

Abstract
The flowering plant family Annonaceae includes important commercially grown tropical crops, but development of promising species is hindered by a lack of genomic resources to build breeding programs. Annonaceae are part of the magnoliids, an ancient lineage of angiosperms for which evolutionary relationships with other major clades remain unclear. To provide resources to breeders and evolutionary researchers, we report a chromosome‐level genome assembly of the soursop (Annona muricata). We assembled the genome using 444.32 Gb of DNA sequences (676× sequencing depth) from PacBio and Illumina short‐reads, in combination with 10× Genomics and Bionano data (v1). A total of 949 scaffolds were assembled to a final size of 656.77 Mb, with a scaffold N50 of 3.43 Mb (v1), and then further improved to seven pseudo‐chromosomes using Hi‐C sequencing data (v2; scaffold N50: 93.2 Mb, total size in chromosomes: 639.6 Mb). Heterozygosity was very low (0.06%), while repeat sequences accounted for 54.87% of the genome, and 23,375 protein‐coding genes with an average of 4.79 exons per gene were annotated using de novo, RNA‐seq and homology‐based approaches. Reconstruction of the historical population size showed a slow continuous contraction, probably related to Cenozoic climate changes. The soursop is the first genome assembled in Annonaceae, supporting further studies of floral evolution in magnoliids, providing an essential resource for delineating relationships of ancient angiosperm lineages. Both genome‐assisted improvement and conservation efforts will be strengthened by the availability of the soursop genome. As a community resource, this assembly will further strengthen the role of Annonaceae as model species for research on the ecology, evolution and domestication potential of tropical species in pomology and agroforestry.
Keywords: Annonaceae, basal angiosperms, crop improvement, high quality draft génome, magnoliids, pomology
1. INTRODUCTION
Since the publication of the first plant genome (Arabidopsis thaliana; Arabidopsis Genome Initiative, 2000), there has been a steady increase in the number of sequenced eudicot and monocot genomes. However, with the exception of the iconic Amborella trichopoda, angiosperm diversity represented by the ancient lineages of Nymphaeales, Austrobaileyales, Chloranthales, and magnoliids has largely been overlooked. After eudicots and monocots, Magnoliidae are the most diverse clade of angiosperms (Massoni et al., 2014) with 9000–10,000 species in four orders (Canellales, Piperales, Laurales and Magnoliales). Despite this diversity and economic value (e.g., avocado, black pepper, cinnamon, soursop), only four genomes in three families have been published to date (Chaw et al., 2019; Chen et al., 2019; Hu et al., 2019; Rendón‐Anaya et al., 2019). Analysis of such genomic data was expected to resolve the still unclear relationships of magnoliids with the rest of angiosperms (Soltis & Soltis, 2019). However, recently published results strongly disagree on the position of magnoliids, supporting either a sister relationship to eudicots and monocots (Chen et al., 2019; Hu et al., 2019; Rendón‐Anaya et al., 2019), or to eudicots alone (Chaw et al., 2019).
Here, we report the genome sequence of Annona muricata (the soursop, guanábana [Spanish], graviola [Portuguese]) which is one of the c. 2500 species of the custard apple family (Annonaceae) (Rainer & Chatrou, 2014), the second most species‐rich family of magnoliids (Chatrou et al., 2012). Its species are frequent components of tropical rain forests worldwide (Gentry, 1993; Punyasena et al., 2008; Sonké & Couvreur, 2014; Tchouto et al., 2006). Widely known examples include ylang‐ylang (Cananga odorata), used for its essential oils, and species of the Neotropical/African genus Annona, cultivated for their edible fruits, medicinal and pharmaceutical properties.
Annona muricata originated in the Neotropics, but is now widely cultivated in tropical and subtropical regions around the world. In some of the main producing nations (Brazil, Mexico and Venezuela), cultivation of soursop resulted in annual revenues of 5.4 million USD (8000 t; 1997), 23.7 million USD (349,000 t; 1996) and 6.9 million USD (10,100 t; 1987), respectively (Pinto et al., 2005). Annona muricata is a small (up to 9 m), evergreen tree, typically with hairy branches when young. Leaves are oblong to oval, with a glossy green surface, while flowers are simple, with green sepals and thick yellowish petals (Figure 1a,b). The fruits are ovoid, dark green and tuberculate (Figure 1c,d), and can be up to 30 cm long, with a moderately firm texture. Their whitish flesh is juicy, acidic, whitish and aromatic. The fruit contains significant amounts of vitamins (e.g., vitamin C, vitamin B1 and B2), but also the neurotoxine annocianin. Both fruits and leaves as well as seeds have long been used to treat a wide range of ailments owing to their pharmacological activities (e.g., antimicrobial, leishmanial, hyperglycemic, parasitic, inflammatory, neuralgic, rheumatic a.o.). More recently, research has focussed increasingly on the potential of using compounds extracted from Annona muricata to treat carcinogenic cell lines.
FIGURE 1.
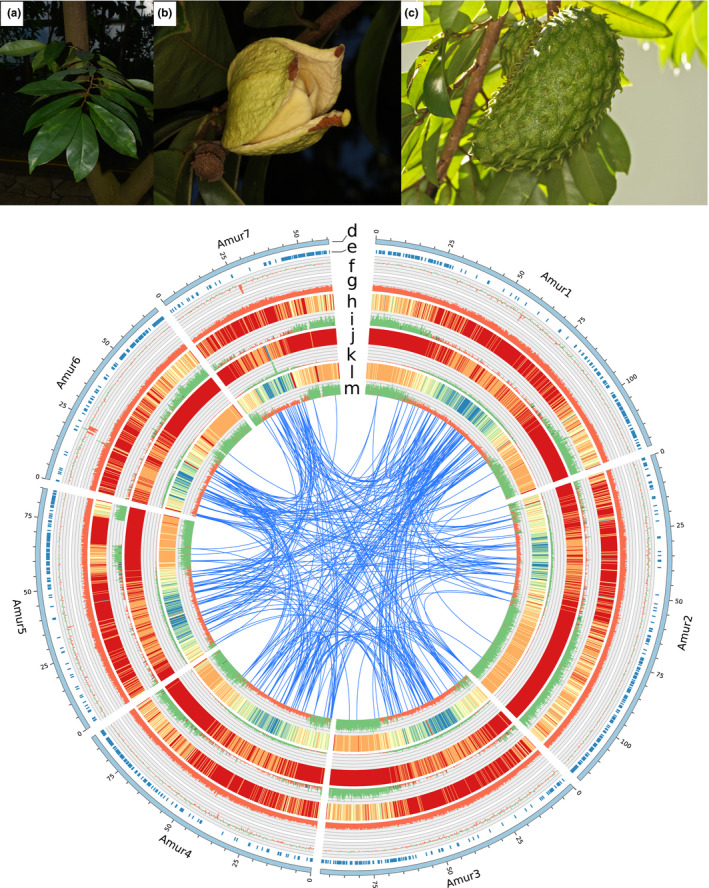
Annona muricata description and genomic landscape. Top: (a) leaves, (b) mature flower, (c) mature fruit. Bottom: Circular view of the chromosome organization of Annona muricata, with genomic features indicated from outer to inner layers in sequence windows of 200 kb, (d) Structural organisation of the chromosomes arranged by size, indicated in Mb, (e) loci density from Couvreur et al., 2019, (f) GC déviation, (g) GC content (percentage), (h) gene breadth (i.e., the percentage of the sequence window occupied by coding regions) heatmap, (i) gene density (i.e., the number of genes found in one sequence window) histogram, (j) TE protein breadth heatmap, (k) TE protein density histogram, (l) transposon breadth heatmap; (m) transposon density histogram. In (i), (k) and (m), values above and below the mean are indicated in green and red, respectively.
2. MATERIALS AND METHODS
2.1. Genomic DNA extraction, Illumina sequencing and genome size estimation
Fresh leaves were collected from the living collections in Xishuangbanna Tropical Botanical Garden (Menglun, China) and frozen on site. High‐quality genomic DNA was extracted from freshly frozen leaf tissue of one individual of A. muricata using the Plant Genomic DNA Kit (Tiangen), following the manufacturer's specification. After purification, a short‐insert library (300–350 base pairs [bp]) was constructed using a Illumina TruSeq library construction kit according to the manufacturer's instructions (Illumina), and sequenced on the Illumina HiSeq 2500 platform (Illumina Inc.), according to the manufacturer's specifications (paired‐end 2 × 150 bp). A total of ~65.47 Gb of raw data were generated. Sequencing adapters were then removed from the raw reads and reads from non‐nuclear origin (chloroplast, mitochondrial, bacterial and viral sequences, etc.), screened by aligning them to the nr database (NCBI, http://www.ncbi.nlm.nih.gov, accessed on 12/07/2017) using megablast v2.2.26 with the parameters “‐v 1 ‐b 1 ‐e 1e‐5 ‐m 8 ‐a 13”; The script duplication_rm.v2 (Strijk et al., 2019) was used to remove the duplicated read pairs; low‐quality reads were filtered as follows: (i) reads with ≥10% unidentified nucleotides (N) were removed, (ii) reads with adapters were removed, and finally (iii) reads with >20% bases having Phred quality <5 were removed. After the removal of low‐quality and duplicated reads, ~65 Gb of clean data (Table 1) were used for the genome size estimation, based on the 17‐mer frequency of Illumina short reads. The formula – “genome size = (total number of 17‐mer)/(position of peak depth)” ‐ was used to obtain an estimate of 799.11 Mb. An additional library was built (250 bp), sequenced as above and combined with the 350 bp library to generate approximately 900 million reads to provide a first estimation of the GC content, heterozygosity rate and repeat content (as outlined in Li et al., 2019, as well as with GenomeScope 2) based on the k‐mer analyses.
TABLE 1.
Sequencing strategy and statistics used for the A. muricata genome assembly and annotation
| Step | Technology | Tissue | Insert size | Bases generated (Gb) | Sequence coverage (x) |
|---|---|---|---|---|---|
| Genome assembly | Illumina reads | Leaves | 250 bp | 65.96 | 82.54 |
| 350 bp | 65.47 | 81.93 | |||
| PacBio reads | Leaves | 20 kb | 36.95 | 46.24 | |
| 10× | Leaves | 180.04 | 225.3 | ||
| Bionano | Leaves | 95.9 | 120.01 | ||
| Total | 444.32 | 556.02 | |||
| Chromosome scaffolding | Hi‐C | Leaves | N.A. | 66.17 | N.A. |
| Genome annotation | Illumina reads | Flowers (several developmental stages) | 350 bp | 5.52 | N.A |
| Young fruit | 350 bp | 9.93 | N.A | ||
| Ripening fruit | 350 bp | 5.73 | N.A | ||
| Bark | 350 bp | 4.80 | N.A | ||
| Leaves | 350 bp | 5.04 | N.A | ||
| Total | 25.51 |
2.2. PacBio, 10× Genomics and Bionano library preparation and sequencing
A 20 kb insert size PacBio library was built as previously described (Strijk et al., 2019). This library was sequenced on the PacBio RS II platform (Pacific Biosciences), yielding about 37 Gb of data (read quality ≥0.80, mean read length ≥7 Kb). 10× Genomics DNA sample preparation, indexing, and barcoding were done using the GemCode Instrument (10× Genomics). About 0.7 ng of very high molecular weight DNA (N50 ~165 kb, 3% of the DNA >500 kb) was used for GEM reaction procedure during PCR, and 16 bp barcodes were introduced into droplets. Then, the droplets were fractured following the purifying of the intermediate DNA library. Next, we sheared the DNA into 500 bp for fragments constructing libraries, which were then sequenced on the Illumina HiSeq X platform (Illumina Inc.), according to the manufacturer's instructions. The same high molecular weight DNA was used to construct a Bionano optical map using an Irys platform (BioNano Genomics) with the Nt. BspQ1 (8.19 labels/100 kb), of which 95.9 Gb (120.01×, Table 1) data were generated.
2.3. De novo genome assembly, 10× and optical scaffolding
We used SOAPdenovo2 (Luo et al., 2012) and obtained a preliminary assembly (using Illumina reads) of A. muricata with a scaffold N50 size of 19,908 kb and corresponding contig size of 8.26 Kb. We then used PBjelly (English et al., 2012) to fill gaps with the PacBio data. The options were set to “<blasr>‐minMatch 8 ‐sdpTupleSize 8 ‐minPctIdentity 75 ‐bestn 1 ‐nCandidates 10 ‐maxScore −500 ‐nproc 10 ‐noSplitSubreads</blasr>”. Then, we used Pilon (Walker et al., 2014) with default settings to correct assembled errors. For the input BAM file, we used bwa‐mem v0.7.17 with default parameters (Li & Durbin, 2009) to align all the Illumina short reads to the assembly and SAMtools to sort and index the BAM file. This second assembly reached a contig N50 of approximately 700 kb. It was then combined with a de novo optical genome map (assembled with irysview [version 2.3, BioNano Genomics] from the generated Bionano data (see above]) using Hybrid Scaffold scripts (BioNano Genomics). We used fragScaff (Adey et al., 2014) to generate scaffolds from this scaffolded assembly using the 10× Genomics data (180.04 Gb – 225.30×, Table 1) with default parameters.
2.4. Hi‐C scaffolding
We constructed two Hi‐C libraries from flash‐frozen soursop leaves by cross‐linking HMW gDNA in a 4% formaldehyde solution at room temperature in a vacuum for 30 min. Then, 2.5 M glycine was added to stop the crosslinking reaction for 5 min, and the sample was kept on ice for 15 min. The sample was centrifuged at 1,000 g at 4°C for 10 min, and the pellet was washed with 500 μl PBS, then centrifuged for 5 min at 1,000 g. The pellet was resuspended with 20 μl of lysis buffer (1 M Tris‐HCl, pH 8, 1 M NaCl, 10% CA‐630, and 13 units protease inhibitor), and the supernatant was centrifuged at 4,000 g at room temperature for 10 min. The pellet was washed twice in 100 μl ice cold 1× NEB buffer and then centrifuged for 5 min at 4,000 g. The nuclei were resuspended by 100 μl NEB buffer and solubilized with dilute SDS followed by incubation at 65°C for 10 min. The SDS was neutralized by Triton X‐100, and an overnight digestion was applied to the samples with a 4‐cutter restriction enzyme MboI (400 units) at 37°C on a rocking platform.
This was followed by marking the DNA ends with biotin‐14‐dCTP and blunt‐end ligation of the cross‐linked fragments. The proximal chromatin DNA was religated by ligation enzymes. The nuclear complexes were reverse cross‐linked by incubation with proteinase K at 65°C. DNA was purified using a standard phenol‐chloroform extraction protocol (Sambrook & Russell, 2006). Biotin was removed from nonligated fragment ends using T4 DNA polymerase. Sonication‐sheared fragment ends (200–600 bp) were repaired using a mixture of T4 DNA polymerase, T4 polynucleotide kinase and Klenow DNA polymerase. Biotin‐labeled Hi‐C samples were specifically enriched using streptavidin C1 magnetic beads. After adding A‐tails to the fragment ends and following ligation by the Illumina paired‐end (PE) sequencing adapters, Hi‐C sequencing libraries were amplified by PCR (12–14 cycles) and sequenced on an Illumina NovaSeq platform (PE 150 bp).
After quality assessment, 66.1 Gb of Illumina reads were retained and assessed for Hi‐C cross‐linking efficiency using HiCUP (included in Juicer tools 1.5). The Hi‐C clean data were aligned against the scaffold assembly using bwa‐mem v0.7.17 with default parameters (Camacho et al., 2009). Only the read pairs with both reads aligned to contigs and within 500 bp from a restriction site, uniquely mapped and valid di‐tags paired‐end reads were used to build the pseudo‐chromosome sequences. We used LACHESIS (Lowe & Eddy, 1996) and JUICEBOX (Durand et al., 2016; Robinson et al., 2018) to assemble, order and orientate the scaffolds of our draft genome into the seven chromosomes of the soursop. Previous IPCN entries for Annona muricata listed eight chromosomes (Sarkar et al. 1980; Sobha & Ramachandran, 1980), but both are from Indian introduced and cultivated sources, and based on singular collections.
We assessed the quality of the soursop genome assembly by mapping both the Illumina (with bwa‐mem v0.7.17, default parameters) and PacBio reads (using minimap2 2.17r941, default parameters) back against the assembly. We also evaluated both the quality of our gene predictions and completeness of our assembly using busco v4.0.2 (Simão et al., 2015). Finally, a k‐mer analysis was performed with the k‐mer Analysis Toolkit (KAT) v2.2.0 (Mapleson et al., 2017), comparing k‐mers present in the raw sequencing reads to k‐mers found in the genome assembly with KAT comp.
2.5. Repeat sequences in the soursop genome
Transposable elements in the assembly were identified both at DNA and protein levels. We used RepeatModeler (Smit & Hubley, 2008; Flynn et al., 2020) to de novo identify and classify repeated sequences in the soursop genome, including microsatellites, tandem repeats and transposable elements. RepeatMasker (Smit et al., 2017) was applied for DNA‐level identification using Repbase and the de novo transposable element library. At the protein level, RepeatProteinMask was used to conduct WU‐BLASTX (Camacho et al., 2009) searches against the transposable element protein database. Overlapping transposable elements belonging to the same type of repeats were merged.
The tRNA genes were identified by tRNAscan‐SE (Lowe & Eddy, 1996) with eukaryote parameters. The rRNA fragments were predicted by aligning them with Arabidopsis thaliana and Oryza sativa template rRNA sequences using BlastN (Camacho et al., 2009) at E‐value of 1E‐10. The miRNA and snRNA genes were predicted using infernal (Nawrocki and Eddy, 2013) by searching against the Rfam database (Nawrocki et al., 2015).
2.6. Gene annotation
2.6.1. RNA preparation sequencing and transcriptome assembly
Total RNA was extracted from leaves, flowers, bark and both young and ripe fruits (Table 1) using the RNAprep Pure Plant Kit, and genomic DNA contamination was removed using RNase‐Free DNase I (both from Tiangen, Beijing, China). The integrity of RNA was evaluated on a 1% agarose gel, and its quality and quantity were assessed using a NanoPhotometer spectrophotometer (IMPLEN) and an Agilent 2100 Bioanalyzer (Agilent Technologies). RNA sequencing (RNA‐Seq) libraries were constructed using the NEBNext mRNA Library Prep Master Mix Set for Illumina (New England Biolabs) following the manufacturer's instructions. The PCR products obtained were purified (AMPure XP system; Beckman Coulter Inc.) and library quality was assessed on the Agilent Bioanalyzer 2100 system. Library preparations were sequenced on an Illumina HiSeq 2000 platform (Illumina Inc), generating 100 bp paired‐end reads. Raw reads were filtered by removing those containing undetermined bases (N′) or excessive numbers of low‐quality positions (>10 positions with quality scores <10).
2.6.2. Annotation
Protein coding genes were predicted through a combination of de novo, homology and transcriptome‐based predictions, using the repeat‐masked genome sequence: (i) Structural annotation of protein coding genes and domains was performed by aligning the protein sequences of the soursop against a representative set of angiosperms (Amaranthus hypochondriacus, Amborella trichopoda, Aquilegia coerulea, Arabidopsis thaliana, Coffea canephora, Musa acuminata, Nelumbo nucifera, Oryza sativa, Vitis vinifera) using Tblas tN (Camacho et al., 2009) with an E‐value cutoff of 1E‐5. Blast hits were conjoined by Solar (Yu et al., 2006) and Genewise (Birney et al., 2004) was used for each to predict the exact gene structure in the corresponding genomic régions. (ii) Five ab initio gene prediction programmes, including augustus v3.0.2 (Stanke et al., 2006), genscan v1.0 (Burge & Karlin, 1997), glimmerhmm v3.0.2 (Majoros et al., 2004), geneid (Blanco & Abril, 2009) and snap (Korf, 2004) were used to predict coding genes on the repeat‐masked genomes. Each program was trained using the Solar and Genewise ge ne set with default parameters. (iii) Finally, RNA‐seq‐based predictions were performed using two methods. First, RNA‐seq data were mapped to the A. muricata genome using tophat v2.0.9 (Kim et al., 2013) with the parameters of "‐p 10 ‐N 3 ‐‐read‐edit‐dist 3 ‐m 1 ‐r 0 ‐‐coverage‐search ‐‐microexon‐search”, and then cufflinks (Trapnell et al., 2012) was used to assemble transcripts to gene models. Second, Trinity (Grabherr et al., 2011) was used to assemble the RNA‐seq data keeping the longest transcript, followed by PASA (http://pasapipeline.github.io/) to improve the gene structures.
All gene models predicted from the above three approaches were combined into a weighted nonredundant set of gene structures with EVidenceModeler (EVM) (Haas et al., 2008). Default parameters (notably a minimum intron size = 20 bp and a minimum length of 100 amino acids for a coding region to be reported) were used, and weights for each type of evidence were set as follows: PASA‐derived genes >Homology‐derived genes >Cufflinks‐derived genes > Augustus > GeneID = SNAP = GlimmerHMM = Genscan. Then we filtered out low quality gene models using two criteria: (i) coding region lengths of ≤150 bp and (ii) those supported only by ab initio methods and with FPKM<1 (fragments per kilobase of transcript per million mapped reads). In addition, multiexonic genes were kept only if rpkm >1 and homolog >1, while monoexonic genes with rpkm <6 and homolog <3 were filtered out. Functional annotation of protein coding genes was carried out using BLASTP (Camacho et al., 2009) (e‐value 1E‐05) against two integrated protein sequence databases – SwissProt and TrEMBL (Boeckmann et al., 2003). The annotation information of the best blast hit derived from the database, was transferred to our gene set. Protein domains were annotated by searching interpro (Hunter et al., 2009) and Pfam (El‐Gebali et al., 2019) databases, using interproscan (Quevillon et al., 2005) and hmmer (Finn et al., 2011), respectively. Gene ontology (GO) terms for each gene were obtained from corresponding InterPro or Pfam entries. Gene pathways were assigned by blasting against the KEGG database (https://www.genome.jp/kegg/), with an E‐value cutoff of 1E‐05. These results were then used to investigate functional roles of genes and to compare these patterns with those found in closely related species.
2.7. Positive selection
To detect positive selection on protein‐coding sequences, we calculated the number of synonymous substitutions per site (Ks) and nonsynonymous substitutions per site (Ka) for a set of angiosperms (Amborella trichopoda, Arabidopsis thaliana, Helianthus annuus, Nelumbo nucifera and Oryza sativa) in addition to A. muricata. A ratio Ka/Ks >1 is an indication of positive selection. We used MUSCLE (Edgar, 2004) to generate MSA for the protein and nucleotide sequences, and Gblocks (Castresana, 2000, 2002) with default parameters (‐b3 8;‐b4 10;‐b5 n) to remove poorly aligned positions of alignments. The maximum likelihood‐based branch test (with default settings) implemented in the paml package (Yang, 2007) was used to produce an estimate of the genic Ka/Ks ratio, calculated from the entire length of the protein sequences.
2.8. Population size changes inference
We used PSMC (Liu & Hansen, 2017) to infer the variation in population size of the soursop based on the observed heterozygosity in the diploid genome. As PSMC was shown to perform reliably for scaffolds >100 kb, we removed shorter scaffolds from the assembly. 312 scaffolds >100 kb were kept, totalling 646.64 Mb (98.46% of the total assembly). We assumed a generation time of 15 years (Collevatti et al., 2014) and a per‐generation mutation rate of 7 × 10−9. PSMC was otherwise conducted using default parameters.
2.9. Organellar genome reconstruction
The chloroplast of Annona muricata was reconstructed using getorganelle (Jin et al., 2019), with a subset of the Illumina paired reads (~18 million reads) and default parameters. The three Illumina libraries were then mapped against the resulting circular contig to detect any misassemblies. The draft plastome was annotated using cpgavas 2 (Shi et al., 2019), using default settings and deposited in GenBank (GB number pending).
2.10. Mapping of hybridization capture data
To exemplify usefulness of the v2 assembly, we mapped the data from Couvreur et al. (2019), consisting of hundreds of nuclear loci obtained by targeted enrichment followed by high throughput sequencing, onto our final data set. First, we mapped the reference sequences of the 469 exon regions identified by Couvreur et al. (2019) on the v2 assembly. The sequences of these 469 exon regions were derived from species belonging to different subfamilies in Annonaceae and tribes in Annonoideae, but did not include any species from the tribe Annoneae. To counterbalance the phylogenetic distance between these reference sequences and the soursop genome, we set a low stringency for mapping. We used the Geneious mapper implemented in Geneious R9.1.8, with 5 iterations (default parameters, except minimum mapping quality = 20, word length = 18, maximum mismatch per read = 30%). Second, as Annona muricata was not represented by Couvreur et al. (2019), we retrieved the raw reads resulting from their hybridization sequence capture of closely related Annona glabra, filtered them by removing any position from both ends with a quality <Q20, and mapped them against our v2 assembly using Bowtie2, with default parameters. Regions with a mapping depth >30 were annotated in Geneious Prime (Biomatters, Ltd.), and displayed on the chromosomes using circos 0.69–9 (Krzywinski et al., 2009).
3. RESULTS
3.1. High quality Annona genome
Following the method of Li et al. (2019), we estimated genome size and heterozygosity to be 799.11 Mb and 0.08%, respectively (GenomeScope analysis: genome length = 654 Mb; heterozygosity = 0.038%, Figure S1b), with a repeat content of 59.76% (GenomeScope: 36.2%). The GC content ranged from 35.46% (350 bp library) to 37.64% (250 bp library). The first genome assembly using only Illumina data and the assembly program SOAPdenovo2 (Luo et al., 2012) was approximately 595.5 Mb, with a contig N50 of 8258 bp, a scaffold N50 of 19,908 bp (620.3 Mb total length).
A total of 444.32 Gb of data were produced using Illumina, PacBio, 10× Genomics and Bionano technologies, corresponding to 556× coverage of the soursop genome. This strategy provided sequencing depths of 163×, 46×, 225× and 120× for Illumina, PacBio, 10× Genomics and Bionano libraries sequencing, respectively (Table 1).
Scaffolding with Bionano resulted in a v1 assembly comprising 949 scaffolds, with a scaffold N50 length of 3.43 Mb (Table 2) for a total assembly length of 656.78 Mb. The longest scaffold was 20.46 Mb (GC content of 34.35%) and 29 scaffolds were longer than 5 Mb. Scaffolds longer than 100 kb totalled 646.64 Mb (98.45% of the total length). This level of contiguity is similar to that obtained in Liriodendron chinensis (N50 = 3.5 Mb (Chen et al., 2019)) smaller than obtained in Cinnamomum kanehirae (N50 = 50.4 Mb after Hi‐C scaffolding (Chaw et al., 2019)) but better than that of other genomes assembled at scaffold‐level (Arimoto et al., 2019; Wei et al., 2018; Zhang et al., 2019). A total of 97.16% Illumina reads can be mapped, covering >99.92% of the genome, excluding gaps, while 96.7% of the PacBio reads can be mapped back to the v2 assembly (considering only pseudomolecules). 99.81% of the genome was covered with a depth >20×, which guaranteed the high accuracy of the assembly for SNPs detection (Table S1). SNP calling on the final assembly yielded a heterozygosity rate of 0.032%, lower than 0.08% as estimated by the K‐mer analysis (Figure S1). We then used Hi‐C scaffolding to improve the v1 assembly and produce a chromosome‐level assembly, hereafter referred as “v2 assembly”. Assembly statistics after Hi‐C scaffolding are summarized in Table 2. The Annona muricata genome information after HI‐C scaffolding is summarized in Table 3. Sequencing quality assessment is shown in Table S2. Statistics for the final soursop genome assembly are as follows: the total length of contig is 652,885,881 bp, the length of contig N50 reaches 743,350 bp; the total length of scaffold is 656,813,740 bp, and the length of scaffold N50 reaches 93,205,713 bp. 97.38% of the contigs from the v1 assembly were included in the v2 assembly.
TABLE 2.
Assembly properties
| Length | Number | |||
|---|---|---|---|---|
| Contig a (bp) | Scaffold (bp) | Contig a | Scaffold | |
| Assembly v1 (Illumina +PacBio +10X + BioNano) | ||||
| Total | 652,885,881 | 656,774,640 | 2066 | 949 |
| Max | 4,254,538 | 20,459,086 | ‐ | ‐ |
| Number > =2000 | ‐ | ‐ | 1990 | 873 |
| N50 | 784,561 | 3,429,555 | 250 | 52 |
| N60 | 632,116 | 2,673,626 | 342 | 73 |
| N70 | 483,912 | 2,112,119 | 459 | 101 |
| N80 | 346,983 | 1,573,287 | 618 | 137 |
| N90 | 207,456 | 964,101 | 856 | 189 |
| Assembly v2 (Assembly v1 + Hi‐C) | ||||
| Total | 652,885,881 | 656,813,740 | 2262 | 755 |
| Max | 4,254,538 | 122,620,176 | ‐ | ‐ |
| Number > =2000 | ‐ | ‐ | 2186 | 679 |
| N50 | 743,350 | 93,205,713 | 264 | 3 |
| N60 | 578,736 | 89,409,058 | 364 | 4 |
| N70 | 451,341 | 85,026,703 | 492 | 5 |
| N80 | 320,782 | 69,840,041 | 665 | 6 |
| N90 | 184,498 | 60,483,854 | 929 | 7 |
Contig after scaffolding.
TABLE 3.
Chromosome properties of the v2 assembly
| Chromosome name | Cluster number | Sequences length | |
|---|---|---|---|
| Hic_asm_0 | Amur4 | 49 | 89,409,058 |
| Hic_asm_1 | Amur1 | 68 | 122,620,176 |
| Hic_asm_2 | Amur3 | 57 | 93,205,713 |
| Hic_asm_3 | Amur2 | 75 | 118,991,926 |
| Hic_asm_4 | Amur7 | 34 | 60,483,854 |
| Hic_asm_5 | Amur5 | 62 | 85,026,703 |
| Hic_asm_6 | Amur6 | 53 | 69,840,041 |
3.1.1. Quality assessment
KAT k‐mer‐based analysis of the assembly indicated that 98.01% of the sequence diversity found in Illumina reads was integrated in the v2 assembly (i.e., estimated assembly completeness = 98.01%, Figure S1c). Assessment of the quality of our gene predictions and assembly completeness show that of the universal BUSCO orthologous single copy genes, 235 (92.14%) were retrieved from the soursop assembly v2 (Table S3), while 85.8% of the eudicot specific orthologues were retrieved using the automatic lineage selection in BUSCO. However, this lower value may be an underestimate due to the likely imperfect fit between the selected lineage (eudicots) and the actual lineage (magnoliids).
3.2. Annotation of the soursop genome
3.2.1. Repeat sequences in the soursop genome
Repeats accounted for 54.87% of the soursop genome, a value intermediate between those of Cinnamomum kanehirae (48%) and Liriodendron chinense (63.81%), species of Lauraceae and Magnoliaceae, respectively, which together with Annonaceae represent the three largest plant families in the Magnoliidae. Long terminal repeat (LTR) retrotransposons were the most abundant forms of TE, representing 41.28% of the genome (56.25% in Liriodendron chinense), followed by DNA repeats (7.29%) (Table S4, Figure 2a). The stout camphor tree genome exhibited a different balance between types, with LTR (25.53%) and DNA transposable elements (12.67%) being less dominant. No significant recent accumulation of LTRs and 2LINEs was found in the interspersed repeat landscape, but a concordant accumulation around 40 units was detected (Figure 2b). Assuming a substitution rate similar to the one found in Liriodendron (1.51 × 10−9 subst./site/year), we estimate this burst of transposable elements to have occurred 130–150 Ma ago. By far the main contribution to this old expansion of repeat copy‐numbers were the LTRs, with an increase of up to approximately 1% at 42 units. We identified 1201 microRNA, 560 transfer RNA (tRNA), 315 ribosomal RNA (rRNA), and 3198 small nuclear RNA (snRNA) genes (Table S5).
FIGURE 2.
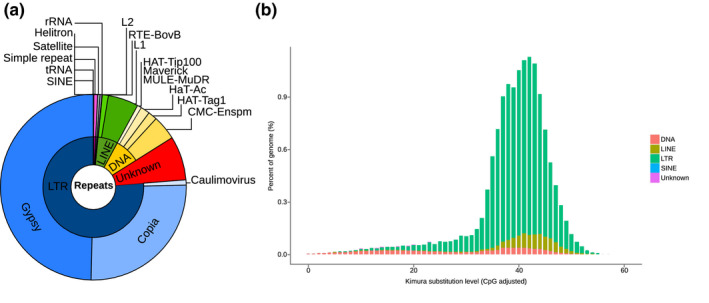
TE characteristics in the soursop genome. (a) Distribution of repeat classes in the soursop génome, (b) divergence distribution of transposable elements in the genome of Annona muricata. Both Kimura substitution level (CpG adjusted) and absolute time are given.
3.2.2. Genes in the soursop genome
We identified 23,375 genes, 21,036 of them supported by at least two of the predictive methods described above (Figure S2), with an average coding‐region length of 1.1 kb and 4.79 exons per gene (Table S6), similar to other angiosperms (Table S7). 22,769 (97.4%) genes were annotated and GO‐terms were retrieved for 20,595 (88.1%) genes (Table S8).
3.3. Genes involved in plant defence and disease resistance
Comparing gene content in Annona with the stout camphor tree, we found a striking difference in diversity of resistance genes. Of 387 resistance genes in Cinnamomum, 82% were nucleotide‐binding site leucine‐rich repeat (NBS‐LRR) or with a putative coiled‐coil domain (CC‐NBS‐LRR). By contrast, the soursop genome contains a similar number of resistance genes (301 annotations), but only 0.66% (2 genes) of them are NBS‐LRR or CC‐NBS‐LRR genes. These results suggest the presence of different evolutionary strategies within magnoliids with respect to pathogen resistance.
We explored the expansion of gene families in magnoliid lineages by adding Cinnamomum and Liriodendron to the quartet (Annona muricata, Arabidopsis thaliana, Amborella trichopoda and Oryza sativa). GO‐terms from annotations of these gene families show that the lineage of Annona experienced a fast expansion of both the MAD1 protein family (+6 copies, involved in flowering time ‐GO:0009908‐ and cold adaptation ‐GO:0009409), and metabolism through mitochondrial fission (GO:0000266), regulation of transcription (GO:0006383, GO:0006366, GO:0045892) and organism development (GO:0007275). Half of the expanded gene families with annotations in the branch of Magnoliales (Liriodendron, Annona) were involved in disease or pathogen resistance. By contrast, gene families experiencing fast expansion on the magnoliids branch ([Liriodendron, Annona], Cinnamomum) were mainly involved in growth function, such as cell wall biogenesis (GO:0042546) and membrane fission (GO:0090148), and metabolism of peptides (GO:0006518), proteins (GO:0046777) and mitosis cytokinesis (GO:0000281). Notably, gene family expansion is consistently lower along internal branches (approximately 1/10th of the gene family expansion is found on their sister branches).
We identified 77 genes putatively under positive selection (p‐value <0.01, FDR <0.05). We identified the 10 most enriched gene families and retrieved their GO‐terms. Two families have GO‐terms (Table S9) and none of these families have a defined KEGG pathway.
3.4. Historical fluctuations in population size
We determined that Annona muricata exhibits heterozygous and homozygous SNP ratios of 0.0032% and 0.0001%, respectively. This very low heterozygosity, usually found in cultivated species that experienced strong bottlenecks during domestication (Doebley et al., 2006; Eyre‐Walker et al., 1998; Zhu et al., 2007), was not due to an intense, recent decrease in population size, as shown by our PSMC analysis. Instead, the very low heterozygosity observed in soursop was due to a slow and regular reduction of the species population sizes (Figure 3). The slow but regular reduction in population size of A. muricata is compatible with the Quaternary contraction of tropical regions in several parts of the world, and suggests that the soursop may have been severely affected by climate changes, as many other tropical taxa (Barlow et al., 2018). The very low heterozygosity in soursop could make future genetic improvement difficult, and will probably require outcrossing with wild relatives (Zamir, 2001).
FIGURE 3.
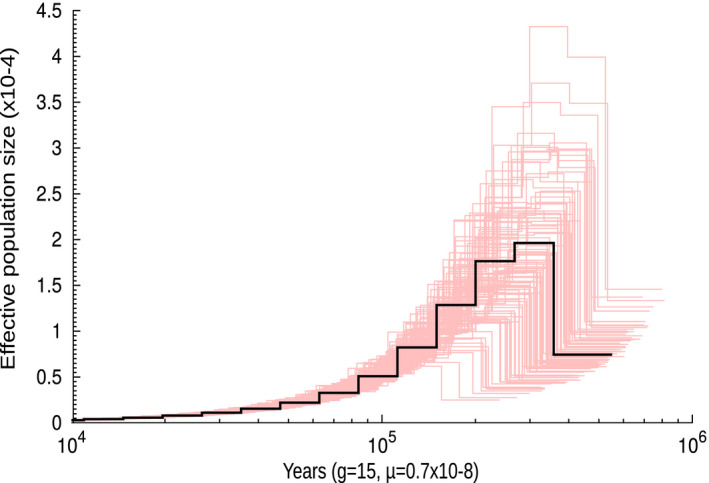
Population size variation in soursop. Effective population size history inferred by the PSMC method (black line), with 100 bootstraps shown (red lines).
3.5. Mapping of genes from hybridization capture
One‐third (154) of the loci used in Couvreur et al. (2019) were successfully mapped on the seven soursop chromosomes, with 21.7 loci per chromosome on average (médian = 19, min = 11 on Amur3, max = 36 on Amur2). The mean and median distance between loci were 3.41 Mb and 5.6 Mb, respectively. Additionally, we mapped the reads from Annona glabra (obtained previously using targeted enrichment of nuclear genes from the study of Couvreur et al. [2019]) to the v2 assembly, and superimposed their position and density onto the circular chromosome map (Figure 1) using circos 0.69–9 (Krzywinski et al., 2009). Mapping was significantly lower in the regions with high numbers of repeat sequences. A total of 1472 regions with coverage (i.e., mapping depth) higher than 30× and length >100 bp were identified across the genome. This is greater than the number of retrieved loci in the original study (469 genes, Couvreur et al., 2019), suggesting that a proportion of off‐target sequences were retrieved during hybridization sequence capture in this species, potentially expanding the resulting phylogenomic data set for inferring relationships between closely related species (e.g., of the large genus Annona). The mapped regions were spread across all chromosomes, with 210 mapped regions per chromosome on average (min: 123 on Amur7 ‐ the shortest chromosome; max 274 loci on Amur1 ‐ the longest chromosome), and a mean coverage of 192.6× in these regions (max coverage: 1475× found in Amur1). The loci recovered were 369 bp long in average, relatively constant across the chromosomes. However, we found some variation in the longest locus found on each chromosome, ranging from 1305 bp (Amur5) to 3432 bp (Amur4). The mean and median distance between two loci were 433,177 bp and 10,989 bp respectively. This discrepancy is due to the lower density of loci in centromeric regions, where repeats occur more frequently. We used the soursop v2 genome to improve the reliability and utility of genomic data generated for Annonaceae, but further investigation is needed to estimate if our results, which focus on the Annona species included in Couvreur et al. (2019), can be applied to other genera in the family.
4. CONCLUSIONS
This study presents the first high‐quality genome assembled for a plant in the Annonaceae ‐ a large tropical tree family of global ecological and economic importance. The Annona muricata genome provides an important resource for research on the evolution of magnoliids and on the conservation of this tropical tree species. It also provides support for further studies of floral evolution and floral morphological diversity in magnoliids (Sauquet et al., 2017). As such, it is an essential resource for delineating relationships of major lineages at the base of the angiosperms, furthering our understanding of the role that past genomic changes have had on the evolution of the early angiosperm flower and the later appearance of clade specific features of genomes and flower morphology in contemporary clades. The soursop genome is not only of importance for the scientific community, but also for breeders of other tropical trees (e.g., avocado, Annona species, pepper, Magnolia) as it provides novel data on disease resistance and plant defence. Of particular relevance is the positional information inherent in genome data, which is absent from transcriptomes. This allowed us to discover the distribution and proximity of phylogenomic markers used in Annonaceae (Couvreur et al., 2019) across all the chromosomes, and will allow breeders to use linkage disequilibrium estimation in their programmes (Barabaschi et al., 2016).
AUTHOR CONTRIBUTIONS
Joeri S. Strijk designed the study and funded genome sequencing. Damien D. Hinsinger sampled the sequenced specimen and performed computational analyses. Mareike M. Roeder provided sampling assistance. Lars W. Chatrou, Thomas L. P. Couvreur, Roy H. J. Erkens, Hervé Sauquet, Michael D. Pirie, Daniel C. Thomas provided advice on the experimental design. Kunfang Cho contributed reagents and resources. Joeri S. Strijk and Damien D. Hinsinger wrote and edited the manuscript with contributions from all authors.
Supporting information
Fig S1
Fig S2
Table S1‐S9
ACKNOWLEDGEMENTS
Genome sequencing, assembly and annotation were conducted by Novogene Bioinformatics Institute, Beijing, China; Project No. P2016112416. We kindly acknowledge Ghent University Botanical Garden for granting access to their living collections of A. muricata.
This work was supported through the Guangxi Province One Hundred Talent program and the Bagui Scholarship team funding under Grant No. C33600992001 to JSS, and the China Postdoctoral Science Foundation (grant number 2015M582481 and 2016T90822) to DDH. The basis for this manuscript was laid down during the 2015 Dialogue seminar on Annonaceae under the Joint Scientific Thematic Research Programme (JSTP) funded by the Netherlands Organisation for Scientific Research and the Chinese Academy of Sciences (grant number 045.011.020). TLPC was supported by the Agence Nationale de la Recherche (grant AFRODYN: ANR‐15‐CE02‐0002‐01). MDP was supported by the Heisenberg programme of the Deutsche Forschungsgemeinschaft (PI 1169/3‐1).
Strijk and Hinsinger equally contributed to this study.
DATA AVAILABILITY STATEMENT
Raw reads produced in this study were deposited at EBI under project number PRJEB30626.
REFERENCES
- Adey, A. , Kitzman, J. O. , Burton, J. N. , Daza, R. , Kumar, A. , Christiansen, L. , Ronaghi, M. , Amini, S. , Gunderson, K. L. , Steemers, F. J. , & Shendure, J. (2014). In vitro, long‐range sequence information for de novo genome assembly via transposase contiguity. Genome Research, 24(12), 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature, 408, 796. [DOI] [PubMed] [Google Scholar]
- Arimoto, A. , Nishitsuji, K. , Higa, Y. , Arakaki, N. , Hisata, K. , Shinzato, C. , Satoh, N. , & Shoguchi, E. (2019). A siphonous macroalgal genome suggests convergent functions of homeobox genes in algae and land plants. DNA Research, 26(2), 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabaschi, D. , Tondelli, A. , Desiderio, F. , Volante, A. , Vaccino, P. , Valè, G. , & Cattivelli, L. (2016). Next generation breeding. Plant Science, 242, 3–13. [DOI] [PubMed] [Google Scholar]
- Barlow, J. , França, F. , Gardner, T. A. , Hicks, C. C. , Lennox, G. D. , Berenguer, E. , Castello, L. , Economo, E. P. , Ferreira, J. , Guénard, B. , & Leal, C. G. (2018). The future of hyperdiverse tropical ecosystems. Nature, 559(7715), 517–526. [DOI] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. , & Durbin, R. (2004). GeneWise and genomewise. Genome Research, 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, E. , & Abril, J. F. (2009). Computational gene annotation in new genome assemblies using GeneID. In: Bioinformatics for DNA sequence analysis (pp. 243–261). Humana Press. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B. , Bairoch, A. , Apweiler, R. , Blatter, M. C. , Estreicher, A. , Gasteiger, E. , Martin, M. J. , Michoud, K. , O'Donovan, C. , Phan, I. , & Pilbout, S. (2003). The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research, 31(1), 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C. , & Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology, 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics, 10(1), 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17(4), 540–552. [DOI] [PubMed] [Google Scholar]
- Castresana, J. (2002). Gblocks, v. 0.91 b. Retrieved form http://molevol.cmima.csic.es/castresana.Gblocks_server.html (accessed 2 February 2020).
- Chatrou, L. W. , Pirie, M. D. , Erkens, R. H. , Couvreur, T. L. , Neubig, K. M. , Abbott, J. R. , Mols, J. B. , Maas, J. W. , Saunders, R. M. , & Chase, M. W. (2012). A new subfamilial and tribal classification of the pantropical flowering plant family Annonaceae informed by molecular phylogenetics. Botanical Journal of the Linnean Society, 169(1), 5–40. [Google Scholar]
- Chaw, S. M. , Liu, Y. C. , Wu, Y. W. , Wang, H. Y. , Lin, C. Y. , Wu, C. S. , Ke, H. M. , Chang, L. Y. , Hsu, C. Y. , Yang, H. T. , Sudianto, E. , Hsu, M. H. , Wu, K. P. , Wang, L. N. , Leebens‐Mack, J. H. , & Tsai, I. J. (2019). Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nature Plants, 5(1), 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Hao, Z. , Guang, X. , Zhao, C. , Wang, P. , Xue, L. , Zhu, Q. , Yang, L. , Sheng, Y. , Zhou, Y. , & Xu, H. (2019). Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nature Plants, 5(1), 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collevatti, R. G. , Telles, M. P. C. , Lima, J. S. , Gouveia, F. O. , & Soares, T. N. (2014). Contrasting spatial genetic structure in Annona crassiflora populations from fragmented and pristine savannas. Plant Systematics and Evolution, 300, 1719–1727. [Google Scholar]
- Couvreur, T. L. , Helmstetter, A. J. , Koenen, E. J. , Bethune, K. , Brandão, R. D. , Little, S. A. , Sauquet, H. , & Erkens, R. H. (2019). Phylogenomics of the major tropical plant family Annonaceae using targeted enrichment of nuclear genes. Frontiers in Plant Science, 9, 1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley, J. F. , Gaut, B. S. , & Smith, B. D. (2006). The molecular genetics of crop domestication. Cell, 127, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Durand, N. C. , Robinson, J. T. , Shamim, M. S. , Machol, I. , Mesirov, J. P. , Lander, E. S. , & Aiden, E. L. (2016). Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell systems, 3(1), 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5(1), 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Gebali, S. , Mistry, J. , Bateman, A. , Eddy, S. R. , Luciani, A. , Potter, S. C. , Qureshi, M. , Richardson, L. J. , Salazar, G. A. , Smart, A. , & Sonnhammer, E. L. (2019). The Pfam protein families database in 2019. Nucleic Acids Research, 47(D1), D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, A. C. , Richards, S. , Han, Y. , Wang, M. , Vee, V. , Qu, J. , Qin, X. , Muzny, D. M. , Reid, J. G. , Worley, K. C. , & Gibbs, R. A. (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long‐read sequencing technology. PLoS One, 7(11), e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre‐Walker, A. , Gaut, R. L. , Hilton, H. , Feldman, D. L. , & Gaut, B. S. (1998). Investigation of the bottleneck leading to the domestication of maize. Proceedings of the National Academy of Sciences, USA, 95, 4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D. , Clements, J. , & Eddy, S. R. (2011). HMMER web server: Interactive sequence similarity searching. Nucleic Acids Research, 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, J. M. , Hubley, R. , Goubert, C. , Rosen, J. , Clark, A. G. , Feschotte, C. , & Smit, A. F. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences, 117(17), 9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry, A. H. (1993). Four neotropical rainforests. Yale University Press. [Google Scholar]
- Grabherr, M. G. , Haas, B. J. , Yassour, M. , Levin, J. Z. , Thompson, D. A. , Amit, I. , Adiconis, X. , Fan, L. , Raychowdhury, R. , Zeng, Q. , & Chen, Z. (2011). Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B. J. , Salzberg, S. L. , Zhu, W. , Pertea, M. , Allen, J. E. , Orvis, J. , White, O. , Buell, C. R. , & Wortman, J. R. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology, 9(1), R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L. , Xu, Z. , Wang, M. , Fan, R. , Yuan, D. , Wu, B. , Wu, H. , Qin, X. , Yan, L. , Tan, L. , & Sim, S. (2019). The chromosome‐scale reference genome of black pepper provides insight into piperine biosynthesis. Nature Communications, 10(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, S. , Apweiler, R. , Attwood, T. K. , Bairoch, A. , Bateman, A. , Binns, D. , Bork, P. , Das, U. , Daugherty, L. , Duquenne, L. , & Finn, R. D. (2009). InterPro: the integrative protein signature database. Nucleic Acids Research, 37(suppl_1), D211–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J. J. , Yu, W. B. , Yang, J. B. , Song, Y. , dePamphilis, C. W. , Yi, T. S. , & Li, D. Z. (2019). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. BioRxiv. 256479. [DOI] [PMC free article] [PubMed]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. , & Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S. J. , & Marra, M. A. (2009). Circos: an information aesthetic for comparative genomics. Genome Research 19(9), 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. Q. , Song, L. X. , Jin, C. Q. , Li, M. , Gong, S. P. , & Wang, Y. F. (2019). Genome survey and SSR analysis of Apocynum venetum . Bioscience Reports, 39(6), BSR20190146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , & Hansen, M. M. (2017). PSMC (pairwise sequentially Markovian coalescent) analysis of RAD (restriction site associated DNA) sequencing data. Molecular Ecology Resources, 17, 631–641. [DOI] [PubMed] [Google Scholar]
- Lowe, T. M. , & Eddy, S. R. (1996). TRNAscan‐SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , He, G. , Chen, Y. , Pan, Q. , Liu, Y. , & Tang, J. (2012). SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. GigaScience, 1(1), 2047–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros, W. H. , Pertea, M. , & Salzberg, S. L. (2004). TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Mapleson, D. , Garcia Accinelli, G. , Kettleborough, G. , Wright, J. , & Clavijo, B. J. (2017). KAT: a K‐mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics, 33(4), 574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoni, J. , Forest, F. , & Sauquet, H. (2014). Increased sampling of both genes and taxa improves resolution of phylogenetic relationships within Magnoliidae, a large and early‐diverging clade of angiosperms. Molecular Phylogenetics and Evolution, 70, 84–93. [DOI] [PubMed] [Google Scholar]
- Nawrocki, E. P. , Burge, S. W. , Bateman, A. , Daub, J. , Eberhardt, R. Y. , Eddy, S. R. , Floden, E. W. , Gardner, P. P. , Jones, T. A. , Tate, J. , & Finn, R. D. (2015). Rfam 12.0: Updates to the RNA families database. Nucleic Acids Research, 43(D1), D130–D137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E. P. , & Eddy, S. R. (2013). Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, A. D. , Cordeiro, M. C. , De Andrade, S. R. , Ferreira, F. R. , Filgueiras, H. D. , Alves, R. E. , & Kinpara, D. I. (2005). Annona species. International Centre for Underutilised Crops. University of Southampton. [Google Scholar]
- Punyasena, S. W. , Eshel, G. , & McElwain, J. C. (2008). The influence of climate on the spatial patterning of Neotropical plant families. Journal of Biogeography, 35, 117–130. [Google Scholar]
- Quevillon, E. , Silventoinen, V. , Pillai, S. , Harte, N. , Mulder, N. , Apweiler, R. , & Lopez, R. (2005). InterProScan: protein domains identifier. Nucleic Acids Research, 33(suppl_2), W116–W120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer, H. , & Chatrou, L. W. (2014). AnnonBase: World species list of Annonaceae. https://www.catalogueoflife.org/col/details/database/id/40
- Rendón‐Anaya, M. , Ibarra‐Laclette, E. , Méndez‐Bravo, A. , Lan, T. , Zheng, C. , Carretero‐Paulet, L. , Perez‐Torres, C. A. , Chacón‐López, A. , Hernandez‐Guzmán, G. , Chang, T. H. , & Farr, K. M. (2019). The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen‐influenced gene space adaptation. Proceedings of the National Academy of Sciences, 116(34), 17081–17089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Turner, D. , Durand, N. C. , Thorvaldsdóttir, H. , Mesirov, J. P. & Aiden, E. L. , (2018). Juicebox. js provides a cloud‐based visualization system for Hi‐C data. Cell systems, 6(2), 256–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , & Russell, D. W. (2006). Purification of nucleic acids by extraction with phenol: chloroform. Cold Spring Harbor Protocols, 2006(1), pdb‐rot4455. [Google Scholar]
- Sarkar, A. K. , Chakraverty, M. , Das, S. K. , Pal, C. R. , & Hazara, D. (1980). Annona muricata L. Chromosome Number Reports LXVII. Taxon, 29, 358–360. [Google Scholar]
- Sauquet, H. , von Balthazar, M. , Magallón, S. , Doyle, J. A. , Endress, P. K. , Bailes, E. J. , de Morais, E. B. , Bull‐Hereñu, K. , Carrive, L. , Chartier, M. , & Chomicki, G. (2017). The ancestral flower of angiosperms and its early diversification. Nature Communications, 8(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L. , Chen, H. , Jiang, M. , Wang, L. , Wu, X. , Huang, L. , & Liu, C. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Research, 47(W1), W65–W73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smit, A. F. , & Hubley, R. (2008). RepeatModeler Open‐1.0. http://www.repeatmasker.org.
- Smit, A. , Hubley, R. , & Green, P. (2017). RepeatMasker Open‐4.0.6. http://www.repeatmasker.org.
- Sobha, V. , & Ramachandran, K. (1980). Annona muricata L. IOPB Chromosome number reports LXVI. Taxon, 29, 165–166. [Google Scholar]
- Soltis, D. E. , & Soltis, P. S. (2019). Nuclear genomes of two magnoliids. Nature Plants, 5, 6. [DOI] [PubMed] [Google Scholar]
- Sonké, B. , & Couvreur, T. (2014). Tree diversity of the Dja Faunal Reserve, southeastern Cameroon. Biodiversity Data Journal, 2, e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. , & Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research, 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijk, J. S. , Hinsinger, D. D. , Zhang, F. , & Cao, K. (2019). Trochodendron aralioides, the first chromosome‐level draft genome in Trochodendrales and a valuable resource for basal eudicot research. GigaScience, 8(11), giz136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchouto, M. G. P. , Yemefack, M. , De Boer, W. F. , De Wilde, J. J. F. E. , Van Der Maesen, L. J. G. , & Cleef, A. M. (2006). Biodiversity hotspots and conservation priorities in the Campo‐Ma’an rain forests. Cameroon. Biodiversity Conservation, 15, 1219–1252. [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D. R. , Pimentel, H. , Salzberg, S. L. , Rinn, J. L. , & Pachter, L. (2012). Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B. J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C. A. , Zeng, Q. , Wortman, J. , Young, S. K. , & Earl, A. M. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9(11), e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, C. , Yang, H. , Wang, S. , Zhao, J. , Liu, C. , Gao, L. , Xia, E. , Lu, Y. , Tai, Y. , She, G. , & Sun, J. (2018). Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proceedings of the National Academy of Sciences, 115(18), E4151–E4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24(8), 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu, X.‐J. , Zheng, H.‐K. , Wang, J. , Wang, W. , & Su, B. (2006). Detecting lineage‐specific adaptive evolution of brain‐expressed genes in human using rhesus macaque as outgroup. Genomics, 88, 745–751. [DOI] [PubMed] [Google Scholar]
- Zamir, D. (2001). Improving plant breeding with exotic genetic libraries. Nature Reviews Genetics, 2, 983. [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Qiao, Q. , Novikova, P. Y. , Wang, Q. , Yue, J. , Guan, Y. , Ming, S. , Liu, T. , De, J. , Liu, Y. , & Al‐Shehbaz, I. A. (2019). Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proceedings of the National Academy of Sciences, 116(14), 7137–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Q. , Zheng, X. , Luo, J. , Gaut, B. S. , & Ge, S. (2007). Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: Severe bottleneck during domestication of rice. Molecular Biology and Evolution, 24, 875–888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1‐S9
Data Availability Statement
Raw reads produced in this study were deposited at EBI under project number PRJEB30626.