites/degradation products cortexolone 21‐propionate (M1, Figure S2) and cortexolone (M2, Figure S3) were analyzed by MicroConstants Inc (San Diego, California) employing a validated liquid chromatology‐tandem mass spectrometry method with liquid‐liquid extraction. This was developed and satisfactorily validated for the measurements of cortexolone 17α‐propionate and M1 over a calibration range of 0.250 to 250 ng/mL and for M2 over the calibration range of 0.500 to 500 ng/mL. Chromatographic separation was performed through a Kinetex XB‐C18 (50 × 2.1 mm, Phenomenex, Macclesfield, Cheshire, UK) analytical column with a mobile phase nebulized using heated nitrogen in electrospray positive ionization mode (see Supplement 1). Precision, accuracy, and selectivity of the method were found to be within satisfactory bounds using validated methods, and no issues were encountered during the execution of the sample analysis.
Pharmacokinetic analyses were performed using a noncompartmental method with SAS software, version 9.4 (SAS Institute Inc., Marlow, Buckinghamshire, UK).
ECG Cardiac Assessments and QTc Evaluation
Intensive cardiac assessments were performed on days 1 and 4 to achieve our primary objectives. All ECG recordings were obtained in triplicate at each time (at 1‐minute intervals over 3 minutes, lasting 10 seconds each) and were compliant with the correct recording and manual adjudication of ECG in thorough QT/QTc (TQT) studies in accordance with ICH guidelines. 14
Twelve‐lead ECGs were acquired using a GE Marquette MAC1200/MAC1200ST (GE Healthcare, Chicago, Illinois) and stored electronically on the MUSE information system. Only ECGs recorded electronically at a stable heart rate (HR) were valid for QT‐interval measurements. ECG recordings were collected on days 1 and 4 at 2, 1.5, and 1 hours predose and 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 hours postdose; on days 2 and 3 at 1 hour predose and on day 5 at 24 hours postdose. All ECGs were recorded after the volunteers had been resting in a supine position for at least 10 minutes. To obtain consistent ECG recordings, the clinical staff ensured that the volunteers were awake and avoided any postural changes.
Each ECG data file contained the ECG data and the result of the automated ECG analysis performed by the Marquette 12SL ECG Analysis Program. All ECGs and their associated automated interval measurements were subsequently reviewed by qualified cardiologists in accordance with the ICH Guidance for Industry document and ICH E14 Implementation Working Group Questions‐and‐Answers document 14 before any of the ECGs were used for the subsequent statistical analysis.
The uncorrected QT interval, the RR interval, the HR (derived according to the formula HR = 60 000/RR), the PR interval, QRS duration, the presence or absence of U waves, and quantitative and qualitative ECG variations were assessed by the cardiologist who had extensive experience with manual on‐screen overreading using electronic calipers in MUSE. All ECGs were overread by the same cardiologist, who was blinded to the time, treatment of the recording being evaluated, and any data identifying an individual, in accordance with principles set out in the ICH E14 guidelines. If manual adjustments of the automated measurement became necessary and the first overreader requested adjudication, then a second cardiologist performed overreading and assessment. Similarly, if the second cardiologist requested further adjudication, then the third most senior cardiologist performed the assessment. Corrected QT interval (QTcF) was used for all analyses, as relevant HR changes were not expected.
Both the matching vehicle placebo and active compound dosing data were included in the analysis. A total of 817 ECGs (26%) were corrected after adjudication from a total of 3151 ECGs. Predose baseline values were obtained from 3 predose times (2, 1.5, and 1 hour before drug administration); the mean of the values obtained at these times was used as baseline.
Statistical Analysis
Analysis of drug‐related QT/QTc interval changes relative to plasma PK concentrations was conducted using concentration‐effect modeling. The principles of this analysis followed statistical methods previously described. 15 , 16 Specifically, a primary linear mixed‐effects model was used for the concentration‐QTc analysis based on the change from average predose baseline that included a fixed time effect and a treatment effect. Time was presented as a fixed effect, considering that dosing was repeated on several days. The model had a centered baseline (ie, baseline minus mean of baseline across volunteers), in which the mean of baseline across participants was zero. No fixed intercept was allowed. Baseline was included in the model as a covariate. The model had random effects per volunteer for the intercept and the concentration.
The Kenward‐Roger approximation (a linear mixed model that allows for better estimation of precision and interference for fixed effects in small sample sets) 17 was used to calculate degrees of freedom and any t test‐based quantities, specifically 2‐sided 90% confidence intervals for the model parameters. Based on this model, predictions of the effect of the investigational medicinal product on QTcF at concentrations seen in the study were made. Specifically, predictions of the effect at the geometric mean of the individual Cmax values were given together with 2‐sided 90% confidence intervals.
A series of models using all possible combinations of the 3 moieties analyzed, as covariates did not show any improvement in fit when compared with the model using only cortexolone 17α‐propionate as a covariate, as measured by the Akaike information criterion. Models with moieties other than cortexolone 17α‐propionate were, thus, not further explored.
Assay Sensitivity
The effects of a meal on the ECG were used to establish assay sensitivity, that is, to confirm that the study was capable of detecting small changes in QTc. Tests for assay sensitivity were performed on the basis of the estimates of the time course of ΔQTcF obtained from the primary model described above. For the 2‐ and 3‐hour times on days 1 and 4, the change from predose baseline for day 1 and the change from the 1‐hour predose values on day 4, respectively, were tested at the 1‐sided 5% level. The study was declared to be adequately sensitive to show a small change in mean QTc if a shortening significant at the 1‐sided 5% level could be shown.
Safety Assessments
Adverse events (AEs) were continuously monitored throughout the study from the date of informed consent until the end of each individual's participation. The intensity and potential relationship with the study drug of each of the reported AEs were assessed. Volunteers underwent physical examinations and clinical laboratory tests (hematology, coagulation, biochemistry, and urinalysis). Telemetry, 12‐lead ECGs, blood pressure (systolic and diastolic), HR, and temperature were regularly evaluated during the study. Any clinically significant abnormalities were reported as AEs. All AEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 (CTCAE).
Results
Participant Disposition and Demographics
Of the 82 volunteers screened, 50 did not meet the screening criteria. A total of 32 volunteers fulfilled the eligibility criteria and were randomized to treatment. All completed the study and were included in the analysis sets. One volunteer withdrew consent from the study on day 2 after receiving 1 dose. Demographic data and volunteer disposition are summarized in Table 1.
Table 1.
Summary of Volunteer Demographic Characteristics
| Parameter | Statistics | Cortexolone (n = 24) | Placebo (n = 8) |
|---|---|---|---|
| Sex | Male, n (%) | 19 (79.2%) | 8 (100.0%) |
| Female, n (%) | 5 (20.8%) | 0 (0%) | |
| Race | White, n (%) | 17 (70.8%) | 3 (37.5%) |
| Hispanic, n (%) | 0 (0%) | 0 (0%) | |
| Black or African American, n (%) | 3 (12.5%) | 3 (37.5%) | |
| Asian, n (%) | 1 (4.2%) | 0 (0%) | |
| Indian/Pakistani, n (%) | 0 (0%) | 0 (0%) | |
| Other, n (%) | 3 (12.5%) | 2 (25.0%) | |
| Age (y) | n | 24 | 8 |
| Mean ± SD | 26.3 ± 6.0 | 25.4 ± 4.7 | |
| Range | 18‐38 | 18‐31 | |
| Weight (kg) | n | 24 | 8 |
| Mean ± SD | 70.0 ± 9.7 | 73.5 ± 9.1 | |
| Range | 53.2‐89.1 | 61.4‐86.7 | |
| BMI (kg/m2) | n | 24 | 8 |
| Mean ± SD | 22.4 ± 1.9 | 22.4 ± 1.5 | |
| Range | 19.3‐25.0 | 20.6‐24.5 |
SD, standard deviation.
Single‐Dose Pharmacokinetics
After application of single doses, plasma concentration of cortexolone 17α‐propionate rose gradually, which is likely a reflection of topical absorption, reaching Tmax at 5 hours (Figure 2). Levels then declined before plateauing at 6 hours, with no evidence of elimination during the 12‐hour single‐dosing period. Plasma concentrations of metabolites/degradation products were not detected (M1) or only marginally rose above the lower limit of quantification (M2).
Figure 2.

Mean (SD) of plasma concentrations of cortexolone 17α‐propionate—single dose (A) and multiple dose (B)—and cortexolone (M2)—single dose (C) and multiple dose (D).
Multiple‐Dose Pharmacokinetics
Following multiple dosing, plasma concentrations of cortexolone 17α‐propionate rose most markedly between 12 and 23 hours (representing the second and third doses, respectively). Concentrations then plateaued with Tmax at 77 hours before declining to near zero at 120 hours (Figure 2). M1 was detected in concentrations marginally above the lower limit of quantification in only 5 participants, at 1 point each. No other PK parameters were calculated for M1 because of negligible plasma concentrations. M2 concentrations rose the most between 12 and 23 hours, reaching Tmax at 48 hours, then fluctuated before plateauing between 96 and 120 hours.
For both cortexolone 17α‐propionate and M2, the ratio of AUC0‐24 on day 4 to AUC0‐24 on day 1 and the ratio of Cmax on day 4 to Cmax on day 1 were greater than 1.0, indicating an accumulation during multiple dosing. In addition, geometric mean trough values for M2 rose on day 5 following an initial decline, pointing to the continued metabolism of cortexolone 17α‐propionate into M2 after the end of dosing.
Descriptive statistics of PK parameters of cortexolone 17α‐propionate and M2, comparing mean values and standard deviations between day 1 and day 4 are presented in Table 2.
Table 2.
Descriptive Statistics of PK Parameters of Cortexolone 17α‐Propionate and Cortexolone (M2)
| Cortexolone 17α‐Propionate | Cortexolone (M2) | |||
|---|---|---|---|---|
| Parameter | Day 1 (Hours 0‐24) | Day 4 (Hours 72‐96) | Day 1 (Hours 0‐24) | Day 4 (Hours 72‐96) |
| Cmax (ng/mL), mean ± SD |
4.81 ± 2.64 (n = 24) |
7.65 ± 4.35 (n = 22) |
1.16 ± 0.43 (n = 5) |
1.27 ± 0.59 (n = 21) |
| AUC0‐12 (ng·h/mL), mean ± SD |
19.96 ±7.70 (n = 24) (hours 0‐12) |
59.86 ± 22.25 (n = 21) (hours 72‐84) |
NC |
11.24 ± 3.31 (n = 8) |
| AUC0‐24 (ng·h/mL), mean ± SD |
57.28 ± 16.53 (n = 23) |
113.94 ± 53.63 (n = 22) |
15.52 ± 0.08 (n = 2) |
20.40 ± 8.19 (n = 19) |
| AUC0‐last (ng·h/mL), mean ± SD |
55.15 ± 19.24 (n = 24) |
181.87 ± 97.87 (n = 22) |
15.52 ± 0.08 (n = 2) |
31.65 ± 23.11 (n = 21) |
| AUC0‐inf (ng·h/mL), mean ± SD | NC |
219.34 ± 92.03 (n = 20) |
NC |
110.83 ± 42.64 (n = 7) |
| Tmax (h) |
21.00 ± 5.70 (n = 24) |
6.20 ± 6.60 (n = 22) |
18.40 ± 10.30 (n = 5) |
10.90 ± 15.50 (n = 21) |
| Half‐life (h), mean ± SD | NC |
26.80 ± 14.60 (n = 20) |
NC |
90.10 ± 53.70 (n = 7) |
NC, not calculated; SD, standard deviation.
If data did not rise above the lower limit of quantitation, it was not included in the calculation of descriptive statistics.
Cardiac Assessments
The primary analysis was conducted following the statistical methods previously described 18 and employed the change from average baseline of the QT interval corrected for HR using Fridericia's formula (QTcF).
All the volunteers in the safety data set who had valid ECG data for at least 1 postdosing time were included in the primary analysis set. A total of 11 values had to be excluded because the time difference between the ECG and the blood sampling exceeded the predefined limit.
An increase in HR under active treatment was observed within 5 hours postdose on day 1 and for a number of times on day 4. There was no indication that this difference was because of differences at baseline or a causal relationship with the drug concentration. Concentration‐effect modeling performed on HR data yielded a negative slope, suggesting that the model did not fully capture the effect of the drug on heart rate (see Supplement 2).
Based on the model, predictions of the effect of cortexolone 17α‐propionate on QTcF at concentrations seen in the study were made. The prediction of the effect at the geometric mean of the individual Cmax values is shown in Table 3, highlighting a negative estimate.
Table 3.
Prediction of the Effect at the Geometric Mean Cmax of Cortexolone 17α‐Propionate
| Concentration | Predicted Effect on QTcF (ms) | |||||
|---|---|---|---|---|---|---|
| (ng/mL) | Estimate | SE | df | t | 90% Confidence Interval | |
| 6.602 | −0.45 | 2.11 | 35.2 | −0.21 | −4.0 | 3.1 |
df, degrees of freedom; SE, standard error.
The estimates of the time course analysis derived from the primary model are shown in Figure 3. “Time effect” refers to variations in duration of QTc throughout the day and represents fluctuations not caused by drug administration. All estimates were negative, ranging from −13.6 to −1.1 milliseconds. All postdose 90% confidence intervals were negative based on the model.
Figure 3.
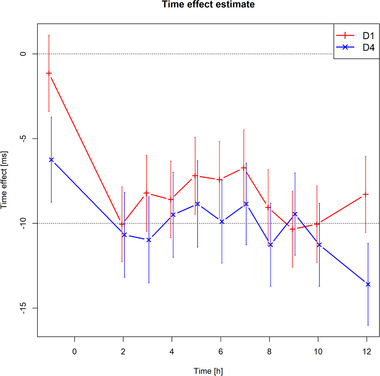
Estimates of the time course analysis derived from the primary model for day 1 (red) and day 4 (blue).
The relationship between QTcF and the cortexolone 17α‐propionate plasma concentration showed that the majority of the effect observed was because of the meal effect. There was a small but nonsignificant shortening effect at some points because of the effect of the drug (Figure 4).
Figure 4.

Raw values of ΔQTcF versus plasma concentration of cortexolone 17α‐propionate with regression line (slope, 0.13 adjusted for the average time effect to match the data displayed). The slight, nonsignificant positive correlation shows cortexolone 17α‐propionate has no effect on QTc.
Sensitivity of the Assay
The sensitivity of the assessment to consistently detect changes of QTcF was established by observing the meal effect. Providing a meal and then observing a small shortening of QTc 2 to 4 hours later demonstrate the ability of the experiment to detect small changes in QTc duration. The change from baseline of the time effect in the primary linear model for day 1 and from 1 hour predose on day 4, provided the opportunity to evaluate assay sensitivity on each of the 2 ECG assessment days (Table 4). All 95% confidence intervals were negative, with point estimates around −5 milliseconds or below and the 95%CI < 0. This corresponds to the upper limit of the 2‐sided 90%CI, and therefore assay sensitivity was considered shown.
Table 4.
Change From Baseline of the Time Effect in the Primary Linear Model for Day 1 and From −1 Hour on Day 4 to Demonstrate Assay Sensitivity
| Time | Estimate | SE | df | t | 90% Confidence Interval | |
|---|---|---|---|---|---|---|
| D1 2 h | −10.1 | 1.35 | 83.9 | −7.44 | −12.3 | −7.8 |
| D1 3 h | −8.2 | 1.37 | 87.9 | −5.98 | −10.5 | −5.9 |
| D4 2 h | −4.4 | 1.32 | 725.0 | −3.37 | −6.6 | −2.3 |
| D4 3 h | −4.7 | 1.31 | 725.1 | −3.60 | −6.9 | −2.6 |
D, day; df, degrees of freedom; SE, standard error.
Safety Assessments
A total of 12 adverse events (AEs) were reported in 9 participants who had all received the study medication. No volunteers given the vehicle placebo reported any AEs. Of the 12 AEs reported, 6 were considered related to study medication.
The most common system organ class for AEs relating to study medication was skin and subcutaneous tissue disorders and nervous system disorders. There was 1 pretreatment AE (skin exfoliation). All other AEs were treatment‐emergent.
All AEs were graded as mild (CTCAE grade I), except for a migraine, which was graded as moderate (CTCAE grade II) and was considered related to study medication. All AEs had resolved without sequelae by the end of the study. No serious AEs were reported in this study, and no AEs that led to an individual withdrawing from the study.
Discussion
Cortexolone 17α‐propionate has been designed for the topical treatment of AGA and acne—both of which are conditions that typically require long‐term use of medication under minimal medical supervision. This study was prompted by in vitro preclinical studies showing a mild hERG inhibition. Because cortexolone 17α‐propionate is a topical preparation, volunteers were able to apply the medication more frequently or to a larger area than prescribed, causing supratherapeutic exposures.
Because the in vitro effect on hERG inhibition was mild, and no QT effect was observed in prior clinical studies, 2 the initial likelihood of QT prolongation for cortexolone 17α‐propionate was low. However, because of the cosmetic indications of cortexolone 17α‐propionate and the opportunity for long‐term use in the absence of medical monitoring, we sought to assess QT prolongation at supratherapeutic systemic exposures to determine beyond a reasonable doubt, that the medication is safe for long‐term use at home without supervision. By the upper end of the 2‐sided 90% confidence interval for the cortexolone 17α‐propionate concentration‐effect model falling below 10 milliseconds, 17 this study has established the cardiac safety of cortexolone 17α‐propionate.
Given that the tested medication resulted in minimal systemic exposure when applied at therapeutic doses, one of our major considerations in designing this study was to achieve the necessary supratherapeutic exposures. Therefore, a stronger 7.5% solution formulation was used and applied to both thighs in addition to the scalp to maximize the surface area for absorption. Using a higher‐concentration solution, the results of this study also apply to the standard cream formulation.
To achieve regulatory acceptance as a QT study for both the cream and solution formulations, we also needed to demonstrate assay sensitivity or the ability to detect small changes in QT intervals. To this end, a standardized meal was used because the food effect has been shown to shorten the QT interval and is an established alternative to a pharmacological positive control arm—usually moxifloxacin. 19 , 20 , 21 At all times, the 2‐sided 90% confidence interval was negative, meaning the study methodology could detect the QT‐shortening effect of a meal on volunteers, thereby confirming assay sensitivity. The results demonstrate this study's ability to detect QTcF changes at the 5‐millisecond threshold of regulatory interest. 17 The results also support the use of a meal as a nontoxic alternative to a pharmacological positive control to demonstrate assay sensitivity in QTc assessments in line with results from previous studies. 21 , 22 , 23
By achieving a greater than 2‐fold increase in exposures relative to the anticipated therapeutic dose, the study meets ICH E14 requirements for acceptance as an alternative to a TQT study. 23 In the absence of procedural differences between this study and the previous one, 1 doses of cortexolone 17α‐propionate being 4.5‐fold greater than previously tested 1 may have been achieved by requiring volunteers to keep the solution on their skin for a longer time before washing. Typically, fewer than 2% of similar topical preparations are absorbed systemically when applied for a whole day. 24 To allow for a more homogeneous administration and more reproducible PK assessments, the heads and legs (if hairy) of volunteers were shaved. This presents a possible limitation, as previous research has shown that hair follicles are an important route of absorption for many topical compounds and can act as reservoirs for target drugs. 25 , 26 Conversely, some authors have argued that shaving may enhance percutaneous penetration of select compounds because it removes the stratum corneum, 27 which, when intact, acts as a rate‐limiting barrier to topical drug absorption. 28 Indeed, shaving may have enhanced the reproducibility of our findings, yet future assessments are needed to verify the consistency of our results when applied to nonshaven sites.
There are also limits to the use of a supratherapeutic dose for maximizing systemic exposures, as the doses administered in this study may have reached the maximum threshold for percutaneous absorption of cortexolone 17α‐propionate, meaning that the skin was saturated with study medication and that higher doses would not have resulted in higher exposures.
The absence of QT prolongation for cortexolone 17α‐propionate seen in this study is consistent with existing research on antiandrogenic therapy 11 and glucocorticoids, which conversely suggest a QT‐shortening effect of glucocorticoids at therapeutic doses because of the role of glucocorticoid signaling in cardiac myocytes, 12 , 13 albeit at therapeutic glucocorticoid doses with systemic exposures that were likely far greater than those achieved in this study. Our findings suggest that although antiandrogenic drugs such as cortexolone 17α‐propionate are physiologically capable of exerting effects on the duration of QTc, a standard topical application would not achieve a plasma concentration high enough to induce any cardiac effect.
Conclusion
This study met the regulatory requirements for a negative QT study, with the upper end of the 2‐sided 90% confidence interval for a change in QTcF falling below 10 milliseconds at the supratherapeutic doses tested. The effect of a meal on the QTc interval was successfully employed as a positive control to demonstrate assay sensitivity. Cortexolone 17α‐propionate has previously been shown to be generally well tolerated in healthy male and female volunteers, with the most common AEs being of mild severity and local to the application site. In conclusion, our results show that cortexolone 17α‐propionate and its metabolites/degradation products are tolerated at supratherapeutic doses and are not associated with clinically meaningful QTcF prolongation.
Conflicts of Interest
J.T., W.B., S.F., and A.P. are employees of Richmond Pharmacology Ltd. G.F. is an employee of Statistik Georg Ferber GmbH who has received honoraria for consulting from Richmond Pharmacology. C.S. and A.F. are employees of the Richmond Research Institute. A.M. is an employee of Cassiopea SpA.
Funding
Clascoterone is being developed by Cassiopea SpA. Cassiopea SpA funded this clinical trial to investigate Clascoterone.
Contributors
A.M. and J.T. conceived the study. W.B., A.M., and J.T. were involved in the data interpretation. C.S. and A.F. prepared and edited the manuscript. All authors were involved in the drafting of the manuscript. All authors approved the final submitted version and agreed to the publication.
Data Sharing
Requests for access to data should be addressed to the corresponding author.
Supporting information
Supplementary information
Supplementary information
Acknowledgments
The authors thank all the volunteers participating in this study. The authors thank Cassiopea SpA for funding the study and providing feedback and experimental conception. Special thanks are extended to the clinical and support staff at Richmond Pharmacology Ltd for their invaluable assistance in the execution of the clinical study.
References
- 1. Ferraboschi P, Legnani L, Celasco G, Moro L, Ragonesi L, Colombo D. A full conformational characterization of antiandrogen cortexolone‐17α‐propionate and related compounds through theoretical calculations and nuclear magnetic resonance spectroscopy. MedChemComm. 2014;5(7):904‐914. [Google Scholar]
- 2. Jonklaas J, Holst J, Verbalis J, Pehlivanova M, Soldin S. Changes in steroid concentrations with the timing of corticotropin stimulation testing in participants with adrenal sufficiency. Endocr Pract. 2012;18(1):66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen C, Puy LA, Simard J, Li X, Singh SM, Labrie F. Local and systemic reduction by topical finasteride or flutamide of hamster flank organ size and enzyme activity. J Invest Dermatol. 1995;105(5):678‐682. [DOI] [PubMed] [Google Scholar]
- 4. Sintov A, Serafimovich S, Gilhar A. New topical antiandrogenic formulations can stimulate hair growth in human bald scalp grafted onto mice. Int J Pharm. 2000;194(1):125‐134. [DOI] [PubMed] [Google Scholar]
- 5. Marks DH, Prasad S, De Souza B, Burns LJ, Senna MM. Topical antiandrogen therapies for androgenetic alopecia and acne vulgaris. Am J Clin Dermatol. 2020;21(2):245‐54. [DOI] [PubMed] [Google Scholar]
- 6. Trifu V, Tiplica GS, Naumescu E, Zalupca L, Moro L, Celasco G. Cortexolone 17α‐propionate 1% cream, a new potent antiandrogen for topical treatment of acne vulgaris. A pilot randomized, double‐blind comparative study vs. placebo and tretinoin 0·05% cream. Br J Dermatol. 2011;165(1):177‐183. [DOI] [PubMed] [Google Scholar]
- 7. Priest B, Bell IM, Garcia M. Role of hERG potassium channel assays in drug development. Channels. 2008;2(2):87‐93. [DOI] [PubMed] [Google Scholar]
- 8. Vink AS, Clur S‐AB, Wilde AA, Blom NA. Effect of age and gender on the QTc‐interval in healthy individuals and patients with long‐QT syndrome. Trends Cardiovasc Med. 2018;28(1):64‐75. [DOI] [PubMed] [Google Scholar]
- 9. Abehsira G, Bachelot A, Badilini F, et al. Complex influence of gonadotropins and sex steroid hormones on QT interval duration. J Clin Endocrinol Metab. 2016;101(7):2776‐2784. [DOI] [PubMed] [Google Scholar]
- 10. Lubart E, Yarovoy A, Gal G, Krakover R, Leibovitz A. QT interval length in elderly prostatic cancer patients on anti‐testosterone treatment. Isr Med Assoc J. 2015;17(6):356‐359. [PubMed] [Google Scholar]
- 11. Suzuki R, Hayashi T, Asano H, Ohashi K, Sakamoto M. Prolonged QT intervals in isolated ACTH deficiency: Case Report and Mini Review of Literature. J Steroids Horm Sci. 2016;7(176):2. [Google Scholar]
- 12. Saribayev M, Tufan F, Oz F, et al. Corticosteroid treatment normalizes QTc prolongation and improves heart block in an elderly patient with anti‐Ro‐positive systemic lupus erythematosus. Aging Clin Exp Res. 2014;26(3):337‐339. [DOI] [PubMed] [Google Scholar]
- 13. Celasco G, Piacquadio D, Moro L. Cortexolone 17a‐propionate: preclinical profile of a new topical, skin selective, steroidal antiandrogen. J Am Acad Dermatol. 2012;66(4). [Google Scholar]
- 14. ICH E14 Guideline: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs Questions & Answers (R3) 2015 [2/6/2016]. 61134]. In:2015.
- 15. Garnett C, Needleman K, Liu J, Brundage R, Wang Y. Operational characteristics of linear concentration‐QT models for assessing QTc interval in the thorough QT and phase I clinical studies. Clin Pharmacol Ther. 2016;100(2):170‐178. [DOI] [PubMed] [Google Scholar]
- 16. Ferber G, Wang D, Täubel J. Concentration–effect modeling based on change from baseline to assess the prolonging effect of drugs on QTc together with an estimate of the circadian time course. J Clin Pharmacol. 2014;54(12):1400‐1406. [DOI] [PubMed] [Google Scholar]
- 17. Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53(3):983‐997. [PubMed] [Google Scholar]
- 18. Täubel J, Fernandes S, Ferber G. Stability of the effect of a standardized meal on QTc. Ann Noninvasive Electrocardiol. 2017;22(1):e12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cirincione B, Sager PT, Mager DE. Influence of meals and glycemic changes on QT interval dynamics. J Clin Pharmacol. 2017;57(8):966‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration‐QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383‐397. [DOI] [PubMed] [Google Scholar]
- 21. Täubel J, Ferber G, Lorch U, Wang D, Sust M, Camm AJ. Single doses up to 800 mg of E‐52862 do not prolong the QTc interval–a retrospective validation by pharmacokinetic‐pharmacodynamic modelling of electrocardiography data utilising the effects of a meal on QTc to demonstrate ECG assay sensitivity. PLoS One. 2015;10(8):e0136369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Täubel J, Ferber G, Fernandes S, Santamaria E, Izquierdo I. Cardiac safety of rupatadine in a single‐ascending‐dose and multiple‐ascending‐dose study in healthy Japanese subjects, using intensive electrocardiogram assessments—comparison with the previous white Caucasian thorough QT study. Clin Pharmacol Drug Dev. 2018;7(1):67‐76. [DOI] [PubMed] [Google Scholar]
- 23. Täubel J, Lorch U, Coates S, et al. Confirmation of the cardiac safety of PGF2α receptor antagonist OBE022 in a first‐in‐human study in healthy subjects, using intensive ECG assessments. Clin Pharmacol Drug Dev. 2018;7(8):889‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dhar S, Seth J, Parikh D. Systemic side‐effects of topical corticosteroids. Indian J Dermatol. 2014;59(5):460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Otberg N, Patzelt A, Rasulev U, et al. The role of hair follicles in the percutaneous absorption of caffeine. Br J Clin Pharmacol. 2008;65(4):488‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leite‐Silva VR, de Almeida MM, Fradin A, Grice JE, Roberts MS. Delivery of drugs applied topically to the skin. Expert Rev Dermatol. 2012;7(4):383‐397. [Google Scholar]
- 27. Baker H, Kligman AM. Technique for estimating turnover time of human stratum corneum. Arch Dermatol. 1967;95(4):408‐411. [PubMed] [Google Scholar]
- 28. Ng K, Penetration enhancement of topical formulations. Pharmaceutics. 2018;10(2):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information