

Abstract
Background and purpose
Peripapillary retinal nerve fiber layer (pRNFL) and macular ganglion cell plus inner plexiform layer (GCIPL) thinning are markers of neuroaxonal degeneration in multiple sclerosis (MS), which is reduced by disease‐modifying treatment (DMT). We aimed to investigate the potential of pRNFL and GCIPL thinning for prediction of DMT failure in relapsing MS (RMS).
Methods
In this 4‐year prospective observational study on 113 RMS patients, pRNFL and GCIPL were measured at DMT initiation and after 12 months (M12) and 24 months (M24). Treatment failure was defined as 6‐month confirmed Expanded Disability Status Scale (EDSS) progression and/or Symbol Digit Modalities Test (SDMT) worsening. Optimal cutoff values for predicting treatment failure were determined by receiver operating characteristic analyses and hazard ratios (HRs) by multivariable Cox regression adjusting for age, sex, disease duration, EDSS/SDMT, and DMT class.
Results
Thinning of GCIPL >0.5 μm/year at M24 showed superior value for treatment failure prediction (HR: 4.5, 95% confidence interval [CI]: 1.8–7.6, p < 0.001; specificity 91%, sensitivity 81%), followed by GCIPL >0.5 μm at M12 (odds ratio [OR]: 3.9, 95% CI: 1.4–6.9, p < 0.001; specificity 85%, sensitivity 78%), and pRNFL ≥2 μm/year at M24 (OR: 3.7, 95% CI: 1.1–6.5, p = 0.023; specificity 84%, sensitivity 69%), whereas pRNFL at M12 was not predictive.
Conclusions
GCIPL, and to a lesser degree pRNFL, thinning predicts disability progression after DMT initiation and may be a useful and accessible biomarker of treatment failure in RMS.
Keywords: disease‐modifying treatment, GCIPL, multiple sclerosis, OCT, retinal thinning
In this 4‐year prospective observational study on 113 RMS patients, retinal thinning was investigated for prediction of disease‐modifying treatment failure. Thinning of GCIPL and to a lesser degree pRNFL were found to reliably predict disability progression after DMT initiation and may be a useful and accessible biomarker of treatment failure in RMS.

INTRODUCTION
Multiple sclerosis (MS) is a chronic immune‐mediated inflammatory neurological disease carrying the risk of physical and cognitive disability [1]. Over the last quarter century, an ever‐increasing number of immunomodulating or immunosuppressive disease‐modifying treatments (DMTs) have proven to effectively reduce the number of relapses and, to a lesser extent, disability progression in relapsing MS (RMS).
However, DMTs strongly differ in both the degree of efficacy and the severity of associated potential risks [2]. MS treatment has changed toward early treatment, thus aiming to suppress disease activity below the level of detectability [3]. However, RMS displays an extremely variable clinical course ranging from highly active disease despite DMT to patients stable over long periods under moderately effective or even without DMT [4].
Defining the response to DMT is very challenging in RMS and considers various surrogates including relapses, progression of physical disability, and magnetic resonance imaging (MRI) activity. All of these surrogates harbor significant limitations, and their determination occupies time and resources [5]. This urges the need for further biomarkers that allow a reliable and easy evaluation of the impact of therapeutic interventions.
Peripapillary retinal nerve fiber layer (pRNFL) and macular ganglion cell plus inner plexiform layer (GCIPL) thinning, measured by optical coherence tomography (OCT), are markers of neuroaxonal damage in MS [6]. pRNFL and GCIPL thinning is more pronounced in patients with physical or cognitive disability progression, whereas it is reduced by DMT [7, 8, 9, 10, 11. Still, retinal thinning has not yet been studied prospectively as a marker of DMT response. Thus, in the present study, we aimed to investigate the potential of retinal layer thinning for prediction of treatment failure in RMS.
METHODS
Patients
In this 4‐year prospective observational study, we included 150 patients between September 2013 and August 2015, diagnosed with RMS according to the 2010 McDonald criteria, and aged between 18 and 65 years [12]. The baseline visit was performed within 14 days after DMT initiation. Study visits were conducted quarterly during the follow‐up of at least 4 years. Demographic data and neurological and treatment history, including DMT, were obtained from each participant at every visit.
DMT was grouped as moderately effective DMT (M‐DMT), including interferon β preparations, glatiramer acetate, dimethyl fumarate, or teriflunomide, or highly effective DMT (H‐DMT) comprising natalizumab, fingolimod, or alemtuzumab.
Study endpoints
The primary endpoint was disability worsening defined as a combined endpoint of physical disability worsening, assessed by the Expanded Disability Status Scale (EDSS) score, and/or cognitive decline during the observation period. Physical disability worsening was defined as a confirmed EDSS increase of ≥1.5 points in patients with a baseline score of 0, ≥1.0 points in patients with a baseline score of 1.0 to 5.5, or ≥0.5 points in patients with a baseline score of >5.5 sustained for at least 6 months as compared to baseline [5]. Cognitive function was assessed by the Symbol Digit Modalities Test (SDMT), because it is particularly suitable for longitudinal assessment of MS‐related cognitive changes [13, 14. A cognitive decline was defined as an absolute decrease of ≥4 points or a relative decrease of ≥10% in the SDMT score compared to baseline sustained for at least 6 months [14].
Secondary endpoints were the occurrence of relapse and a combined endpoint labeled as clinical disease activity defined as occurrence of disability worsening and/or relapse. A relapse was defined as patient‐reported symptoms with objectively observed signs typical of an acute central nervous system inflammatory demyelinating event, current or prior to the visit, with a duration of at least 24 hours in the absence of fever or infection, separated from the last relapse by at least 30 days [12].
If the newly initiated DMT was discontinued because of insufficient efficacy as defined by the treating neurologist, patients were considered to have reached the primary and secondary endpoints at the time of discontinuation. If the newly initiated DMT was discontinued because of reasons other than efficacy (e.g., adverse events, tolerability, pregnancy or desire to have children, insufficient compliance/adherence) within 2 years after initiation, patients were excluded from analysis. Thus, per definition, no switch of DMT occurred during the observation period in the patients included.
Optical coherence tomography
OCT imaging was performed at DMT initiation (M0) and after 12 months (M12) and 24 months (M24) of follow‐up by two experienced technicians using the same spectral‐domain OCT (Heidelberg Eye Explorer software, version 5.4.8.0; Heidelberg Engineering, Heidelberg, Germany) without pupil dilatation in a dark room on both eyes of each patient. For GCIPL measurement, a 20° × 20° macular volume scan (512 A‐scans, 257 B‐scans, vertical alignment, automatic real time [ART] 16) automatically centered around the fovea was performed. The follow‐up function was activated to ensure that longitudinal scans were obtained at the same locations. GCIPL thickness was calculated as the mean thickness of the inner and outer four quadrants of the grid (corresponding to the 3‐mm and 6‐mm rings as defined by the Early Treatment Diabetic Retinopathy Study) [15]. For pRNFL measurement, a custom 3.4‐mm ring scan (12°) centered on the optic nerve head was used (automatic real time ART 100). Semiautomated image processing was conducted with manual correction of obvious errors. All examinations were checked for sufficient quality using OSCAR‐IB criteria [16, 17.
Thicknesses of GCIPL and pRNFL were calculated as the mean of the values for both eyes. Patients with a history of unilateral optic neuritis (ON) <6 months before baseline were excluded from the study. Eyes with a history of ON ≥6 months before baseline were eligible for inclusion, because further retinal thinning does not seem to differ between eyes with and without a history of ON [18]. Eyes suffering ON during the observation period were excluded from the study, and only the values of eyes without ON during the observation period were used for calculation of retinal thinning in the analyses [7, 8. To identify subclinical ON during the course of the study, we used interocular asymmetry in retinal thinning (i.e., intereye difference in GCIPL/pRNFL thickness reduction compared to the prior OCT), with cutoff values of ≥4 µm for GCIPL and ≥5 µm for pRNFL [19, 20. In these cases, we used only the eye with the higher value. Thus, all parameters used for statistical analyses are not underlying intereye interactions.
Other exclusion criteria were previous diagnoses of ophthalmological (i.e., myopia greater than −4 diopters, optic disc drusen), neurological, systemic (such as previous diagnoses of diabetes mellitus or arterial hypertension), or drug‐related causes of vision loss or retinal damage not attributable to MS [16]. The investigators performing OCT were blinded to clinical parameters and vice versa (Figure 1).
FIGURE 1.
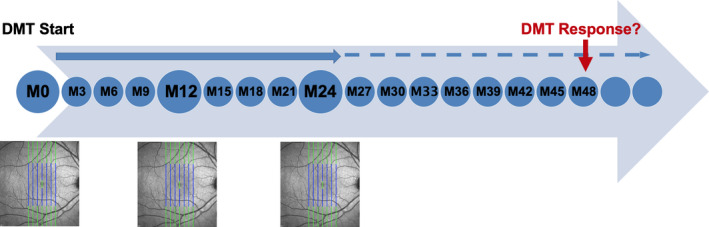
Schematic overview of study design. Small circles: relapse, Expanded Disability Status Scale/Symbol Digit Modalities Test rating. Big circles: Relapse, Expanded Disability Status Scale/Symbol Digit Modalities Test rating, and optical coherence tomography. DMT, disease‐modifying treatment; M, month. [Colour figure can be viewed at wileyonlinelibrary.com]
Statistics
Statistical analysis was performed using R statistical software (Version 4.0.0; R Foundation for Statistical Computing, Vienna, Austria). Retinal thinning was expressed as annualized loss of GCIPL (aLGCIPL) and pRNFL (aLpRNFL) determined as the difference of thickness at baseline and M12 or M24. We performed receiver operating characteristic (ROC) analyses to identify the optimal cutoff value of aLGCIPL and aLpRNFL for determining patients reaching the primary endpoint and for calculating sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV). Areas under the curve (AUC) were compared using variance estimates recovery on the basis of inverse hyperbolic sine transformations [21].
Univariate comparisons of outcome variables (disability worsening, EDSS progression, cognitive decline, relapse activity, clinical disease activity) according to retinal thinning above and below the respective cutoffs for prediction of treatment failure were done by χ2 test or independent t test (with Welch correction in case of unequal standard deviations between the groups) as appropriate. Time to outcome variables were univariately compared by log‐rank test.
Finally, for analyzing the value of retinal thinning parameters for prediction of treatment failure, we performed multivariate Cox proportional hazard models with disability worsening/relapse activity/clinical disease activity as the dependent variable and retinal thinning parameters (aLGCIPL M12, aLGCIPL M24, aLpRNFL M12, aLpRNFL M24) as the independent variable adjusted for age, sex, disease duration, baseline pRNFL/GCIPL, number of relapses in year before baseline, EDSS at baseline, SDMT at baseline, and DMT group (M‐DMT or H‐DMT).
Missing values were handled by multiple (20 times) imputation using the missing not at random (MNAR) approach with pooling of estimates according to Rubin's rules [22]. A two‐sided p value <0.05 was considered statistically significant.
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the ethics committees of the Medical Universities of Vienna and Innsbruck (EK Nr: 2323/2019 and AM3743‐281/4.). Written informed consent was obtained from all study participants.
RESULTS
Characteristics of the study cohort are given in Table 1. Of 150 patients recruited, 37 patients had exclusion criteria or were lost to follow‐up (Figure 2). The remaining 113 (75.3%) patients were available for statistical analysis. Of those, four EDSS ratings and three SDMT scores were missing and subsequently imputed as described above. Thirty‐six patients (31.9%) had disability worsening, 52 (46.0%) had a relapse, and 68 (60.2%) had clinical disease activity as described previously during the observation period.
TABLE 1.
Demographic and clinical characteristics of the cohort
| Whole cohort, n = 113 | |
|---|---|
| Females a | 91 (80.5) |
| Age, b years | 34.2 (8.6) |
| MS disease duration, b years | 7.0 (6.4) |
| OCB positivity a | 110 (97.3) |
| Relapse in year before baseline a | 112 (99.1) |
| No. of relapses in year before baseline c | 1 (0–3) |
| EDSS at baseline c | 1.0 (0–6.5) |
| EDSS progression in year before baseline a | 11 (9.7) |
| SDMT b | 55.3 (9.8) |
| Received DMT before baseline a | 81 (71.7) |
| No. of DMTs received before baseline c | 1 (0–4) |
| Newly initiated DMT | |
| Interferon β a | 32 (28.3) |
| Glatiramer acetate a | 32 (28.3) |
| Fingolimod a | 24 (21.2) |
| Natalizumab a | 25 (22.1) |
| Previous optic neuritis a | 22 (19.5) |
| pRNFL thickness,μm, at baseline b | 92.4 (12.2) |
| GCIPL thickness, μm, at baseline b | 81.1 (12.0) |
Abbreviations: DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; GCIPL, macular ganglion cell and inner plexiform layer; MS, multiple sclerosis; OCB, oligoclonal bands; pRNFL, peripapillary retinal nerve fiber layer; SDMT, Symbol Digit Modalities Test.
Number (percentage).
Mean and standard deviation.
Median and range.
FIGURE 2.
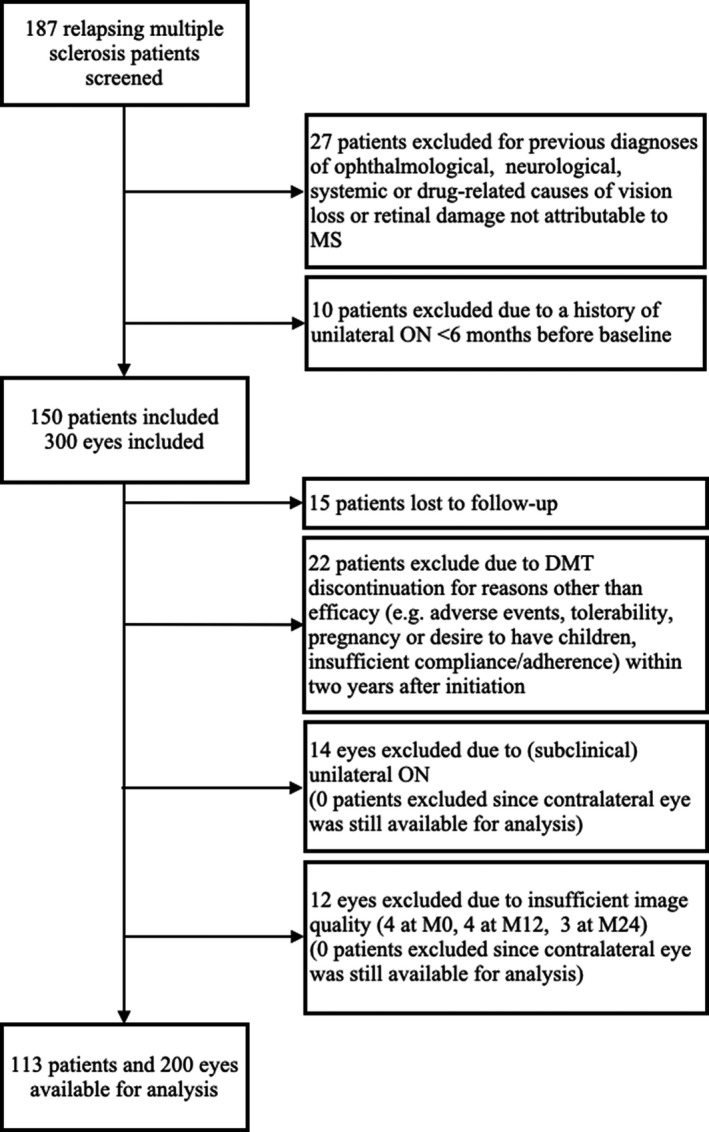
Inclusion flowchart. DMT, disease‐modifying treatment; M0, baseline; M12, month 12; M24, month 24; MS, multiple sclerosis; ON, optic neuritis.
Patients with disability worsening displayed significantly more marked annualized retinal thinning compared to patients without disability worsening in GCIPL at both M12 (1.6 µm vs. 0.4 µm, p < 0.001) and M24 (1.6 µm vs. 0.3 µm, p < 0.001) as well as in pRNFL (M12: 2.3 µm vs. 1.2 µm, p < 0.001; M24: 2.6 µm vs. 1.3 µm, p < 0.001). These results did not change significantly when comparing patients according to EDSS progression or cognitive decline instead of the combined endpoint of disability worsening (Table S1). Annual loss of GCIPL and pRNFL were also significantly higher at both M12 and M24 in patients with relapse and clinical disease activity (Table S1). Disease duration was weakly inversely correlated with annual loss of both GCIPL (M12: ρ = −0.114; M24: ρ = −0.125; p < 0.001, respectively) and pRNFL (M12: ρ = −0.132; M24: ρ = −0.148; p < 0.001, respectively), whereas both baseline GCIPL and pRNFL were weakly correlated with aLGCIPL (M12: ρ = 0.165; M24: ρ = 0.173; p < 0.001, respectively) and aLpRNFL (M12: ρ = 0.157; M24: ρ = 0.163; p < 0.001, respectively).
The best possible cutoff values for identifying patients with disability worsening were >0.5 µm for aLGCIPL (p < 0.001) and ≥2.0 µm for aLpRNFL (p < 0.001). Measured at M24, aLGCIPL above the cutoff of 0.5 µm provided discrimination with 81% sensitivity and 86% specificity, with a PPV of 71% and NPV of 97% (Figure 3). Diagnostic accuracy of aLGCIPL >0.5 µm at M24 (AUC: 0.86, 95% confidence interval [CI]: 0.78–0.93) was slightly superior to aLGCIPL >0.5 µm at M12 (sensitivity: 78%, specificity: 86%, PPV: 66%, NPV: 95%, AUC: 0.85, 95% CI: 0.78–0.93) and significantly superior to aLpRNFL ≥2.0 µm at M24 (sensitivity: 69%, specificity: 69%, PPV: 56%, NPV: 90%, AUC: 0.74, 95% CI: 0.63–0.85, p < 0.001 compared to aLGCIPL) and at M12 (sensitivity: 58%, specificity: 78%, PPV: 47%, NPV: 85%, AUC: 0.74, 95% CI: 0.64–0.85, p < 0.001).
FIGURE 3.
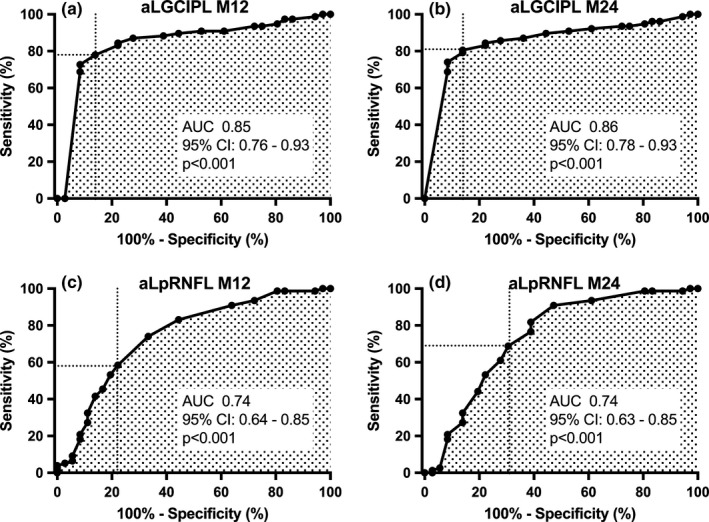
Accuracy of disease‐modifying treatment failure prediction by annualized retinal layer thinning and as determined by receiver operating characteristic analyses of aLGCIPL measured after 12 and 24 months (a, b) and aLpRNFL after 12 and 24 months (c, d). aLGCIPL, annualized loss of macular ganglion cell and inner plexiform layer; aLpRNFL, annualized loss of peripapillary retinal nerve fiber layer; AUC, area under the curve; CI, confidence interval; M12, measured between baseline and 12 months after DMT initiation; M24, measured between baseline and 24 months after DMT initiation.
Annualized loss of GCIPL >0.5 µm at M12 and M24 also performed similarly well at identifying patients with clinical disease activity (sensitivity: 85%/87%, specificity: 69%/70%, PPV: 88%/92%, NPV 62%/63%, AUC: 0.76/0.78), but significantly better than aLpRNFL ≥2.0 µm (p < 0.001, Table S2).
Regarding relapse, overall accuracy was significantly lower with aLGCIPL >0.5 µm at M24 displaying the best accuracy among all of the parameters (sensitivity: 77%, specificity: 52%, PPV 63%, NPV 66%, AUC: 0.66, p = 0.003, Table S2).
Combining aLGCIPL >0.5 µm and aLpRNFL ≥2.0 µm did not result in an improved diagnostic accuracy of variables defining treatment failure, neither at M12 nor at M24 (Table S2).
Univariate analyses of associations between retinal thinning and variables defining treatment failure are shown in detail in Table S3. In multivariate models, aLGCIPL >0.5 µm at M24 was found as the strongest predictor of disability worsening (hazard ratio [HR]: 4.5, p < 0.001) closely followed by aLGCIPL >0.5 µm at M12 (HR: 3.9, p < 0.001). Although aLpRNFL ≥2.0 µm at M24 showed a weaker but still significant association with disability worsening (HR: 2.7, p < 0.001), aLpRNFL ≥2.0 µm at M12 did not (Table 2, Figure 4). Annualized loss of GCIPL >0.5 µm at M12 and M24 were also significant predictors of secondary endpoints clinical disease activity (HR: 3.4 and HR: 3.7, p < 0.001, respectively) and relapse (HR: 1.9, p = 0.031 and HR: 2.0, p = 0.027, respectively). Annualized loss of pRNFL ≥2.0 µm at M24 was significantly associated with clinical disease activity (HR: 2.4, p = 0.025) but not with relapse, whereas pRNFL ≥2.0 µm at M12 did not predict either (Table 2, Figure 4).
TABLE 2.
Cox regression models for prediction of treatment failure by retinal layer thinning
| aLGCIPL M12 >0.5 µm | aLGCIPL M24, >0.5 µm | aLpRNFL M12 ≥2.0 µm | aLpRNFL M24 ≥2.0 µm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR a | 95% CI | p value | HR a | 95% CI | p value | HR a | 95% CI | p value | HR a | 95% CI | p value | |
| Disability worsening | 3.9 | 1.7–8.6 | <0.001 | 4.5 | 1.8–7.6 | <0.001 | 2.1 | 0.8–5.6 | 0.214 | 2.7 | 1.3–7.5 | <0.001 |
| EDSS progression | 3.8 | 1.8–8.5 | <0.001 | 4.1 | 1.9–7.9 | <0.001 | 2.5 | 0.9–7.2 | 0.285 | 3.1 | 1.4–7.2 | <0.001 |
| Cognitive decline | 3.2 | 1.5–7.3 | <0.001 | 3.6 | 1.8–8.1 | <0.001 | 1.9 | 0.7–8.0 | 0.361 | 2.2 | 1.2–8.5 | 0.018 |
| Relapse | 1.9 | 1.1–3.9 | 0.031 | 2.0 | 1.1–3.7 | 0.027 | 2.1 | 0.7–6.1 | 0.363 | 2.2 | 0.9–4.8 | 0.143 |
| Clinical disease activity | 3.4 | 1.9–7.0 | <0.001 | 3.7 | 2.1–6.8 | <0.001 | 2.0 | 0.8–5.2 | 0.209 | 2.4 | 1.1–6.5 | 0.025 |
Calculated by Cox regression models with disability worsening/EDSS progression/cognitive decline/relapse/clinical disease activity as the dependent variable and aLGCIPL/aLpRNFL as the independent variable adjusted for age, sex, disease duration, baseline pRNFL/GCIPL, number of relapses in the year before baseline, EDSS at baseline, Symbol Digit Modalities Test at baseline, and DMT group (moderately effective DMT or highly effective DMT).
Abbreviations: 95% CI, 95% confidence interval; aLGCIPL, annualized loss of ganglion cell and inner plexiform layer; aLpRNFL, annualized loss of peripapillary retinal nerve fiber layer; DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; HR, hazard ratio.
Higher values indicate higher probability of disability worsening/EDSS progression/cognitive decline/relapse/clinical disease activity.
FIGURE 4.
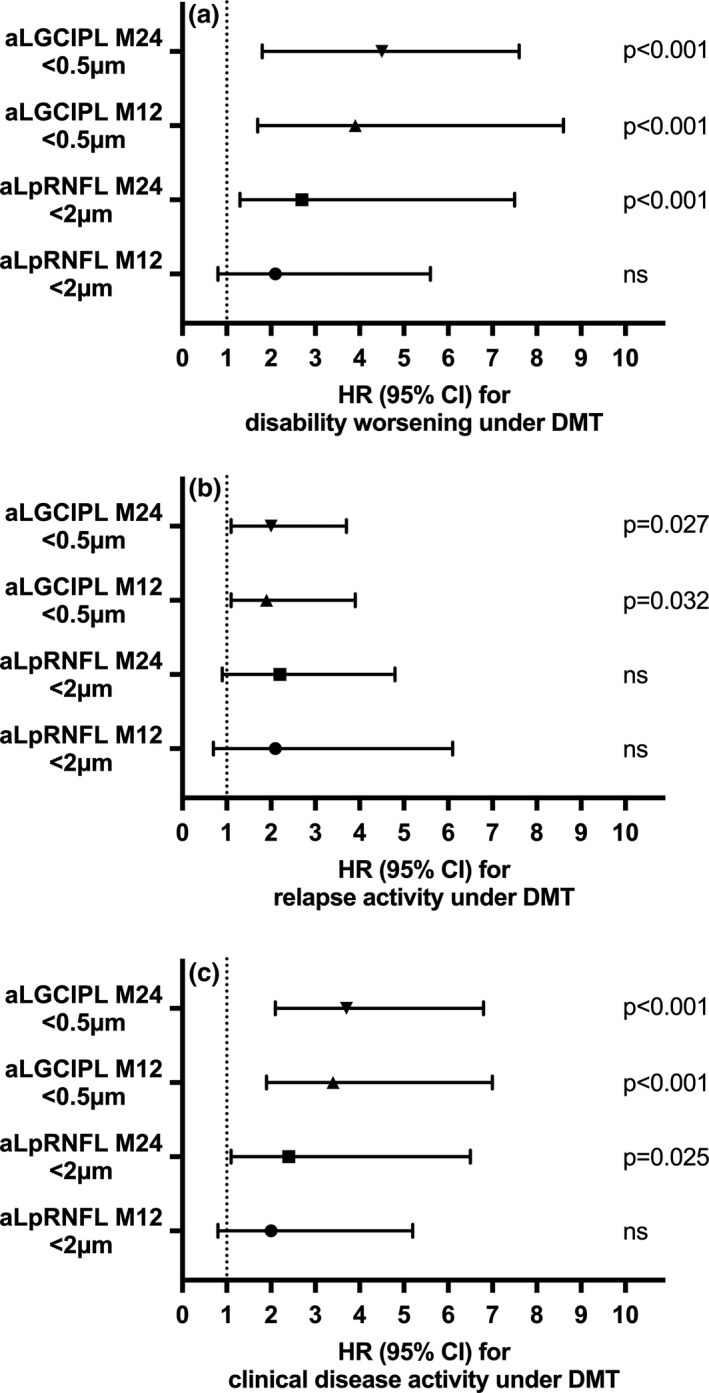
Annualized retinal layer thinning predicts DMT failure. Probability of DMT failure regarding disability worsening (a), relapse (b), and clinical disease activity (c) as predicted by annualized loss of GCIPL and pRNFL measured 12 and 24 months after DMT initiation. aLGCIPL, annualized loss of macular ganglion cell and inner plexiform layer; aLpRNFL, annualized loss of peripapillary retinal nerve fiber layer; CI, confidence interval; DMT, disease‐modifying treatment; HR, hazard ratio; M12, measured between baseline and 12 months after DMT initiation; M24, measured between baseline and 24 months after DMT initiation; ns, not significant.
DISCUSSION
Defining response to DMT (i.e., recognizing failure of DMT), is one of the biggest challenges in managing patients with RMS, because it is the key for tailoring treatment according to individual disease activity.
Here, we show that DMT failure, defined as physical and/ or cognitive disability worsening, can be predicted by measuring GCIPL thinning over 12 months (specificity 85%, sensitivity 78%, PPV 66%, NPV 95%) with a small additional gain in accuracy when extending measurement to 24 months (specificity 91%, sensitivity 81%, PPV 71%, NPV 97%), whereas pRNFL is considerably less accurate and necessitates 24 months of observation (specificity 84%, sensitivity 69%, PPV 56%, NPV 90%).
Although it is already established that loss of both GCIPL and pRNFL indicates neuroaxonal damage and that is slowed in patients treated with high‐efficacy DMT (natalizumab or alemtuzumab) compared to patients with moderate‐efficacy DMT (interferon β, glatiramer acetate), the present study is the first prospectively investigating retinal layer thinning as a marker of future DMT failure [7, 8, 9, 10, 11 Consistent with previous results, the effect size was larger measuring GCIPL than pRNFL, underlining that GCIPL is better suited for measuring DMT response [7, 8, 9, 10 GCIPL thinning is more specific for MS, displays superior structure‐function correlation, and reflects neuroaxonal damage faster than pRNFL, while being less prone to a flooring effect in more advanced MS or to confounding by swelling from acute ON [6] In accordance with earlier studies, we found disease duration weakly inversely correlated with annual loss of both GCIPL and pRNFL, whereas baseline GCIPL and pRNFL thickness were weakly correlated with the degree of annual GCIPL and pRNFL thinning [6] This is likely because patients with shorter disease duration and higher baseline retinal thickness have more tissue to lose.
Currently, DMT response is primarily measured by counting relapses and assessing physical disability, usually by the EDSS [5] Occurrence of a relapse or EDSS progression carries significant prognostic value, and thus, a patient can be labeled a nonresponder to a DMT if sufficient adherence is given triggering consideration of changing to a more effective DMT [5] However, both relapses and EDSS progression are rare events; therefore, sensitivity for detecting DMT failure is low, and long observation periods of at least 2 years are needed to establish whether a DMT is effective [5].
In addition to relapses and EDSS progression, MRI‐based monitoring of newly occurring T2‐hyperintense lesions (T2Ls) and contrast‐enhancing lesions (CELs) is used widely for assessing DMT response. New T2Ls and CELs are significantly more sensitive indicators of MS disease activity than clinical measures and correlate with future disability [23] However, MRI metrics mediate only about 50% of the treatment effect on relapses and EDSS progression, and the effect size is even smaller when excluding patients with clinically isolated syndrome, who in at least 30% do not have MS [23] There is also no consensus on the relevant number of new T2Ls or CELs to predict disability progression with proposed cutoffs ranging from two to more than five T2Ls, limiting the use of MRI metrics as a basis for treatment decisions [24, 25, 26.
As a result, there is currently a considerable period of uncertainty of whether the DMT is actually effective for both patients and treating neurologists after initiating a DMT. Even if a patient appears to respond to a DMT by having neither relapse nor EDSS progression or new CELs/T2Ls within 2 years after DMT initiation, 20% to 50% of those patients will still have disability progression within 2 to 5 years [27, 28 Thus, additional and more precise biomarkers are needed to evaluate the individual response to DMT.
Among other potential markers of DMT failure, neuropsychological assessment of cognitive decline might be more sensitive than EDSS, but is limited by a lack of a consensual and standardized testing approach and, most importantly, by requiring high expenditure of personnel and time as well as a lack of availability in clinical routine [29] Quantification of brain atrophy by MRI‐based measurement of whole brain volume loss (BVL) has been suggested to complement relapses, EDSS progression, and new T2Ls/CELs as a marker of (subclinical) neuroaxonal damage. Because long‐term prognosis is essentially determined by the amount of accumulating loss of neuroaxonal tissue, this is certainly a valid approach. A cutoff of BVL exceeding 0.4% per year has been proposed for identifying pathological brain atrophy [30] However, BVL is limited in distinguishing healthy controls and RMS patients (sensitivity: 26%, specificity: 90%) and is estimated to mediate about only 50% of DMT response regarding long‐term disability progression [31, 32 This is likely explained by methodological limitations of BVL quantification such as the pseudoatrophy phenomenon, susceptibility to confounding by non–MS‐associated variation in intra‐ and extracellular compartments and interrating variability, which exceeds the expected change over 2 years even in monocentric studies using the same scanner [33, 34 Thus, longitudinal BVL assessment is deemed unreliable in individual patients [34].
Contrary to BVL, GCIPL, and in absence of acute ON also pRNFL, thickness is not directly affected by inflammation, hydration, comedication, or alcohol use because the compartment measured has a less complex constitution [6] OCT also has some advantages over MRI because it is faster, less expensive, easily accessible, and produces standardized, reliable quantitative measures [35] Thus, we are convinced that retinal thinning might be a better surrogate marker of neuroaxonal damage than BVL for determining DMT response. However, our study did not include MRI, and we cannot draw direct comparisons between GCIPL/pRNFL and BVL, which should be performed in a large prospective cohort of MS patients.
Apart from the unavailability of MRI parameters for correlation, there are some limitations to this study. The validity of our results depends on meticulous quality control of OCT scans, rigorously ruling out confounding factors (e.g., severe myopia, optic disc drusen), and most importantly, accounting for history and timing of ON. Biological variability and measurement errors are also minimized by a homogeneous single‐center dataset. These sources of errors might be increased in a real‐world setting when OCT protocols and devices may vary, and multicenter datasets are used. Also, our results cannot be applied to patients with primary or secondary progressive MS, because they were excluded from our study. Finally, the prognostic accuracy of the reported cutoff values might be enhanced, because they were generated and assessed in the same cohort. Thus, validation in a separate external cohort is required, which is an important future direction.
In summary, we present evidence that retinal layer thinning predicts physical and cognitive disability progression upon DMT initiation. This underscores the potential of retinal layer thinning as a biomarker for monitoring neuroaxonal damage in MS, and if validated, it may also be a useful and easily accessible biomarker of treatment response/failure in RMS.
Measuring response to DMT will certainly require a multimodal approach, in which GCIPL thinning can contribute as a highly accurate marker of MS‐associated neurodegeneration, potentially combined with MRI markers and/or body fluid markers such as neurofilament light chains. In clinical practice, we advocate that OCT measuring both GCIPL and pRNFL should be performed within 3 months after initiating DMT as a (re)baseline and should then be repeated annually in routine monitoring, where sufficient quality of imaging and layer segmentation is available.
DISCLOSURES
G.B. has participated in meetings sponsored by, received speaker honoraria or travel funding from Biogen, Celgene, Merck, Novartis, Sanofi‐Genzyme, and Teva, and received honoraria for consulting from Biogen, Roche, and Teva. H.H. has participated in meetings sponsored by, received speaker honoraria or travel funding from Bayer, Biogen, Merck, Novartis, Sanofi‐Genzyme, Siemens, and Teva, and received honoraria for consulting from Biogen and Teva. P.A. has participated in meetings sponsored by, received speaker honoraria or travel funding from Biogen, Merck, Roche, Sanofi‐Genzyme, and Teva, and received honoraria for consulting from Biogen. He received a research grant from Quanterix International and was awarded a combined sponsorship from Biogen, Merck, Roche, Sanofi‐Genzyme, and Teva for a clinical study. M.A. received speaker honoraria and/or travel grants from Biogen, Merck, Novartis, and Sanofi‐Genzyme. K.B. has participated in meetings sponsored by and received travel funding from Roche. F.D.P. has participated in meetings sponsored by, received honoraria (lectures, advisory boards, consultations) or travel funding from Bayer, Biogen, Celgene, Merck, Novartis, Roche, Sanofi‐Genzyme, and Teva. F.L. has participated in meetings sponsored by or received honoraria for acting as an advisor/speaker for Bayer, Biogen, Celgene, MedDay, Merck, Novartis, Roche, Sanofi‐Genzyme, and Teva. P.R. has received honoraria for consultancy/speaking from AbbVie, Alexion, Almirall, Biogen, Merck, Novartis, Roche, Sandoz, Sanofi‐Genzyme, and has received research grants from Amicus, Biogen, Merck, and Roche. S.W. has participated in meetings sponsored by, received honoraria or travel funding from Biogen, Merck, Novartis, Sanofi‐Genzyme, Teva, Allergan, Ipsen Pharma, and Roche. A.Z. has participated in meetings sponsored by, received speaking honoraria or travel funding from Biogen, Merck, Sanofi‐Genzyme, and Teva. T.Z. has participated in meetings sponsored by or received travel funding from Biogen, Merck, Novartis, Roche, Sanofi‐Genzyme, and Teva. F.D. has participated in meetings sponsored by or received honoraria for acting as an advisor/speaker for Alexion, Almirall, Biogen, Celgene, Merck, Novartis, Roche, and Sanofi‐Genzyme. His institution received scientific grants from Biogen and Sanofi‐Genzyme. T.B. has participated in meetings sponsored by and received honoraria (lectures, advisory boards, consultations) from pharmaceutical companies marketing treatments for MS: Allergan, Almirall, Bayer, Biogen, Biologix, Bionorica, Celgene, MedDay, Merck, Novartis, Octapharma, Roche, Sanofi‐Genzyme, Teva, and TG Pharmaceuticals. His institution has received financial support in the past 12 months by unrestricted research grants (Biogen, Merck, Novartis, Sanofi‐Genzyme, Teva) and for participation in clinical trials in MS sponsored by Alexion, Biogen, Merck, Novartis, Octapharma, Roche, Sanofi‐Genzyme, and Teva.
AUTHOR CONTRIBUTIONS
Gabriel Bsteh: Conceptualization (lead); data curation (lead); formal analysis (lead); methodology (lead); writing–original draft (lead); writing–review & editing (lead). Harald Hegen: Conceptualization (lead); methodology (lead); writing–original draft (lead); writing–review & editing (lead). Patrick Altmann: Methodology (equal); writing–original draft (supporting). Michael Auer: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Klaus Berek: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Franziska Di Pauli: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Fritz Leutmezer: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Paulus Rommer: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Sebastian Wurth: Investigation (supporting); writing–original draft (supporting). Anne Zinganell: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Tobias Zrzavy: Investigation (supporting); methodology (supporting); writing–original draft (supporting); writing–review & editing (supporting). Florian Deisenhammer: Investigation (supporting); methodology (supporting); supervision (supporting); writing–original draft (supporting); writing–review & editing (supporting). Thomas Berger: Conceptualization (supporting); investigation (supporting); methodology (supporting); supervision (lead); writing–original draft (equal); writing–review & editing (equal).
Supporting information
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
The authors want to explicitly thank Yvonne Wehle and Daniela Schneider who diligently performed OCT scans for this study.
Gabriel Bsteh and Harald Hegen contributed equally
Funding information
There was no funding for this study.
DATA AVAILABILITY STATEMENT
Anonymized data will be shared by reasonable request from any qualified investigator.
REFERENCES
- 1. Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221‐1231. [DOI] [PubMed] [Google Scholar]
- 2. Cree BAC, Mares J, Hartung H‐P. Current therapeutic landscape in multiple sclerosis. Curr Opin Neurol. 2019;32:365‐377. [DOI] [PubMed] [Google Scholar]
- 3. Giovannoni G. Disease‐modifying treatments for early and advanced multiple sclerosis. Curr Opin Neurol. 2018;31:1‐11. [DOI] [PubMed] [Google Scholar]
- 4. Bsteh G, Hegen H, Dosser C, et al. To treat or not to treat – sequential individualized treatment evaluation in relapsing multiple sclerosis. Mult Scler Relat Dis. 2019;101908. [DOI] [PubMed] [Google Scholar]
- 5. Hegen H, Bsteh G, Berger T. ‘No evidence of disease activity’ – is it an appropriate surrogate in multiple sclerosis? Eur J Neurol. 2018;359:1221‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petzold A, Balcer LJ, Calabresi PA, et al. Retinal layer segmentation in multiple sclerosis: a systematic review and meta‐analysis. Lancet Neurol. 2017;16:797‐812. [DOI] [PubMed] [Google Scholar]
- 7. Bsteh G, Berek K, Hegen H, et al. Macular ganglion cell–inner plexiform layer thinning as a biomarker of disability progression in relapsing multiple sclerosis. Mult Scler J. 2021;27:684–694. [DOI] [PubMed] [Google Scholar]
- 8. Bsteh G, Hegen H, Teuchner B, et al. Peripapillary retinal nerve fibre layer as measured by optical coherence tomography is a prognostic biomarker not only for physical but also for cognitive disability progression in multiple sclerosis. Mult Scler J. 2019;25:196–203. [DOI] [PubMed] [Google Scholar]
- 9. Bsteh G, Hegen H, Teuchner B, et al. Peripapillary retinal nerve fibre layer thinning rate as a biomarker discriminating stable and progressing relapsing–remitting multiple sclerosis. Eur J Neurol. 2019;26:865‐871. [DOI] [PubMed] [Google Scholar]
- 10. Button J, Al‐Louzi O, Lang A, et al. Disease‐modifying therapies modulate retinal atrophy in multiple sclerosis. Neurology. 2017;88:525‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You Y, Barnett MH, Yiannikas C, et al. Interferon‐β is less effective than other drugs in controlling the rate of retinal ganglion cell loss in MS. Neurol Neuroimmunol Neuroinflamm. 2021;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benedict R, Duquin J, Jurgensen S, et al. Repeated assessment of neuropsychological deficits in multiple sclerosis using the Symbol Digit Modalities Test and the MS Neuropsychological Screening Questionnaire. Mult Scler J. 2008;14:940‐946. [DOI] [PubMed] [Google Scholar]
- 14. Benedict RH, DeLuca J, Phillips G, et al. Validity of the Symbol Digit Modalities Test as a cognition performance outcome measure for multiple sclerosis. Mult Scler J. 2017;27:1352458517690821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Group ETDRSR . Photocoagulation for diabetic macular edema. Early treatment diabetic retinopathy study report number 1. Arch Ophthalmol‐chic. 1985;103:1796. [PubMed] [Google Scholar]
- 16. Tewarie P, Balk L, Costello F, et al. The OSCAR‐IB consensus criteria for retinal OCT quality assessment. PLoS One. 2012;7:e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schippling S, Balk L, Costello F, et al. Quality control for retinal OCT in multiple sclerosis: validation of the OSCAR‐IB criteria. Mult Scler J. 2014;21:163‐170. [DOI] [PubMed] [Google Scholar]
- 18. Britze J, Pihl‐Jensen G, Frederiksen JL. Retinal ganglion cell analysis in multiple sclerosis and optic neuritis: a systematic review and meta‐analysis. J Neurol. 2017;264:1837‐1853. [DOI] [PubMed] [Google Scholar]
- 19. Nolan‐Kenney RC, Liu M, Akhand O, et al. Optimal intereye difference thresholds by optical coherence tomography in multiple sclerosis: an international study. Ann Neurol. 2019;85:618‐629. [DOI] [PubMed] [Google Scholar]
- 20. Bsteh G, Hegen H, Altmann P, et al. Validation of inter‐eye difference thresholds in optical coherence tomography for identification of optic neuritis in multiple sclerosis. Mult Scler Relat Dis. 2020;102403. [DOI] [PubMed] [Google Scholar]
- 21. Zou GY, Yue L. Using confidence intervals to compare several correlated areas under the receiver operating characteristic curves. Stat Med. 2013;32:5077‐5090. [DOI] [PubMed] [Google Scholar]
- 22. Council of National Research. The prevention and treatment of missing data in clinical trials . National Academies Press (US), 2010. Epub ahead of print 2010. DOI: 10.17226/12955. [PubMed]
- 23. Sormani MP, Gasperini C, Romeo M, et al. Assessing response to interferon‐β in a multicenter dataset of patients with MS. Neurology. 2016;87:134‐140. [DOI] [PubMed] [Google Scholar]
- 24. Río J, Castilló J, Rovira A, et al. Measures in the first year of therapy predict the response to interferon beta in MS. Mult Scler J. 2009;15:848‐853. [DOI] [PubMed] [Google Scholar]
- 25. Sormani M, Rio J, Tintorè M, et al. Scoring treatment response in patients with relapsing multiple sclerosis. Mult Scler J. 2012;19:605‐612. [DOI] [PubMed] [Google Scholar]
- 26. Freedman MS, Selchen D, Arnold DL, et al. Treatment optimization in MS: Canadian MS Working Group Updated Recommendations. Can J Neurological Sci. 2013;40:307‐323. [DOI] [PubMed] [Google Scholar]
- 27. Uher T, Havrdová E, Sobisek L, et al. Is no evidence of disease activity an achievable goal in MS patients on intramuscular interferon beta‐1a treatment over long‐term follow‐up? Mult Scler J. 2017;23:242‐252. [DOI] [PubMed] [Google Scholar]
- 28. Prosperini L, Mancinelli CR, Giglio LD, et al. Interferon beta failure predicted by EMA criteria or isolated MRI activity in multiple sclerosis. Mult Scler J. 2014;20:566‐576. [DOI] [PubMed] [Google Scholar]
- 29. van Munster CEP, Uitdehaag BMJ. Outcome measures in clinical trials for multiple sclerosis. CNS Drugs. 2017;31:217‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stefano ND, Stromillo ML, Giorgio A, et al. Establishing pathological cut‐offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psych. 2016;87:93‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uher T, Vaneckova M, Krasensky J, et al. Pathological cut‐offs of global and regional brain volume loss in multiple sclerosis. Mult Scler J. 2017;25:541‐553. [DOI] [PubMed] [Google Scholar]
- 32. Sprenger T, Kappos L, Radue E‐W, et al. Association of brain volume loss and long‐term disability outcomes in patients with multiple sclerosis treated with teriflunomide. Mult Scler J. 2019;26:1207‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bermel RA, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. 2006;5:158‐170. [DOI] [PubMed] [Google Scholar]
- 34. Biberacher V, Schmidt P, Keshavan A, et al. Intra‐ and interscanner variability of magnetic resonance imaging based volumetry in multiple sclerosis. NeuroImage 2016;142:188‐197. [DOI] [PubMed] [Google Scholar]
- 35. Ontaneda D, Fox RJ. Imaging as an outcome measure in multiple sclerosis. Neurotherapeutics. 2017;14:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Data Availability Statement
Anonymized data will be shared by reasonable request from any qualified investigator.