

Abstract
Mesenteric lymph node (mLN) T cells undergo tissue adaptation upon migrating to intestinal lamina propria (LP) and intraepithelial (IE) compartments, ensuring appropriate balance between tolerance and resistance. By combining mouse genetics with single-cell and chromatin analyses, we uncovered the molecular imprinting of gut epithelium on T cells. Transcriptionally, conventional and regulatory (Treg) CD4+ T cells from mLN, LP and IE segregate based on the gut layer they occupy; trajectory analysis suggests a stepwise loss of CD4-programming and acquisition of an intraepithelial profile. Treg fate-mapping coupled with RNA- and ATAC-sequencing revealed that the Treg program shuts down before an intraepithelial program becomes fully accessible at the epithelium. Ablation of CD4 lineage-defining transcription factor ThPOK results in premature acquisition of an IEL profile by mLN Tregs, partially recapitulating epithelium imprinting. Thus, coordinated replacement of circulating lymphocyte program with site-specific transcriptional and chromatin changes is necessary for tissue imprinting.
Introduction
The plethora of food and bacteria derived antigens in the intestinal lumen is separated from the lamina propria (LP) by a single layer of epithelial cells, comprising a highly stimulating environment for the underlying immune system 1, 2, 3. A central mechanism for intestinal tolerance is mediated by Foxp3+ regulatory T cells (Tregs)4, 5, 6. Tregs can be induced by dietary and microbiota antigens in a TGF-β and retinoic acid dependent manner in the gut-draining mesenteric lymph nodes (mLN), and primarily reside in the LP7, 8, 9, 10. Likewise, TGF-β and retinoic acid, along with T-bet, are required for the differentiation of CD8αα-expressing CD4+ intraepithelial lymphocytes (also referred as CD4-IELs, DP-IELs or CD4+CD8αα+ IELs) from both conventional CD4+ T cells (Tconv) and Tregs upon migration to the intestinal epithelium (IE) in a microbiota-dependent manner8, 10, 11, 12, 13, 14. CD4-IELs (CD4+CD8αα+) have also been implicated in controlling local inflammation8, 11. The hallmarks of CD4-IEL differentiation are induced by a switch from conventional CD4-to-CD8 lineage-defining transcription factors. ThPOK (encoded by Zbtb7b), expressed by all conventional mature CD4+ T cells, is downregulated whereas the long form of Runx3, expressed by mature CD8+ T cells8, 11, 15, is upregulated in CD4-IELs. While environmental cues and transcription factors involved in the differentiation of naïve CD4+ T cells into gut-adapted T cell subsets have been described in the past decade, the transcriptional and chromatin changes that accompany such tissue-specific imprinting have not been elucidated.
We used genetic fate-mapping and gene ablation mouse models coupled with RNA-, ATAC- and ChIP-sequencing approaches to uncover the molecular mechanisms of how the intestinal tissue can imprint T-cell fate decisions on migrating CD4+ T cells. We found that gut-associated CD4+ T cell transcriptional profiles largely segregate by tissue location, indicating that upon leaving the gut-draining LNs, migrating cells adapt to either LP or IE compartments; IE-adapted cells followed a stepwise acquisition of an IEL profile through a distinct pre-IEL stage. The Treg program is first downregulated at this pre-IEL stage before a cytotoxic IEL program is made accessible at the chromatin level and then subsequently transcribed. Finally, we showed that while natural ThPOK downmodulation marks the pre-IEL stage, premature ThPOK loss in Tregs allows for the expression of the IEL profile before the Treg program is fully shut down. Our studies uncovered wide, tissue-specific, and stepwise chromatin and transcriptional changes in T cells upon transitioning from draining LNs to intestinal tissue sites and revealed specific roles for lineage-defining transcription factors in driving this process.
Results
Intestinal tissue sites imprint a unique program on migrating CD4+ T cells
Both peripheral Tregs and Tconv CD4+ T cells can further differentiate into CD4-IELs upon migrating to the intestinal epithelium8, 11, 15. To characterize CD4+ T cell heterogeneity in the gut along the draining lymph nodes (LNs)-tissue axis, we combined single-cell RNA-Sequencing (scRNA-Seq) with a fate-mapping approach. First, we crossed the Foxp3CreER:Rosa26lsl-tdTomato (iFoxp3Tomato) Treg-fate mapping strain16 with Zbtb7bGFP (ThPOKGFP) reporter mice17 to simultaneously study the transcriptional changes of Tregs and Tconvs as they differentiate to CD4-IELs. We administered tamoxifen to label Tregs and follow their fates (as Tomato+) in three different sites along with the LN-tissue axis: gut-draining mesenteric lymph nodes (mLN), lamina propria (LP) and epithelium (IE). We sorted Tconv-derived tomato negative or Treg-derived tomato positive CD4+ T cells from the three locations and pooled the cells per tissue at approximately a 2:1 (Tomato−: Tomato+) ratio in order to achieve a better representation of current Tregs (Tomato+ GFP+), former Tregs (Tomato+ GFP−) and non-Tregs (Tomato−). We performed droplet-based scRNA-Seq using the Chromium 10X (10X Genomics) platform on the three libraries, allowing us to explore the tissuedependent relative heterogeneities of 6,668 cells (Supplementary Fig. 1a–c). As expected from our previous studies11, 15, CD4+ T cell populations in the epithelium showed low levels of ThPOK primarily in the IE, which correlated with the acquisition of CD8α (Supplementary Fig. 1a, b).
Cells were positioned based on gene expression similarities, visualized by uniform manifold approximation and projection (UMAP),18 and assigned to 25 unbiased clusters (0–24, numbered in order by size), including a small cluster of contaminating B cells (cluster 24). The vast majority of cells were segregated by tissue, suggesting that different gut microenvironments play a major role in defining the gene expression programs (Fig. 1a–c). We defined the clusters based on their top differentially expressed genes (Fig. 1b, c, Supplementary Fig. 1d, Supplementary Table 1). Four clusters (11, 16, 19, and 20) contained cells from at least two different locations (Fig. 1a, b), and we classified them as mixed clusters (MX). Cluster 20 was defined by genes encoding mitochondria localized proteins (Atad3a, Cox7b), and genes related to ER stress such as heat shock proteins (Hspd1, Hspa9) (Supplementary Table 1), and was thus defined as “stressed” cells. Cluster 16 was characterized by the expression of genes related to cell cycle, including Cdc’s, Cdk’s, and Ccn’s and was inferred to contain cycling cells. Cells of cluster 19 expressed high levels of integrins (Itga4, Itgb1, Itgb7) as well as Vim and Actg1, suggesting these are motile cells (Supplementary Table 1, Supplementary Fig. 1e). The largest mixed tissue cluster, cluster 11, was partly defined by effector Treg genes (Izumo1r, Pdcd1, Lag3, Icos, Tnfrsf4) (Fig. 1d, Supplementary Fig. 1d, Supplementary Table 1). The three largest mLN clusters (MN_0,9,10) were defined by naïve markers (Sell, Ccr7, Klf2), as expected given this location (Supplementary Fig. 1f, Supplementary Table 1). One of the smallest mLN clusters, cluster 17, was defined by expression of Actb, whereas cluster 23 consists of multiple genes encoding interferon-induced proteins (Ifits) (Supplementary Fig. 1d, Supplementary Table 1). In general, cells isolated from mLN expressed more ribosomal protein-encoding genes in comparison to LP and IE cells, possibly due to their more naïve state and thus lower expression of cell state-defining markers (Fig. 1c). As a whole, T cells from the LP expressed genes related to activation such as those in the Jun, Dusp, and NF-κB families (Fig. 1c, d, Supplementary Fig. 1d). Clusters 3 and 21 consisted of cells with relatively higher levels of tdTomato, Ctla4, Icos, and other effector Treg genes19, 20 (Fig. 1d, Supplementary Fig. 1g, Supplementary Table 1). Cells in LP clusters 2 and 15 expressed Th1 (Il12rb2, Il18r, Stat4) and Th17 (Il17a, Il22, Rora)-associated genes, respectively (Fig. 1b, Supplementary Fig. 1d, h, Supplementary Table 1). Clusters 1 and 7 contained cells from LP that did not express the typical Th1 or Th17 signatures but had the effector phenotype. Cluster 7 was polarized towards an IEL-profile, with higher expression of Nkg7, Gzmk, and Ccl5 (Fig. 1b, e, Supplementary Fig. 1d, h, Supplementary Table 1). We considered all clusters in the IE as tdTomato-expressing Tregs (clusters 5,18), Cd8a-expressing CD4-IELs (clusters 4, 6, 13, 22), or cells that shared multiple genes with CD4-IELs but lacked Cd8a expression (clusters 8, 12, 14) (Fig. 1b, e, f, Supplementary Fig. 1i, j, Supplementary Table 1).
Figure 1. Intestinal epithelium imprints a cytotoxic program on migrating CD4+ T cells.
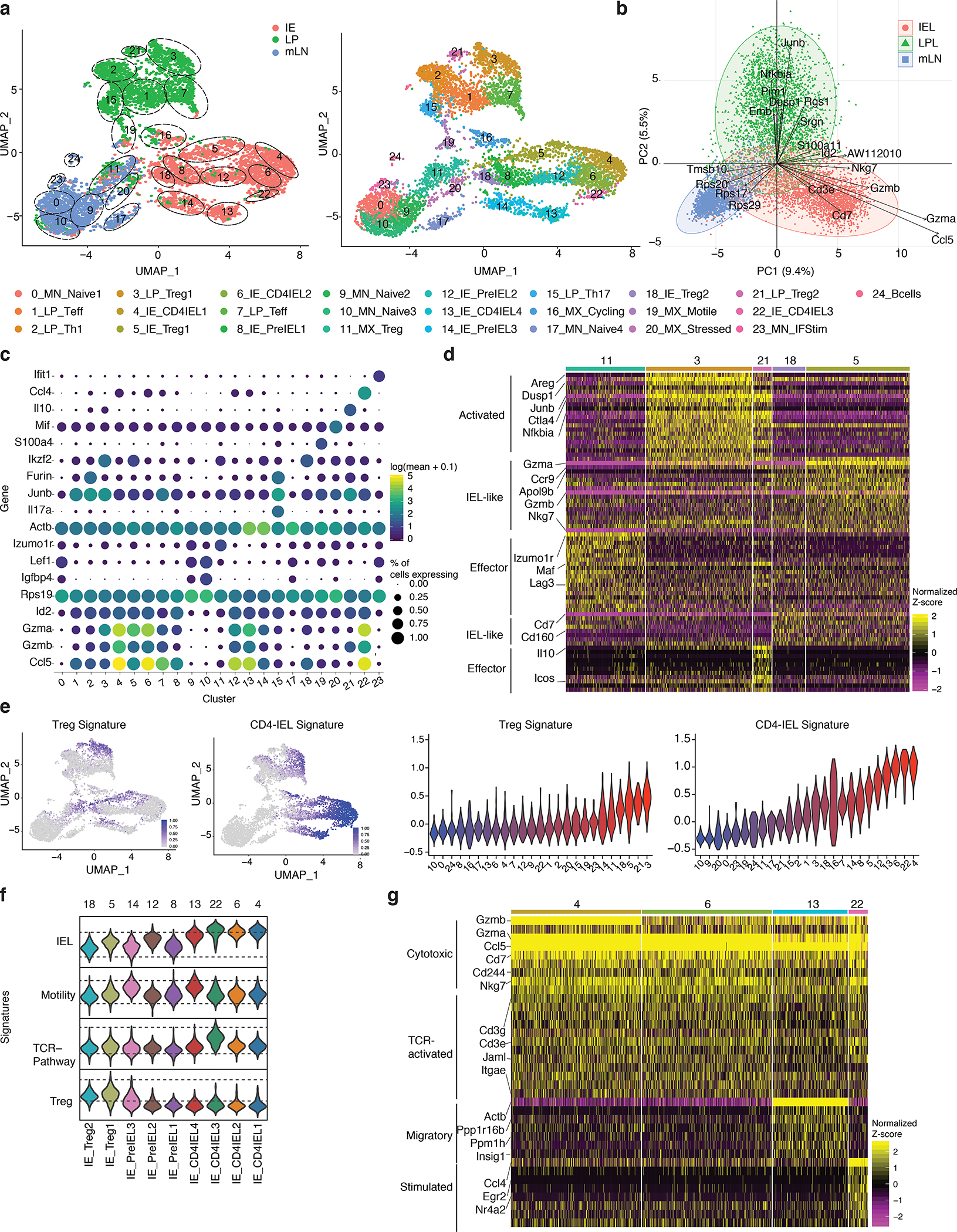
(a-g) Foxp3eGFP-Cre-ERT2xRosa26lsl-tdTomatoxZbtb7bGFP(iFoxp3TomThPOKGFP) mice were treated with tamoxifen for 10 weeks, Tomato− and Tomato+ CD4+ T cells from mesenteric lymph nodes (mLN), lamina propria (LP) and intestinal epithelium (IE) were sorted for scRNA-Seq using 10X Genomics platform from one mouse. Sorted Tomato− and Tomato+ cells were pooled in a 2:1 ratio per tissue, resulting in 3 separate libraries. (a-b) Uniform manifold approximation and projection (UMAP) representation of all sequenced cells (6,668), color coded by cluster and separated by tissue of origin (a) or mixed (b). Cluster names and numbers correspond to colors throughout the figure as indicated. (c) Principle component analysis of cells per tissue, exhibiting top 30 contributing variables variable features. (d) Heatmap of the top differentially expressed genes in Treg clusters represented by the normalized Z-score. (e) Expression levels of Treg and CD4-IEL signatures in sequenced cells (left) and quantified per cluster (right). (f) Expression levels of indicated signatures among IE clusters as indicated. (g) Heatmap of top differentially expressed genes in CD4-IEL clusters by the normalized Z-score. Significant genes were filtered based on adjusted pvalues less than 0.01 and log2fold changes higher than 0.3.
We further compared all Treg clusters (11, 3, 21, 18, 5) within the different gut-associated sites (Fig. 1d, e, Supplementary Table 1). The two Treg clusters (3, 21) present in the LP displayed a more activated profile (Ctla4, Areg, Dusp1, Junb, Nfkbia). Cluster 21 consisted of Tregs expressing Icos and Il10, while those of cluster 3 expressed higher levels of Gzma. Both IE Treg clusters (5, 18) displayed an activated and/or effector-like phenotype (Cd3e, Cd3g, Tnfrsf9, Tnfrsf18), and were also more CD8-like (Cd160, Cd7). Additionally, cells in cluster 5 expressed elevated levels of granzymes and Nkg7, suggestive of cytotoxic potential. Cells in cluster 11, the only cluster to include mLN Tregs, had lower expression of effector Treg markers and the gut homing receptor Ccr9 compared to enteric tissue Tregs (Fig. 1d). These results point to a previously unappreciated level of Treg heterogeneity between closely related, yet distinct tissues, with a skew towards the IEL/cytotoxic program within the epithelium.
We next characterized the heterogeneity of epithelial CD4+ T cells. We defined gene-expression signatures for Tregs, TCR-stimulated cells, motile cells, and CD4-IELs based on the expression profiles of the different clusters, and mapped them on IE cells (Fig. 1e, f, Supplementary Fig. 1e, j). All CD4-IELs expressed Cd8a, Cd244, Gzma, Cd7, Nkg7, Jaml as well as Ccl5 (Fig. 1g, Supplementary Fig. 1i, j). CD4-IEL clusters were separated by minor differences, including upregulation of genes in the TCR pathway in cluster 22 (Egr2 and Nr4a2) and of a motility signature in cluster 13 (Actb and Ppp1r16b), suggesting increased migratory capacity (Fig. 1e–g, Supplementary Fig. 1e, i, j, Supplementary Table 1). Finally, while three out of the four CD4-IEL clusters (4, 6 and 22) displayed equally high CD4-IEL signatures, cluster 13 was slightly less polarized towards the CD4-IEL phenotype (Fig. 1 e–g, Supplementary Fig. 1i, j). The three non-CD4-IEL and non-Treg clusters in the IE (8, 12, 14) nevertheless displayed a high level of the CD4-IEL signature (Fig. 1e, f). These clusters were characterized by the presence of cells expressing equal or slightly lower levels of IEL markers such as Itgae, Cd160, Nkg7 compared to bona fide IELs (Supplementary Fig. 1j, k, Supplementary Table 1), suggesting that they constitute a pre-IEL population. Among these pre-IEL clusters, cluster 12 was the most similar to CD4-IELs, expressing higher levels of Ccl5 and Gzmb (Fig. 1e, f, Supplementary Fig. 1j, k). Of note, tomato positive and negative cells were dispersed among all CD4-IEL clusters, suggesting that both Tregs or Tconvs can differentiate into transcriptionally similar CD4-IELs (Fig. 1e, Supplementary Fig. 1g); data corroborated by our parallel study analyzing the TCR repertoire of differentiating CD4-IELs21. Together, our results indicate that CD4+ T cells in the IE are placed on a gradient leading to the expression of a CD4-IEL signature, while cells from lamina propria are composed of distinct CD4+ T cell subsets.
To understand how the gradient of transcriptional changes culminates in tissue adaptation of CD4+ T cells within the lymphoid to non-lymphoid tissue axis, we performed pseudotime analyses that produce cell trajectory inferences using two independent, but complementary methods, Monocle3 and Slingshot (Fig. 2a). While Monocle3 constructs a minimum spanning tree utilizing the individual cells and projects it onto the UMAP embedding, Slingshot uses the cluster centers making it less sensitive to outliers. Color–coding cells along differentiation paths showed that cells making up the clusters along the edges of the LP and IE compartments in the UMAP, including all CD4-IEL clusters, are the most differentiated (higher pseudotime scores) compared to mLN cells. (Fig. 2a). The trajectory branches leading to LP versus IE clusters bifurcated within the mixed-tissue cluster 11 and no further connections were detected between them (Fig. 2a).
Figure 2. Tissue segregation precedes acquisition of an IEL profile.
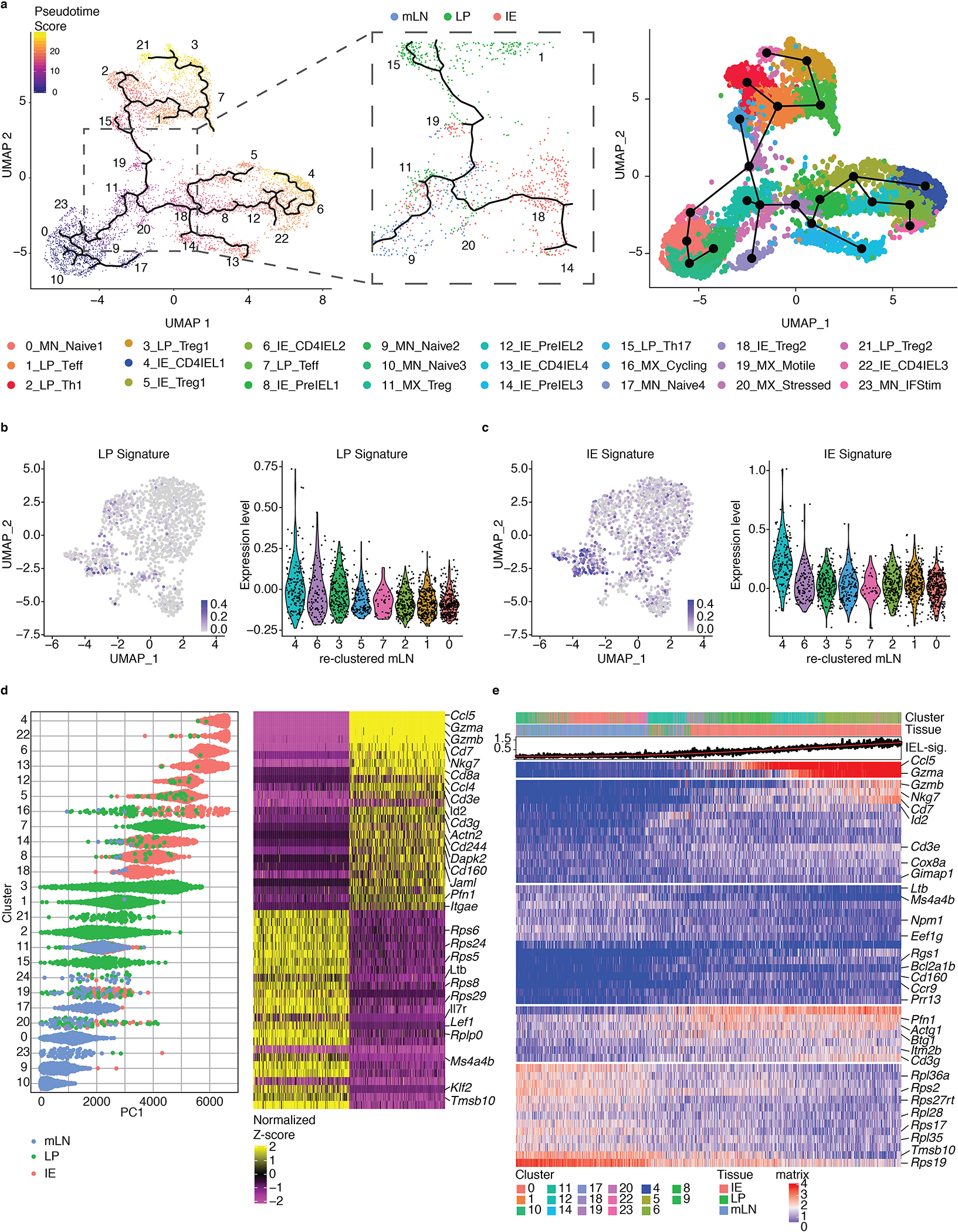
(a-e) Foxp3eGFP-Cre-ERT2xRosa26lsl-tdTomatoxZbtb7bGFP(iFoxp3TomThPOKGFP) mice were treated with tamoxifen for 10 weeks, Tomato− and Tomato+ CD4+ T cells from mesenteric lymph nodes (mLN), lamina propria (LP) and intestinal epithelium (IE) were sorted for scRNA-Seq using 10X Genomics platform. Sorted Tomato− and Tomato+ cells were pooled in a 2:1 ratio per tissue, resulting in 3 separate libraries, with 6,668 sequenced cells analyzed. (a) Pseudotime analysis of sequenced cells using Monocle3 with cells color-coded by pseudotime gradient (left) and by Slingshot with cells color-coded by cluster (right). Numbers indicate the UMAP clusters. Middle panel displays a point of bifurcation in the Monocle3 trajectory analysis, cells colored by tissue. Numbers indicate the UMAP clusters. (b, c) Expression levels overlayed on re-clustered mLN-exclusive cells (left) or quantified per cluster (right) of the LP signature (b) and the IE signature (c). (d) Cells ranked according to the first principal component (PC1) and color-coded according to tissue of origin (left) with the top 25 genes contributing to each side of the PC1 axis, and the 250 cells with the most extreme PC1 coordinates (right). (e) Gene expression levels of cells along one trajectory from a naive mLN cluster to a CD4-IEL cluster.
Next, we asked whether potential precursors of LP or IE cells within the mLN clusters could be identified. We mapped a shared “LP signature” and a shared “IE signature” onto re-clustered mLN cells and found a population (cluster 4 of re-clustered mLN, representing a mix of clusters 11 and 19 of the original UMAP) that highly express both signatures relative to other mLN cells (Fig. 2b, c, Supplementary Fig. 2a–c). While we did not observe strong biases towards LP- versus IE-signatures in these cells, they might contain “tissue-committed precursors” within the mLN, possibility further supported by the presence of LP- and IE-derived cells among them (Fig. 2b, c, Supplementary Fig. 2a–c). Additionally, since some LP-derived cells in clusters 3 and 7 express a low level of the CD4-IEL signature as shown in Fig. 1e, it is possible that a subset of CD4+ T cells may acquire IE features in the LP. Upon ordering all cells by the 1st principal component (PC1), we found that relative to IE cells and most mLN cells, LP cells distributed more diffusely, suggesting that the majority of clusters containing these cells follow a transcriptional progression distinct from cells that would end up in the IE (Fig. 2d). Moreover, ordering cells according to one of the pseudotime trajectories revealed that the mLN signature gradually decreases as the IEL signature increases, with very few LP cells from mixed-tissue cluster 11 in between (Fig. 2e). These analyses further suggest that cells with an IE profile do not first acquire a LP profile. Finally, in agreement with the UMAP, pre-IELs were positioned as intermediate stages in the trajectories from naïve CD4+ T cells of the mLN to CD4-IELs (Fig. 2a, d, e). This progression consisted of the gradual upregulation of genes such as Ccl5, granzymes, Nkg7 and Cd8a, until their IEL differentiation is complete (Fig. 2e, Supplementary Fig. 2d). These genes were also the main drivers of the IE signature described in figure 1, reinforcing that the IEL profile defines epithelial imprinting. Hence, after exiting the mesenteric lymph nodes, localization in the epithelium is largely responsible for acquisition of the IEL profile.
Coordinated transcriptional and chromatin changes during T cell adaptation to the epithelium
We next sought to couple the stepwise changes in transcription with those of chromatin accessibility as Tregs destabilized to become CD4-IELs. Our scRNA-Seq analysis using the ThPOKGFP reporter mouse revealed intermediate stages within the IE, and thus we subsequently refer to ThPOKlowCD8α− cells as pre-IELs. We performed assay for transposase-accessible chromatin followed by sequencing (ATAC-Seq) and bulk RNA-Seq on peripherally induced Tregs (iTreg; neuropilin1low Tomato+ GFPhigh CD8α−), pre-IELs derived from Tregs (Tomato+ GFPlow CD8α−), Treg-derived CD4-IELs (exTreg-IEL; Tomato+ GFPlow CD8α+) and Tconv-derived CD4-IELs (Tomato− GFPlow CD8α+) sorted from the IE of iFoxp3TomatoThPOKGFP mice (Supplementary Fig. 3a–c).
In agreement with previous ATAC-Seq data22, most accessible chromatin peaks occurred close to promoters (Supplementary Fig. 3d, e). However, among the genes with differentially accessible chromatin regions (DACR) between the populations surveyed, the majority fell within gene bodies, not promoter regions, suggesting non-random chromatin changes (Supplementary Fig. 3f). The likelihood ratio test (LRT) showed that DACRs were divided into 6 clusters; regions in clusters 1–4 gradually decreased accessibility, while those in clusters 5–6 become more accessible as Tregs differentiate into CD4-IELs (Fig. 3a). Both activation (Cd28, Dusp and NF-κB families) and naïve-related genes (Sell, Klf2, Ccr7, Il7r) contained chromatin regions which decreased accessibility as Tregs destabilized, suggesting a shift in the activation state during cell-type progression. DACRs in this progression also included Ifng and Stat4, indicating a role for interferon regulation in the Treg to IEL progression, as previously suggested12. Classic Treg (Foxp3, Il10, Cd83, Il2ra, Areg) and effector (Lag3, Pdcd1, Ctla4, Il10, Icos) genes decreased in accessibility as Tregs develop into IELs. Of note, while most Treg markers appeared in multiple clusters (1–4), Foxp3 was detected only in the 4th cluster of regions that decreased in accessibility as pre-IELs transitioned to the IEL phenotype. This suggests that regulation of other Treg-signature genes may occur before Foxp3, in agreement with previous reports19. Runx3, which increases in expression in differentiating CD4-IELs,11 has also been reported to stabilize Foxp323, and may play a role in the Treg program after Foxp3 expression shuts down. Chromatin regions in clusters 5 and 6, which began to become more accessible at the pre-IEL stage, and peaked in accessibility in IELs, included typical CD8-IEL (Runx1/2/3, Gzma, Litaf, Fasl) and homing (Ccl5, Itga4) genes. Cluster 5 contained regions that remained equally accessible in IELs derived from Tregs (exTreg-IEL) and Tconv (CD4-IEL), further suggesting that the IEL signature overrides previous lineage or stage characteristics. Chromatin regions in cluster 6, which become more accessible at the exTreg-IEL stage, were more open in CD4-IELs. In addition to regions in typical IEL genes, cluster 6 included Cd8a, suggesting that it is not until the IEL program is made accessible that Cd8a is upregulated (Fig. 3a, Supplementary Table 2). Moreover, pairwise comparisons (Fig. 3b, c, Supplementary Table 3) as well as Euclidean distance analysis (Supplementary Fig. 3g) showed that the largest changes in chromatin accessibility occurred as iTregs destabilized towards the ThPOK-low pre-IEL stage. The majority of the DACRs in this step fell on gene bodies (Fig. 3b). A relatively minor number of regions, including that of Cd8a, became more accessible as pre-IELs transitioned into CD8α-expressing exTreg-IELs, which were the most similar to CD4-IELs; however, the former did retain a number of accessible regions at Treg genes, including Foxp3, when compared to CD4-IELs (Fig. 3c). Overall, it is not until the Treg program is set to become less accessible that the chromatin of the IEL program becomes more accessible, suggesting that ThPOK loss at the pre-IEL stage, or the events that lead to it, initiate the Treg plasticity observed at the intestinal epithelium.
Figure 3: Treg program shutdown precedes IEL programming through a pre-IEL stage.
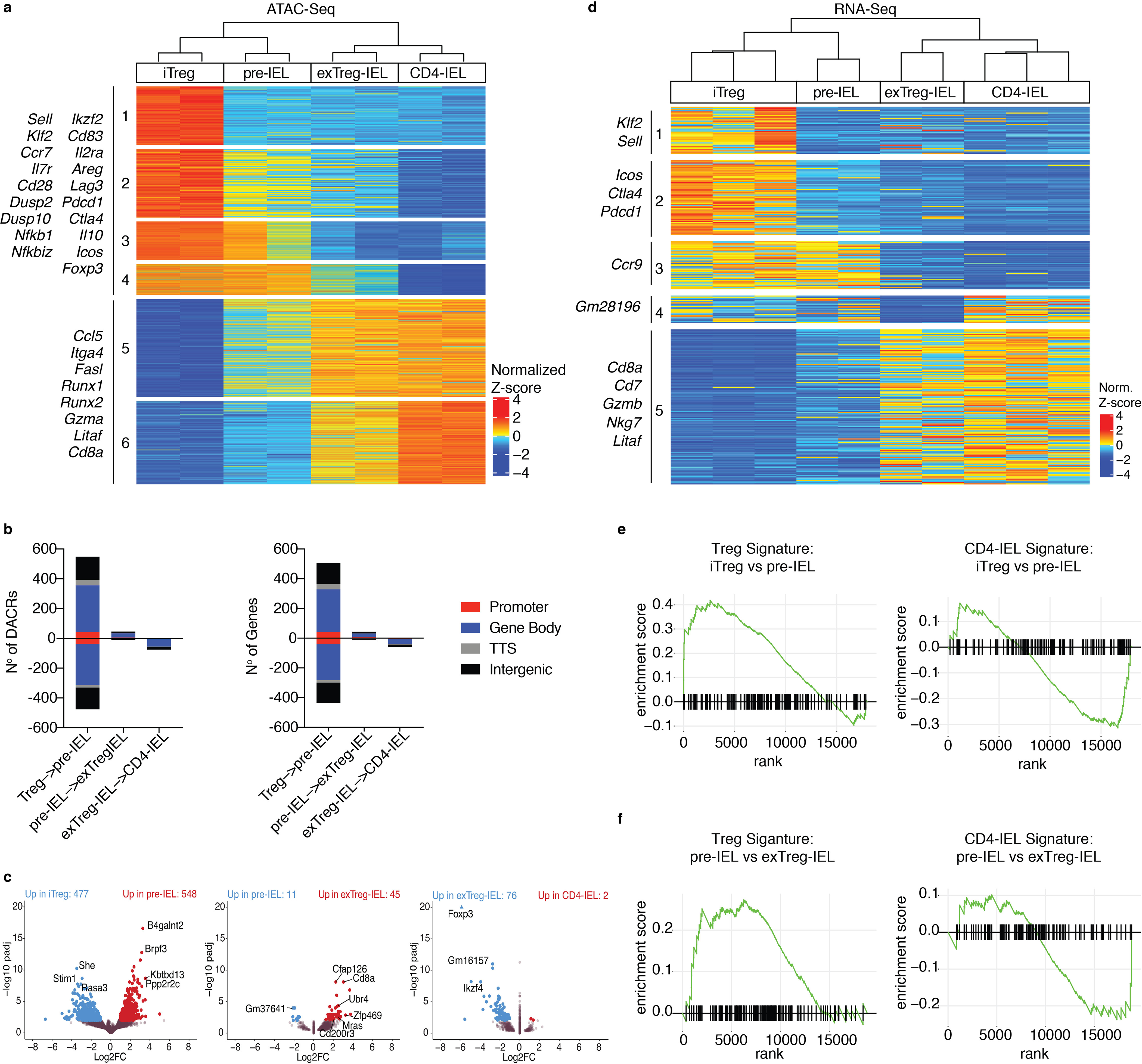
(a-f) Foxp3eGFP-Cre-ERT2xRosa26lsl-tdTomatoxZbtb7bGFP(iFoxp3TomThPOKGFP) mice were treated with tamoxifen for 10 weeks and induced Tregs (iTreg; CD4+ Tomato+ GFPHigh neuropilin-1− CD8α−), pre-IELs (CD4+ Tomato+ GFPLow CD8α−), exTreg-IELs (CD4+ Tomato+ GFPLow CD8α+), and CD4-IELs (CD4+ Tomato− GFPLow CD8α+) were sorted in bulk from the IE. Assay for transposase-accessible chromatin (ATAC) or RNA libraries were prepared followed by sequencing of indicated populations. (a) Heatmap of likelihood ratio test (LRT) of all differentially accessible chromatin regions (DACR) of indicated bulk populations. (b) Number of DACR (left) and the genes on which they are positioned on (right) between cell types in sequential progression as performed by Wald pairwise test. Colored by chromosome region as follows: 5’UTR and promoters (Promoter; red), 3’UTR with exons and introns (Gene Body; blue), transcriptional termination site (TTS; gray) and intergenic (black). (c) Volcano plots representing DACR between cell types in sequential progression as performed by Wald pairwise test. (d) Heatmap of LRT of differentially expressed genes (DEG) between indicated populations. (e, f) Gene set enrichment analysis of Treg and CD4-IEL signatures from scRNA-Seq (cluster 21 and cluster 6, respectively as shown in Figure 1) in iTreg to pre-IEL (e) and pre-IEL to exTreg-IEL (f) progressions. Significant DACR p<0.01 and significant DEG p<0.05 in RNA-Seq. Each sample for ATAC-Seq consisted of 5,000–40,000 cells from 6 or 9 pooled mice, n=2 samples. Each sample for RNA-Seq consisted of 300–800 cells per mouse, n=2–3 mice.
To define how changes in chromatin accessibility relate to differentially expressed genes in IE populations, we performed bulk RNA-Seq on the same IE subsets. We also sequenced mLN iTregs in order to compare their transcriptional profile to that of iTregs in the IE (Supplementary Fig. 3h). Comparing mLN to IE iTregs confirmed that transcriptional differences segregated by tissue, the latter expressing more activation, intestinal and CD8-associated genes (Runx3, Cd160, Cbfb, Itgae), whereas the former expressed genes associated with resting LN Tregs (Ccr7, Sell, Klf2) (Supplementary Fig. 3h, Supplementary Table 4). Differentially expressed genes between all sequenced IE populations revealed 5 clusters, highlighting the changes in transcriptional profile as Tregs convert to IELs (Fig. 3d). At the pre-IEL stage when ThPOK is downmodulated, expression of effector Treg genes (Cluster 2; Icos, Ctla4, and Pdcd1) was decreased. The clusters of genes down-regulated at the exTreg-IEL stage (Clusters 3 and 4) included multiple pseudogenes and lincRNAs, which could be implicated in cell maintenance and identity integrity, serving as a hallmark of cell differentiation at the exTreg-IEL stage. Finally, cluster 5, which is upregulated by both Treg- and Tconv-derived IELs (exTreg-IELs and CD4-IELs), includes many CD8- and IEL-associated genes (Cd8a, Cd7, Gzmb, Nkg7, Litaf). Although these genes begin to be slightly upregulated in the pre-IEL stage, they reach higher levels only when T cells become IELs, as marked by CD8α protein and gene expression (Fig. 3d, Supplementary Fig. 3b, Supplementary Table 5). Whereas the signatures of clusters 1–3 map to the Treg clusters in all tissues in the scRNA-Seq data set, the cluster 5 signature is primarily expressed by IE cells, particularly CD4-IELs (Supplementary Fig. 3i). Gene set enrichment analysis (GSEA) using Treg and IEL signatures from our scRNA-Seq (Clusters 21 and 6, respectively) revealed that expression of the Treg signature decreases as the IEL signature increases and cells progress from the Treg to pre-IEL stage (Fig. 3e). In contrast, the pre-IEL to exTreg-IEL progression displayed only minimal changes in the Treg signature (Fig. 3f), since Treg-derived pre-IELs no longer expressed most of the bona-fide Treg genes. To further elucidate the pathways leading to the down-modulation of ThPOK and subsequent differentiation of Tregs into pre-IELs in the epithelium, we performed Hallmark, Immune and Chemical and Genetic perturbation GSEAs comparing Tregs isolated from mLN versus the epithelium, and also Treg-derived pre-IELs versus IE Tregs, and obtained a curated list of pathways. In addition to differences related to Treg and CD8+ T cell programs, we found pathways related to inflammatory, costimulatory and antigen responses, interferon, and environmental factors such as TGF-β and retinoic acid (Supplementary Fig. 3j), pathways and genes previously linked to IEL development and functional differentiation8, 10, 11, 12, 13, 14, 15. Overall, the gene expression changes during IEL differentiation suggest that the IEL program begins during the pre-IEL stage, before the full Treg program is shut down but after the downregulation of ThPOK.
Our characterization using single cell and bulk approaches indicated that ThPOK downmodulation occurs at the crucial step of Treg destabilization to the pre-IEL stage. To mechanistically understand how ThPOK modulation leads to Tregs to proceed to the pre-IEL stage, we assessed possible genes modulated by ThPOK binding using ThPOK chromatin immunoprecipitation followed by sequencing (ChIP-Seq). Due to antibody and cell number limitations for this assay, we performed ThPOK ChIP-Seq on in vivo expanded splenic Tregs. We identified a binding motif, used in our downstream analysis, and found that ThPOK preferentially bound to promoter regions, followed by gene bodies (Fig. 4a, Supplementary Table 6). The mouse ThPOK binding motif we identified was similar to the published human and mouse motifs24, and upon re-analyzing a published ThPOK-ChIP dataset using in vitro activated CD4+ T cells25, we observed a 66% base pair overlap with our dataset.
Figure 4: Runx binding motifs are increasingly accessible during the iTreg to IEL progression.
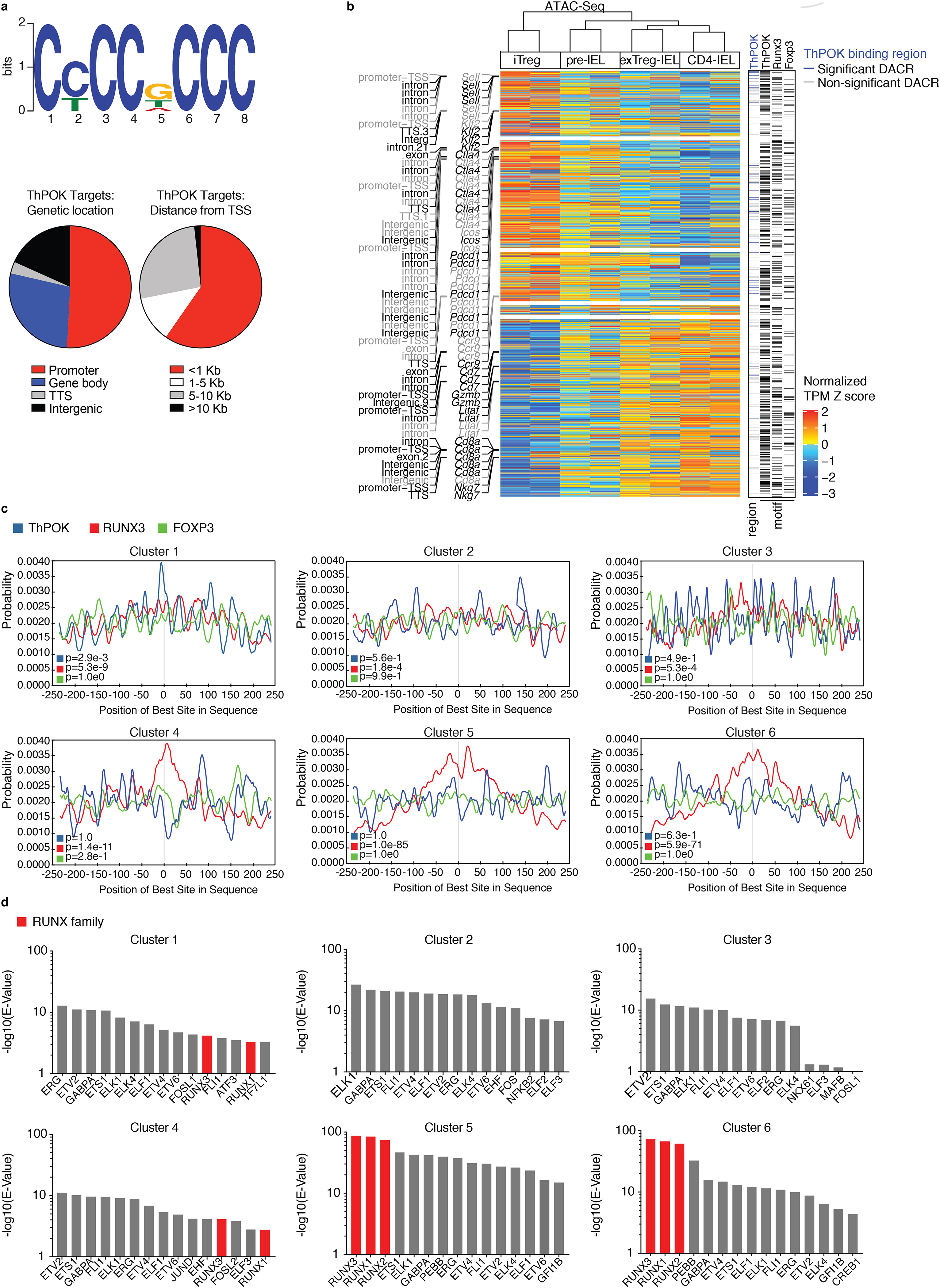
(a-d) ThPOK ChIP-Seq analysis of in vivo–expanded splenic Tregs from Foxp3RFP mice coupled with RNA- and ATAC-Seq of iTregs (CD4+ Tomato+ GFPHigh neuropilin-1−CD8α−), pre-IELs (CD4+ Tomato+ GFPLow CD8α−), exTreg-IELs (CD4+ Tomato+ GFPLow CD8α+), and CD4-IELs (CD4+ Tomato− GFPLow CD8α+) from the IE of iFoxp3TomThPOKGFP mice after tamoxifen treatment (as in Figure 3). (a) A de novo ThPOK binding motif as determined by MEME-ChIP (top) and frequency of ThPOK targets at indicated regions (bottom left) and at indicated distances from transcriptional start sites (TSS, bottom right). (b) Heatmap showing the levels of chromatin accessibility among differentially expressed genes with selected annotations of chromatin (region and gene of chromatin location, left). Black indicates regions of genes with significant differential chromatin accessibility, gray indicates no significant change in chromatin accessibility. Chromatin regions with ThPOK binding regions among significant (blue) and non-significant (grey) differentially accessible chromatin regions (DACRs) and transcription factor (TF) motifs of ThPOK, Runx3, and Foxp3 marked with black lines (right). (c, d) TF motif analysis per cluster of ATAC-Seq LRT heatmap of Figure 3a, performed by MEME-ChIP. (c) Transcription factor (TF) motif analysis of indicated TFs, collapsed around transcriptional start site. (d) Top 15 TF motifs per cluster, Runx family TF’s indicated in red. Significant differentially accessible regions padj<0.01 in ATAC-Seq and significant differently expressed genes padj<0.05 in RNA-Seq. 20×106 cells were used for ChIP-Seq (10% for input, 90% for ThPOK-binding). E-value=1.6e-142 for the ChIP-Seq ThPOK motif. Foxp3 and Runx3 binding motifs are from the JASPAR database.
To further understand the relationship between changes in transcription, chromatin accessibility, and lineage-defining transcription factors (TF), we plotted the levels of chromatin accessibility of genes that changed in transcription in the same order of the heatmap representing differences in gene expression (RNA-Seq from Fig. 3d) along with ThPOK binding regions derived from our ChIP-Seq and TF-binding motifs of ThPOK, Runx3, and Foxp3. This analysis also allowed us to visualize changes in chromatin accessibility at specific regions among differentially expressed genes (DEGs) during the iTreg to IEL transition (Fig. 4b). Genes that were downregulated after the iTreg stage, such as Sell, Klf2, and Ctla4, remained more accessible in the pre-IEL stage and their chromatin gradually closed as they differentiated into IELs. On the other hand, genes that were upregulated at the IEL stage, such as Cd7, Gzmb, and Cd8a, were more accessible already at the pre-IEL stage. ExTreg-IELs display a very similar transcriptional profile to that of Tconv-derived CD4-IELs, although they retain a small level of accessible chromatin of the Treg program (Fig. 4b). We then measured the association between the transcription and chromatin accessibility changes in the Treg to CD4-IEL progression by Pearson’s correlation coefficient. We found a positive correlation between the differences in transcription and chromatin accessibility at the promoters or at combined regions of differentially expressed genes during the Treg to IEL progression. ThPOK binding regions and the putative targets of ThPOK, Runx3, and Foxp3 were dispersed throughout the Treg to IEL progression among DEGs (Supplementary Fig. 4a). We also assessed the extent to which ThPOK binding regions identified in our ChIP-Seq overlapped with the accessible chromatin regions of DEGs. The ThPOK binding regions among DACRs were primarily observed in the first 3 clusters of regions associated with Tregs, but not with IELs: clusters 1–3 accounted for 92% of the total (24% in cluster 1, 50% in cluster 2, 18% in cluster 3) (Fig. 4b, Supplementary Fig. 4b, Supplementary Table 7,). As suggested by our previous analyses, changes in transcription and chromatin accessibility occurred primarily at the iTreg-to-pre-IEL stage. After ThPOK down-regulation, transcriptional changes were not further accompanied by alterations in chromatin accessibility (Supplementary Fig. 4c).
Next, we assessed the incidence of Runx3, ThPOK and Foxp3 binding motifs in differentially accessible chromatin regions during the iTreg to IEL progression. We separately performed this analysis on DACRs from each cluster of the ATAC-Seq heatmap (from Fig. 3a). We found that the Runx3 binding motif was centrally enriched in all clusters, with a marked increase during the acquisition of the IEL program (clusters 5 and 6, Fig. 4c). While the ThPOK binding motif was centrally enriched in differentially accessible chromatin regions only in cluster 1, it was accessible in other clusters, suggesting that ThPOK may play a role in both the expression of Treg and repression of IEL genes, as suggested by previous studies targeting ThPOK in mature CD4+ T cells8, 11, 25. The Foxp3 binding motif was not centrally enriched in any cluster, implying that it may no longer play a functional role in transitioning epithelial Tregs (Fig. 4c). We also performed TF motif analysis using MEME-ChIP26 on each cluster of the ATAC-Seq data (from Fig. 3a) (Fig. 4d). Out of the top 15 significantly enriched TF motifs per cluster, those bound by the RUNX family stood out. Accessible chromatin regions with the RUNX binding motif increased during the iTreg to IEL progression, reaching peak significance in clusters 5 and 6. ThPOK downmodulation may leave space for other TFs to bind in adjacent DNA regions, a possibility analogous to previously described roles of Runx3 to the CD8 lineage27, 28. For instance, Runx3 can bind to ThPOK silencer regions that in turn allows for the expression of a CD8 program in thymocytes29.
ThPOK downmodulation in the epithelial environment is required for IEL differentiation
We hypothesized that ThPOK and Runx3 respective modulation could initiate the transcriptional and chromatin changes that ultimately lead to CD4+ T cell differentiation towards the IEL fate. We thus asked whether disrupting ThPOK and Runx3 could affect the Treg to IEL progression. To simultaneously fate-map Tregs and abrogate their ThPOK and/or Runx3 expression, we crossed iFoxp3Tomato mice with Zbtb7bfl/fl and/or Runx3fl/fl mice30, 31 and administered tamoxifen to the iFoxp3Tomato (ΔThPOK, ΔRunx3, and ΔThPOK ΔRunx3 vs. WT) mice over 10 weeks. As previously suggested by our study using a similar strategy8, removal of ThPOK promoted the destabilization of Tregs and their acquisition of the CD4-IEL phenotype (Supplementary Fig. 5a, b). Concomitant ThPOK and Runx3 ablation in part abrogated the effect of ThPOK deletion, confirming a positive role of Runx3 in CD4-IEL differentiation11. Runx3 ablation in Tregs alone did not play a significant role in Treg stability or CD8α-acquisition (Supplementary Fig. 5a, b), implying that it acts primarily in the absence of ThPOK at this stage. In contrast to cells in the epithelium, all mLN Tregs remained stable, highlighting the finding that Treg plasticity is tissue-dependent (Supplementary Fig. 5a, b). Furthermore, we did not observe any overt signs of disease, suggesting that the remaining stable Tregs, together with the augmented CD4-IELs, were sufficient to maintain intestinal homeostasis.
One key difference between mLN and IE Tregs is that while the former mostly consists of natural Tregs (nTregs), the latter is predominantly composed of less stable peripherally-induced Tregs (iTregs)5 (Supplementary Fig. 5c). We next examined whether potential functional roles mediated by ThPOK and Runx3 on Treg plasticity were masked by differences in nTreg versus iTreg stability. We then used the model of T cell-transfer colitis in Rag1-deficient mice32 in which mice were co-transferred with naïve CD45.1 CD4+ T cells along with peripheral CD45.2 Tomato+ nTregs or iTregs (neuropilin-1 high or low, respectively) sorted from iFoxp3Tomato (ΔThPOK, ΔRunx3, ΔThPOK ΔRunx3 vs WT) mice (Supplementary Fig. 5d). In the epithelium, iTregs lost more Foxp3 expression than nTregs (p=0.0013) (Supplementary Fig. 5e). ThPOK deletion in iTregs led to an increased frequency of exTreg-IELs but without a significant loss of Foxp3-expressing cells compared to mice transferred with WT iTregs (Supplementary Fig. 5e, Fig. 5a). This result is likely related to the increase in Foxp3-CD8α double positive cells (Fig. 5b). Moreover, concomitant deletion of Runx3 and ThPOK did not result in increased accumulation of CD8α-expressing cells (Fig. 5a). Furthermore, we did not observe significant weight loss or intestinal inflammation as shown by low fecal lipocalin-2 levels (Supplementary Fig. 5f–g). Thus, both remaining Tregs and/or Tregs that differentiated into CD4-IELs were able to confer protection against colitis. Overall, while the deletion of ThPOK was not sufficient to destabilize nTregs, in iTregs it led to the development of CD4-IELs (Fig. 5a, Supplementary Fig. 5e).
Figure 5: Abrogation of ThPOK in Tregs anticipates progression to IELs at the transcriptional level.
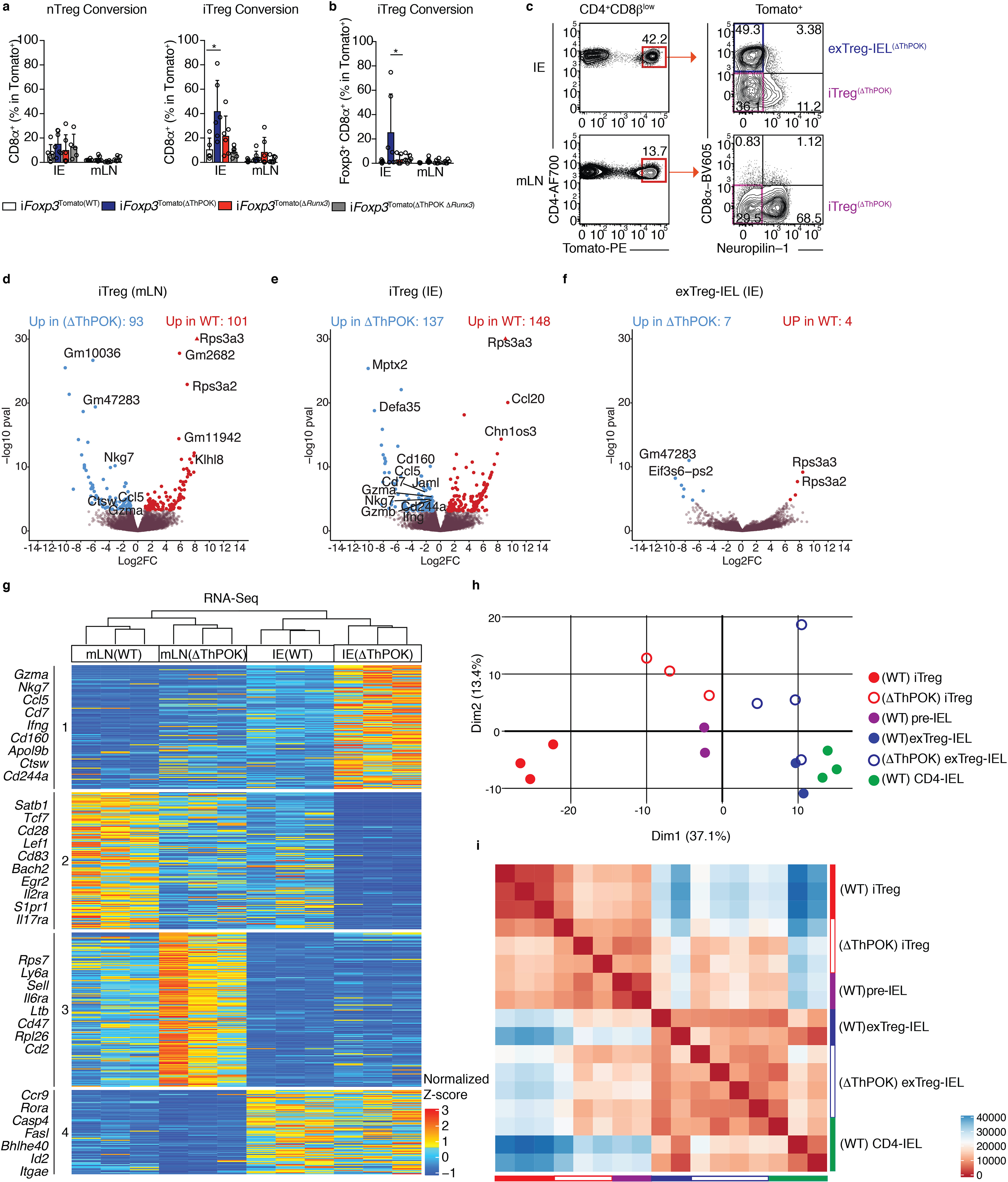
(a-b) nTreg (neuropilin-1−) or iTregs (neuropilin-1+) were sorted from spleens and mLNs of CD45.2 Zbtb7bfl/+xRunx3fl/+xRosa26|s|tdTomatoxFoxp3CreER (iFoxp3), iFoxp3xRunx3fl/+ (iFoxp3(ΔThPOK)), iFoxp3xZbtb7bfl/+xRunx3fl/fl (iFoxp3(ΔRunx3)), iFoxp3xZbtb7bfl/flxRunx3fl/fl (iFoxp3(ΔThPOK ΔRunx3)) mice after tamoxifen administration and co-transferred with CD45.1 naïve CD4+ T cells to Rag1−/− hosts. CD45.2+TCRβ+CD4+CD8β−Tomato+ lymphocytes from the IE and mLN were analyzed 10 weeks after transfer. (a) Frequencies of total surface CD8α+ cells after nTreg (left) or iTreg (right) transfer. (b) Frequency of Foxp3+CD8α+ cells after iTreg transfer. (c-i) Induced Tregs (iTregs; Tomato+CD8α−neuropilin-1−) and Treg-derived CD4-IELs (exTreg-IELs; Tomato+CD8α+) were sorted from Zbtb7bfl/flxRunx3fl/+xRosa26|s|tdTomato xFoxp3CreER (iFoxp3(ΔThPOK)) mice after 10 weeks of tamoxifen administration followed by RNA-Sequencing from IE or mLN. (c) ΔThPOK T cell populations sequenced. (d-f) Volcano representation of differentially expressed genes (DEG) between WT and ΔThPOK of the same cell type as indicated, performed by Wald pairwise comparison test, padj< 0.05. (g) Heatmap of DEGs between indicated bulk populations. Expression values represents the normalized Z-score of gene abundances (TPM). (h, i) Principle component analysis (h) and corresponding Euclidean distance analysis (i) of DEG of all WT and ΔThPOK cell types from the IE. (n=3–4 iFoxp3(ΔThPOK)). (c-i) Each sample consisted of 300–800 cells per mouse, n=2–4 mice. (a-c) Data are expressed as mean +/− SEM of individual mice (n=5–6 per genotype, 3 separate experiments). *p<0.05, **p<0.01, ***p<0.001 [one-way ANOVA and Bonferonni test].
Our sequencing data revealed that the Treg and IEL programs begin to turn off and on, respectively, at the pre-IEL stage, a transition marked by ThPOK loss. Likewise, our in vivo data showed that ablation of ThPOK at the Treg stage led to premature conversion of Tregs to CD4-IELs. To understand the underlying mechanisms set by ThPOK loss during this progression, we performed RNA and ATAC sequencing on iTregs and exTreg-IELs after tamoxifen-induced deletion of ThPOK in Tregs from iFoxp3Tomato(ΔThPOK) mice (Fig. 5c). In agreement with our in vivo data, expression of Foxp3 and related Treg genes was not affected by ThPOK deletion (Fig. 5d–f, Supplementary Table 8). Moreover, ThPOK deletion did not affect the distribution of the putative targets of ThPOK and Runx3 (Supplementary Fig. 6 a, b). Transcriptional profiling of WT and ΔThPOK mLN Tregs revealed that ThPOK loss anticipated the expression of a number of IEL-related transcripts (Ccl5, Nkg7, Ctsw) outside the gut environment (Fig. 5d, g, Supplementary Table 8, 9). ThPOK-deficient iTregs within the epithelium displayed increased expression of additional IEL genes (Ccl5, Cd7, Jaml, Cd244a, Nkg7, Cd160, Gzma, Gzmb) relative to their WT counterparts. However, only a few gene expression changes between WT and ΔThPOK exTreg-IELs were detected, an expected result given that ThPOK is naturally downregulated in WT exTreg-IELs (Fig. 5e–g, Supplementary Table 8). Principle component analysis (PCA) and Euclidean distances comparing the transcriptional profiles of all sequenced WT and ΔThPOK cell types in the IE confirmed that abrogation of ThPOK has the most effect at the iTreg stage (Fig. 5h, i). The profiles of both iTregs and exTreg-IELs in the IE of ΔThPOK mice more closely resembled the WT pre-IELs than their WT counterparts, indicating that forced ThPOK loss at the Treg stage prematurely pushes cells to differentiation to IELs before the Treg program is shut down (Fig. 5h, i). This was also suggested by the increase in Foxp3+ CD8α+ CD4+ T cells we observed in the IE of mice transferred with ΔThPOK iTregs.
Pairwise comparisons of accessible chromatin between WT and ΔThPOK counterparts revealed minimal effects of ThPOK ablation at the Treg stage, corroborating our in vivo data and suggesting that the effects of ThPOK loss primarily result in transcriptional changes relative to chromatin accessibility (Fig.6a, b, Supplementary Table 10). About 30–40% of all chromatin accessibility differences between the genotypes occur at promoter regions (Fig. 6c). Similar to the RNA-Seq analysis, PCA and corresponding Euclidean distances of the ATAC-Seq data also revealed that the ablation of ThPOK at the Treg stage results in both iTregs and exTreg-IELs from ΔThPOK mice to more closely resemble WT pre-IELs (Fig. 6d, e). Taken together, our RNA and ATAC-sequencing data show that the untimely downmodulation of ThPOK at the Treg stage results in increased chromatin accessibility and transcription of the IEL program before shutting down the Treg program, resulting in IELs that resemble destabilized Tregs at the pre-IEL stage. This suggests that the natural IE–induced downmodulation of ThPOK allows for the complete loss of the Treg program and acquisition of the IEL program in succession. Taken together, these analyses suggest that while ThPOK downmodulation initiates the acquisition of an IEL program already outside of the gut, the epithelial environment is still critical for full IEL differentiation.
Figure 6: Abrogation of ThPOK in Tregs enhances progression to IELs at the chromatin level.
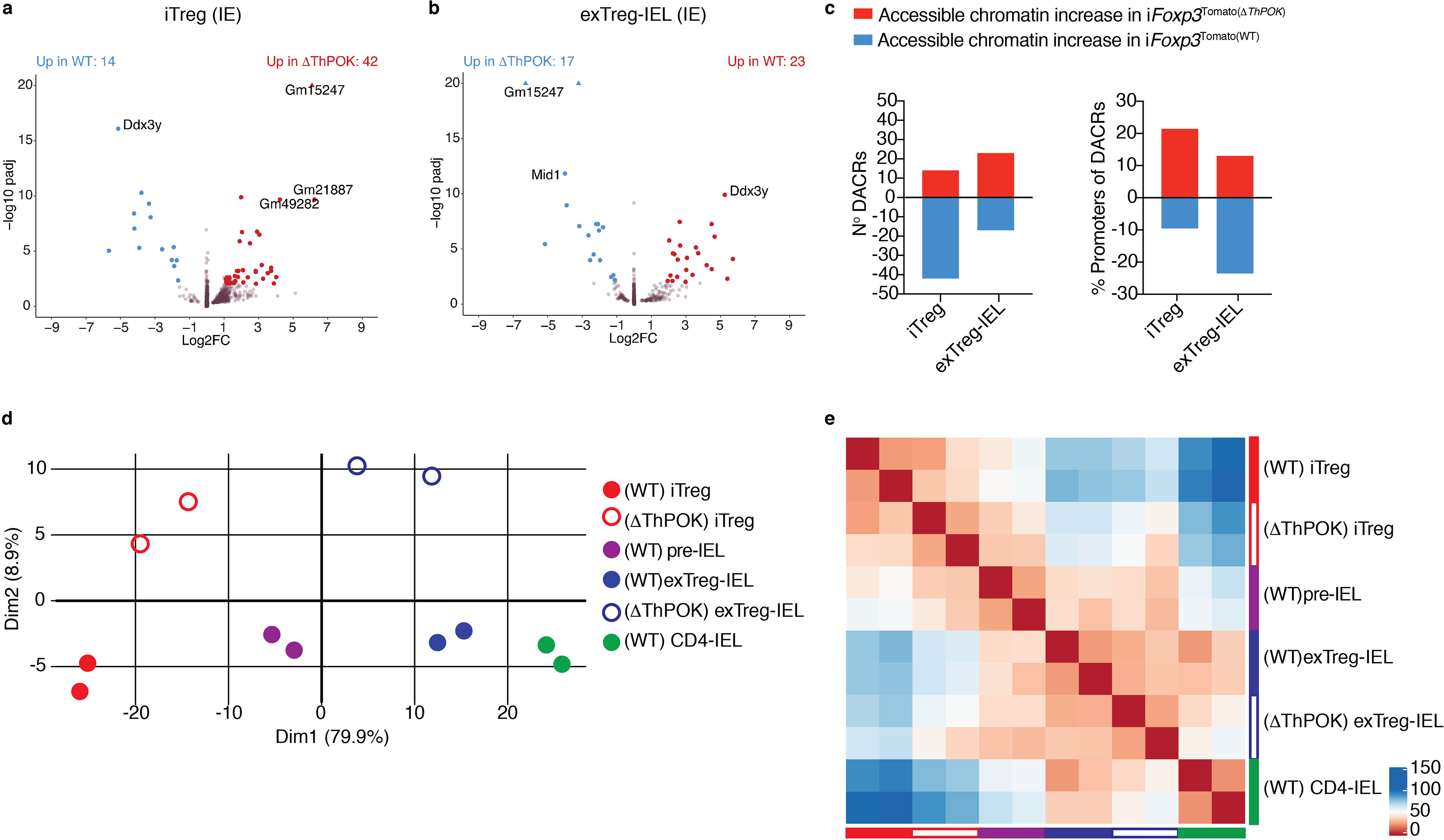
(a-e) Induced Tregs (iTregs; Tomato+CD8α−neuropilin-1−) and Treg derived CD4-IELs (exTreg-IELs; Tomato+CD8α+) were sorted from Zbtb7bfl/flxRunx3fl/+xRosa26|s|tdTomato xFoxp3CreER (iFoxp3(ΔThPOK)) mice after 10 weeks of tamoxifen administration followed by ATAC-Sequencing from IE. (a, b) Volcano representation of differentially accessible chromatin regions (DACR) between WT and ΔThPOK of the same cell type as indicated, performed by Wald pairwise comparison test, padj<0.01. (c) Data of accessible chromatin increase (red) and decrease (blue) between WT and ΔThPOK of the same cell type as indicated. Numbers of DACR (left) and percent of DACRs at promoter regions (right). (d-e) Principle component analysis (d) and corresponding Euclidean distance heatmap (e) of DACR of all WT and ΔThPOK cell types from the IE. Each sample consisted of 5,000–40,000 cells from 2 or 3 pooled mice, n=2 samples).
Discussion
We uncovered a stepwise process imprinted on T cells upon migrating to the epithelium, regardless of the subset of origin. Despite a very similar transcriptional profile between IELs derived from Tregs and conventional CD4+ T cells, exTreg-IELs retained low chromatin profile levels from their Treg precursors. This finding corroborates a previous study showing that chromatin accessibility changes and transcription did not always match as Tregs adapted to different tissues33. Nevertheless, our identification of a pre-IEL signature, marked by the loss of key features of conventional CD4 and/or Treg programs, suggests that during tissue adaptation, a cell must first shut down transcriptional programs in place (or decouple TFs bound to its DNA targets) that may prevent tissue imprinting, in agreement with previous studies describing the role of Runx3 in the CD8-lineage or tissue-resident memory T cells (TRM)27, 28. A recent study also described a stepwise process taken by Tregs, in which peripheral precursors gradually acquire chromatin accessibility and reprograming towards nonlymphoid-tissue Tregs, and the transcription factor BATF was identified as the main driver of the molecular tissue programing34. These observations parallel our findings regarding epithelial imprinting on Tregs; while Runx3 and ThPOK seemed largely dispensable for Treg function in the periphery or tissues, ThPOK downmodulation was a driver of induced Treg plasticity towards the IEL fate at the epithelium. The inflammatory potential of IE CD4+ T cells is dampened upon loss of ThPOK or exacerbated in the absence of Runx311. Conversely, the anti-inflammatory function of CD4-IELs depends on ThPOK loss and acquisition of Runx38. Upon intestinal infection by Citrobacter rodentium, the inflammatory potential of Runx3-deficient CD4+ T cells was enhanced while ThPOK loss was impaired11. Nevertheless, IEL hallmarks are maintained after infection with Listeria monocytogenes expressing an IEL cognate antigen21. Because our studies presented here, and our recent TCR repertoire analysis of intraepithelial CD4+ T cells performed in parallel21, revealed that Treg and Tconv derived CD4-IELs are very transcriptionally similar, it is plausible that they share a similar function.
Our previous studies pointed to an important role for ThPOK in Treg stability as assessed by the continuous expression of Foxp3 despite ThPOK ablation8. If so, prolonged ThPOK inactivation in Tregs should result in overt autoimmunity. Our in vivo experiments presented here refute this possibility, as Treg suppressive function appeared unaltered, while Foxp3 expression was only modestly affected. Similarly, while ThPOK deletion in activated CD4+ T cells does not affect viral clearance or the expansion of antigen-specific cells following initial LCMV infection, it plays a role in the regulation of CD4+ T cell differentiation, particularly towards memory or Th1 cells, which share some expression profile with IFNγ-producing CD4-IELs25. One caveat of broad conclusions regarding the role of ThPOK in Treg programming is that our sequencing analyses revealed that Foxp3 is one of the last canonical Treg genes to be downmodulated during the IEL transition, which reinforces previous studies suggesting that Foxp3 is a late-acting transcription factor which binds to pre-established accessible enhancer regions35. Therefore, it is plausible that the Treg program may be partially modulated independently of Foxp3; in the case of tissue adaptation events addressed here, this may preclude the use of Foxp3 as a Treg marker without considering the entire Treg profile. Additionally, it is possible that ThPOK acts in conjunction with other TFs, including Runx323, 36, 37, to regulate Treg function. By inactivating both ThPOK and its homolog LRF in differentiated Tregs, a previous study concluded that these transcription factors redundantly support Foxp3 function38. Finally, the differential methylation patterns in the regulatory regions on the Foxp3 locus between natural and induced Tregs39 raises the possibility that they display distinct dependence on ThPOK, as the latter are more prone to losing Foxp3 expression in vivo. Previous work has helped to elucidate environmental cues leading to ThPOK downmodulation, including T-bet inducing factors such as IFNγ, IL-27, and IL-15, in combination with Treg-inducing factors retinoic acid and TGF-β, as well as microbiota-derived metabolites such as ligands for aryl-hydrocarbon receptor11, 12, 15. Moreover, our T cell receptor repertoire analyses showed that upon migration to epithelium, peripheral CD4+ T cells undergo a “bottleneck” and only those with selected TCRs can expand as pre-IELs and subsequently into CD4-IELs 21, highlighting the role of local antigen stimulation via TCR a major factor contributing to differentiation of peripheral CD4+ T cells into ThPOKlow IELs. However, it remains to be defined how these cues, and additional factors yet to be identified, synergize to trigger the consorted molecular mechanisms described here.
Our studies point to a major role of specific intestinal layers in the functional programming of migrating T cells. Indeed, lymphocytes are thought to display common features inherent to the tissue in which they reside20, 33, 40, 41. Migrating immune cells must be able to readily adapt to their new environment at the transcriptional level, although the type and speed of tissue-imprinting will vary with the tissue and the immune cell of interest19, 33, 40, 41, 42, 43, 44. For instance, it was proposed that while Tregs from skin and colon retain a common Treg program, they concomitantly acquire tissue-specific profiles20. Conversely, studies characterizing TRM, which share several characteristics with IEL subsets, also observed a progressive acquisition of a tissue-specific transcriptional profile, distinct of circulating memory cells41. Our studies add to this line of investigation by defining a relationship between two distinct but adjacent intestinal compartments and their draining LNs; peripheral T cells are likely to either go to the intestinal lamina propria and reside there as distinct CD4+ subsets or proceed to the epithelial compartment without being imprinted with a LP signature but rather become polarized with an IEL profile. Our inter-tissue scRNA-Seq analysis points to a specific subset of mLN cells with LP and IE-precursor characteristics, although cell transfer or fate-mapping approaches will be required to confirm it. These observations point to important distinctions between these two closely juxtaposed intestinal layers and may call for specific approaches to target cells located in each site. Additionally, they raise the possibility that similar reciprocal trajectories take place in other sites, such as the skin, between dermal and epidermal compartments42, or the lung, between the stroma and epithelium43, 44. We conclude that T cells undergo wide, tissue-specific and stepwise chromatin and transcriptional changes from lymphoid to non-lymphoid tissue sites, highlighting the existence of a late stage of lymphocyte plasticity likely required for the maintenance of tissue homeostasis.
Methods
Animals
Animal care and experimentation were consistent with the NIH guidelines and were approved by the Institutional Animal Care and Use Committee at the Rockefeller University. Rag1−/− (002216), Zbtb7beGFP(027663), Zbtb7bfl/fl30(009369), Rosa26|s|tdTomato(007914), Foxp3eGFP-Cre-ERT2(016961), Foxp3IRES-mRFP(008374), Cd4Cre-ERT2(022356) and B6.SJL-PtprcaPepcb(002014) mice were purchased from the Jackson Laboratories and maintained in our facilities. Runx3fl/fl (008773) mice were kindly provided by T. Egawa (Wash. U.). Several of these lines were interbred in our facility to obtain the final strains described in the text. Genotyping was performed according to the protocols established for the respective strains by Jackson Laboratories. Mice were maintained at the Rockefeller University animal facilities under specific pathogen-free (SPF) conditions.
Antibodies and flow cytometry analysis
Fluorescent dye–conjugated antibodies were purchased from BD Biosciences, Biolegend or Ebioscience (Thermofisher). The following clones were used: anti-CD45.1, A20; anti-Foxp3, FJK-16s; anti-CD4, RM4–5; anti-CD45.2, 104; anti-CD8α, 53–6.7; anti-CD8β, YTS 156.7.7; anti-CD44, IM7; anti-CD45, 30-F11; anti-CD62L, MEL-14; G8.8; anti-TCRβ, H57–597; anti-TCRγδ, eBioG23; anti-CD25, PC61.5;. Live/dead fixable dye Aqua (ThermoFisher Scientific) was used according to manufacturer’s instructions. Intracellular staining of Foxp3 was conducted using Foxp3 Mouse Regulatory T Cell Staining Kit (eBioscience, USA). Flow cytometry data was acquired on a LSR-II flow cytometer (Becton Dickinson, USA) and analyzed using FlowJo software package (Tri-Star, USA). Anti-ThPOK ChIP antibody was kindly provided by T. Egawa (Wash. U.).
Isolation of intestinal T cells
Intraepithelial and lamina propria lymphocytes were isolated as previously described 11, 14. Briefly, small intestines were harvested and washed in PBS and 1mM dithiothreitol (DTT) followed by 30 mM EDTA. Intraepithelial cells were recovered from the supernatant of DTT and EDTA washes and mononuclear cells were isolated by gradient centrifugation using Percoll. Lamina propria lymphocytes were obtained after collagenase digestion of the tissue. Single-cell suspensions were then stained with fluorescently labeled antibodies for 20min at 4°C prior to downstream flow cytometry (analysis or sorting) as specified in figure legends.
Tamoxifen treatment
For in vivo treatment, mice were administered by intragastric gavage with 5 mg of tamoxifen (Sigma) dissolved in corn oil (Sigma) and 10% ethanol at 50 mg/ml. Tamoxifen was administered to mice starting at 6–7 weeks old, 4 times in the first week and then 2 times every week (3 days apart) every other week for 8–10 weeks. To label cells for transfer, tamoxifen was administered only twice, 2 days apart, 2 days prior to sorting. Two days after transfer into Rag1−/− mice, animals were administered 100μL of 10mg/mL Tamoxifen intraperitonially.
Cell sorting
Lymphocytes were sorted on a FACS Aria II instrument as indicated in the figure legends.
Cell transfer
Induced (neuropilin-1−) and natural (neuropilin-1+) Tregs (CD45.2+TCRβ+CD4+CD8−CD25+Tomato+) from spleen and mLN of donor CD45.2 mice were sorted 2 days after mice were given 10mg of tamoxifen (5mgday for 2 days), by intragastric gavage, as described. Rag1−/− hosts were then intravenously co-transferred with 100,000 CD45.2+ Tregs and 400,000 naïve CD4+ T cells (CD45.1+TCRβ+CD4+CD8−CD62LhighCD44low) sorted from CD45.1 donor mice. Body weight and fecal lipocalin-2 levels were monitored until terminal analysis.
ELISA for Lipocalin-2
Lipocalin-2 was analyzed by using Lcn-2 ELISA kit (R&D, MN) as described45.
ATAC-Sequencing
ATAC-Seq was performed as previously described22 on 5,000–40,000 FACS-purified cells from 2–9 mice. In brief, cells were lysed in lysis buffer for 1 minute and transposed with Tagment DNA Enzyme 1 (Illumina) for 30 minutes. DNA was cleaned up using a MinElute DNA purification Kit (Qiagen), followed by barcoding and library preparation by the Nextera DNA Library preparation kit (Illumina) according to manufacturer’s guidelines and sequenced on an Illumina NextSeq500.
ChIP-Sequencing
Cells were fixed in 1% formaldehyde for 20min, quenched with 0.15M glycine and washed in PBS. 2×106 were then sorted and lysed for 30min at 4°C. Cells were then sonicated for 22 minutes at 30 seconds on/off using the Bioruptor sonicator (Diagenode) and spun down. 10% was frozen for input, the remaining 90% was incubated with anti-ThPOK antibody bound to goat anti-rabbit M280 magnetic beads (Invitrogen) overnight at 4°C prior to washing with RIPA buffer and overnight decrosslinking at 65°C. DNA was then eluted off beads into TE buffer and, along with input, purified using the Zymogen DNA clean & concentrator kit. DNA was sequenced using NextSeq2500. To obtain Tregs, Foxp3RFP reporter mice were injected with IL-2/αIL-2 complex as previously described for 3 consecutive days for Treg expansion46, 47. CD4+ T cells were isolated from the spleen and mLNs and sorted as Thy1+TCRβ+CD4+CD8α−RFP+.
Bulk RNA-Sequencing
Sorted cells (300–800) were lysed in TCL buffer (Qiagen, 1031576) supplemented with 1% β-mercaptoethanol. RNA was isolated using RNAClean XP beads (Agentcourt, A63987), reversibly transcribed, and amplified as described48. Uniquely barcoded libraries were prepared using Nextera XT kit (Illumina) following manufacturer’s instructions. Sequencing was performed on an Illumina NextSeq550.
Single cell RNA-Seq library preparation
Lymphocytes were sorted, counted for viability and immediately subjected to library preparation. The scRNA-Seq library was prepared using the 10x Single Cell Chromium system, according to the manufacturer’s instructions at the Genomics core of Rockefeller University and was sequenced on an Illumina NextSeq550 to a minimum sequencing depth of 50,000 reads per cell using read lengths of 26 bp read 1, 8 bp i7 index, 98 bp read 2.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Data was analyzed by applying Student’s t test, Mann-Whitney test, one-way ANOVA with Tukey post-test, Log-rank test or Kruskal-Wallis with Dunns post-test whenever necessary. For analysis of non-parametric histological scores, a Mann-Whitney test was used. A P value of less than 0.05 was considered significant. * P<0.05, ** P<0.01, *** P<0.001.
Single Cell RNA-Seq analysis
The scRNA-Seq fastq files originated from the Chromium 10x libraries were processed with Cell Ranger (v. 3.0.2) for cell barcode aggregation, genome alignment, and UMI transcript quantification. The matrix of gene counts was analyzed with the Seurat (v. 3.1.2) package for the R environment 49. For downstream analysis, the top 3000 genes containing the highest standardized variance and expression were pre-selected and used for dimensionality reduction by Principal Component Analysis. Next, cell clusters were detected by constructing a Shared Nearest Neighbor (SNN) graph. The detected cell clusters were visualized by performing Uniform Manifold Approximation and Projection (UMAP) algorithm 50. Differentially expressed genes among clusters were determined by the Wilcoxon Rank Sum test and genes containing adjusted p-values smaller than 0.01 were considered significant. Heatmap visualization were constructed either using the ComplexHeatmap package or the convenience function implemented in Seurat 49, 51.
Pseudotime trajectory analyses
For pseudotime trajectory analysis, cells were first ranked accordingly to the first and second PCA dimension coordinates. Next we converted the Seurat dataset into a ‘SingleCellExperiment’ object and used as input for the Slingshot and Monocle3 packages for the R enviroment52, 53. Both Slingshot and Monocle3 algorithm were performed by setting the cluster 10 (naïve cells from mLN) as the root. To visualize the top genes correlated to the slingshot pseudotime trajectory we performed a generalized additive model (GAM) algorithm with a loess fit for the pseudotime using the gam package for R https://cran.rproject.org/web/packages/gam/index.html.
Bulk RNA-Seq data analysis
The raw fastq sequencing files were processed by using the mouse transcriptome provided by the gencode consortium (v. M21), with kallisto (v. 0.46.0) to calculate the transcript abundances 54, 55[PMID: 30357393]. Next, the abundance files were submitted to the sleuth (v. 0.30.0) pipeline based on R, for transcript abundance normalization, gene expression analysis and statistics56, 57. The quantified transcripts were combined into genes for the downstream analysis. Batch effects were evaluated through principal component analysis and removed with Limma58. To capture significantly expressed genes we performed a likelihood ratio test between a null model and our experimental design using an adjusted p-value of 0.05 as threshold for downstream analysis. The selected features were clustered using the k-means algorithm and represented as a heatmap to define group gene signatures. Pairwise comparisons between groups were performed using the Wald-test and significant genes were considered for downstream analysis by having an adjusted p-value smaller than 0.05 and a log2 fold-change of 1. Pre-ranked gene set enrichment analysis (GSEA) were performed with the fgsea package59 by comparing gene lists sorted by their log2 fold-change with gene signatures from the clusters 21 (Activated-Treg) and 6 (CD4-IEL) of the single cell dataset (10x Genomics) or gene sets from the MSigDB collections; h: hallmark gene sets, c2.cpg: chemical and genetic pertubations, c5.go: full gene ontology.
ATAC-Seq analysis
The raw bulk ATAC-Seq files were processed using the ENCODE-DCC pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline) automated through Cromwell (https://github.com/broadinstitute/cromwell). Shortly, adapter sequences were trimmed (Cutadapt) prior to genome mapping (Bowtie2)60 and filtered subsequently filtered (Samtools)61. Next, sequencing enriched regions, associated to chromatin accessible peaks were called (MACS2) and filtered (IDR) by running Irreproducible Discovery Rate algorithm with a 10% threshold, meaning 10% chance of being an irreproducible peak. Later, filtered peak regions were annotated accordingly to the mouse transcriptome (gencode v. M21) and Foxp3 (MA0850.1), Runx3 (MA0684.1) and Zbtb7b (as suggested by our ChIP) transcriptional factors motifs were used to determine potential binding sites by using HOMER software toolset62, 63. Comprehensive motif screening was performed with MEME-ChIP26 by using promoter originated peaks and, length-normalized reads to a fixed window of 500bp. Enriched transcriptional factor motifs on ATAC-Seq peaks were detected with Centrimo64 by providing known Human Runx3 (MA0684.1) and Foxp3 (MA0850.1) as published in JASPAR 2020 62, 65. As indicative of the presence of Zbtb7b (Thpok) we used the enriched motif detected in our ThPOK ChIP-Seq peaks.
ChIP-Seq analysis
The raw bulk ChIP-Seq files were processed using the ENCODE-DCC pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2) automated through Cromwell (https://github.com/broadinstitute/cromwell). Shortly, adapter sequences were trimmed (Cutadapt) prior to genome mapping (BWA) and filtered subsequently (Samtools)61. Next, sequencing enriched regions, associated to chromatin accessible peaks were called (SPP) and filtered (IDR) by running Irreproducible Discovery Rate algorithm with a 5% threshold, meaning 5% chance of being an irreproducible peak66, 67. Later, filtered peak regions were annotated by the HOMER software suite of tools63. Optimal peak regions with a peak score higher than 20 and located at the promoter site were later used for the construction of Zbtb7b motif. Denovo motif discovery was performed by MEME-ChIP26.
Data availability
All generated raw and processed sequencing data for this study (all main figures) can be accessed on GEO: GSE146292, Bioproject: PRJNA610060.
Supplementary Material
Acknowledgements
We are grateful to A. Rogoz, and S. Gonzalez for exceptional animal care, mouse colony management and genotyping and the Rockefeller University employees for continuous assistance. We thank K. Gordon and K. Chhosphel for assistance with cell sorting. We thank C. Zhao and the entire Genomics Core of Rockefeller University for library preparation for 10X Genomics and assistance with all sequencing platforms used in this paper. We are grateful to U. Schaefer (The Rockefeller University) for help with ChIP protocols and guidance and S. Larsen for help with the ATAC-Seq. We thank B. Reis and G. Victora for suggestions and critical reading of the manuscript, and all the members of the Mucida lab for fruitful discussions.
Funding
This work was supported by the Leona M. and Harry B. Helmsley Charitable Trust, the Black Family Metastasis Center, the Kavli Foundation, the Burroughs Wellcome Fund PATH Award, and National Institute of Health grants AI144827, DK113375 and DK093674 (D.M.).
Footnotes
Competing financial interests
The authors declare no conflict of interest.
References
- 1.Olivares-Villagomez D & Van Kaer L Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends in immunology 39, 264–275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDonald BD, Jabri B & Bendelac A Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nature reviews. Immunology 18, 514–525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vantourout P & Hayday A Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nature reviews. Immunology 13, 88–100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mucida D et al. Oral tolerance in the absence of naturally occurring Tregs. The Journal of clinical investigation 115, 1923–1933 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bilate AM & Lafaille JJ Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annual review of immunology 30, 733–758 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Hadis U et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Sujino T et al. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 352, 1581–1586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atarashi K et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atarashi K et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I & Mucida D Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nature immunology 14, 271–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reis BS, Hoytema van Konijnenburg DP, Grivennikov SI & Mucida D Transcription Factor T-bet Regulates Intraepithelial Lymphocyte Functional Maturation. Immunity 41, 244–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konkel JE et al. Control of the development of CD8alphaalpha+ intestinal intraepithelial lymphocytes by TGF-beta. Nature immunology 12, 312–319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilate AM et al. Tissue-specific emergence of regulatory and intraepithelial T cells from a clonal T cell precursor. Sci Immunol 1, eaaf7471 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mucida D et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nature immunology 14, 281–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubtsov YP et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Setoguchi R et al. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science 319, 822–825 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nature biotechnology (2018). [DOI] [PubMed] [Google Scholar]
- 19.Zemmour D et al. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nature immunology 19, 291–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miragaia RJ et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 50, 493–504 e497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bilate AM et al. T Cell Receptor Is Required for Differentiation, but Not Maintenance, of Intestinal CD4(+) Intraepithelial Lymphocytes. Immunity 53, 1001–1014 e1020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klunker S et al. Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. J Exp Med 206, 2701–2715 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Badis G et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciucci T et al. The Emergence and Functional Fitness of Memory CD4(+) T Cells Require the Transcription Factor Thpok. Immunity 50, 91–105.e104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machanick P & Bailey TL MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milner JJ et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shan Q et al. The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nature immunology 18, 931–939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muroi S et al. Cascading suppression of transcriptional silencers by ThPOK seals helper T cell fate. Nature immunology 9, 1113–1121 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Egawa T & Littman DR ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nature immunology 9, 1131–1139 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naoe Y et al. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med 204, 1749–1755 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powrie F, Leach MW, Mauze S, Caddle LB & Coffman RL Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. International immunology 5, 1461–1471 (1993). [DOI] [PubMed] [Google Scholar]
- 33.DiSpirito JR et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delacher M et al. Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF. Immunity (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samstein RM et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruno L et al. Runx proteins regulate Foxp3 expression. J Exp Med 206, 2329–2337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitoh A et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31, 609–620 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Carpenter AC et al. Control of Regulatory T Cell Differentiation by the Transcription Factors Thpok and LRF. Journal of immunology 199, 1716–1728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanamori M, Nakatsukasa H, Okada M, Lu Q & Yoshimura A Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends in immunology 37, 803–811 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Masopust D & Soerens AG Tissue-Resident T Cells and Other Resident Leukocytes. Annual review of immunology 37, 521–546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fonseca R et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nature immunology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gebhardt T et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477, 216–219 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Ricardo-Gonzalez RR et al. Tissue signals imprint ILC2 identity with anticipatory function. Nature immunology 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen M et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 175, 1031–1044 e1018 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Chassaing B et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PloS one 7, e44328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyman O, Kovar M, Rubinstein MP, Surh CD & Sprent J Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Webster KE et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 206, 751–760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trombetta JJ et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Current protocols in molecular biology 107, 4 22 21–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McInnes L, Healy J, Saul N & Grossberger L UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861 (2018). [Google Scholar]
- 51.Gu Z, Eils R & Schlesner M Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Street K et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic acids research 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pimentel H, Bray NL, Puente S, Melsted P & Pachter L Differential analysis of RNA-seq incorporating quantification uncertainty. Nature methods 14, 687–690 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Team, R.C. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; (2020). [Google Scholar]
- 58.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sergushichev SA An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv (2016). [Google Scholar]
- 60.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fornes O et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic acids research 48, D87–D92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bailey TL & Machanick P Inferring direct DNA binding from ChIP-seq. Nucleic acids research 40, e128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jolma A et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Kharchenko PV, Tolstorukov MY & Park PJ Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26, 1351–1359 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Q, Brown JB, Huang H & Bickel PJ Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All generated raw and processed sequencing data for this study (all main figures) can be accessed on GEO: GSE146292, Bioproject: PRJNA610060.