

Abstract
Brasilicardin A (1) consists of an unusual anti/syn/anti‐perhydrophenanthrene skeleton with a carbohydrate side chain and an amino acid moiety. It exhibits potent immunosuppressive activity, yet its mode of action differs from standard drugs that are currently in use. Further pre‐clinical evaluation of this promising, biologically active natural product is hampered by restricted access to the ready material, as its synthesis requires both a low‐yielding fermentation process using a pathogenic organism and an elaborate, multi‐step total synthesis. Our semi‐synthetic approach included a) the heterologous expression of the brasilicardin A gene cluster in different non‐pathogenic bacterial strains producing brasilicardin A aglycone (5) in excellent yield and b) the chemical transformation of the aglycone 5 into the trifluoroacetic acid salt of brasilicardin A (1 a) via a short and straightforward five‐steps synthetic route. Additionally, we report the first preclinical data for brasilicardin A.
Keywords: brasilicardin A, heterologous expression, natural products, semi-synthesis, terpenoids
The development of a heterologous producer strain that enables the sustainable production of the stereochemically complex natural products brasilicardin C and E at excellent rates is presented. The semi‐synthetic approach allows an efficient gram‐scale conversion of brasilicardin E into the potent immunosuppressant brasilicardin A for which the first preclinical data are reported.
The application of the first immunosuppressive drugs, such as azathioprine and corticosteroids, in the 1950s heralded a new age in organ‐transplantation. Despite the subsequent development of further immunosuppressants, including foremost the natural products cyclosporin A, tacrolimus and mycophenolic acid, there are still two major unmet clinical needs that are required to be addressed by new immunosuppressive drugs: Firstly, most organ transplants continue to have low rates of long‐term graft and patient survival. [1] The second issue is that current immunosuppressive drugs still cause serious side effects. Since the adverse effects of a single immunosuppressant is often too severe, commonly an individualized combination therapy regimen consisting of 2–4 immunosuppressants, each with a reduced dose, is applied to balance out the side effects and raise efficacy. [2] Thus, the development of new immunosuppressants that show improved long‐term survival in patients and lack undesirable side effects remains a high priority. [1]
Brasilicardin A (BraA, 1) was isolated, along with its biosynthetic intermediates brasilicardins B, C and D (BraB‐D, 2–4), from the human pathogenic bacterium Nocardia terpenica IFM 0406 (Figure 1). [3] The natural product brasilicardin A (1) [3] represents a promising drug candidate since it possesses strong immunosuppressive potency, low toxicity and a mode of action that differs from all drugs which are currently in clinical use. [4] Among the brasilicardin family, 1 was shown to be highly potent in a mouse mixed‐lymphocyte reaction (MLR) assay and surpassed with an IC50 value of 0.057 μg mL−1 (63.8 nM) the potency of cyclosporin (0.15 μg mL−1) by factor ≈3. [3c] In human T cells, BraA displayed in vitro a similar immunosuppressive activity (IC50 65 nM). [5] Usui and co‐workers [4] demonstrated that 1 mediates this activity by inhibition of the amino acid transporter system L; the major transporter for essential amino acid uptake in activated human T cells. More precisely, it represents an obligatory 1:1 amino acid exchanger that can couple the cellular uptake of branched‐chained and aromatic amino acids with the efflux of cytoplasmic amino acids, such as Gln. Inhibition of the transporter system leads to a cellular depletion of essential amino acids within activated lymphocytes and causes GCN2‐dependent integrated stress responses, which results in the inhibition or retardation of cell proliferation. [4] With this novel mode of action, BraA is assumed to be less toxic than cyclosporin and tacrolimus. It was reported that intravenous administration of BraA caused no sign of toxicity at a dose of 100 mg kg−1 in mice. [3a] Thus, BraA (1) was expected to be a promising new immunosuppressive drug. However, development of BraA has been hampered due to the scarcity of available material as the original producer is classified as a biosafety level (BSL) 2 strain and shows a very low production yield (0.2 mg L−1). [3b] Recently, the technical feasibility of the multi‐step total synthesis of brasilicardin A (1) was demonstrated. [6] Nonetheless, when aiming for an environmentally friendly, sustainable and, most importantly, cost‐efficient production, a biotechnological production is more desirable. Encouraged by the promising preliminary data and with ideas for improving the given production bottleneck, we embarked on the development of a safe (BSL‐1) and economical biotechnological production platform by employing heterologous expression.
Figure 1.
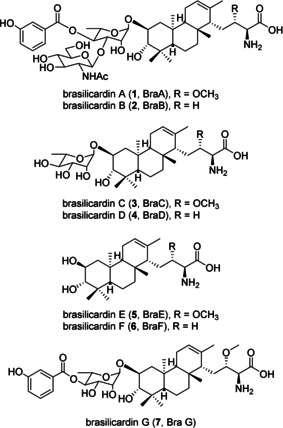
Chemical structures of brasilicardins A–G (1–7).
The biosynthetic gene cluster (BGC) of brasilicardin A (1) was identified by the pioneering work of Dairi and co‐workers and reported to consist of 11 genes (bra1‐bra11, Figure 2). [7] Recently, we expanded on this work and were able to redefine the borders of the BGC, employing genome sequencing and heterologous expression in Amycolatopsis japonicum. [8] Briefly, we provided evidence that the BGC had to be expanded by the adjacent genes bra0 and bra12, encoding a dioxygenase and a transcriptional activator, respectively. However, this proof‐of‐concept study did not lead to the heterologous production of 1, instead yielding the biosynthetic intermediates BraC (3) and BraD (4) and their corresponding aglycons, for which we proposed the trivial names brasilicardins E (BraE, 5) and F (BraF, 6) (Figure 1).
Figure 2.
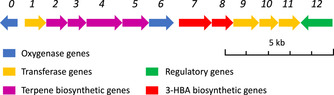
Biosynthetic gene cluster encoding brasilicardins.
In order to achieve production of the complete BraA (1) molecule and raise the yield of brasilicardins in general, we investigated the heterologous expression of the brasilicardin BGC in more than 70 actinomycetes (Table S1). For maximum flexibility, we established a phage P1‐derived artificial chromosome (PAC)‐based methodology alongside the fosmid‐based heterologous expression system. [9]
As well as standard Streptomyces model host strains, a set of 45 attini ant associated Streptomyces strains [10] were included. Furthermore, strains from the genera Pseudonocardia, Amycolatopsis, Prauseria, Saccharothrix and Rhodococcus were selected as phylogenetically close relatives of the original producer strain, N. terpenica IFM 0406. It was observed that nine actinomycetes hosts produced solely BraE (5), while further 10 strains secreted BraE (5) and BraF (6), respectively. Another set of 10 host strains exhibited a shifted or extended production spectrum and were enabled to produce BraC (3), BraD (4), and BraF (6) (Table S1). The sole production of the aglycons 5 and 6 was observed foremost in Streptomyces strains, coherent with the absence of the TDP‐L‐rhamnose biosynthetic genes in the corresponding host strains. Upon introduction of the plasmid (pRHAMO), which contains the full biosynthetic cassette to generate TDP‐L‐rhamnose, the production of the mono‐glycosylated BraC (3) and BraD (4) was detected in the majority of the strains (Table S1). Only a few actinomycetes, such as the attini ant‐derived S. griseus:bcaAB01 (pRHAMO) and R. erythropolis:2G3 additionally produced a new brasilicardin derivative 7 (Figure 1), albeit in a lower amount. The new derivative was isolated, and its structure fully elucidated by NMR analysis (Figures S12 and S13, Table S4). As a result, the compound represents a new brasilicardin congener, solely lacking the GlcNAc moiety when compared to BraA (1) (Figure 1), for which we proposed the trivial name brasilicardin G (BraG, 7). However, BraA (1) or BraB (2) could not be discovered in any of the heterologous hosts. In terms of production yield, the S. griseus:bcaAB01 (pRHAMO) strain was superior to all other tested strains (Table S1). Under non‐optimized brasilicardin production conditions, [3a] this host produced 305 mg L−1 of 3, 168 mg L−1 of 5 and 15 mg L−1 of 7 after 12 days of cultivation (Figure S1). Despite the fact that we did not achieve the heterologous production of the target compound BraA (1), the significant production of the stereochemically complex intermediates 3 and 5 we attained opened up the avenue for a semi‐synthetic route to BraA. In order to develop an efficient process, we next aimed for further optimization of the production yield of either 3 or 5. These improvements were solely focused on the best performing strain, S. griseus:bcaAB01 (pRHAMO).
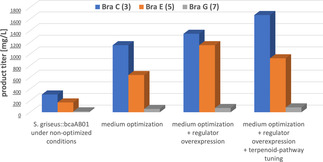
We improved the production of 3 and 5 with several modifications, applied in three steps. One critical parameter for the quantity of a target secondary metabolite produced by a microorganism are the cultivation conditions. [11] Therefore, a screening with 34 different media was performed, which revealed a medium with a high glucose and amino acid content as the optimal choice. BraC (3), and BraE (5) production yields of S. griseus:bcaAB01 (pRHAMO) in the optimized medium were increased up to 1151 mg L−1 and 639 mg L−1, respectively (Figures 3, S2 and S3).
Figure 3.
Summary of the stepwise improvement of the brasilicardin production titer in S. griseus 65.
In a second modification, we implemented genetic engineering measures. Since our previous biosynthetic studies revealed that bra12 is a positive regulator of the brasilicardin BGC, [8b] we hypothesized that its overexpression in the heterologous host S. griseus:bcaAB01 (pRHAMO) might increase the production yield. Indeed, the introduction of bra12 under the control of the constitutive ermEp* promoter led to a further increase in production of the compounds 3 (1347 mg L−1) and 5 (1151 mg L−1) (Figures 3 and S4).
Finally, metabolic flow considerations were inspected. We scrutinized the brasilicardin biosynthesis pathway for bottlenecks in biosynthetic substrates and hypothesized that the supply of terpenoid building blocks might be a limitation in production. The diterpenoid backbone of 1 is synthesized with isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) units. In a bacterial setting, these precursors are usually synthesized via the methylerythritol 4‐phosphate (MEP) pathway. In addition to the MEP pathway, only a few actinomycetes possess the mevalonate (MVA) pathway, which is often located next to an isoprenoid biosynthetic gene cluster. Employing 13C‐isotopic labeling experiments, Kobayashi and co‐workers demonstrated that the diterpenoid moiety of 1 is, as expected, biosynthesized from D‐glucose via the MEP pathway. [12] Since the MEP pathway undergoes a strong and complex feedback regulation, the MVA pathway is easier to manipulate and therefore more suitable for a metabolic engineering approach. Therefore, we integrated the MVA pathway as a second terpene pathway into S. griseus:bcaAB01 (pRHAMO) to enrich the DMAPP/IPP precursor pool.
Furthermore, it had been reported that the overexpression of the gene isopentenyl diphosphate isomerase (idi), whose product converts IPP into DMAPP, led to an increased production of the target terpenoid. [13] Likewise, the overexpression of the native geranylgeranyl and farnesyl diphosphate synthase genes (ggpps, fpps), whose products utilize DMAPP/IPP units to form farnesyl‐PP and geranyl‐geranyl‐PP, were shown to exert a positive effect on the production yield.
To examine whether these genes would also further increase production in S. griseus:bcaAB01 (pRHAMO), the idi gene from the MVA pathway and the genes bra12 and ggpps/fpps were heterologously expressed in S. griseus:bcaAB01 (pRHAMO). As a result, the strain produced 1669 mg L−1 BraC (3) and 926 mg L−1 BraE (5) (Figures 3 and S5), representing a ≈5‐fold improvement of the yield when compared with the first experiments in both cases. In summary, the stepwise optimization of the medium composition, BGC regulation and precursor supply resulted in the generation of a safe and sustainable platform for the production of BraC (3) and BraE (5) on gram‐scale (Figure 3).
Employing semi‐synthesis, the now readily available aglycone 5 was used for conversion into the biologically highly active BraA (1). Inspired by previous synthetic efforts by us and others,[ 5 , 6 , 14 ] we developed a synthesis comprising five linear steps (Scheme 1). In the first steps of the synthesis, the primary amino function and carboxyl group of aglycone 5 were protected as a carboxybenzyl (Cbz) and benzyl ester, respectively.
Scheme 1.
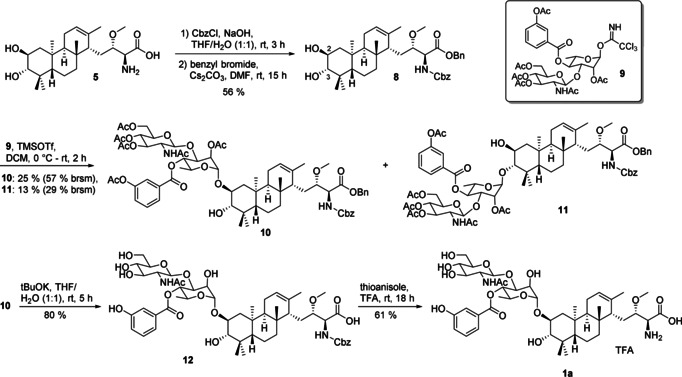
Semi‐synthesis of the TFA salt of brasilicardin A (1 a) starting from aglycone 5.
In order to minimize the length of the synthesis, we opted to use protected aglycone 8, bearing two hydroxyl groups, in the glycosylation step, even though glycosylation reaction could take place at positions C‐2 and/or C‐3. We reasoned that the hydroxy group at position C‐3 would be sterically more hindered compared to the hydroxy group at position C‐2, as position C‐3 is situated between one hydroxyl and two methyl groups and glycosylation reaction should preferentially occur at the hydroxy group at position C‐2.
The key step of this synthesis, the coupling of 8 with the acetyl protected carbohydrate side chain of BraA (compound 9), [14] was performed using Schmidt's glycosylation conditions in the presence of TMSOTf as promoter. Applying these conditions, reported by Jung and Koch,[ 5 , 14 ] using 1.4 equiv of donor 9 and 0.2 equiv of promoter, resulted in low conversion of the reaction. After a reaction time of 5 h, regioisomers 10 and 11 were obtained in 20 % and 10 % yield, respectively, whereas a substantial amount of acceptor 8 could be recovered. The use of an excess of imidate 9 (3 equiv), a decreased amount of the promoter (0.05 equiv) and a shorter reaction time (2 h), resulted in a slight improvement of the yield of fully protected brasilicardin A (compound 10, 25 %; 57 % based on recovered starting material (brsm)). However, the ratio of glycosylation at position C‐2 (compound 10) and position C‐3 (compound 11) was unchanged (2:1). Double glycosylation at positions C‐2 and C‐3 on compound 8 was not observed.
In the last steps of the synthesis, all protective groups present in 10 were cleaved off. The simultaneous removal of all O‐acetyl groups and the benzyl ester proceeded smoothly under mild basic conditions without affecting the hydroxy‐ benzoate moiety at rhamnose C‐4′ position. Finally, the Cbz protecting group present in 12 had to be cleaved off without degrading any other functionality in the molecule. Due to the presence of a double bond in the perhydrophenanthrene skeleton, the Cbz group could not be cleaved off using hydrogenation conditions. After testing different conditions on model compounds, the method described by Kiso and co‐workers [15] using thioanisole in trifluoroacetic acid (TFA) was selected. Treatment of 12 under mild conditions with 50 equiv of thioanisole in TFA gave the target molecule BraA as the TFA‐salt (1 a) in 61 % yield.
The overall yield of the synthetic sequence was 7 % and 16 % brsm. The spectroscopic analysis revealed that the 1H and 13C NMR spectra of synthetic 1 a were superimposable with those of 1, obtained from strain IFM 0406 and reported by Shigemori et al. [3b] (Figures S28 and S29).
In addition, the immunosuppressive activity of natural BraA (1), its biosynthetic intermediates BraC (3), BraE (5) and BraG (7) and synthetic BraA (1 a) were evaluated in a human CD3+ cell proliferation assay, and compared with the standard drug cyclosporin A (Figures S30 and S31). BraA (1) exhibited the most potent immunosuppressive activity and showed a greater reduction in proliferation than the marketed drug cyclosporin A, which was in a good agreement with previous studies.[ 3c , 5 ] Synthesized BraA (1 a) showed the same inhibition profile and potency as isolated BraA (1), corroborating that our synthesis produced the correct structure. Notably, the antiproliferative activity of BraG (7), lacking the N‐acetyl‐glucosamine moiety found in BraA (1), was still comparable with cyclosporin, while BraC (3) prevented proliferation only at higher concentrations (5 μM). BraE (5) did not inhibit T cell proliferation at the tested concentrations. These results suggest the presence of the benzoyl group in the carbohydrate side chain is important for the immunosuppressive activity of BraA.
Since BraA is a potent inhibitor of the amino acid transport system L, [4] BraA (1 a) and natural BraC (3) were further evaluated in an antiproliferation assay using the human malignant glioma cell line LN229 (Table S6). The selective L‐amino acid transporter 1 (LAT1) inhibitor JPH203 (IC50 15.0 μM) served as a positive control in this study. BraC (3, IC50 15.7 μM) showed a similar antiproliferation activity as JPH203. Intriguingly, BraA (1 a, IC50 0.13 μM) was >100‐fold more active in this assay than the standard LAT1 inhibitor JPH203, which entered very recently phase I clinical studies. [16]
The ability of BraA (1 a), BraC (3), BraE (5) and BraG (7) to interact with LAT1 in LN229 cells was further determined using a radiolabel uptake assay (Table S7). All tested compounds showed very good to moderate affinity for [3H]‐gabapentin, a LAT1 selective substrate, in an uptake competition assay. BraA (1 a), BraC (3) and JPH203 displayed IC50 values of 40, 80 and 29 nM, respectively, and represent the most potent molecules of this series.
The cytotoxic activities of BraA (1), C (3), E (5) and G (7) were tested in tumor cell lines and 3T3 fibroblasts after 72 h incubation. IC50 values were higher than 5 μM for all the cell lines and brasilicardins tested, except for the Jurkat cell line, which is a lymphocyte cell model. Again, the effect of BraA (1) in Jurkat cells was more potent than those of the other brasilicardins (Table S8). The new availability of the compounds BraA (1 a) and C (3) also permitted the first in vitro ADME evaluation. Firstly, the stability of both compounds was tested in gastrointestinal fluid, hepatocytes and plasma. The stability of 1 a and 3 at 1 μM in simulated gastric fluid (SGF) containing pepsin and simulated intestinal fluid (SIF) containing pancreatin was assessed at 37 °C. Both compounds were stable for more than 4 h under those conditions (Table S12), and their calculated half‐lives (t 1/2) were greater than 8 h. Next the stability and intrinsic clearance of both compounds at 1 μM was assayed using human and mouse hepatocytes (5×105 cells mL−1) (Table S14). In the presence of human hepatocytes, both compounds were stable with calculated t 1/2s greater than 6 h. In the presence of mouse hepatocytes, 3 was also stable with a calculated t 1/2>6 h. In contrast, BraA (1 a) exhibited a calculated t 1/2 of 296±97 min, and an intrinsic clearance (CLint) of 4.686±1.539 μL min−1/106 cells, suggesting some hepatic metabolism. In stability tests using human and mouse plasma, both compounds were stable with calculated t 1/2s>8 h. Finally, a Caco‐2 cell monolayer assay was conducted to determine the intestinal permeability, and identify specific transport and intestinal metabolization events (Table S13). In the Caco‐2 monolayer assay, at a concentration of 5 μM, BraA (1 a) exhibited mean apical‐to‐basolateral permeability (Papp(A→B)) of 0.01×106 cm s−1 and Papp(B→A) of 0.05×106 cm s−1, while BraC (3) exhibited a Papp(A→B) of 0.06×106 cm s−1 and Papp(B→A) of 0.07×106 cm s−1. The intrinsic Caco‐2 transwell permeability was low for both compounds, and the ratio (B→A)/(A→B) for 1 supports the notion that 1 may be a substrate for P‐gp efflux transporters. [17]
In conclusion, we have reported the identification and consecutive development of a heterologous producer strain that enables the sustainable production of the stereochemically complex natural products BraC (3) and BraE (5) at excellent rates. In addition, the new derivative BraG (7) was isolated and structurally elucidated. Furthermore, the biotechnological approach was complemented with a short 5‐step semi‐synthesis, which enabled the conversion of BraE (5) to the target compound BraA (1 a). This coupled system provides a) novel, economically viable access to BraA (1 a) itself, and b) the opportunity to readily generate derivatives, either from 3 or 5, in order to conduct SAR studies and optimize its pharmacologic, pharmacokinetic and biopharmaceutical properties.
In addition, we conducted the first ADME studies of these compounds, which provide a promising basis for further drug development since no red flag is raised to hamper the further development of brasilicardins. The therapeutic potential of this compound class became even more significant since it became apparent that the target LAT1 is also related to other diseases, particularly cancer. [18] Therefore, the established platform will be useful to develop derivatives for this indication.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The work of Carmen Méndez was funded by Ministerio de Economía y Competitividad (MINECO) (PCIN‐2014‐066). The work from Francisco Morís were funded by Ministerio de Economía y Competitividad (MINECO) (PCIN‐2014‐097). The results from Harald Gross and Pierre Koch were funded by Bundesministerium für Bildung und Forschung (BMBF) (FKZ 031A568A). The work of Wolfgang Wohlleben was funded by Bundesministerium für Bildung und Forschung (BMBF) (FKZ 031A568B). The results from Jolanta Zakrzewska‐Czerwińska were funded by MNiSW Narodowe Centrum Badań i Rozwoju (NCBR) (ERA‐NET‐IB/NeBrasCa/10/2015). Open access funding enabled and organized by Projekt DEAL.
A. Botas, M. Eitel, P. N. Schwarz, A. Buchmann, P. Costales, L. E. Núñez, J. Cortés, F. Morís, M. Krawiec, M. Wolański, B. Gust, M. Rodriguez, W.-N. Fischer, B. Jandeleit, J. Zakrzewska-Czerwińska, W. Wohlleben, E. Stegmann, P. Koch, C. Méndez, H. Gross, Angew. Chem. Int. Ed. 2021, 60, 13536.
Contributor Information
Prof. Evi Stegmann, Email: evi.stegmann@biotech.uni-tuebingen.de.
Prof. Pierre Koch, Email: pierre.koch@uni-tuebingen.de.
Prof. Carmen Méndez, Email: cmendezf@uniovi.es.
Prof. Harald Gross, Email: harald.gross@uni-tuebingen.de.
References
- 1. Stegall M. D., Morris R. E., Alloway R. R., Mannon R. B., Am. J. Transplant. 2016, 16, 1094–1101. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Srinivas T. R., Meier-Kriesche H.-U., Clin. J. Am. Soc. Nephrol. 2008, 3, S101–S116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Söderlund C., Radegran G., Transplant. Rev. 2015, 29, 181–189; [DOI] [PubMed] [Google Scholar]
- 2c.J. Andrade-Sierra, P. A. Vazquez-Galvan, H. Hernandez-Reyes, L. A. Mercado-Jáuregui, J. S. Chávez-Iñiguez, E. González-Espinoza, B. Gómez-Navarro, J. I. Cerrillos-Gutiérrez, Current Status and Future Challenges (Ed.: G. Tsoulfas), IntechOpen, 10.5772/intechopen.77292, 2018. [DOI]
- 3.
- 3a. Komaki H., Nemoto A., Tanaka Y., Takagi H., Yazawa K., Mikami Y., Shigemori H., Kobayashi J., Ando A., Nagata Y., J. Antibiot. 1999, 52, 13–19; [DOI] [PubMed] [Google Scholar]
- 3b. Shigemori H., Komaki H., Yazawa K., Mikami Y., Nemoto A., Tanaka Y., Sasaki T., In Y., Ishida T., Kobayashi J., J. Org. Chem. 1998, 63, 6900–6904; [DOI] [PubMed] [Google Scholar]
- 3c. Komatsu K., Tsuda M., Shiro M., Tanaka Y., Mikami Y., Kobayashi J., Bioorg. Med. Chem. 2004, 12, 5545–5551. [DOI] [PubMed] [Google Scholar]
- 4. Usui T., Nagumo Y., Watanabe A., Kubota T., Komatsu K., Kobayashi J., Osada H., Chem. Biol. 2006, 13, 1153–1160. [DOI] [PubMed] [Google Scholar]
- 5. Jung M. E., Chamberlain B. T., Koch P., Niazi K. R., Org. Lett. 2015, 17, 3608–3611. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Anada M., Hanari T., Kakita K., Kurosaki Y., Katsuse K., Sunadoi Y., Jinushi Y., Takeda K., Matsunaga S., Hashimoto S., Org. Lett. 2017, 19, 5581–5584; [DOI] [PubMed] [Google Scholar]
- 6b. Yoshimura F., Itoh R., Torizuka M., Mori G., Tanino K., Angew. Chem. Int. Ed. 2018, 57, 17161–17167; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17407–17413. [Google Scholar]
- 7. Hayashi Y., Matsuura N., Toshima H., Itoh N., Ishikawa J., Mikami Y., Dairi T., J. Antibiot. 2008, 61, 164–174. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Buchmann A., Eitel M., Koch P., Schwarz P. N., Stegmann E., Wohlleben W., Wolański M., Krawiec M., Zakrzewska-Czerwinska J., Méndez C., Botas A., Núñez L. E., Morís F., Cortés J., Gross H., Genome Announc. 2016, 4, e01391-01316; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Schwarz P. N., Buchmann A., Roller L., Kulik A., Gross H., Wohlleben W., Stegmann E., Biotechnol. J. 2018, 13, 1700527; [DOI] [PubMed] [Google Scholar]
- 8c. Schwarz P. N., Roller L., Kulik A., Wohlleben W., Stegmann E., Synth. Sys. Biotechnol. 2018, 3, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones A. C., Gust B., Kulik A., Heide L., Buttner M. J., Bibb M. J., PLoS One 2013, 8, e69319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malmierca M. G., González-Montes L., Pérez-Victoria I., Sialer C., Braña A. F., García-Salcedo R., Martin J., Reyes F., Méndez C., Olano C., Salas J. A., Front. Microbiol. 2018, 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bode H. B., Bethe B., Hofs R., Zeeck A., ChemBioChem 2002, 3, 619–627. [DOI] [PubMed] [Google Scholar]
- 12. Shigemori H., Komaki H., Yazawa K., Mikami Y., Nemoto A., Tanaka Y., Kobayashi J., Tetrahedron Lett. 1999, 40, 4353–4354. [Google Scholar]
- 13.
- 13a. Du F.-L., Yu H.-L., Xu J.-H., Li C.-X., Bioresour. Bioprocess. 2019, 1, 10; [Google Scholar]
- 13b. Wang Q., Quan S., Xiao H., Bioresour. Bioprocess. 2019, 6, 6; [Google Scholar]
- 13c. Lim H., Park J., Woo H. M., J. Agric. Food Chem. 2020, 68, 10780–10786. [DOI] [PubMed] [Google Scholar]
- 14. Jung M. E., Koch P., Org. Lett. 2011, 13, 3710–3713. [DOI] [PubMed] [Google Scholar]
- 15. Kiso Y., Ukawa K., Akita T., J. Chem. Soc. Chem. Commun. 1980, 101–102. [Google Scholar]
- 16. Okano N., Naruge D., Kawai K., Kobayashi T., Nagashima F., Endou H., Furuse J., Invest. New Drugs 2020, 38, 1495–1506. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Choi Y. H., Yu A. M., Curr. Pharm. Des. 2014, 20, 793–807; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Liu X., Adv. Exp. Med. Biol. 2019, 1141, 13–100. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Enomoto K., Hotomi M., Endocrinol. Metab. 2020, 35, 227–236; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Zhang J., Xu Y., Li D., Fu L., Zhang X., Bao Y., Zheng L., Front. Chem. 2020, 8, 564809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary