

Abstract
Objectives
Little is known about the evolution of epilepsy in individuals with tuberous sclerosis complex (TSC) in adulthood. This study aims at describing the characteristics of epilepsy in adult TSC patients attending a single multidisciplinary clinic.
Materials and Methods
We collected data about epilepsy (age at onset, seizure types, history of infantile spasms (IS), epilepsy diagnosis and outcome), genetic and neuroradiological findings, cognitive outcome and psychiatric comorbidities.
Results
Out of 257 adults with TSC, 183 (71.2%) had epilepsy: 121 (67.2%) were drug‐resistant; 59 (32.8%) seizure‐free, at a median age of 18 years. 22% of the seizure‐free patients (13/59) discontinued medication.
Median age at seizure onset was 9 months. Seventy‐six patients (41.5%) had a history of IS. TSC2 pathogenic variants (p = 0.018), cortical tubers (p < 0.001) and subependymal nodules (SENs) (p < 0.001) were more frequent in those who developed epilepsy. Cognitive functioning was lower (p < 0.001) and psychiatric disorders more frequent (p = 0.001). We did not find significant differences regarding age, gender, mutation and tubers/SENs in seizure‐free vs drug‐resistant individuals. Intellectual disability (p < 0.001) and psychiatric disorders (p = 0.004) were more common among drug‐resistant patients.
Conclusions
Epilepsy in TSC can be a lifelong disorder, but one‐third of individuals reach seizure freedom by early adulthood. In the long term, age at epilepsy onset has a crucial role in drug resistance and in developing intellectual disability, both in drug‐resistant and drug‐sensible patients. Patients with drug‐refractory seizures tend to develop psychiatric issues, which should be recognized and adequately treated.
Keywords: adulthood, epilepsy, seizure, TSC, tuberous sclerosis complex
1. INTRODUCTION
Tuberous sclerosis complex (TSC) is a rare autosomal dominant disorder characterized by the development of benign hamartomas in multiple organ systems. The incidence is estimated to be 1:6,000–10,000 live births, and the prevalence around 1:20,000 in the general population. 1 Inactivating mutations of TSC1 (chr 9p34) or TSC2 (chr 16p13) can be identified in up to 90% of the clinically affected people. TSC1 and TSC2 encode for hamartin and tuberin, which regulate the mTOR pathway, implicated in cortical development and cellular proliferation. 2 Only 30% of the cases are inherited, whereas the majority of affected individuals has a de novo pathogenic variant. In simplex cases, TSC2 mutations are detected 4–5 times more than TSC1 mutations, whereas in inherited cases TSC1 and TSC2 mutations are equally identified. 3 , 4 The identification of a pathogenic variant is a stand‐alone diagnostic criterion. 1 However, no mutations are identified (NMI) in 10–25% of affected people using conventional testing methods and in 5–10% when next generation sequencing techniques are applied. It is therefore important to keep in mind that negative genetic results do not exclude the diagnosis when clinical criteria are met. 5 , 6
Typically diagnosed in early childhood or adolescence, TSC is a lifelong complex multisystemic disorder with age‐dependent manifestations. 7 The neurological manifestations include structural brain lesions, neurodevelopmental and psychiatric disorders, and epilepsy. 8
Epilepsy in TSC remains a major challenge, since >60% of the patients have drug‐refractory seizures. 9 Epilepsy usually begins in the first months of life with onset in the first year in 62.5–73% of cases and 80% within three years of age, although seizures may occur at any age. 10 The early onset of epilepsy often occurs with focal seizures (67.5%) and/or infantile spasm (IS) (38.9%). 11 Patients with TSC can present all seizure types such as tonic, atonic, myoclonic, atypical absences or tonic‐clonic seizures during their lifetime. 12 , 13
Like in other rare neurological diseases with childhood onset, little is known about the evolution of epilepsy in TSC patients in adulthood. 9 , 14 , 15 In a previous paper, 10 we reviewed the clinical characteristics of 160 individuals with TSC, regularly followed since 2000 at the San Paolo Multidisciplinary TSC Centre in Milan, Italy. One hundred and twenty‐seven patients, who were adults at the time of the previous paper, were included in this study. During the last years, an increasing number of patients attended our clinic, transitioning from childhood or being referred from other Italian centres. 16 Therefore, in the present study, we sought to evaluate the clinical aspects, including epilepsy features, genetics, neuroimaging, response to pharmacological and non‐pharmacological therapies, and neuropsychiatric comorbidities of a large cohort of adult individuals with TSC.
2. METHODS
We performed a retrospective single centre study. From the database of patients followed at the TSC Center, ASST Santi Paolo e Carlo, Milan, we selected those who were >18 years old as of end of 2019. The diagnosis of TSC was based on the 2012 criteria. 1
Family background and the complete history of epilepsy (including age of onset, history of infantile spasms, history of status epilepticus, seizure types and their frequency, therapy, epilepsy surgery and vagus nerve stimulation (VNS)) up to the last follow‐up visit were collected for each patient. Drug‐resistant epilepsy was defined as the failure of adequate trials of two tolerated and appropriately chosen and used anti‐epileptic drug (AED) schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom. Drug‐responsive epilepsy was considered when the patient receiving the current AED regimen had been seizure‐free for a minimum of three times, the longest pre‐intervention inter seizure interval or 12 months, whichever was longer. 17
Genetic analyses of TSC1 and TSC2 were performed. The neurological evaluation also included the review of video‐electroencephalograms performed during wakefulness and sleep. Brain MRI (at least T1‐, T2‐, FLAIR‐weighted sequences in the 3 planes without and with contrast, in some cases also DWI‐weighted sequences) was obtained for each patient and reviewed carefully to identify cerebral features of TSC (cortical tubers, SENs, SEGAs, white matter radial migration lines, other abnormalities). Timing of neuroradiological follow‐up varied based on clinical indication and age, according to the current recommendations. 1 In addition, as data were available, the patients were assigned a neurologic severity score (NSS), described by Wong et al., and based on the most relevant clinical neurological features affecting quality of life in this population. 18 The following components were considered to determine the score: developmental or intellectual disability (ID), seizures (controlled or intractable), autism and other neuropsychiatric disorders (behavioural issues, language and learning disorders, other psychiatric symptoms). ID was assigned 3 points, autism 2 and other neuropsychiatric disorders 1. Seizure was assigned 2 points in case of intractable seizures, and 1 point in controlled seizures.
The cognitive level was assessed at least once during the follow‐up using the Wechsler Intelligence scale (WAIS‐R) for adult patients when possible, or Raven's standard progressive matrices based on the applicability of the test.
When a formal test could not be performed due to severely impaired mental status, ID was evaluated clinically. As part of their clinical management, a team of psychiatrists with wide expertise in TSC interviewed all the patients in order to assess psychiatric disorders, following the DSM‐5 diagnostic criteria. The multi‐system involvement and the possible influence of some manifestations—such as the presence of facial angiofibromas—on depression and other psychiatric symptoms have not been explored in this article as we focused our attention on the epileptic phenotype. Education was measured in years of schooling.
All the patients included had a follow‐up of at least 12 months, and data were updated at the last follow‐up visit.
Informed consent was obtained, and this study was approved by our Institution's Ethics Committee (9570/2013/rev 2020).
2.1. Statistical analysis
Clinical and demographic data were transferred into an electronic database and processed using the Statistical Package for the Social Sciences (SPSS, IBM) for Windows software, version 26.0. Qualitative data were described as numbers and percentages and quantitative data as median and interquartile range (IQR).
The comparison between proportions was made using the chi‐square. We performed the Shapiro‐Wilk test to check the normality of the distributions, which showed that the distribution of variables was never normal. The demographic and clinical characteristics of TSC patients were compared using the Mann‐Whitney U test (two groups).
We considered a two‐tailed p‐value of ≤0.05 statistically significant.
3. RESULTS
3.1. Clinical data of adult patients as a whole
We selected 257 adult patients (111 males [43.2%], 146 females [56.8%]), aged 18–87 years (median age 37 years). Genetic data were available for 239/257 patients (93%): 76/239 (31.8%) had a TSC1 pathogenic variant, 140/239 had a TSC2 pathogenic variant (58.6%), and in 23/239 patients no mutation was identified (9.6%). Somatic mosaicism was detected in 11/239 (4.6%) patients: 10 were TSC2 mutation carriers, while one patient carried a mutation in TSC1. On the basis of the anamnestic interview, only 71/257 were inherited cases (27.6%), whereas 171/257 patients had no affected relatives (66.5%). In familiar cases, we found TSC1 mutations in 37/71 patients (52.1%), and TSC2 mutations in 29/71 (40.8%); in sporadic cases, TSC1 mutations were seen in 36/171 patients (21%), and TSC2 mutations in 105/171 (61.4%).
With regard to cognitive functioning, 131/257 individuals had a normal intelligence quotient (IQ) (51%), 20/257 had borderline intellectual functioning (7.8%), and 99/257 patients had ID (38.5%). In particular, 21/257 had mild ID (8.2%), 22/257 had moderate ID (8.6%), 52/257 had severe ID (20.2%), whereas for 4 patients the level of ID was not specified and for 7 patients the cognitive level was not available. The IQ median values, available for 90 patients, are indicated in Table 1.
TABLE 1.
Demographic, molecular and clinical characteristics of the study cohort
|
Total sample N = 257 |
% | |
|---|---|---|
| Median age, y (IQR) | 37.00 (26.5–48) | |
| Gender, n (%) | ||
| Male | 111 | 43.2 |
| Female | 146 | 56.8 |
| Familial cases | 71 | 27.6 |
| Level of education, median years of schooling (IQR) | 11.00 (8–13) | |
| Genetic analysis, n (%) | ||
| TSC1 | 76 | 29.6 |
| TSC2 | 140 | 54.5 |
| NMI | 23 | 8.9 |
| NA | 18 | 7.0 |
| Severity score, median (IQR) | 2.00 (1–5) | |
| IQ, n (%) | ||
| Normal IQ | 131 | 51.0 |
| BIF | 20 | 7.8 |
| Mild ID | 21 | 8.2 |
| Moderate ID | 22 | 8.6 |
| Severe ID | 52 | 20.0 |
| ID not otherwise specified | 4 | 1.5 |
| NA | 7 | 2.7 |
| IQ, median (IQR) | ||
| Full scale IQ | 74.00 (52.75–90) | |
| Verbal IQ | 74.00 (57–88) | |
| Performance IQ | 79.00 (54–96) | |
| Cortical tubers, n (%) | 245 | 95.3 |
| Subependymal nodules, n (%) | 198 | 77.0 |
| SEGA, n (%) | 48 | 18.7 |
| Psychiatric disorders, n (%) | 113 | 44.0 |
| Epilepsy, n (%) | ||
| Yes | 183 | 71.2 |
| Single seizure | 4 | 1.6 |
| No | 70 | 27.2 |
|
Patients with Epilepsy N = 183 |
% |
|
|---|---|---|
| Epilepsy onset, median age in months (IQR) | 9.00 (9–60) | |
| Seizure type at onset, n (%) | ||
| Focal onset seizures | 105 | 57.3 |
| IS | 53 | 29.0 |
| Focal onset seizures and IS | 10 | 5.5 |
| Absences | 4 | 2.2 |
| Generalized tonic‐clonic seizures | 3 | 1.6 |
| Tonic seizures | 1 | 0.5 |
| NA | 7 | 3.8 |
| History of IS, n (%) | 76 | 41.5 |
| Epilepsy syndrome | ||
| Focal/multifocal epilepsy | 137 | 74.9 |
| Generalized epilepsy | 8 | 4.4 |
| Lennox‐Gastaut syndrome | 10 | 5.5 |
| Not determined | 28 | 15.3 |
| Stop seizures, n (%) | 59 | 32.2 |
| Median age at seizure freedom (IQR) | 18.00 (10–29) | |
| Drug resistance | 121 | 66.1 |
| Antiepileptic therapy, n (%) | ||
| Polytherapy | 86 | 47.0 |
| Monotherapy | 60 | 32.8 |
| None | 15 | 8.2 |
| NA | 22 | 12.0 |
| Everolimus | 5 | 2.7 |
| VNS | 4 | 2.2 |
| Epilepsy surgery | 11 | 6.0 |
| Febrile seizures, n (%) | 21 | 11.5 |
| Status (not febrile), n (%) | 24 | 13.1 |
Abbreviations: BIF, borderline intelligence functioning; ID, intellectual disability; IQ, intelligence quotient; IQR, interquartile range; IS, Infantile Spasms; NA, not available; NMI, no mutation identified; SEGA, subependymal giant cell astrocytoma; VNS, vagus nerve stimulation.
Neuroradiological investigations revealed cortical tubers in 245/257 patients (95.3%), subependymal nodules (SENs) in 198/257 patients (77%) and subependymal giant cell astrocytoma (SEGA) in 48/257 (18.7%).
One hundred and thirteen (113/257) patients developed psychiatric issues or received a psychiatric diagnosis (44%), including mood, anxiety, psychotic disorders or symptoms, ASD, ADHD, behavioural problems (aggression, self‐injury and impulsivity), eating disorder, alcohol abuse disorder and personality disorder.
Table 1 describes the characteristics of the whole sample of adult patients with TSC.
Regarding the severity of the neurological issues, we managed to obtain data to calculate the NSS in 220/257 (85.6%) patients: the score clearly reflected the impact of epilepsy on all the variables examined (Table 1, Supplementary materials). In the remaining patients, the neuropsychiatric evaluation performed in adulthood was inconclusive and did not provide sufficient data to calculate the score appropriately.
3.2. Different features in patients with Epilepsy vs without Epilepsy
In our cohort, 183/257 patients had epilepsy (71.2%), and 4 patients had had only one seizure in their lifetime, until our last follow‐up. Age at epilepsy onset varied from the first day of life to 48 years, with a median age of 9.0 months (Table 1). The seizure type at onset was focal onset seizure in 105/183 patients (57.3%), IS in 53/183 (29.0%), both focal seizures and IS in 10/183 (5.5%), and other seizure types in 8/183 patients (4.3%). Seventy‐six patients (76/183; 41.5%) had a history of IS. Most patients in our cohort had a focal or multifocal epilepsy (74.9%); 10/183 developed Lennox‐Gastaut syndrome (5.5%). Considering treatment, 47% of the patients with epilepsy (86/183) were receiving multiple AEDs at the last follow‐up. Everolimus was given as antiepileptic therapy to 5/183 patients (2.7%). Among them, 4 patients were taking everolimus also for SEGA and one also for renal angiomyolipoma.
Eleven patients in our cohort received surgical treatment for epilepsy: 6 out of the 11 (54.5%) were free from disabling seizures (Engel class I), and 3/11 (27.3%) were seizure‐free since surgery (Engel Ia). Four patients with drug‐refractory seizures not eligible for epilepsy surgery were implanted with VNS, and none of them became seizure‐free.
Fifty‐nine patients (59/183, 32.2%) obtained complete seizure control at a median age of 18 years, and 13 out of the 59 (22%) withdrew from the antiepileptic therapy, at a median age of 7 years. Among these patients, 5/13 (38.5%) presented with IS, 3/13 (23.1%) with focal onset seizures, 2/13 (15.4%) with focal onset seizures in addition to IS and 2/13 (15.4%) with generalized seizures as seizure type at onset. Median age at epilepsy onset was 13 months.
Comparing patients with epilepsy and patients without epilepsy, we found statistically significant differences between the two groups with regard to mutational analysis (p = 0.018), presence of cortical tubers (p < 0.001) and SENs (p < 0.001) (Table 2). The cognitive and academic level was significantly lower in individuals with epilepsy (p < 0.001). The median years of education were 4 years higher in patients without epilepsy.
TABLE 2.
Comparison between TSC patients with and without epilepsy
|
Epilepsy N = 183 (72.3%) |
No Epilepsy N = 70 (27.7%) |
p‐value | |
|---|---|---|---|
| Median age, y (IQR) | 35.00 (26–46) | 45.00 (29.75–54.25) | 0.007 |
| Level of education, median years of schooling (IQR) | 9.00 (8–13) | 13.00 (9.5–13) | <0.001 |
| Genetic analysis, n (%) | |||
| TSC1 | 48 (26.2) | 26 (37.1) | 0.018 |
| TSC2 | 109 (59.6) | 29 (41.4) | |
| NMI | 12 (6.5) | 11 (15.7) | |
| NA | 14 (7.7) | 4 (5.7) | |
| Familial cases, n (%) | 45 (24.6) | 24 (34.3) | 0.282 |
| Cognitive level, n (%) | |||
| Normal IQ | 65 (35.5) | 62 (88.6) | <0.001 |
| BIF | 16 (8.7) | 4 (5.7) | |
| Mild ID | 18 (9.8) | 3 (4.3) | |
| Moderate ID | 22 (12.0) | 0 | |
| Severe ID | 52(28.4) | 0 | |
| ID not otherwise specified | 4 (2.2) | 0 | |
| NA | 6 (3.3) | 1 (1.4) | |
| Median IQ (IQR) | |||
| Full Scale IQ | 69.00 (47.75–82.75) | 93.00 (73–109) | <0.001 |
| Verbal IQ | 68.00 (53–83.5) | 88.50 (69.75–104.75) | 0.009 |
| Performance IQ | 69.50 (52.5–87) | 100.00 (72.75–112.25) | 0.001 |
| Cortical tubers, n (%) | |||
| Yes | 179 (97.8) | 63 (90.0) | <0.001 |
| Subependymal nodules, n (%) | |||
| Yes | 152 (83.1) | 44 (62.9) | <0.001 |
| Psychiatric disorders, n (%) | |||
| Yes | 95 (51.9) | 16 (22.9) | 0.001 |
| Psychiatric disorders, n (%) | N = 95 | N = 16 | |
|---|---|---|---|
| ASD | 37 (38.9) | 0 | 0.001 |
| Anxiety disorders | 17 (17.9) | 9 (56.3) | |
| Behavioural difficulties | 15 (15.8) | 1 (6.2) | |
| Depressive/bipolar disorders | 6 (6.3) | 2 (12.5) | |
| Schizofrenia spectrum and other psychotic disorders | 7 (7.4) | 0 | |
| Anxiety and depressive disorders | 4 (4.2) | 2 (12.5) | |
| ADHD | 4 (4.2) | 0 | |
| Personality disorders | 2 (2.1) | 1 (6.2) | |
| Conversion disorder | 2 (2.1) | 0 | |
| Eating disorder | 0 | 1 (6.2) | |
| Alcohol use disorder | 1 (1.1) | 0 |
Abbreviations: ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; BIF, borderline intelligence functioning; ID, intellectual disability; IQ, intelligence quotient; IQR, interquartile range; NA, not available; NMI, no mutation identified.
In addition, 62/70 patients without epilepsy (88.6%) had a normal cognitive functioning level, whereas only 65/183 epileptic patients (35.5%) had a normal IQ (p < 0.001). Furthermore, 96/99 patients with ID (97%) suffered from epilepsy, and all individuals presenting moderate or severe ID (74) had epilepsy. People with epilepsy had a full scale, verbal and performance median IQ of 69, 68 and 69.5, respectively, significantly lower than the corresponding values of 93, 88.5 and 100 in patients without epilepsy (Figure 1).
FIGURE 1.
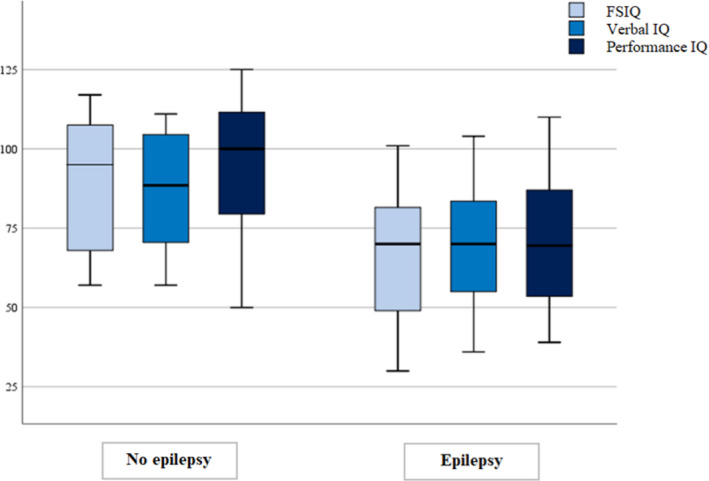
Distribution of full scale IQ (FSIQ), verbal IQ and performance IQ: patients with epilepsy showed significantly lower total, verbal and performance median IQ than patients without epilepsy.
Psychiatric disorders were more frequent in the epileptic patients (51.9% vs 22.9%, p = 0.001). Autism spectrum disorder (ASD) was the most frequent disorder in the epileptic patients (37/95, 38.9%), and all the individuals with ASD had epilepsy. On the contrary, anxiety was the most frequent psychiatric disorder in the patients without epilepsy: affecting 9 out of the 16 patients with psychiatric disorders. In the epilepsy group, the other most frequent psychiatric disorders were anxiety (21/95, 22.1%) and behavioural issues (15/95, 15.8%).
3.3. Drug‐responsive vs drug‐resistant Epilepsy
We then compared the clinical, genetic and neuropsychological data of seizure‐free individuals with drug‐resistant patients (Table 3). Gender, genetic mutation and cortical tubers/SENs did not differ between the two groups. Interestingly, sporadic patients were more frequently drug‐resistant (100/121, 82.6%), whereas seizure‐free patients had more frequently affected relatives (23/59, 39.0%, p = 0.010). Seizure‐free patients had epilepsy onset at a median age of 27 months, significantly later than drug‐resistant patients, who presented their first seizure at a median age of 6 months (p = 0.001). Furthermore, seizure‐free patients had more frequently focal epilepsy (49/59, 83.1% vs 86/121, 71.1%, p = 0.029) and a lower rate of history of spasms (16/59, 27.1% vs 59/121, 48.8%, p = 0.007), when compared to drug‐resistant patients.
TABLE 3.
Comparison between seizure‐free and drug‐resistant patients
|
Seizure‐free N = 59 (32.8%) |
Drug resistance N = 121 (67.2%) |
p‐value | |
|---|---|---|---|
| Median age, y (IQR) | 35.00 (26–48) | 34.00 (26–44) | 0.547 |
| Gender, n (%) | |||
| Male | 26 (44.1) | 59 (48.8) | 0.54 |
| Female | 33 (55.9) | 62 (51.2) | |
| Level of education, median years of schooling (IQR) | 13.00 (8.25–13) | 8.00 (8–13) | 0.002 |
| Genetic analysis, n (%) | |||
| TSC1 | 19 (32.2) | 29 (24.0) | 0.538 |
| TSC2 | 32 (54.2) | 77 (63.6) | |
| NMI | 5 (8.5) | 7 (5.8) | |
| NA | 3 (5.1) | 8 (6.6) | |
| Familial cases, n (%) | 23 (39.0) | 21 (17.4) | 0.010 |
| Epilepsy type, n (%) | |||
| Focal | 49 (83.1) | 86 (71.1) | 0.029 |
| Spasms | 0 | 2 (1.7) | |
| Generalized | 2 (3.4) | 3 (2.5) | |
| LGS | 0 | 10 (8.3) | |
| Combined | 2 (3.4) | 15 (12.4) | |
| Unknown | 6 (10.2) | 5 (4.1) | |
| Epilepsy onset, median age in months (IQR) | 27.00 (6–108) | 6.00 (3–30) | 0.001 |
| Age at diagnosis, years, median (IQR) | 9.50 (4–26) | 3.00 (0.725–12) | <0.001 |
| Seizure type at onset, n (%) | |||
| Focal | 38 (64.4) | 65 (53.7) | 0.211 |
| IS | 12 (20.3) | 40 (33.0) | |
| Focal and IS | 2 (3.4) | 8 (6.6) | |
| Absences | 2 (3.4) | 2 (1.7) | |
| Generalized tonic‐clonic seizures | 0 | 2 (1.7) | |
| Tonic seizures | 1 (1.7) | 0 | |
| NA | 4 (6.8) | 4 (3.3) | |
| History of IS, n (%) | |||
| Yes | 16 (27.1) | 59 (48.8) | 0.007 |
| Febrile seizures, n (%) | |||
| Yes | 4 (6.8) | 15 (12.4) | 0.236 |
| AEDs therapy, n (%) | |||
| None | 13 (22) | 0 | <0.001 |
| Monotherapy | 29 (49.2) | 35 (28.9) | |
| Polytherapy | 8 (13.6) | 78 (64.5) | |
| NA | 9 (15.2) | 8 (6.6) | |
| IQ, n (%) | |||
| Normal IQ | 35 (59.3) | 29 (23.9) | <0.001 |
| BIF | 5 (8.5) | 10 (8.3) | |
| Mild intellectual disability | 8 (13.6) | 10 (8.3) | |
| Moderate intellectual disability | 3 (5.1) | 19 (15.7) | |
| Severe intellectual disability | 6 (10.2) | 45 (37.2) | |
| ID not otherwise specified | 0 | 4 (3.3) | |
| NA | 2 (3.3) | 4 (3.3) | |
| IQ, median (IQR) | |||
| Full Scale IQ | 67.00 (47.25–85) | 68.00 (46.5–81.25) | 0.876 |
| Verbal IQ | 72.00 (59–86) | 67.50 (50.25–81.5) | 0.427 |
| Performance IQ | 68.00 (54–96) | 70.00 (51–87) | 0.907 |
| Cortical tubers, n (%) | |||
| Yes | 57 (96.6) | 119 (98.3) | 0.489 |
| Subependymal nodules, n (%) | |||
| Yes | 43 (72.9) | 106 (87.6) | 0.158 |
| Psychiatric disorders, n (%) | |||
| Yes | 22 (37.3) | 72 (59.5) | 0.013 |
| Psychiatric disorders, n (%) | |||
| ASD | 4 (18.2) | 32 (44.4) | 0.004 |
| Anxiety disorders | 7 (31.8) | 10 (13.9) | |
| Behavioural difficulties | 0 | 15 (20.8) | |
| Depressive/bipolar disorders | 3 (13.6) | 3 (4.2) | |
| Schizofrenia spectrum and other psychotic disorders | 2 (9.1) | 5 (6.9) | |
| Anxiety and depressive disorders | 3 (13.6) | 1 (1.4) | |
| ADHD | 2 (9.1) | 2 (2.8) | |
| Personality disorders | 0 | 2 (2.8) | |
| Conversion disorder | 1 (4.5) | 1 (1.4) | |
| Alcohol use disorder | 0 | 1 (1.4) | |
Abbreviations: ADHD, attention deficit hyperactivity disorder; AED, antiepileptic drug; ASD, autism spectrum disorder; BIF, borderline intelligence functioning; ID, intellectual disability; IQ, intelligence quotient; IQR, interquartile range; IS, infantile spasm; LGS, Lennox‐Gastaut syndrome; NA, not available; NMI, no mutation identified.
We also found a significant difference in the level of education between the two groups, with a median scholar age in seizure‐free patients being 5 years higher than in drug‐resistant individuals (p = 0.002). Moreover, seizure‐free patients had more frequently normal cognitive functioning (35/59, 59.3%, p < 0.001); on the contrary, patients with drug‐resistant seizures had more frequently moderate or severe ID (64/121, 52.9% vs 9/59, 15.3%). We did not find any differences between the two groups when comparing IQ values.
When evaluating age at epilepsy onset and ID, earlier seizures were associated with the development of ID in both drug‐resistant patients (p < 0.001) and drug‐sensible patients (p = 0.05); moreover, the earlier the onset, the worst the cognitive outcome (Figure 2).
FIGURE 2.
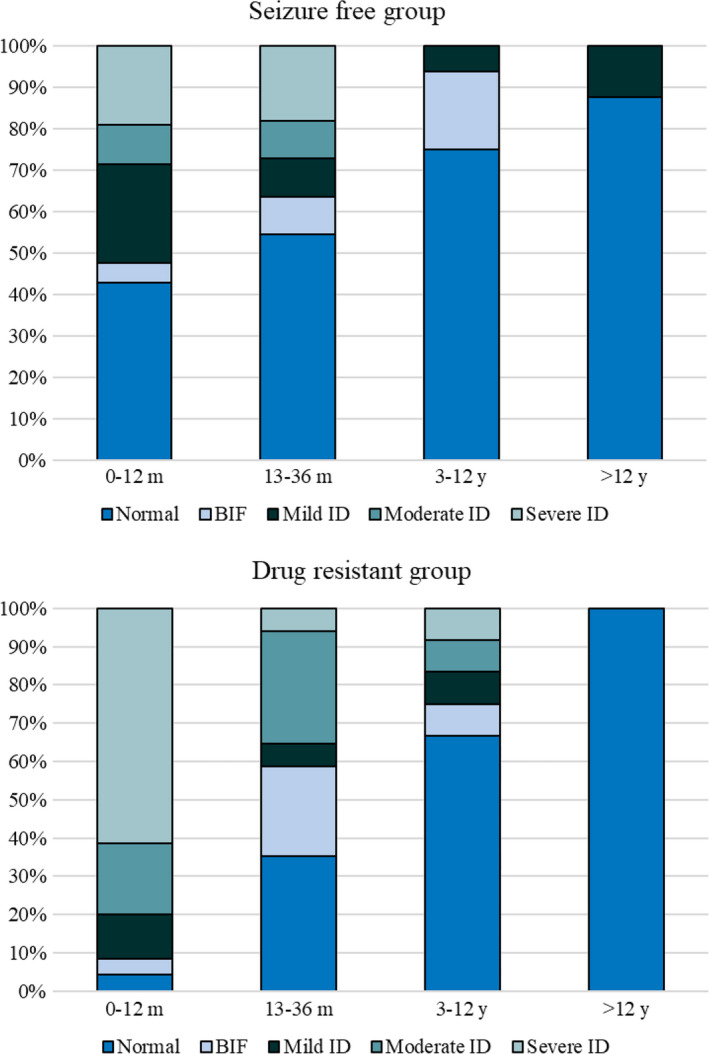
Cognitive functioning according to age at epilepsy onset in the drug‐responsive and the drug‐resistant group.
We also considered the potential role of the duration of epilepsy (< or >5 years) on the cognitive outcome: patients experiencing a shorter time of active seizures had a statistically significant better intellectual functioning (Figure 3, p < 0.024).
FIGURE 3.
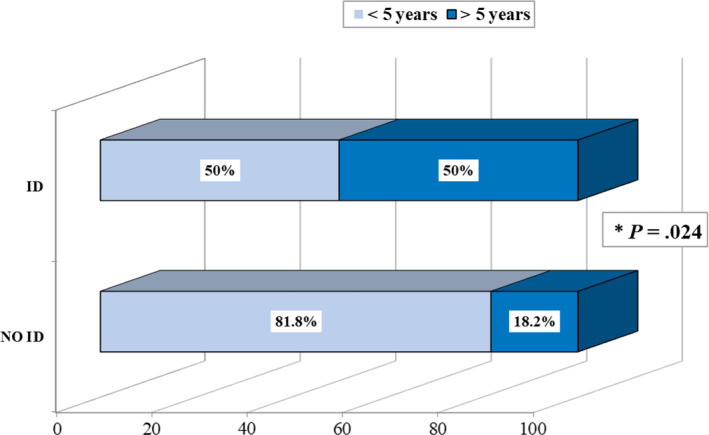
The duration of active epilepsy has an impact on cognitive outcome in TSC: Patients without intellectual disability (NO ID) had a shorter history of active seizures (< 5 years) compared to those who developed intellectual disability (ID).
Psychiatric disorders were more common in the drug‐resistant cohort (72/121, 59.5%) than in seizure‐free patients (22/59, 37.3%) (p = 0.013). Moreover, ASD was the most common diagnosis in drug‐resistant patients, affecting 44.4% of patients (32/72), a significantly higher rate than the ASD rate in seizure‐free patients (4/22, 18.2%, p = 0.004). In seizure‐free patients, the most common psychiatric disorder was anxiety, followed by ASD, mood, and combined mood and anxiety disorder. Besides ASD, drug‐resistant people were affected by behavioural issues, anxiety and psychotic disorder.
Thirteen seizure‐free patients (22%) had stopped the antiepileptic treatment, and 29 were taking only one AED (49.2%). The patients who stopped antiepileptic therapy had seizure onset at a median age of 13 months, and 6/13 (46.1%) had seizure onset in the first year of life. Seven patients in this group had IS (7/13, 53.8%), and 9/13 had focal epilepsy (69.2%). Median age at antiseizure medication withdrawal was 7 years.
More than half (55.2%) of the seizure‐free patients in monotherapy were receiving carbamazepine (16/29, 55.2%), which was the most effective drug in our cohort, followed by valproic acid (6/29, 20.7%) and oxcarbazepine (3/29, 10.3%), while the other 4 patients received different monotherapies. On the contrary, 64.5% of drug‐resistant patients (78/121) were assuming multiple AEDs. Among drug‐resistant patients in monotherapy, 37.1% were receiving carbamazepine (13/35), 11.4% lamotrigine (4/35) and other drugs at a similar rate in the other cases.
4. DISCUSSION
Epilepsy is a major burden in people with TSC, and seizure control is therefore a critical issue in their clinical management. 13
In TSC, seizures usually start at an early age, and their characteristics and treatment options have been widely investigated. 19 , 20 , 21 Clinical recommendations have also been given 12 especially in children and adolescents. Nevertheless, we know that seizures tend to be lifelong persistent, and there is an urgent need for indications for the challenging epilepsy management in adult patients with TSC. 15 , 16 , 22 , 23
In the last years, we have had the opportunity to take care of patients with TSC of all ages at our multidisciplinary clinic. Indeed, we have seen patients with mild to severe phenotype, frequently getting to know their history from the very beginning of the disease.
After our previous paper on epilepsy prognosis in patients with TSC including both children and adults, 10 in this study we evaluated the characteristics and the evolution of epilepsy in 257 adult individuals, regularly followed at our TSC Clinic in Milan.
In our cohort, the prevalence of epilepsy is 73%, which is in line with other population‐based studies 22 and with our previous findings, 10 thus indicating that epilepsy can be overestimated due to referral bias in tertiary care centres. Indeed, seizures in TSC are very frequently drug‐resistant also in adulthood (67.2% in our cohort), twice as frequent as currently reported for epilepsy in general. 24
Comparing the prevalence of drug‐resistant patients in the present study with our previous cohort, in which also children were included, 10 we found a higher percentage of drug resistance (67.2% vs 36.9%). Indeed, the percentage of seizure‐free patients did not differ between the two populations: 32.8% in the adult cohort against 35.6% in the whole cohort 10 ; but in the present study 22% of the patients have stopped AEDs, against the 12% of the previously reported cohort. 10 This latest finding may reflect the physicians’ attitude towards AEDs withdrawal after a sufficient period of seizure freedom, generally at least five years.
On the other hand, it is worth noting that more than one‐third of adults with TSC had their seizures controlled by the age of 18. This finding is new and may be helpful in counselling patients and their families, although needs to be further confirmed in larger cohorts.
Analysing the characteristics of epilepsy in relation to long‐term prognosis, we found that an earlier seizure onset, the history of IS and the type of epilepsy are significantly associated with drug resistance. These variables have already been evaluated as determinant in other series in the literature, 9 but we confirmed their important role also in adults. Among seizure types, focal onset seizures and epileptic spasms are those most frequently reported, confirming the unusual persistence of the latter in this specific population of patients. 25
When considering the genetic background, individuals carrying TSC2 pathogenic variants are at greater risk of developing epilepsy. However, among patients with epilepsy, genetics is not a risk factor of drug resistance, which is in line with previous findings. 9
Moreover, as indicated by recent literature, 10 , 26 we confirm that inheritance in our series shows a significant protective role on the epilepsy outcome.
Although the presence of cortical tubers and SEN are determinants in developing seizures, intriguingly, the comparison between drug‐sensible vs drug‐refractory patients did not assign any significant role to neuroradiological findings. Therefore, the relationship between the number and location of lesions detected by MRI and the severity of epilepsy remains controversial. 27 , 28 It must be noted that, due to the retrospective nature of this study, timing and methodologies used to obtained brain MRIs over time were heterogeneous. This represents a limitation of this study, and only a prospective study with homogeneous imaging protocols will be able to provide a definitive answer.
Among the patients who achieved seizure freedom, the great majority was on monotherapy—mainly on carbamazepine or valproic acid—a small number on polytherapy, and some patients discontinued AEDs without seizure recurrence.
Comparing our data with other series, in the TSC Natural History Study, 99.5% of the patients with epilepsy were prescribed AEDs, 25.3% underwent surgery, 7.9% were prescribed special diets, and 1% was prescribed mTOR inhibitors. Of the patients receiving AEDs, over half (64.5%) used ≥3 different AEDs, due to drug resistance. 21
Considering AEDs prescription patterns in patients with TSC, some AEDs were identified as useful 10 , 13 , 22 but data regarding the adult population are relatively limited. Clobazam was noted to be effective in treating refractory epilepsy in 29 adult individuals with TSC (69% had >50% seizure reduction), 29 and topiramate and lamotrigine were demonstrated to be effective, respectively, in 64% and 37% obtaining >50% reduction in seizures, in both children and adults with TSC. 30 Lacosamide was effective in 48% of patients having >50% reduction in seizures, irrespectively of age. 31
Cannabidiol has been recently reported to be effective in reducing seizure frequency in up to 50% of patients, including adults. 32
In the present cohort of adult individuals, we cannot confirm the promising therapeutic role of Everolimus for epilepsy in TSC patients, although younger than ours. 33 So far, only five adults of our series have been on Everolimus, due to the recent introduction of this drug in the Italian market, which is in line with the data of the TSC Natural History Study, 21 but does not permit us to evaluate the efficacy of this medicine on seizure control.
Epilepsy surgery offers a 50–60% chance of seizure freedom in individuals with TSC, 34 also in our cohort. In fact, out of the patients who underwent epilepsy surgery, 54.5% became seizure‐free, even on a long‐term follow‐up. The rate of patients who had access to epilepsy surgery was relatively high (6%, 11/183) compared to the recently reported overall proportions of patients with epilepsy who received epilepsy surgery among those with medically refractory epilepsy (around 1%). 35 With regard to the potential benefit of epilepsy surgery, we think that early pre‐surgical workup should be recommended in all TSC patients with drug‐resistant seizures.
Considering the impact of epilepsy on cognitive functions and academic achievement, individuals with TSC having experienced seizures show a significantly lower IQ (both on verbal and performance scales) and reach a lower level of education compared with individuals without epilepsy. It is also noteworthy that, when seizure freedom can be achieved, a statistically significant part of patients has a normal cognitive level, while, on the other hand, patients with drug‐resistant seizures exhibit moderate/severe ID significantly more frequently. The relationship between seizures and neurodevelopmental outcome in childhood has been demonstrated in both retrospective 24 and prospective studies. 36 , 37 This finding was replicated in our adult cohort, since both early seizure onset, especially in the first year of life, and seizure drug resistance are predictive of moderate/severe ID, also in a long‐term follow‐up.
Indeed, Tye et al. 37 showed that early‐onset and severe epilepsy in the first 2 years of life are associated with an increased risk of long‐term ID in children with TSC, while the occurrence of epileptic spasms may play a role in adaptive functioning and ASD.
Finally, we should underline the emerging issue of psychiatric disorders in people with TSC. In our cohort, several disorders were diagnosed, among which the most frequent were ASD, behavioural problems and anxiety/mood disorders, as previously reported. 38 Having epilepsy generally increases the risk of developing psychiatric problems, especially when seizures are drug‐resistant. 39
Although not evaluated in our study, it would be interesting to investigate how systemic manifestations (i.e., the presence of facial angiofibromas) could possibly influence the development of mood disorders, anxiety and other psychiatric disorders, and quality of life.
Intriguingly, we can confirm in the long term that people with TSC and epilepsy are clearly at greater risk of developing ASD, especially when seizures remain drug‐resistant. 26 , 37 On the other hand, anxiety and mood disorders are experienced also by individuals without epilepsy or patients who are seizure‐free and could be interpreted as typical of the syndrome itself. These findings emphasize the urgent need for diagnosis and care of psychiatric comorbidities in TSC individuals, as addressed by the international TANDem project (https://tandconsortium.org). Indeed, a psychiatric evaluation is time‐consuming and needs specialized staff. As a result, neuropsychiatric complications may remain underdiagnosed even in expert centres. 40 For these purposes, a wider diffusion of easy to use tools, such as the TAND‐Checklist (https://www.tscinternational.org/wp‐content/uploads/2018/11/TAND_checklist‐2014.pdf), is mandatory in this population. Our results strongly support the need for specialized teams who can diagnose and treat such comorbidities, and warrant the implementation of programmes dedicated to giving the opportunity to treat such comorbidities.
5. CONCLUSIONS/STRENGTHS AND LIMITATIONS
This study, although retrospective, adds data about the management of epilepsy in a large cohort of adult individuals with TSC. Our experience confirms the previously reported findings in children with TSC and supports the role of early seizure control—among other yet to be discovered factors—in preventing the development of ID, psychiatric comorbidities and drug resistance itself in the long term. 41
While analysing data about our adult population, we should consider that the outcome of these individuals in terms of seizure control and cognitive functioning surely reflects the therapeutic attitudes that were in use 20–40 years ago. Of note, Vigabatrin and mTOR inhibitors, which are now known to significantly improve the outcome of young patients with TSC, were not widely used at that time. Therefore, we think that the current management of epilepsy in newborns and children with TSC has strongly improved with the introduction of international guidelines and new therapeutic strategies (e.g., early epilepsy surgery, Vigabatrin, Everolimus). For these reasons, we hope that individuals with TSC may have a better outcome in the upcoming years. In the meantime, we encourage clinicians to consider the global care of people with TSC, and to use current guidelines and useful diagnostic tools to detect psychiatric comorbidities, and to program therapeutic interventions.
CONFLICTS OF INTEREST
AV, AP, FLB and MPC received consulting fees from Italfarmaco outside the submitted work. AV and FLB received consulting fees from GW Pharmaceuticals outside the submitted work. The remaining authors have no conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. We ensure that we have liaised with all co‐authors to confirm agreement with the final statement.
Supporting information
Table S1
ACKNOWLEDGEMENTS
The authors would like to thank all the other healthcare professionals involved in the TSC Clinic of the San Paolo Hospital in Milan for their valuable work and participation in the care of our patients.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Northrup H, Krueger DA, Northrup H, et al. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49(4):243‐254. 10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franz DN, Capal JK. MTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders. Orphanet J Rare Dis. 2017;12(1):15. 10.1186/s13023-017-0596-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype‐phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13(6):731‐741. 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- 4. Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics. 2008;9(7):475‐487. 10.2174/138920208786241243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caban C, Khan N, Hasbani DM, Crino PB. Genetics of tuberous sclerosis complex: implications for clinical practice. Appl Clin Genet. 2017;10:1‐8. 10.2147/TACG.S90262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peron A, Northrup H. Tuberous sclerosis complex. Am J Med Genet Part C Semin Med Genet. 2018;178(3):274‐277. 10.1002/ajmg.c.31657. [DOI] [PubMed] [Google Scholar]
- 7. Rupert M, Katie R. Reproduced with permission of the copyright owner. Further reproduction prohibited without. J Allergy Clin Immunol. 2012;130(2):556. 10.1016/j.jaci.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 8. Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14(7):733‐745. 10.1016/S1474-4422(15)00069-1. [DOI] [PubMed] [Google Scholar]
- 9. Chu‐Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236‐1241. 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vignoli A, Briola FL, Turner K, et al. Epilepsy in TSC: certain etiology does not mean certain prognosis. Epilepsia. 2013;54(12): 10.1111/epi.12430. [DOI] [PubMed] [Google Scholar]
- 11. Nabbout R, Belousova E, Benedik MP, et al. Epilepsy in tuberous sclerosis complex: findings from the TOSCA study. Epilepsia Open. 2019;4(1):73‐84. 10.1002/epi4.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Curatolo P, Nabbout R, Lagae L, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol. 2018;22(5):738‐748. 10.1016/j.ejpn.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 13. Canevini MP, Kotulska‐Jozwiak K, Curatolo P, et al. Current concepts on epilepsy management in tuberous sclerosis complex. Am J Med Genet Part C Semin Med Genet. 2018;178(3):299‐308. 10.1002/ajmg.c.31652. [DOI] [PubMed] [Google Scholar]
- 14. Wei CC, Sheu JN, Liu JT, Yang SH, Chou IC, Tsai JD. Trend of seizure remission in patients with tuberous sclerosis complex: a retrospective medical review. J Chinese Med Assoc. 2018;81(8):724‐728. 10.1016/j.jcma.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 15. Hamer HM, Pfäfflin M, Baier H, et al. Characteristics and healthcare situation of adult patients with tuberous sclerosis complex in German epilepsy centers. Epilepsy Behav. 2018;82:64‐67. 10.1016/j.yebeh.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 16. Peron A, Canevini MP, Ghelma F, Di Marco F, Vignoli A. Healthcare transition from childhood to adulthood in tuberous sclerosis complex. Am J Med Genet Part C Semin Med Genet. 2018;178(3):355‐364. 10.1002/ajmg.c.31653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51(6):1069‐1077. 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 18. Wong AM, Wang H‐S, Schwartz ES, et al. Cerebral diffusion tensor MR tractography in tuberous sclerosis complex: correlation with neurologic severity and tract‐based spatial statistical analysis. Am J Neuroradiol. 2013;34(9):1829‐1835. 10.3174/ajnr.A3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krsek P, Jahodova A, Kyncl M, et al. Predictors of seizure‐free outcome after epilepsy surgery for pediatric tuberous sclerosis complex. Epilepsia. 2013;54(11):1913‐1921. 10.1111/epi.12371. [DOI] [PubMed] [Google Scholar]
- 20. Overwater IE, Bindels‐de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia. 2015;56(8):1239‐1245. 10.1111/epi.13050. [DOI] [PubMed] [Google Scholar]
- 21. Song J, Swallow E, Said Q, et al. Epilepsy treatment patterns among patients with tuberous sclerosis complex. J Neurol Sci. 2018;391:104‐108. 10.1016/j.jns.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 22. Welin KO, Carlqvist P, Svensson A, Althin R, Eklund E, Rask O. Epilepsy in tuberous sclerosis patients in Sweden – Healthcare utilization, treatment, morbidity, and mortality using national register data. Seizure. 2017;53:4‐9. 10.1016/j.seizure.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 23. Bar C, Ghobeira R, Azzi R, et al. Experience of follow‐up, quality of life, and transition from pediatric to adult healthcare of patients with tuberous sclerosis complex. Epilepsy Behav. 2019;96:23‐27. 10.1016/j.yebeh.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 24. Kalilani L, Sun X, Pelgrims B, Noack‐Rink M, Villanueva V. The epidemiology of drug‐resistant epilepsy: a systematic review and meta‐analysis. Epilepsia. 2018;. 10.1111/epi.14596. [DOI] [PubMed] [Google Scholar]
- 25. Savini MN, Mingarelli A, Vignoli A, et al. Ictal signs in tuberous sclerosis complex: clinical and video‐EEG features in a large series of recorded seizures. Epilepsy Behav. 2018;85:14‐20. 10.1016/j.yebeh.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 26. Gupta A, de Bruyn G, Tousseyn S, et al. Epilepsy and neurodevelopmental comorbidities in tuberous sclerosis complex: a natural history study. Pediatr Neurol. 2020;106:10‐16. 10.1016/j.pediatrneurol.2019.12.016. [DOI] [PubMed] [Google Scholar]
- 27. Park SM, Lee YJ, Son YJ, Kim YO, Woo YJ. Clinical progress of epilepsy in children with tuberous sclerosis: prognostic factors for seizure outcome. Chonnam Med J. 2011;47(3):150. 10.4068/cmj.2011.47.3.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jahodova A, Krsek P, Kyncl M, et al. Distinctive MRI features of the epileptogenic zone in children with tuberous sclerosis. Eur J Radiol. 2014;83(4):703‐709. 10.1016/j.ejrad.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 29. Jennesson M, van Eeghen AM, Caruso PA, Paolini JL, Thiele EA. Clobazam therapy of refractory epilepsy in tuberous sclerosis complex. Epilepsy Res. 2013;104(3):269‐274. 10.1016/j.eplepsyres.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 30. Jennesson M, van Eeghen AM, Caruso PA, et al. Lamotrigine therapy of epilepsy in tuberous sclerosis. Epilepsy Res. 2016;57(3):269‐274. 10.1016/j.eplepsyres.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 31. Geffrey AL, Belt OD, Paolini JL, Thiele EA. Lacosamide use in the treatment of refractory epilepsy in tuberous sclerosis complex. Epilepsy Res. 2015;112:72‐75. 10.1016/j.eplepsyres.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 32. Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug‐resistant epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(10):1617‐1624. 10.1111/epi.13499. [DOI] [PubMed] [Google Scholar]
- 33. Krueger DA, Wilfong AA, Mays M, et al. Long‐term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology. 2016;87(23):2408‐2415. 10.1212/WNL.0000000000003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ostrowsky‐Coste K, Neal A, Guenot M, et al. Resective surgery in tuberous sclerosis complex, from Penfield to 2018: a critical review. Rev Neurol (Paris). 2019;175(3):163‐182. 10.1016/j.neurol.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 35. Engel J. Evolution of concepts in epilepsy surgery*. Epileptic Disord. 2019;21(5):391‐409. 10.1684/epd.2019.1091. [DOI] [PubMed] [Google Scholar]
- 36. Capal JK, Bernardino‐Cuesta B, Horn PS, et al. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. 2017;70:245‐252. 10.1016/j.yebeh.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tye C, Mcewen FS, Liang H, et al. Long‐term cognitive outcomes in tuberous sclerosis complex. Dev Med Child Neurol. 2020;62(3):322‐329. 10.1111/dmcn.14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leclezio L, De Vries PJ. Advances in the treatment of tuberous sclerosis complex. Curr Opin Psychiatry. 2015;28(2):113‐120. 10.1097/YCO.0000000000000136. [DOI] [PubMed] [Google Scholar]
- 39. Salpekar JA, Mula M. Common psychiatric comorbidities in epilepsy: how big of a problem is it? Epilepsy Behav. 2019;98:293‐297. 10.1016/j.yebeh.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 40. Cervi F, Saletti V, Turner K, et al. The TAND checklist: a useful screening tool in children with tuberous sclerosis and neurofibromatosis type 1. Orphanet J Rare Dis. 2020;15(1):1‐11. 10.1186/s13023-020-01488-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kotulska K, Kwiatkowski DJ, Curatolo P, et al. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann Neurol. 2021;89(2):304‐314. 10.1002/ana.25956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.