

Abstract
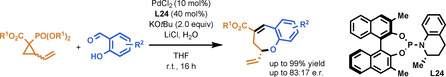
A palladium‐catalyzed intermolecular cascade (4+3) cyclocondensation of salicylaldehydes and vinylcyclopropanes is reported. A key feature of the reaction is the use of a phosphonate group as an acceptor moiety on the cyclopropane, exploiting its propensity to undergo olefination with aldehydes. Subsequent O‐allylation enabled the formation of a range of substituted benzoxepinsWith a novel chiral ligand, the products were obtained in generally good yield and with reasonable enantioselectivity.
Keywords: benzoxepins, cascade reactions, olefination, palladium, seven-membered rings
Enantiomerically enriched benzoxepins were synthesized from phosphonate‐functionalized vinylcyclopropanes and salicylaldehydes by palladium‐catalyzed ring opening followed by Horner–Wadsworth–Emmons olefination and subsequent O‐allylation (see scheme). In contrast to existing cycloaddition reactions of donor–acceptor cyclopropanes, the two bonds are each formed in a separate event, thus allowing greater flexibility in reaction design.
Donor–acceptor cyclopropanes (DACs) are among the most useful and versatile three‐carbon building blocks in organic synthesis. [1] The presence of electron‐withdrawing and electron‐donating groups on vicinal positions of a DAC (I, Scheme 1) allows facile heterolytic ring opening to give a 1,3‐dipolar intermediate (II). Since the discovery and first applications of DACs, [2] these reagents have been widely employed, particularly in cycloaddition reactions. Among these, (3+2) cycloadditions (pathway A in Scheme 1) are by far the most common class owing to the abundance of 1,2‐dipolar reaction partners, including activated olefins, [3] alkynes, [4] (ox)indoles, [5] aldehydes, [6] ketones, [7] imines [8] and ketenes [9] (also in combination with organocatalysis [10] ). Compared to (3+2) cycloadditions, (3+3) [11] and (4+3) [12] cycloadditions (pathways B and C in Scheme 1) are still underdeveloped. In particular, we considered the typically challenging formation of seven‐membered rings worth pursuing. [13]
Scheme 1.
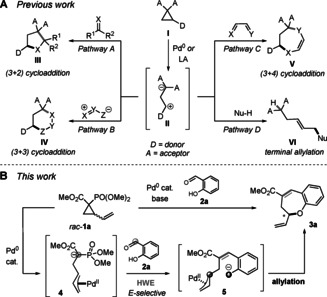
A) Previously reported reaction types of DACs I. B) Cascade ring opening/olefination/O‐allylation (this work). LA=Lewis acid; HWE=Horner–Wadsworth–Emmons reaction.
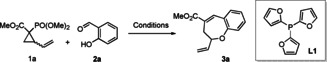
The acceptor moiety of DACs often comprises two ester functionalities. However, several other electron‐withdrawing groups may also activate the DAC, including nitriles, sulfones, ketones, and even arenes. [14] In formal cycloaddition reactions of DACs, electrons typically flow from this moiety bearing the negative charge to the dipolarophile and back to the donor‐activated electrophilic moiety of the DAC in a likely stepwise, but mechanistically intertwined process. [15] Driven by our interest in cascade reactions and palladium‐catalyzed allylic substitution, [16] we envisioned a formal cycloaddition reaction in which the two reactive sites of the DAC undergo two distinct transformations with a bifunctional reaction partner. We focused on using phosphonates as an acceptor moiety on vinylcyclopropanes (VCPs) 1 in combination with palladium‐catalyzed ring opening. The resulting α‐deprotonated phosphonate 4 may undergo Horner–Wadsworth–Emmons (HWE) olefination with aldehydes. Then, the electrophilic π‐allylpalladium moiety of the DAC fragment may react with a pendant nucleophile on the aldehyde reaction partner. To the best of our knowledge, such a cascade reaction of DACs replacing a concerted cycloaddition by two independent transformations has not been reported to date.
The first challenge we faced was finding a suitable latent nucleophile to incorporate in the aldehyde reaction partner. Since aldehydes are also known to react with VCPs as dipolarophiles in (3+2) cycloaddition reactions, elimination of the dialkyl phosphate should outcompete nucleophilic substitution. To showcase our cascade approach, we selected salicylaldehyde 2 a as model substrate under palladium catalysis, which would lead to the formation of the benzoxepin 3 a (Scheme 1). After palladium‐catalyzed formation of intermediate 4, the α‐deprotonated phosphonate can undergo HWE olefination with the aldehyde, leading to intermediate 5. The π‐allylpalladium moiety in 5 can then be attacked by the nucleophilic alkoxide to afford 3 a. Although there are scattered reports of DACs bearing a phosphonate EWG in the literature, [15] their active participation in an ensuing HWE olefination/allylation cascade has not been demonstrated previously. [17]
As benzoxepins of both natural [18] and synthetic [19] origin display diverse biological activities, we decided to further pursue this uncommon scaffold. Preliminary investigation (for full details, see the Supporting Information) showed that the proposed reaction is indeed feasible. We then began the optimization of the reaction conditions using 1 a and 2 a as the benchmark reagents. Under the initial conditions [1.2 equiv 1 a, 1.0 equiv 2 a, 5 mol % Pd2dba3, 1.2 equiv KOtBu, THF, 50 °C], the vinylbenzoxepin product 3 a could be isolated in 43 % yield (Table 1, entry 1). The yield could be marginally improved by adding trifurylphosphine (L1) as an ancillary ligand for Pd (entry 2). Somewhat unexpectedly, we found that two equivalents of KOtBu gave the optimal result (entries 3–5). [20] Moreover, the addition of LiCl (1.2 equiv) proved to be beneficial, significantly increasing the yield (entry 6). Finally, increasing the stoichiometry of 1 a to 2.5 equivalents was determined to be optimal, affording 3 a in 95 % yield (entries 7–10).
Table 1.
Optimization of the reaction conditions.
|
Entry[a] |
1 a (equiv) |
Ligand (mol %) |
Base (equiv) |
Additive (equiv) |
Yield [%][b] |
|---|---|---|---|---|---|
|
1[c] |
1.2 |
– |
KOtBu (1.2) |
– |
43 |
|
2 |
1.2 |
L1 (20) |
KOtBu (1.2) |
– |
47 |
|
3 |
1.2 |
L1 (20) |
KOtBu (1.5) |
– |
53 |
|
4 |
1.2 |
L1 (20) |
KOtBu (2) |
– |
62 |
|
5 |
1.2 |
L1 (20) |
KOtBu (2.5) |
– |
54 |
|
6 |
1.2 |
L1 (20) |
KOtBu (2) |
LiCl (1.2) |
75 |
|
7 |
1.5 |
L1 (20) |
KOtBu (2) |
LiCl (1.2) |
83 |
|
8 |
2 |
L1 (20) |
KOtBu (2) |
LiCl (1.2) |
89 |
|
9 |
2.5 |
L1 (20) |
KOtBu (2) |
LiCl (1.2) |
95 |
|
10 |
3 |
L1 (20) |
KOtBu (2) |
LiCl (1.2) |
95 |
[a] Reaction conditions: 2 a (0.236 mmol), Pd2dba3 (5 mol %), L1 (20 mol %), 1.18 mL of THF (1.18 mL), 50 °C, overnight. [b] Determined by 1H NMR analysis using 2,5‐dimethylfuran as an internal standard. [c] Pd(PPh3)4 was used instead of Pd2dba3 and L1. dba=dibenzylideneacetone.
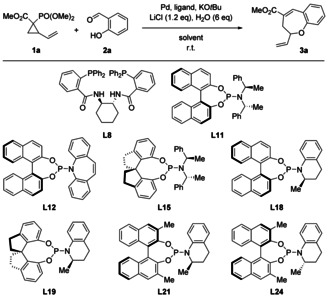
With the optimized racemic conditions at hand, we sought to create a catalytic enantioselective process by employing a chiral ligand. This brings inherent challenges: while typically the two new bond formations in formal cycloadditions of DACs are mechanistically intertwined, our cascade transformation involves two consecutive, independent reactions, with the enantiodetermining step occurring second, potentially reducing the level of stereocontrol. Moreover, while most DACs are symmetrically substituted at the acceptor moiety and hence only incorporate one stereocenter, 1 a is used as a mixture of four stereoisomers. When a chiral catalyst is used, this may lead to a number of catalyst/substrate match–mismatch cases, which may or may not readily interconvert under the reaction conditions. Consequently, it is very difficult to predict whether all diastereomers of 1 are converted with the same efficiency and/or selectivity. Nevertheless, we set out to address these challenges. Firstly, various ligand types were tested, showing that commonly used chiral phosphine ligands such as (S)‐tBuPHOX (L2) and (S)‐BINAP (L6) were unable to effect the transformation (for full details, see the Supporting Information). Similarly, when Trost ligand L8 was used, 3 a was obtained in only 15 % yield and with poor enantioselectivity (entry 1). Since phosphoramidites have also been reported to be effective ligands for Pd in cycloaddition reactions of DACs,[ 5a , 5c ] we shifted our attention to this class of ligands. However, the use of L11, L12 and L15 only moderately improved the yield and/or the enantioselectivity (entries 2–4). Interestingly, we found that different diastereomers of these ligands gave dramatically different results in terms of both yield and e.r. (L9 vs. L11, L14 vs. L15; see the Supporting Information). Therefore, we designed and synthesized both diastereomers of several new phosphoramidite ligands (L19–L26, see the Supporting Information). Gratifyingly, L24 gave 3 a in near quantitative yield and with good enantioselectivity.
The results could only be slightly improved by increasing the stoichiometry of 1 a (entry 9), whereas varying the solvent, catalyst and ligand stoichiometry, cyclopropane substituents, or base did not improve the yield or enantioselectivity (see the Supporting Information). Upon further streamlining of the reaction conditions, we discovered that the presence of traces of water is crucial for the reaction. When solid LiCl was replaced by a 0.5 m solution of dry LiCl in THF, we found that only traces of 3 a were formed. Apparently, weighing in LiCl under non‐inert conditions absorbs sufficient atmospheric moisture to promote the reaction, owing to its hygroscopic properties. Fortunately, complementing LiCl added as a THF solution with 25 μL H2O fully restored the efficiency of the transformation, while allowing more precise and reproducible control over the reaction conditions (for details, see the Supporting Information). Finally, we found that the use of different sources and even batches of Pd2dba3 led to considerable fluctuations in yield. We therefore switched to PdCl2 as a more reliable palladium source, although two additional equivalents of ligand were required in this case to in situ reduce PdII to Pd0 (Table 2, entry 10).
Table 2.
Optimization of the asymmetric synthesis of 3 a.
|
Entry[a] |
Ligand |
Solvent |
Yield [%][b] |
e.r.[c] |
|---|---|---|---|---|
|
1 |
L8 |
THF |
15 |
64:36 |
|
2 |
L11 |
THF |
30 |
51:49 |
|
3 |
L12 |
THF |
31 |
50:50 |
|
4 |
L15 |
THF |
40 |
73:27 |
|
5 |
L18 |
THF |
79 |
56:44 |
|
6 |
L19 |
THF |
20 |
76:24 |
|
7 |
L21 |
THF |
15 |
66:34 |
|
8 |
L24 |
THF |
98 |
77:23 |
|
9[d] |
L24 |
THF |
99 |
80:20 |
|
10[d,e] |
L24 |
THF |
98 |
80:20 |
[a] Reaction conditions: 2 a (0.236 mmol), Pd2dba3 (5 mol %), ligand (10 mol % for bidentate, 20 mol % for monodentate), 1 a (0.590 mmol), LiCl (0.283 mmol), H2O (26 μL), KOtBu (0.472 mmol), solvent (1.18 mL), room temperature, overnight. [b] Determined by 1H NMR analysis using 2,5‐dimethylfuran as an internal standard. [c] Determined by SFC analysis on a chiral stationary phase. [d] 1 a: 0.826 mmol. [e] PdCl2 (10 mol %) and L24 (40 mol %) were used instead of Pd2dba3.
With the optimal conditions in hand, we set out to investigate the scope of the reaction (Scheme 2). In general, the reaction showed high tolerance towards functional groups, affording the desired benzoxepins in good to excellent yield and consistent enantioselectivities. Alkyl groups and esters at the 4‐position of the salicylaldehyde only slightly decreased yields and enantioselectivities of the resulting products (3 b, 3 g) compared to the unsubstituted product 3 a. Similar enantioselectivities but lower yields were observed with halogen substituents, probably due to a competitive insertion of the palladium catalyst (3 e, 3 f). Interestingly, the lowest enantioselectivity was observed for an electron‐donating methoxy substituent (3 c), although the yield was still reasonable. Electron‐donating groups at the 5‐position (Me, MeO) generally afforded the corresponding products in excellent yields and good enantioselectivities (3 g, 3 j). Halogen substituents are also well tolerated (3 k–n) with Cl and F performing best, whereas Br and I led to slightly lower yield and/or enantioselectivity. Aldehydes 2 bearing electron‐deficient (halogens, ester, CF3) or electron‐rich (RO, Me) aromatic substituents were converted very efficiently, affording the corresponding products (3 o–v) with only minor fluctuations in e.r. and in good to very good yield, even with a 2‐furyl substituent (3 v). In general, we observed that electron‐donating substituents on the arene are beneficial for both yield and enantioselectivity. The presence of substituents on the ortho position with respect to both the aldehyde and phenol proved to be less favorable for both yield and enantioselectivity (3 w, 3 x). Finally, VCP 1 b (R1=Et) was tested, affording products 3 y, 3 z and 3 za in similar er but generally lower yields (45–61 %) compared to their methyl ester counterparts (3 a, 3 b and 3 o, respectively).
Scheme 2.
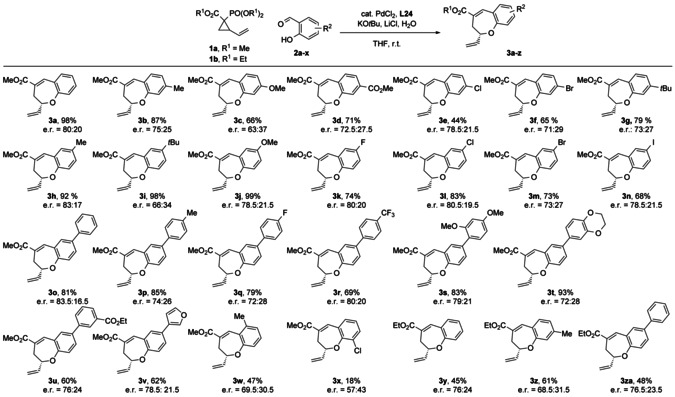
Reaction conditions: 2 a–x (0.236 mmol), PdCl2 (10 mol %), L24 (40 mol %), 1 a,b (0.826 mmol), LiCl (0.283 mmol), H2O (26 μL), KOtBu (0.472 mmol, 1.0 m in THF), solvent (1.18 mL), room temperature, overnight.
We then shifted our interest to the mechanism (Scheme 3), in particular on the role of LiCl. It is well established that halide ions can improve the yield and enantioselectivity [21] of palladium‐catalyzed asymmetric allylations by influencing the rates of syn–anti isomerization and apparent rotation processes of the π‐allyl Pd complex. [22] However, in this reaction the beneficial effect was only observed with Li+ as the counterion (see the Supporting Information for details). We postulate that the Lewis acidic Li+ ion may stabilize the zwitterionic intermediate 4 by coordination between the oxygen atoms of the carbonyl and phosphonate moieties, leading to an overall synergic effect of LiCl. Interestingly, other lithium salts (LiBr and LiI) did not show the same effect, indicating an active role for the chloride ion. Thus far, we have been unable to rationalize the critical role of water in the reaction, although plausibly hydrogen bonding to either the aldehyde or VCP may be involved.
Scheme 3.
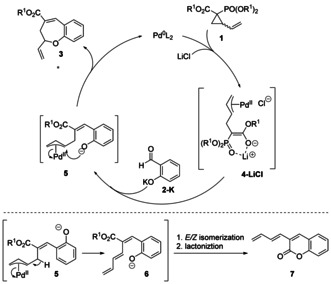
Proposed mechanism.
Regarding the order of events in the mechanism, it was initially unclear whether the HWE olefination or the palladium‐catalyzed O‐allylation occurs first. However, the observation that traces of β‐elimination/transesterification product 7 were formed under racemic conditions in some cases (2 e, 2 k, 2 l) suggests that the O‐allylation step takes place only after the initial HWE olefination. The fact that it is not observed with other, more donating substituents suggests that the nucleophilic attack is kinetically favored over, hence faster than, β‐elimination in the other cases, however the two rates become comparable in case of electron‐withdrawing substituents. Moreover, a control experiment using vinylcyclopropane 1 f (Scheme 4 A), which would generate the Z‐configured alkene intermediate 8 by a Still–Gennari olefination, [23] still afforded 3 a, albeit in only 15 % yield. This observation not only suggests that in the reaction C−C bond formation indeed precedes the O‐allylation, [24] but also that alkene isomerization is possible (likely as a result of the reduced double bond character in the delocalized anionic intermediates 8 and 9) and that the π‐allyl–palladium complex is sufficiently stable to undergo the alkylation step at a later stage. Finally, in order to rule out reversibility of the O‐allylation, we subjected rac‐3 a to the enantioselective condition and enantioenriched 3 a to the racemic conditions (Scheme 4 B). No change in the enantiomeric ratio was observed in either case, suggesting that the alkylation step is irreversible.
Scheme 4.
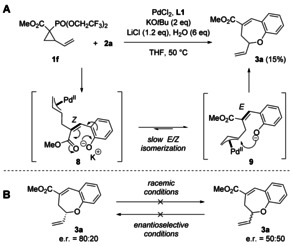
A) Still–Gennari variation. B) Reversibility experiments.
In order to determine the absolute configuration of the newly formed stereocenter, we attempted to grow crystals of the enantioenriched products 3 suitable for X‐ray diffraction, but without success. As an alternative, we measured the circular dichroism (CD) spectrum of enantioenriched 3 a. Comparison with simulated spectra of both enantiomers led to the conclusion that the absolute configuration is S (see the Supporting Information for details).
In conclusion, we developed a formal (3+4) cycloaddition of phosphonate‐functionalized vinylcyclopropanes 1 and salicylaldehydes 2, affording a range of functionalized benzoxepins 3. The reaction proceeds by palladium‐catalyzed ring opening of the vinylcyclopropane followed by HWE olefination and O‐allylation to give the benzoxepin products in high yields (up to 99 %) and with reasonable enantioselectivity (up to 83:17 e.r.) using a new chiral ligand.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by The Netherlands Organization for Scientific Research (NWO). Part of the work was carried out on the Dutch national e‐infrastructure with the support of SURF Cooperative. We kindly acknowledge Sebastian Strähler for practical assistance, Elwin Janssen for NMR measurements and Daniel Preschel for HRMS measurements and technical support (all VUA).
M. Faltracco, K. N. A. van de Vrande, M. Dijkstra, J. M. Saya, T. A. Hamlin, E. Ruijter, Angew. Chem. Int. Ed. 2021, 60, 14410.
References
- 1.For reviews, see:
- 1a. Grover H. K., Emmett M. R., Kerr M. A., Org. Biomol. Chem. 2015, 13, 655–671; [DOI] [PubMed] [Google Scholar]
- 1b. Cavitt M. A., Phun L. H., France S., Chem. Soc. Rev. 2014, 43, 804–818; [DOI] [PubMed] [Google Scholar]
- 1c. Schneider T. F., Kaschel J., Werz D. B., Angew. Chem. Int. Ed. 2014, 53, 5504–5523; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5608–5628; [Google Scholar]
- 1d. Reissig H.-U., Zimmer R., Chem. Rev. 2003, 103, 1151–1196; [DOI] [PubMed] [Google Scholar]
- 1e. Werz D. B., Biju A. T., Angew. Chem. Int. Ed. 2020, 59, 3385–3398; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3410–3424. [Google Scholar]
- 2.
- 2a. Wenkert E., Alonso M. E., Buckwalter B. L., Chou K. J., J. Am. Chem. Soc. 1977, 99, 4778–4782; [Google Scholar]
- 2b. Piers E., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1979, 18, 791–792; [Google Scholar]; Angew. Chem. 1979, 91, 857–858; [Google Scholar]
- 2c. Reissig H.-U., Hirsch E., Angew. Chem. Int. Ed. Engl. 1980, 19, 813–814; [Google Scholar]; Angew. Chem. 1980, 92, 839–840; [Google Scholar]
- 2d. Brückner C., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1985, 24, 588–589; [Google Scholar]; Angew. Chem. 1985, 97, 578–579; [Google Scholar]
- 2e. Shimizu I., Oashi Y., Tsuji J., Tetrahedron Lett. 1985, 26, 3825–3828. [Google Scholar]
- 3.
- 3a. Mei L. Y., Wei Y., Xu Q., Shi M., Organometallics 2012, 31, 7591–7599; [Google Scholar]
- 3b. Ma C., Huang Y., Zhao Y., ACS Catal. 2016, 6, 6408–6412; [Google Scholar]
- 3c. Zhang S. C., Lei X. X., Yang Y., Luo Y. C., Zhang H. H., Xu P. F., Org. Chem. Front. 2019, 6, 2415–2419. [Google Scholar]
- 3d. Li W.-K., Liu Z.-S., He L., Kang T.-R., Liu Q.-Z., Asian J. Org. Chem. 2015, 4, 28–32; [Google Scholar]
- 3e. Wei F., Ren C. L., Wang D., Liu L., Chem. Eur. J. 2015, 21, 2335–2338. [DOI] [PubMed] [Google Scholar]
- 4. Ding W. P., Zhang G. P., Jiang Y. J., Du J., Liu X. Y., Chen D., Ding C. H., Deng Q. H., Hou X. L., Org. Lett. 2019, 21, 6805–6810. [DOI] [PubMed] [Google Scholar]
- 5.For indoles, see:
- 5a. Sun M., Zhu Z. Q., Gu L., Wan X., Mei G. J., Shi F., J. Org. Chem. 2018, 83, 2341–2348; [DOI] [PubMed] [Google Scholar]
- 5b. Laugeois M., Ling J., Férard C., Michelet V., Ratovelomanana-Vidal V., Vitale M. R., Org. Lett. 2017, 19, 2266–2269; [DOI] [PubMed] [Google Scholar]
- 5c. Liu Z. S., Li W. K., Kang T. R., He L., Liu Q. Z., Org. Lett. 2015, 17, 150–153; for oxindoles, see: [DOI] [PubMed] [Google Scholar]
- 5d. Singh K., Pramanik S., Hamlin T. A., Mondal B., Das D., Saha J., Chem. Commun. 2019, 55, 7069–7072. [DOI] [PubMed] [Google Scholar]
- 6. Parsons A. T., Campbell M. J., Johnson J. S., Org. Lett. 2008, 10, 2541–2544. [DOI] [PubMed] [Google Scholar]
- 7. Mei L., Wei Y., Xu Q., Shi M., Organometallics 2013, 32, 3544–3556. [Google Scholar]
- 8.
- 8a. Huang X. B., Li X. J., Li T. T., Chen B., Chu W. D., He L., Liu Q. Z., Org. Lett. 2019, 21, 1713–1716; [DOI] [PubMed] [Google Scholar]
- 8b. Zhou Q., Chen B., Huang X. B., Zeng Y. L., Chu W. D., He L., Liu Q. Z., Org. Chem. Front. 2019, 6, 1891–1894. [Google Scholar]
- 9.
- 9a. Mondal M., Panda M., McKee V., Kerrigan N. J., J. Org. Chem. 2019, 84, 11983–11991; [DOI] [PubMed] [Google Scholar]
- 9b. Liu J., Li M. M., Qu B. L., Lu L. Q., Xiao W. J., Chem. Commun. 2019, 55, 2031–2034. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Laugeois M., Ponra S., Ratovelomanana-Vidal V., Michelet V., Vitale M. R., Chem. Commun. 2016, 52, 5332–5335; [DOI] [PubMed] [Google Scholar]
- 10b. Ma G., Afewerki S., Deiana L., Palo-Nieto C., Liu L., Sun J., Ibrahem I., Cordova A., Angew. Chem. Int. Ed. 2013, 52, 6050–6054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6166–6170; [Google Scholar]
- 10c. Halskov K. S., Næsborg L., Tur F., Jørgensen K. A., Org. Lett. 2016, 18, 2220–2223. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Petzold M., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2019, 58, 6225–6229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6291–6295; [Google Scholar]
- 11b. Chagarovskiy A. O., Vasin V. S., Kuznetsov V. V., Ivanova O. A., Rybakov V. B., Shumsky A. N., Makhova N. N., Trushkov I. V., Angew. Chem. Int. Ed. 2018, 57, 10338–10342; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10495–10499; [Google Scholar]
- 11c. Dhote P. S., Ramana C. V., Org. Lett. 2019, 21, 6221–6224; [DOI] [PubMed] [Google Scholar]
- 11d. Chidley T., Vemula N., Carson C. A., Kerr M. A., Pagenkopf B. L., Org. Lett. 2016, 18, 2922–2925; [DOI] [PubMed] [Google Scholar]
- 11e. Garve L. K. B., Petzold M., Jones P. G., Werz D. B., Org. Lett. 2016, 18, 564–567; [DOI] [PubMed] [Google Scholar]
- 11f. Liu H., Yuan C., Wu Y., Xiao Y., Guo H., Org. Lett. 2015, 17, 4220–4223. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Xu H., Hu J.-L., Wang L., Liao S., Tang Y., J. Am. Chem. Soc. 2015, 137, 8006–8009; [DOI] [PubMed] [Google Scholar]
- 12b. Cheng Q., Xie J. H., Weng Y. C., You S. L., Angew. Chem. Int. Ed. 2019, 58, 5739–5743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5795–5799; [Google Scholar]
- 12c. Wang Z.-H., Zhang H.-H., Wang D.-M., Xu P.-F., Luo Y.-C., Chem. Commun. 2017, 53, 8521–8524; [DOI] [PubMed] [Google Scholar]
- 12d. Augustin A. U., Merz J. L., Jones P. G., Mlostoń G., Werz D. B., Org. Lett. 2019, 21, 9405–9409. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Nguyen T. V., Hartmann J. M., Enders D., Synthesis 2013, 45, 845–873; [Google Scholar]
- 13b. Mortensen K. T., Osberger T. J., King T. A., Sore H. F., Spring D. R., Chem. Rev. 2019, 119, 10288–10317. [DOI] [PubMed] [Google Scholar]
- 14. Wang J., Blaszczyk S. A., Li X., Tang W., Chem. Rev. 2021, 121, 110–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Mita T., Tanaka H., Higuchi Y., Sato Y., Org. Lett. 2016, 18, 2754–2757; [DOI] [PubMed] [Google Scholar]
- 15b. Moran J., Smith A. G., Carris R. M., Johnson J. S., Krische M. J., J. Am. Chem. Soc. 2011, 133, 18618–18621; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Wu J. Q., Qiu Z. P., Zhang S. S., Liu J. G., Lao Y. X., Gu L. Q., Huang Z. S., Li J., Wang H., Chem. Commun. 2015, 51, 77–80. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Braun J., Ariëns M. I., Matsuo B. T., de Vries S., van Wordragen E. D. H., Ellenbroek B. D., Vande Velde C. M. L., Orru R. V. A., Ruijter E., Org. Lett. 2018, 20, 6611–6615; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Faltracco M., Cotogno S., Vande Velde C. M. L., Ruijter E., J. Org. Chem. 2019, 84, 12058–12070; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Faltracco M., Sukowski V., van Druenen M., Hamlin T. A., Bickelhaupt F. M., Ruijter E., J. Org. Chem. 2020, 85, 9566–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For a related reductive ring opening/HWE cascade of cyclopropane–phosphonates, see: Lloyd M. G., Taylor R. J. K., Unsworth W. P., Org. Biomol. Chem. 2016, 14, 8971–8988. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Engler M., Anke T., Sterner O., Brandt U., J. Antibiot. 1997, 50, 325–329; [DOI] [PubMed] [Google Scholar]
- 18b. Pettit G. R., Numata A., Iwamoto C., Usami Y., Yamada T., Ohishi H., Cragg G. M., J. Nat. Prod. 2006, 69, 323–327; [DOI] [PubMed] [Google Scholar]
- 18c. Kashima K., Sano K., Yun Y. S., Ina H., Kunugi A., Inoue H., Chem. Pharm. Bull. 2010, 58, 191. [DOI] [PubMed] [Google Scholar]
- 19. Gao C.-L., Hou G.-G., Liu J., Ru T., Xu Y.-Z., Zhao S.-Y., Ye H., Zhang L., Chen K.-X., Guo Y.-W., Pang T., Li X.-W., Angew. Chem. Int. Ed. 2020, 59, 2429–2439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2450–2460. [Google Scholar]
- 20.We initially anticipated that an excess of a strong base would quantitatively deprotonate 2, rendering it less reactive in the HWE olefination.
- 21.
- 21a. Bovens M., Togni A., Venanzi L. M., J. Organomet. Chem. 1993, 451, C28-C31; [Google Scholar]
- 21b. Togni A., Burckhardt U., Gramlich V., Pregosin P. S., Salzmann R., J. Am. Chem. Soc. 1996, 118, 1031–1037; [Google Scholar]
- 21c. Burckhardt U., Gramlich V., Hofmann P., Nesper R., Pregosin P. S., Salzmann R., Togni A., Organometallics 1996, 15, 3496–3503. [Google Scholar]
- 22. Fagnou K., Lautens M., Angew. Chem. Int. Ed. 2002, 41, 26–47; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 26–49. [Google Scholar]
- 23.
- 23a. Still W. C., Gennari C., Tetrahedron Lett. 1983, 24, 4405–4408; [Google Scholar]
- 23b. Janickia I., Kiełbasinsky P., Adv. Synth. Catal. 2020, 362, 2552–2596. [Google Scholar]
- 24.Although all our observations suggest that C−C bond formation precedes C−O bond formation, we cannot rule out the occurrence of the O-allylation at an intermediate stage of the HWE olefination, that is, the betaine or phosphaoxetane stage. As these intermediates contain an additional element of chirality and are most likely formed with low stereopreference (as their formation occurs independently of the chiral catalyst), this may explain why we were not able to obtain higher enantioselectivity for the cascade reaction, despite exhaustive optimization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary