

Abstract
Objective
The present work was undertaken to study the genetic contribution to the start of Alzheimer's disease (AD) with amyloid and tau biomarkers in cognitively intact older identical twins.
Methods
We studied in 96 monozygotic twin‐pairs relationships between amyloid‐beta (Aβ) aggregation as measured by the Aβ1–42/1–40 ratio in cerebrospinal fluid (CSF; n = 126) and positron emission tomography (PET, n = 194), and CSF markers for Aβ production (beta‐secretase 1, Aβ1–40, and Aβ1–38) and CSF tau. Associations among markers were tested with generalized estimating equations including a random effect for twin status, adjusted for age, gender, and apolipoprotein E ε4 genotype. We used twin analyses to determine relative contributions of genetic and/or environmental factors to AD pathophysiological processes.
Results
Twenty‐seven individuals (14%) had an abnormal amyloid PET, and 14 twin‐pairs (15%) showed discordant amyloid PET scans. Within twin‐pairs, Aβ production markers and total‐tau (t‐tau) levels strongly correlated (r range = 0.73–0.86, all p < 0.0001), and Aβ aggregation markers and 181‐phosphorylated‐tau (p‐tau) levels correlated moderately strongly (r range = 0.50–0.64, all p < 0.0001). Cross‐twin cross‐trait analysis showed that Aβ1–38 in one twin correlated with Aβ1–42/1–40 ratios, and t‐tau and p‐tau levels in their cotwins (r range = −0.28 to 0.58, all p < .007). Within‐pair differences in Aβ production markers related to differences in tau levels (r range = 0.49–0.61, all p < 0.0001). Twin discordance analyses suggest that Aβ production and tau levels show coordinated increases in very early AD.
Interpretation
Our results suggest a substantial genetic/shared environmental background contributes to both Aβ and tau increases, suggesting that modulation of environmental risk factors may aid in delaying the onset of AD pathophysiological processes. ANN NEUROL 2021;89:987–1000
Aggregation of amyloid‐beta 1–42 (Aβ1–42) in the brain can start up to 20 years before the onset of dementia, 1 followed by tau pathology and cognitive decline. 2 The mechanisms leading to Aβ aggregation and tau pathology are not fully understood, and such knowledge is crucial for the development of primary and secondary prevention strategies. Therefore, it is important to understand the contribution of genetic and environmental factors in the early development of the disease, when biomarkers for Aβ and tau are changing and cognition is still intact. Twin studies suggested that genetic factors may explain 58 to 79% of the variance in Alzheimer's disease (AD)‐type dementia, 3 but it remains largely unknown whether this is also the case for AD biomarkers in elderly cognitively normal individuals. 4
In autosomal dominant variants of AD (ADAD), Aβ aggregation is associated with increased Aβ production. In sporadic AD, however, impaired clearance is suggested to drive the disease. 5 , 6 Nevertheless, induced pluripotent stem cell (iPSC) models in sporadic AD suggest that increased Aβ production and tau secretion are also involved. 7 In people, higher cerebrospinal fluid (CSF) levels of beta‐secretase 1 (BACE1), the rate‐limiting enzyme in Aβ production, have been associated with higher CSF concentrations of Aβ peptides, 8 and subsequent aggregation of Aβ1–42 in individuals with initially normal Aβ CSF levels. 9
It is generally assumed that tau follows Aβ pathology, 10 and CSF levels of total‐tau (t‐tau) and 181‐phosphorylated‐tau (p‐tau) are also increased early in the disease. 11 Possibly, such early increases in CSF tau and Aβ levels are driven by common upstream pathophysiological processes, 12 and to identify such upstream processes it is important to determine the relative contributions of genetic and environmental factors to early changes in these AD pathophysiological processes. A monozygotic twin design provides a unique approach to study the role of genetic and environmental factors in disease development. Because monozygotic twins are genetically identical, a low twin‐pair correlation suggests that the trait is dependent on environmental factors that are unique to each of the twins. Within–twin‐pair difference analysis shows to what extend the correlation between two traits is driven by environmental factors unique to one of the twins. Furthermore, cross‐twin cross‐trait (CTCT) analyses provide the opportunity to investigate whether a relation between traits is driven by shared genetic and environmental influences. 13 , 14
Our objective was to investigate associations between Aβ production, Aβ aggregation, and CSF tau pathology in cognitively intact older monozygotic twins, to determine the role of genetic and environmental factors on these associations. 3 We used twin discordance models to test the dynamic relationship between Aβ production and Aβ aggregation markers. We hypothesized that if higher Aβ production and increased tau levels precede Aβ aggregation, then in twin‐pairs discordant for Aβ aggregation, the discordant twin with normal Aβ levels would already show signs of increased Aβ production and tau markers compared to twin‐pairs in which both twins have normal Aβ aggregation markers. Furthermore, if the increase in Aβ production markers and CSF tau levels before plaque formation are driven by common upstream genetic processes, 12 these markers should correlate with each other across the whole group and higher levels of Aβ markers in one twin should be related to higher levels of tau in the cotwin in CTCT analysis. Furthermore, if the associations between tau and Aβ production markers are driven by environmental factors that are unique for one of the twins, then twin differences in Aβ production and tau markers should be correlated. Finally, we investigated whether the associations between Aβ markers and tau were dependent on the modality used to determine Aβ abnormality (ie, positron emission tomography [PET] or CSF). 15
Subjects and Methods
Participants
Monozygotic twins were invited from the Netherlands Twin Register 16 to participate in the PreclinAD study as part of the Innovative Medicine Initiative European Information Framework for AD (EMIF‐AD) project (http://www.emif.eu/). 17 Inclusion criteria were age 60 years and older, a delayed recall score of >−1.5 standard deviations relative to demographically adjusted normative data from the Consortium to Establish a Registry for Alzheimer's Disease 10‐word list, 18 a Telephone Interview for Cognitive Status–modified score of 23 or higher, 19 a 15‐item Geriatric Depression Scale score of <11, 20 and a Clinical Dementia Rating Scale of 0 with a score on the memory sub domain of 0. 21 The exclusion criterion was any physical, neurological, or psychiatric condition that could lead to interference with normal cognition in aging. Monozygotic twins were asked to collect buccal cell samples for DNA extraction to confirm zygosity. For the present study, we included all individuals who had an amyloid measurement available (n = 197). A total of 194 subjects (94 complete twin‐pairs) had [18F]flutemetamol PET available, and from 126 subjects (54 complete twin‐pairs) CSF samples were collected.
Ethical Considerations
Informed consent was obtained from all participants. The study was approved by the Central Ethics Committee on Research Involving Human Subjects of the VU University Medical Center Amsterdam, an institutional review board (IRB) certified by the US Office of Human Research Protections (IRB number IRB00002991 under Federalwide Assurance FWA00017598). The research was performed according to the principles of the Declaration of Helsinki and in accordance with the Medical Research Involving Human Subjects Act and codes on "good use" of clinical data and biological samples as developed by the Dutch Federation of Medical Scientific Societies. The study was registered in the EU Clinical Trials Register (EudraCT) with number 2014‐000219‐15.
CSF Analysis
To determine Aβ aggregation in CSF, we used the Aβ1–42/1–40 ratio with lower values indicating abnormality. 22 As markers for Aβ production, we used BACE1 concentrations in CSF, as well as concentrations of Aβ1–40 (Aβ40) and Aβ1–38 (Aβ38). 23 For tau pathology we used CSF t‐tau and p‐tau, 24 which reflect different aspects of tau pathology, 24 , 25 with especially higher values for p‐tau being indicative for tau aggregation. CSF samples were collected in 126 (62%) participants through a lumber puncture, performed between 10 am and 2 pm, after at least 2 hours of fasting. Maximal 20ml CSF was collected in Sarstedt polypropylene syringes using a Spinocan 25‐gauge needle in one of the intervertebral spaces between L3 and S1. Samples were centrifuged at 1,300 to 2,000 × g at 4°C for 10 minutes, and supernatants were then stored in aliquots at −80°C until analysis. 26 A maximum of 2 hours was allowed between lumbar puncture and freezing. Levels of Aβ1–38, Aβ1–40, Aβ1–42, BACE1, t‐tau, and p‐tau were analyzed using commercial kits from Euroimmun (Lübeck, Germany) according to manufacturer instructions. 27 All samples were measured in kits from the same lot.
[18F]Flutemetamol PET
To determine Aβ aggregation on PET imaging, we used parametric nondisplaceable binding potential (BPND) of the [18F]flutemetamol tracer, with higher binding of Aβ radioligands indicating the presence of plaques. 28 In general, PET scanning was performed on the same day as the lumbar puncture, except for 26 subjects, due to technical issues (range = 2.2 months before to 6.7 months after lumbar puncture). PET scans were acquired using an Ingenuity TF PET‐MRI camera (Philips Healthcare, Cleveland, OH). All subjects were scanned under standard resting conditions (eyes closed in dimmed ambient light) from 0 to 30 minutes and from 90 to 110 minutes after intravenous injection of 185MBq (±10%) [18F]flutemetamol. 28 After data acquisition, the first emission scan was reconstructed into 18 frames of increasing length (6 × 5, 3 × 10, 4 × 60, 2 × 150, 2 × 300, and 1 × 600 seconds) using the standard LOR‐RAMLA reconstruction algorithm for the brain. Using the same reconstruction algorithm, the second scan was reconstructed into 4 frames of 5 minutes each. Subsequently, data from the 2 scans were combined into a single image dataset after coregistration using Vinci Software 2.56 (Max Planck Institute for Neurological Research, Cologne, Germany). Parametric BPND images were generated from the entire image set using receptor parametric mapping. 29 Cerebellar gray matter, defined on a T1‐weighted structural magnetic resonance imaging scan obtained immediately prior to the PET scan, was used for attenuation correction of the PET data and as reference tissue. 30 T1‐based volumes of interest using the Hammers atlas implemented in PVElab software were projected onto the [18F]flutemetamol parametric images to extract regional values. 31 A global BPND was calculated based on the volume‐weighted average of frontal (ie, superior, middle, and inferior frontal gyrus), parietal (ie, posterior cingulate, superior parietal gyrus, postcentral gyrus, and inferolateral remainder of parietal lobe), and temporal (ie, parahippocampal gyrus, hippocampus, medial temporal lobe, superior, middle, and inferior temporal gyrus) cortical regions. 32 We classified twins as amyloid positive (abnormal) or negative (normal) by visual read of the [18F]flutemetamol scans. Rating was performed on the parametric BPND images by 3 readers (nuclear physician or radiologist) all trained according to General Electric Healthcare (GEHC) guidelines. 33 For 15 cases, not all readers agreed; here, we used the consensus rating of 2 readers. In quantitative analyses, we used the global BPND.
Apolipoprotein E Genotyping
To assess apolipoprotein E (APOE) ε4 allele carriership, the major genetic risk factor for sporadic AD, all subjects were genotyped on the Affymetrix Axiom array and the Affymetrix 6 array. 34 These were first cross chip imputed following the protocols as described by Fedko and colleagues 35 and then imputed to Haplotype Reference Consortium with the Michigan Imputation server. 36 APOE genotype was assessed using imputed dosages of the single nucleotide polymorphism rs429358 (APOE ɛ4, imputation quality = 0.956) and rs7412 (APOE ɛ2, imputation quality = 0.729). 37
Statistical Analysis
We used generalized estimating equations (GEE) to test amyloid PET group differences for demographic variables, including random effect for twin status. We then assessed associations between Aβ and tau markers across the total group, using GEE including random effect for twin status. Analyses were performed unadjusted (Model 1) and adjusted for age, gender, and APOE ε4 genotype (Model 2) when applicable. 38 Next, we performed 3 types of twin analyses. Monozygotic twin‐pair correlations (ie, correlations for a trait between Twin 1 and Twin 2 across the group) for CSF (n = 54 pairs) and PET amyloid (n = 94 pairs) were assessed using Pearson correlations, which provide a proxy of variance explained in a trait by the combination of shared genetic and shared environmental factors. The correlation coefficient‐1 indicates the percentage of the trait explained by unique environmental factors. Partial correlations were also calculated adjusting for age, gender, and APOE ε4 genotype. PET data and CSF tau data were log‐transformed to improve normal distribution of the data. When 2 variables showed a significant association, we performed CTCT analysis to test whether levels of Marker A in one twin were related to levels of Marker B in the cotwin using OpenMx. 39 We also performed a monozygotic within‐pair difference analysis 13 when there was a significant association between variables. This analysis allows examining whether within‐pair differences in Marker A can be explained by within‐pair differences in Marker B. It provides insight into the contribution of unique environmental factors unique to a twin on the observed relation between traits. 13 Statistical analyses were performed in SPSS version 23 for Windows (IBM, Armonk, NY) and R version 3.3.1 (http://www.r‐project.org/).
Results
Sample Description
Twenty‐seven individuals (14%) had an abnormal visual read of the amyloid PET scan. These subjects were older and had lower CSF Aβ1–42/1–40 ratios and higher p‐tau levels compared to individuals with a normal PET scan (Table 1).
TABLE 1.
Cohort Characteristics
| Total Cohort | PET Visual Read Normal | PET Visual Read Abnormal | |
|---|---|---|---|
| n a | 197 | 167 | 27 |
| Singletons b | 7 | 5 | 1 |
| Age, yr, mean (SD) | 70.3 (7.4) | 70.0 (7.2) | 75.0 (7.2) c |
| Female gender, n (%) | 112 (57) | 96 (57) | 16 (59) |
| Education, yr, mean (SD) | 11.5 (2.6) | 11.5 (2.6) | 11.4 (3.0) |
| MMSE, mean (SD) | 29.0 (1.1) | 29.1 (1.1) | 28.6 (1.5) |
| APOE ε4 carrier, n (%) d | 65 (33) | 50 (30) | 14 (52) e |
| CSF markers, n (%) | 126 (64) | 105 (63) | 18 (67) |
| BACE1, pg/ml, mean (SD) | 2,370 (747) | 2,306 (696) | 2,744 (960) |
| Aβ1–40, pg/ml, mean (SD) | 9,592 (2,844) | 9,289 (2,700) | 11,409 (3,114) |
| Aβ1–38, pg/ml, mean (SD) | 2,424 (737) | 2,370 (715) | 2,805 (797) |
| Aβ1–42/1–40 ratio, mean (SD) | 0.10 (0.03) | 0.10 (0.02) | 0.05 (0.02) c |
| t‐tau, pg/ml, mean (SD) | 412 (143) | 379 (111) | 620 (146) |
| p‐tau, pg/ml, mean (SD) | 76 (44) | 64 (20) | 154 (66) c |
| Dynamic PET, n (%) | 188 (95) | 162 (97) | 26 (96) c |
| Global cortical amyloid deposition, mean (SD), BPND | 0.16 (0.12) | 0.12 (0.06) | 0.43 (0.13) c |
| Concordance status visual read amyloid positivity, n (%) f | |||
| Concordant negative pairs | 74 (79) | ||
| Concordant positive pairs | 6 (6) | ||
| Discordant pairs | 14 (15) | ||
PET visual read was based on BPND image.
PET data were missing in 3 subjects.
In 1 twin‐pair, PET was missing for 1 subject; the other twin had a normal scan and is counted as a singleton for PET analysis.
p < 0.01 versus normal PET group for amyloid measures corrected for age, gender, and APOE ε4.
APOE status was missing in 2 subjects.
p < 0.05 versus normal PET group for amyloid measures corrected for age, gender, and APOE ε4.
Ninety‐four twin‐pairs with PET visual read available for both.
Aβ = amyloid‐beta; APOE = apolipoprotein E; BACE1 = beta‐secretase 1; BPND = nondisplaceable binding potential; CSF = cerebrospinal fluid; MMSE = Mini‐Mental State Examination; PET = positron emission tomography; p‐tau = 181‐phosphorylated‐tau; SD = standard deviation; t‐tau = total‐tau.
Twin‐Pair Correlations
Monozygotic twin‐pair correlations (ie, correlation for a trait across paired twins) were strong for Aβ production markers (r ranging between 0.79 and 0.86) and t‐tau (r = 0.73), and moderately strong for Aβ aggregation markers (r range = 0.50–0.52) and p‐tau (r = 0.64; Fig 2A, Table 2). None of the markers tested was correlated across unrelated individuals (r range = 0.05–0.3, all p < 0.99), and effect sizes remained similar when correcting for age, gender, and APOE ε4 genotype (see Table 2).
FIGURE 2.
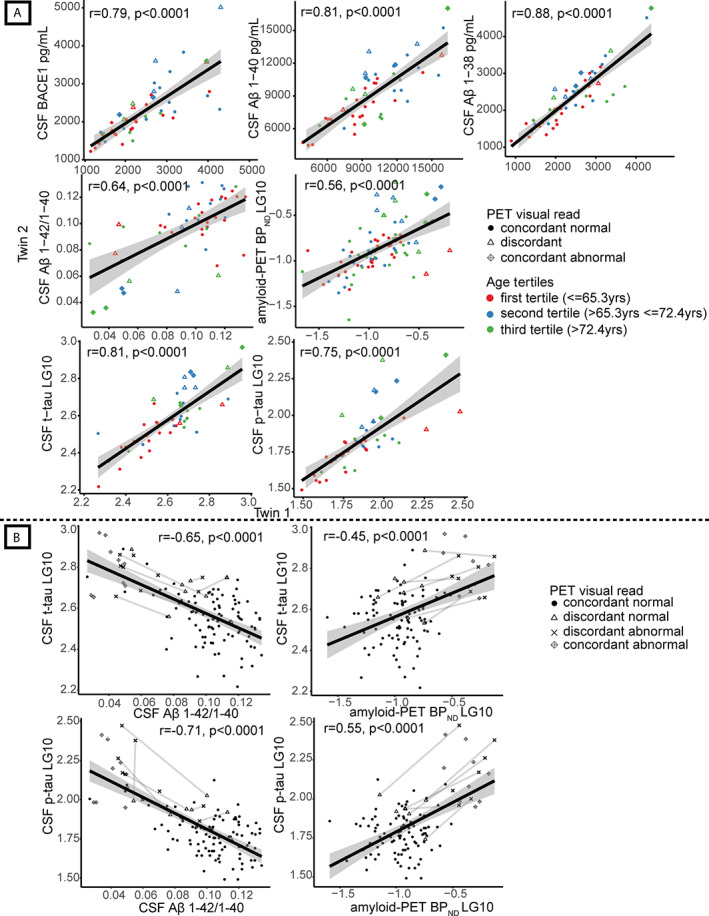
Monozygotic twin‐pair correlations and correlations in amyloid‐beta (Aβ) and tau levels, with discordant pairs connected. (A) Pearson correlation values for association of amyloid markers between one twin and their cotwin (Model 1); all p < 0.0001. Absolute values are shown for cerebrospinal fluid (CSF) beta‐secretase 1 (BACE1), Aβ1–40, Aβ1–38, Aβ1–42/1–40 ratio, and log‐transformed data (LG10) for global cortical positron emission tomography (PET) binding (nondisplaceable binding potential [BPND]) and CSF total‐tau (t‐tau) and 181‐phosphorylated‐tau (p‐tau). Each dot represents one twin‐pair; twin‐pairs who are concordant normal on amyloid PET visual read are shown as dots, and twin‐pairs who are concordant abnormal on amyloid PET visual read are shown as diamonds with a cross inside. Discordant pairs on amyloid PET visual read are shown as open triangles. Age‐groups based on tertiles (60–65.3, 65.3–72.4, >72.4 years) are represented in colors. From left to right: CSF BACE1, CSF Aβ1–40, CSF Aβ1–38, CSF Aβ1–42/1–40 ratio, global cortical PET binding (BPND), CSF t‐tau, CSF p‐tau. (B) Correlations of CSF Aβ1–42/1–40 ratio and CSF t‐tau; global cortical amyloid PET binding (BPND) and CSF t‐tau; CSF Aβ1–42/1–40 ratio and CSF p‐tau; and global cortical amyloid PET binding (BPND) and CSF p‐tau for twins who both have a normal amyloid PET visual read (concordant normal, shown as dots), twins from a discordant pair with a normal amyloid PET visual read (discordant normal, shown as open triangles), twins from a discordant pair with abnormal amyloid PET visual read (discordant abnormal, shown as crosses), and twin‐pairs who both have an abnormal amyloid PET visual read (concordant abnormal, shown as diamonds with a cross inside). Discordant twin‐pairs are connected with lines. LG10 = log‐transformed data.
TABLE 2.
Monozygotic Twin‐Pair Correlations
| Markers | Correlation (95% CI), Model 1 | Correlation (95% CI), Model 2 |
|---|---|---|
| CSF BACE1 | 0.79 (0.66–0.88) | 0.79 (0.65–0.87) |
| CSF Aβ1–38 | 0.88 (0.80–0.93) | 0.86 (0.76–0.92) |
| CSF Aβ1–40 | 0.81 (0.69–0.89) | 0.79 (0.66–0.88) |
| CSF Aβ1–42/1–40 ratio | 0.64 (0.45–0.78) | 0.50 (0.26–0.68) |
| Amyloid PET BPND | 0.56 (0.40–0.69) | 0.52 (0.35–0.66) |
| CSF t‐tau | 0.81 (0.69–0.89) | 0.73 (0.58–0.84) |
| CSF p‐tau | 0.75 (0.60–0.85) | 0.64 (0.44–0.78) |
Correlation values for association of amyloid markers between one twin and their cotwin (Model 1), corrected for gender, age, and apolipoprotein E status (Model 2); all p < 0.0001. Correlations are shown in Figure 2.
Amyloid PET BPND = PET global cortical [18F]flutemetamol nondisplaceable binding potential; Aβ = amyloid beta; BACE1 = beta‐secretase 1; CI = confidence interval; CSF = cerebrospinal fluid; PET = positron emission tomography; p‐tau = 181‐phosphorylated‐tau; t‐tau = total tau.
Association between Aβ and Tau Markers across the Total Group
Across all subjects, higher levels of CSF BACE1 and Aβ38 were associated with lower CSF Aβ1–42/1–40 ratios but not with amyloid PET BPND, and with higher levels of CSF t‐tau and p‐tau. Furthermore, lower CSF Aβ1–42/1–40 ratios and higher amyloid PET BPND were associated with higher levels of t‐tau and p‐tau, with p‐tau showing a higher association with amyloid PET BPND than t‐tau (0.47 vs 0.25; Table 3).
TABLE 3.
Association between Amyloid Production, Amyloid Aggregation, and Tau among Amyloid Production, Amyloid Aggregation, and Tau in Total Cohort
| Predictor | Dependent | Model 1 β (SE) | p | Model 2 β (SE) | p |
|---|---|---|---|---|---|
| Association of Aβ production with Aβ aggregation markers | |||||
| CSF BACE1 | CSF Aβ1–42/1–40 ratio | −0.25 (0.10) | 0.02 | −0.23 (0.09) | 0.008 |
| CSF Aβ1–38 | CSF Aβ1–42/1–40 ratio | −0.35 (0.09) | 0.0003 | −0.27 (0.10) | 0.005 |
| CSF BACE1 | Amyloid PET BPND | 0.13 (0.09) | 0.12 | 0.09 (0.09) | 0.31 |
| CSF Aβ1–38 | Amyloid PET BPND | 0.17 (0.08) | 0.03 | 0.09 (0.08) | 0.23 |
| CSF Aβ1–40 | Amyloid PET BPND | 0.09 (0.09) | 0.34 | 0.02 (0.09) | 0.85 |
| Association between Aβ production markers | |||||
| CSF BACE1 | CSF Aβ1–38 | 0.48 (0.08) | <0.0001 | 0.45 (0.07) | <0.0001 |
| CSF BACE1 | CSF Aβ1–40 | 0.83 (0.07) | <0.0001 | 0.82 (0.06) | <0.0001 |
| CSF Aβ1–38 | CSF Aβ1–40 | 0.80 (0.06) | <0.0001 | 0.82 (0.07) | <0.0001 |
| Association between Aβ aggregation markers | |||||
| CSF Aβ1–42/1–40 ratio | Amyloid PET BPND | −0.58 (0.09) | <0.0001 | −0.57 (0.09) | <0.0001 |
| Association of Aβ aggregation with tau markers | |||||
| CSF Aβ1–42/1–40 ratio | CSF t‐tau | −0.47 (0.07) | <0.0001 | −0.39 (0.08) | <0.0001 |
| Amyloid PET BPND | CSF t‐tau | 0.29 (0.07) | <0.0001 | 0.25 (0.07) | <0.0001 |
| CSF Aβ1–42/1–40 ratio | CSF p‐tau | −0.61 (0.09) | <0.0001 | −0.54 (0.09) | <0.0001 |
| Amyloid PET BPND | CSF p‐tau | 0.50 (0.10) | <0.0001 | 0.47 (0.10) | <0.0001 |
| Association of Aβ production with tau markers | |||||
| CSF BACE1 | CSF t‐tau | 0.63 (0.06) | <0.0001 | 0.61 (0.05) | <0.0001 |
| CSF Aβ1–38 | CSF t‐tau | 0.72 (0.08) | <0.0001 | 0.63 (0.07) | <0.0001 |
| CSF Aβ1–40 | CSF t‐tau | 0.68 (0.08) | <0.0001 | 0.63 (0.08) | <0.0001 |
| CSF BACE1 | CSF p‐tau | 0.60 (0.09) | <0.0001 | 0.60 (0.07) | <0.0001 |
| CSF Aβ1–38 | CSF p‐tau | 0.54 (0.10) | <0.0001 | 0.45 (0.10) | <0.0001 |
| CSF Aβ1–40 | CSF p‐tau | 0.65 (0.08) | <0.0001 | 0.61 (0.08) | <0.0001 |
| Association between tau markers | |||||
| CSF t‐tau | CSF p‐tau | 0.86 (0.06) | <0.0001 | 0.85 (0.08) | <0.0001 |
Generalized estimating equations are shown unadjusted (Model 1) and covariate adjusted for age, apolipoprotein E ε4, and gender (Model 2). All models include random effect for twin status. Standardized betas are shown, calculated with z‐transformed variables. Amyloid aggregation is reflected by higher PET BPND and lower CSF Aβ1–42/1–40 ratio.
Amyloid PET BPND = PET global cortical [18F]flutemetamol nondisplaceable binding potential; Aβ = amyloid‐beta; BACE1 = beta‐secretase 1; CSF = cerebrospinal fluid; PET = positron emission tomography; p‐tau = 181‐phosphorylated‐tau; SE = standard error; t‐tau = total‐tau.
Discordant Twin‐Pair Analyses
Seventy‐four pairs (79% of the 94 complete PET imaging pairs) both had a normal amyloid PET visual read (ie, concordant normal), 14 pairs (15%) were discordant, and 6 pairs (6%) were concordant abnormal (Fig 1). The discordant twin with normal amyloid PET visual reads had higher BPND values compared to concordant twins both having normal amyloid PET visual reads (p < 0.05; see Fig 1H), suggesting that Aβ aggregation has already started in discordant normal twins, although the read is still visually normal. Concordant abnormal twins were older than concordant normal twins, and the age of twins with normal amyloid in discordant pairs was between the ages of concordant normal and abnormal twins (see Fig 1K). We therefore repeated our twin‐pair correlation analyses stratified for age groups according to tertiles. We only observed significant interaction effects with age for Aβ aggregation measures CSF Aβ1–42/1–40 ratio and amyloid PET BPND, with stronger twin‐pair correlations for the intermediate age compared to the youngest age group (both p interaction < 0.05; see Fig 2A), which reflects that the intermediate age group included more discordant pairs.
FIGURE 1.
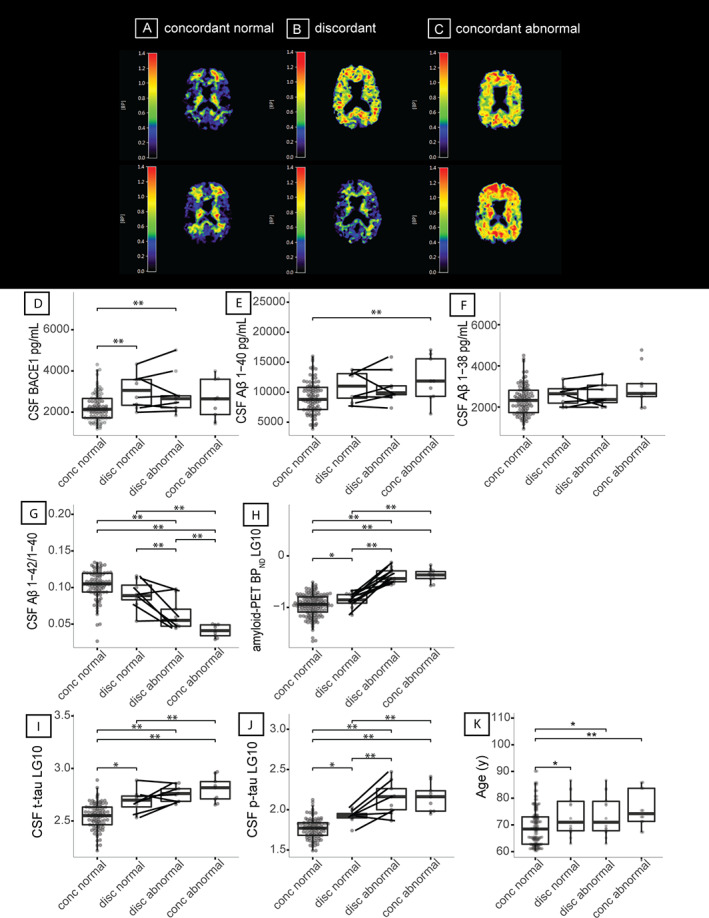
Patterns of amyloid production, amyloid aggregation, and tau for twin discordance and box plots for age and twin discordance. (A–C) [18F]Flutemetamol positron emission tomography (PET) images from a concordant twin‐pair with a normal scan (A), a discordant pair (B), and a concordant pair with an abnormal scan (C). (D–K) Box plots show beta‐secretase 1 (BACE1; D), amyloid‐beta (Aβ) 1–40 (E), Aβ1–38 (F), Aβ1–42/1–40 ratio (G), global cortical PET binding (nondisplaceable binding potential [BPND]) (H), total‐tau (t‐tau; I), 181‐phosphorylated‐tau (p‐tau; J), and age in years (K) for twins who both have a normal amyloid PET visual read (concordant normal, n = 148, of whom 93 have cerebrospinal fluid [CSF] markers), twins from a discordant pair with a normal amyloid PET visual read (discordant normal, n = 14, of whom 8 have CSF markers), twins from a discordant pair with abnormal amyloid PET visual read (discordant abnormal, n = 14, of whom 9 have CSF markers), and twin‐pairs who both have an abnormal amyloid PET vsiual read (concordant abnormal, n = 12, of whom 9 have CSF markers). Discordant twin‐pairs are connected with lines. All analyses for group comparisons were corrected for age, gender, and apolipoprotein E ε4 genotype. *p < 0.05; **p < 0.01. BPND = nondisplaceablebinding potential; conc = concordant; disc = discordant; LG10 = log‐transformed data.
Both amyloid abnormal and amyloid normal twins from discordant twin‐pairs had higher BACE1 concentrations compared to concordant normal amyloid PET twins (see Fig 1D), suggesting that increased BACE1 activity may indicate a very early event in sporadic AD. 40 The twin with normal amyloid from discordant pairs showed higher levels of t‐tau and p‐tau compared to concordant normal twin‐pairs (see Fig 1I, J), suggesting that tau levels are increased very early in the disease. Within discordant twin‐pairs, tau levels increased with increasing Aβ aggregation in a similar way as observed in the total group (see Fig 2B). The twin with abnormal amyloid from discordant pairs showed the highest tau levels, and the twin with normal amyloid from discordant pairs showed CSF Aβ and tau levels near abnormal levels (see Fig 2B).
CTCT and Twin Difference Analyses
CTCT analyses showed that higher BACE1 and Aβ38 concentrations in one twin correlated with lower Aβ1–42/1–40 ratios in the cotwin, and even more strongly with higher t‐tau and p‐tau levels (Aβ aggregation: r = −0.18 to −0.28, p = 0.07–0.007; t‐tau: r = 0.56–0.58, p < 0.0001; p‐tau: r = 0.32–0.54, p < 0.0001–0.002; Fig 4, Supplementary Table S1A). Within‐pair differences in production markers were also related to within‐pair differences in tau levels (t‐tau: r = 0.52–0.61, p < 0.0001; p‐tau: r = 0.49–0.54, p < 0.0001; see Fig 4, Supplementary Table S1B), but not to within‐pair differences in Aβ aggregation markers. CTCT analyses and within‐pair differences further showed that aggregation markers (ie, lower Aβ1–42/1–40 ratios and higher amyloid PET BPND values) in one twin were related to higher t‐tau and p‐tau levels in their cotwin (see Fig 4, Supplementary Table S1). After correction for age, gender, and APOE ε4 genotype (Model 2), CTCT results showed reductions in beta estimates for some markers, that is, Aβ38, amyloid PET BPND, Aβ1–42/1–40 ratio, t‐tau, and p‐tau (eg, Aβ38 uncorrected: r = 0.23; corrected: r = 0.13); this was not the case for the within–twin‐pair difference analysis. We therefore repeated analyses to systematically study the influence of each covariate separately on these results, and observed that the reductions in CTCT estimates for these markers were mostly explained by age (Supplementary Table S2).
FIGURE 4.
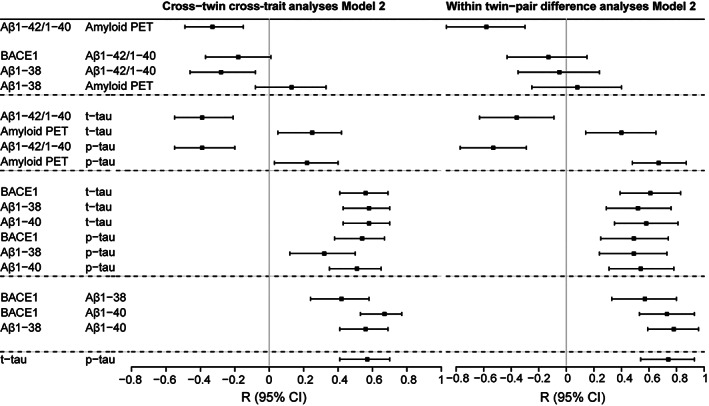
Forest plot of cross‐twin cross‐trait and within–twin‐pair difference analyses. For cross‐twin cross‐trait analyses, data are displayed as correlation coefficient (standard error), comparable to standardized betas given in generalized estimating equations results. Calculated residuals adjusted for age, apolipoprotein E (APOE) ε4, and gender (Model 2). Correlation coefficient indicates the correlation of the production marker in one twin with the aggregation marker in their cotwin. Cross‐twin cross‐trait analyses are shown for variables that had a statistically significant association in the whole cohort (see Table 3). For within–twin‐pair difference analyses, linear regression results are shown for the relation between the standardized difference scores (z scores) within a twin‐pair per amyloid marker adjusted for age, APOE ε4, and gender (Model 2). Beta indicates the association between the within‐pair difference in the production marker and the within‐pair difference in the aggregation marker. Within‐pair difference analyses are shown for variables that had a statistically significant association in the whole cohort (see Table 3). For exact numbers, see Supplementary Table S1. Aβ = amyloid‐beta; Aβ1–38 = CSF Aβ1–38; Aβ1–40 = CSF Aβ1–40; Aβ1–42/1–40 = CSF Aβ1–42/1–40 ratio; Amyloid PET = positron emission tomography global [18F]flutemetamol binding; BACE1 = beta‐secretase 1; CI = confidence interval; CSF = cerebrospinal fluid; p‐tau = CSF 181‐phosphorylated‐tau; t‐tau = CSF total‐tau.
Comparison of CSF and PET Aβ Aggregation Markers, and t‐Tau and p‐Tau Markers
We further studied the relationship between PET and CSF markers for Aβ aggregation, and observed moderately strong correlations between PET BPND and the CSF Aβ1–42/1–40 ratio across the total sample (β = −0.57, standard error [SE] = 0.09, p < 0.0001; see Table 3), with CTCT (r = −0.33, SE = 0.09, p = 0.0005; see Fig 4, Supplementary Table S1A) and within‐pair difference analyses (β = −0.58, SE = 0.14, p < 0.0001; Fig 3, Supplementary Table S1B). For t‐tau and p‐tau, we observed high total sample correlations (β = 0.85, SE = 0.08), which were attenuated in CTCT analyses (r = 0.57, SE = 0.07) and within‐pair difference analyses (β = 0.74, SE = 0.10, all p < 0.0001; see Table 3, Fig 4, Supplementary Table S1).
FIGURE 3.
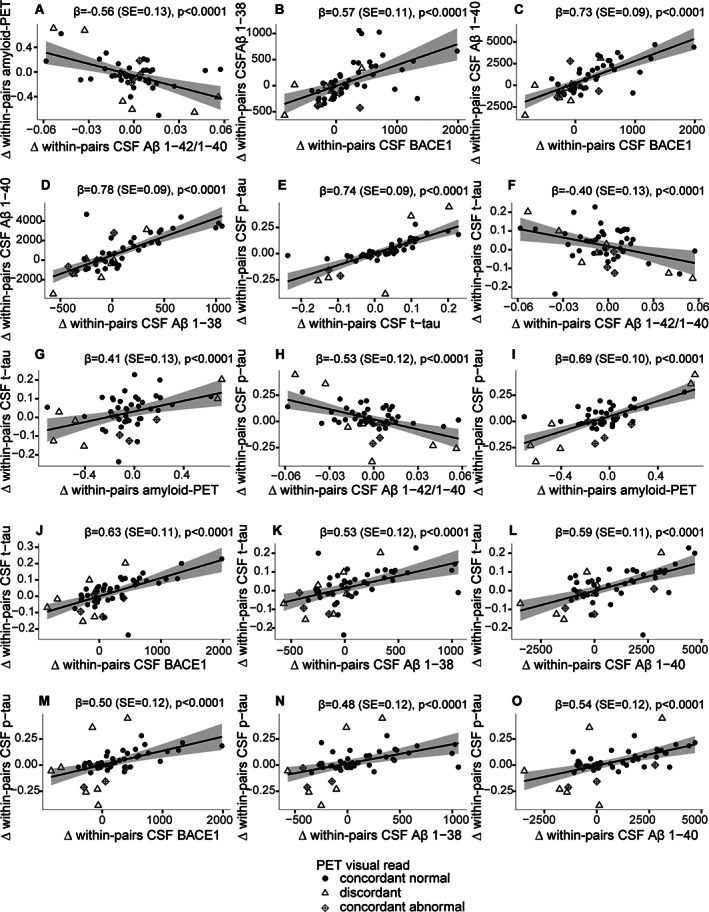
Monozygotic within‐pair difference associations between amyloid aggregation markers, amyloid production markers, and tau. Within‐pair differences of (A) cerebrospinal fluid (CSF) amyloid‐beta (Aβ) 1–42/1–40 ratio with global cortical positron emission tomography (PET) binding (nondisplaceable binding potential [BPND]), (B) CSF beta‐secretase 1 (BACE1) with CSF Aβ1–38, (C) CSF BACE1 with CSF Aβ1–40, (D) CSF Aβ1–38 with CSF Aβ1–40, (E) CSF total‐tau (t‐tau) with CSF 181‐phosphorylated‐tau (p‐tau), (F) CSF Aβ1–42/1–40 ratio with CSF t‐tau, (G) global cortical PET binding (BPND) with CSF t‐tau, (H) CSF Aβ1–42/1–40 ratio with CSF p‐tau, (I) global cortical PET binding (BPND) with CSF p‐tau, (J) CSF BACE1 with CSF t‐tau, (K) CSF Aβ1–38 with CSF t‐tau, (L) CSF Aβ1–40 with CSF t‐tau, (M) CSF BACE1 with CSF p‐tau, (N) CSF Aβ1–38 with CSF p‐tau, (O) CSF Aβ1–40 with CSF p‐tau. Each dot represents one twin‐pair; twin‐pairs who are concordant normal on amyloid PET visual read are shown as black dots, and twin‐pairs who are concordant abnormal on amyloid PET visual read are shown as diamonds with a cross inside. Discordant pairs on amyloid PET visual read are shown as open triangles. Lower CSF Aβ1–42/1–40 ratio and higher global cortical PET binding indicate more amyloid aggregation. SE = standard error.
Discussion
Our study in cognitively normal older monozygotic twins suggests that in the very early stages of sporadic AD, increased levels of Aβ production markers are associated with Aβ aggregation as well as higher levels of t‐tau and p‐tau. CTCT and twin difference analyses indicated that Aβ production and Aβ aggregation, Aβ production and tau markers, and tau and Aβ aggregation share, at least in part, a common underlying pathological mechanism. Furthermore, the moderately strong within–twin‐pair correlations for aggregation markers, combined with the observation of 14 amyloid‐discordant twin‐pairs, suggests that environmental factors influence the start of AD pathological processes.
Our findings suggest a role of increased Aβ production in the very early pathophysiology of sporadic AD. In autosomal dominant AD, certain mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), or PSEN2 lead to increased Aβ production, 6 subsequently causing Aβ aggregation. We further show that increases in Aβ production markers in this very early stage were associated with increased t‐tau and p‐tau levels, suggesting that these pathological hallmarks may be closely coupled in disease pathogenesis. Our observation seems in line with previous studies that used iPSC models from sporadic AD and showed significantly higher levels of Aβ40 41 , 42 and tau 7 compared to controls. In patients, another study that used stable isotope labeling kinetics also observed that the rate of tau production was associated with Aβ burden. 2 The high CTCT and twin difference associations we observe in our study provide a further indication that tau levels and Aβ production markers may share underlying pathophysiological processes. 12 One such mechanism could be neuronal activity, which dynamically regulates APP cleavage and Aβ levels. 43 , 44 In mouse models, aberrant increased neuronal activity has been observed to precede plaque formation in AD, 45 and such hyperactivity has been related to increased neuronal secretion of both Aβ and tau, which compromises the metabolic homeostasis of neurons and contributes to network dysfunction. 43 , 46 Nonetheless, it has to be noted that our analyses were based on cross‐sectional data, and we are currently collecting follow‐up measures to further investigate the relative timing of Aβ aggregation, increased Aβ production, and increased tau secretion longitudinally in this cohort. We observed such changes in tau and amyloid toward abnormality very early in the discordant monozygotic twins who still had normal amyloid PET. We further found that Aβ production markers and tau levels have a shared genetic background. This warrants further investigation into genetic variants associated with these pathophysiological changes in large combined genome‐wide association studies with CSF.
We found different patterns in the discordant twin‐pair analyses between Aβ production markers BACE1, Aβ40, and Aβ38. Possibly, this may indicate functional differences between secretases in production of Aβ peptides, because a previous study using ADAD iPSC‐derived neurons showed that mutations of APP and PSEN1 have distinct effects on Aβ40 and Aβ38 levels by γ‐secretase. 47 Future research should further investigate such effects in more detail, by measuring other markers that could reflect γ‐secretase activity.
Furthermore, monozygotic twin‐pair correlations for Aβ production markers and t‐tau were much higher compared to those of aggregation markers and p‐tau. This suggests that although t‐tau and p‐tau levels are strongly correlated, these markers may in part reflect different aspects of AD pathophysiological processes. Whether the strong correlation between Aβ aggregation and t‐tau resulted from shared genetics or shared environmental factors between twins could not be investigated, as we did not include dizygotic twins in our study. However, previous studies indicated that shared environmental factors have a limited effect on neurological and neurodegenerative traits. 3 , 48 Future studies are needed to clarify the mechanisms that underlie the differences in amyloid production markers between cognitively normal individuals, as this may provide novel clues for reducing Aβ aggregation, t‐tau levels, and tau phosphorylation.
Monozygotic twin‐pair correlations observed for CSF and PET Aβ aggregation markers and p‐tau ranged between 0.50 and 0.64, which is similar to previous CSF and PET concordance estimates reported by previous, smaller studies in cognitively normal individuals, 4 , 49 and lower compared to twin similarity estimates for AD‐type dementia (up to 79%). 3 It is possible that monozygotic twin similarities in Aβ aggregation may be lower because of the early disease stage. As such, it could be hypothesized that in amyloid‐discordant pairs, the twin with a normal amyloid PET visual read may become abnormal in the future. This explanation is supported by the observation that these individuals already showed higher (although still normal) BPND values compared to twin‐pairs both with normal amyloid PET. CTCT and twin difference associations indicated that CSF and PET measures of amyloid aggregation reflect a common underlying pathophysiology. However, these associations were moderate, in line with earlier studies, 50 suggesting that they may also capture different aspects of Aβ aggregation.
We further observed that for some markers, that is, Aβ38, amyloid PET BPND, CSF Aβ1–42/1–40 ratio, and CSF t‐tau, the strength of the CTCT correlation tended to decrease after correction for age, gender, and APOE ε4 genotype, but there was no effect on the twin difference analyses. This finding highlights that CTCT and twin difference analyses capture different aspects. Our additional analyses suggested that a part of the decreases in CTCT associations could be explained by the observation that those markers showed a relationship with age. The twin‐pair differences in these markers, however, were not associated with age, suggesting that although biomarker levels may change with age, they do so in a similar way within a twin‐pair.
Bigger sample sizes for investigating very early pathophysiology of AD that include more individuals with abnormal amyloid would be desirable to increase statistical power. However, our older monozygotic twin approach is unique, and has enabled us to estimate the contribution of genetic and environmental factors to the start of Aβ aggregation. Longitudinal research is needed to investigate whether twin‐pairs discordant for amyloid abnormality will become more similar in their levels of Aβ aggregation and p‐tau over time. Furthermore, although low CSF Aβ1–42/1–40 ratios are established biomarkers for AD pathology, it still remains unclear what differences in CSF Aβ production marker levels represent in sporadic AD. BACE1, Aβ40, and Aβ38 are related to APP metabolism, and although these markers correlate strongly with each other, we cannot exclude the possibility that the levels as measured in CSF may also reflect other aspects of APP metabolism.
Previous population studies have indicated that environmental factors influence dementia risk, 51 and our results show that the effect of such factors, at least partially, could be explained through their impact on Aβ and tau aggregation. It will be of interest to investigate whether twin discordance and their change over time may be explained by risk factors such as educational attainment, hypertension, obesity, alcohol misuse, hearing loss, and smoking, as this may guide prevention studies, 51 and this will be a topic for future research. In addition, identification of genes and mechanisms associated with high amyloid production in cognitively normal individuals may further provide novel leads to reduce Aβ aggregation.
Author Contributions
P.J.V., D.I.B., and P.S. contributed to conception and design of the study; E.K., J.T., A.d.B., M.t.K., M.Y., S.D.M., M.G.N., H.V., A.A.L., C.E.T., B.N.M.v.B., and B.M.T. contributed to acquisition and analysis of data; E.K., J.T., A.d.B., B.M.T., and P.J.V. contributed to drafting the text and/or preparation of figures.
Potential Conflicts of Interest
H.V. is the founder and director of Biomarkable and he is a cofounder of ADx NeuroSciences, where he worked as a consultant through Biomarkable. H.V. has shares in both companies. ADx NeuroSciences provided the enzyme‐linked immunosorbent assays used in this study. The other authors have nothing to report.
Data Availability
The data that support the findings of this study are available from the corresponding author after a material transfer agreement has been signed.
Supporting information
APPENDIX S1. Supporting Information
Acknowledgments
This work has received support from the EU/European Federation of Pharmaceutical Industries and Associations Innovative Medicines Initiative Joint Undertaking (EMIF grant 115372). This work received in‐kind sponsoring of the CSF assay from ADx NeuroSciences and Euroimmun, and the PET tracer [18F]flutemetamol from GE Healthcare.
We thank all participating twins for their dedication.
References
- 1. Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta‐analysis. JAMA 2015;313:1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018;97:1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 4. Johansson V, Jakobsson J, Fortgang RG, et al. Cerebrospinal fluid microglia and neurodegenerative markers in twins concordant and discordant for psychotic disorders. Eur Arch Psychiatry Clin Neurosci 2017;267:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β‐amyloid in Alzheimer's disease. Science 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knupp A, Mishra S, Martinez R, et al. Depletion of the AD risk gene SORL1 selectively impairs neuronal endosomal traffic independent of amyloidogenic APP processing. Cell Rep 2020;31:107719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Timmers M, Barao S, van Broeck B, et al. BACE1 dynamics upon inhibition with a BACE inhibitor and correlation to downstream Alzheimer's disease markers in elderly healthy participants. J Alzheimers Dis 2017;56:1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tijms BM, Vermunt L, Zwan MD, et al. Pre‐amyloid stage of Alzheimer's disease in cognitively normal individuals. Ann Clin Transl Neurol 2018;5:1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jack CRJ, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med 2020;26:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Small SA, Duff K. Linking Abeta and tau in late‐onset Alzheimer's disease: a dual pathway hypothesis. Neuron 2008;60:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Moor MH, Boomsma DI, Stubbe JH, et al. Testing causality in the association between regular exercise and symptoms of anxiety and depression. Arch Gen Psychiatry 2008;65:897–905. [DOI] [PubMed] [Google Scholar]
- 14. Vitaro F, Brendgen M, Arseneault L. The discordant MZ‐twin method: one step closer to the holy grail of causality. Int J Behav Dev 2009;33:376–382. [Google Scholar]
- 15. Zwan MD, Rinne JO, Hasselbalch SG, et al. Use of amyloid‐PET to determine cutpoints for CSF markers: a multicenter study. Neurology 2016;86:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boomsma DI, de Geus EJ, Vink JM, et al. Netherlands twin register: from twins to twin families. Twin Res Hum Genet 2006;9:849–857. [DOI] [PubMed] [Google Scholar]
- 17. Konijnenberg E, Carter SF, ten Kate M, et al. The EMIF‐AD PreclinAD study: study design and baseline cohort overview. Alzheimers Res Ther 2018;10:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morris JC, Heyman A, Mohs RC, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 1989;39:1159–1165. [DOI] [PubMed] [Google Scholar]
- 19. de Jager CA, Budge MM, Clarke R. Utility of TICS‐M for the assessment of cognitive function in older adults. Int J Geriatr Psychiatry 2003;18:318–324. [DOI] [PubMed] [Google Scholar]
- 20. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 1982;17:37–49. [DOI] [PubMed] [Google Scholar]
- 21. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 22. Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Abeta42/40 corresponds better than Abeta42 to amyloid PET in Alzheimer's disease. J Alzheimers Dis 2017;55:813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Portelius E, Westman‐Brinkmalm A, Zetterberg H, Blennow K. Determination of β‐amyloid peptide signatures in cerebrospinal fluid using immunoprecipitation‐mass spectrometry. J Proteome Res 2006;5:1010–1016. [DOI] [PubMed] [Google Scholar]
- 24. Tapiola T, Alafuzoff I, Herukka S‐K, et al. Cerebrospinal fluid β‐amyloid 42 and tau proteins as biomarkers of Alzheimer‐type pathologic changes in the brain. JAMA Neurol 2009;66:382–389. [DOI] [PubMed] [Google Scholar]
- 25. Hampel H, Blennow K, Shaw LM, et al. Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol 2010;45:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. del Campo M, Mollenhauer B, Bertolotto A, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer's and Parkinson's disease cerebrospinal fluid biomarkers: an update. Biomark Med 2012;6:419–430. [DOI] [PubMed] [Google Scholar]
- 27. de Vos A, Jacobs D, Struyfs H, et al. C‐terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer's disease. Alzheimers Dement 2015;11:1461–1469. [DOI] [PubMed] [Google Scholar]
- 28. Nelissen N, van Laere K, Thurfjell L, et al. Phase 1 study of the Pittsburgh compound B derivative 18F‐flutemetamol in healthy volunteers and patients with probable Alzheimer disease. J Nucl Med 2009;50:1251–1259. [DOI] [PubMed] [Google Scholar]
- 29. Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ. Parametric imaging of ligand‐receptor binding in PET using a simplified reference region model. Neuroimage 1998;6:279–287. [DOI] [PubMed] [Google Scholar]
- 30. Hammers A, Allom R, Koepp MJ, et al. Three‐dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp 2003;19:224–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Svarer C, Madsen K, Hasselbalch SG, et al. MR‐based automatic delineation of volumes of interest in human brain PET images using probability maps. Neuroimage 2005;24:969–979. [DOI] [PubMed] [Google Scholar]
- 32. Tolboom N, Yaqub M, van der Flier WM, et al. Detection of Alzheimer pathology in vivo using both 11C‐PIB and 18F‐FDDNP PET. J Nucl Med 2009;50:191–197. [DOI] [PubMed] [Google Scholar]
- 33. Healthcare G. EPAR product information—summary of product characteristics. 2014. https://www.ema.europa.eu/en/documents/assessment-report/vizamyl-epar-public-assessment-report_en.pdf
- 34. Ehli EA, Abdellaoui A, Fedko IO, et al. A method to customize population‐specific arrays for genome‐wide association testing. Eur J Hum Genet 2017;25:267–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fedko IO, Hottenga JJ, Medina‐Gomez C, et al. Estimation of genetic relationships between individuals across cohorts and platforms: application to childhood height. Behav Genet 2015;45:514–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Lee SJ, Wolters FJ, Ikram MK, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer's disease and dementia: a community‐based cohort study. Lancet Neurol 2018;17:434–444. [DOI] [PubMed] [Google Scholar]
- 38. Minică CC, Dolan CV, Kampert MM, et al. Sandwich corrected standard errors in family‐based genome‐wide association studies. Eur J Hum Genet 2015;23:388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boker S, Neale M, Maes H, et al. OpenMx: an open source extended structural equation modeling framework. Psychometrika 2011;76:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gatz M, Fratiglioni L, Johansson B, et al. Complete ascertainment of dementia in the Swedish twin registry: the HARMONY study. Neurobiol Aging 2005;26:439–447. [DOI] [PubMed] [Google Scholar]
- 41. Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 2012;482:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meyer K, Feldman HM, Lu T, et al. REST and neural gene network dysregulation in iPSC models of Alzheimer's disease. Cell Rep 2019;26:1112–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid‐beta deposition. Nat Neurosci 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid‐beta levels in vivo. Neuron 2005;48:913–922. [DOI] [PubMed] [Google Scholar]
- 45. Busche MA, Eichhoff G, Adelsberger H, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 2008;321:1686–1689. [DOI] [PubMed] [Google Scholar]
- 46. Zott B, Simon MM, Hong W, et al. A vicious cycle of β amyloid–dependent neuronal hyperactivation. Science 2019;365:559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arber C, Toombs J, Lovejoy C, et al. Familial Alzheimer's disease patient‐derived neurons reveal distinct mutation‐specific effects on amyloid beta. Mol Psychiatry 2020;25:2919–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polderman TJ, Benyamin B, de Leeuw CA, et al. Meta‐analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 2015;47:702–709. [DOI] [PubMed] [Google Scholar]
- 49. Hinrichs AL, Mintun MA, Head D, et al. Cortical binding of Pittsburgh compound B, an endophenotype for genetic studies of Alzheimer's disease. Biol Psychiatry 2010;67:581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zwan M, van Harten A, Ossenkoppele R, et al. Concordance between cerebrospinal fluid biomarkers and [11C]PIB PET in a memory clinic cohort. J Alzheimers Dis 2014;41:801–807. [DOI] [PubMed] [Google Scholar]
- 51. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020;396:413–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author after a material transfer agreement has been signed.