

Abstract
“There's plenty of room at the bottom” (Richard Feynman, 1959): an invitation for (metalla)carboranes to enter the (new) field of nanomedicine. For two decades, the number of publications on boron cluster compounds designed for potential applications in medicine has been constantly increasing. Hundreds of compounds have been screened in vitro or in vivo for a variety of biological activities (chemotherapeutics, radiotherapeutics, antiviral, etc.), and some have shown rather promising potential for further development. However, until now, no boron cluster compounds have made it to the clinic, and even clinical trials have been very sparse. This review introduces a new perspective in the field of medicinal boron chemistry, namely that boron‐based drugs should be regarded as nanomedicine platforms, due to their peculiar self‐assembly behaviour in aqueous solutions, and treated as such. Examples for boron‐based 12‐ and 11‐vertex clusters and appropriate comparative studies from medicinal (in)organic chemistry and nanomedicine, highlighting similarities, differences and gaps in physicochemical and biological characterisation methods, are provided to encourage medicinal boron chemists to fill in the gaps between chemistry laboratory and real applications in living systems by employing bioanalytical and biophysical methods for characterising and controlling the aggregation behaviour of the clusters in solution.
Keywords: aggregation, (metalla)carboranes, nanomedicine, nanoparticles, protein corona
Before we can catch biological systems in their nanometre working dimension, we have to understand them on the physical (bulk and surface), chemical and biological level. Herein, we comprehensively review the current state of research and approaches in the field of boron cluster medicinal chemistry, highlighting successful approaches, exposing challenges and appealing to standardise the heterogeneous nanomedicine field.
1. Introduction
In the last 20 years, the number of publications on boron‐rich molecular cluster compounds, where specific cluster‐containing systems are investigated for a potential application in medicine, has constantly increased. Thus, reviews and anthologies are regularly published by different research groups, where the single contributions often overlap, both in terms of types of clusters reviewed and perspective of the work.[ 1 , 2a , 2b , 2c , 3 , 4 ] Despite this rich literature, the medicinal boron community is still a rather closed circle of scientists, and in the broader panorama of biological inorganic chemistry, boron‐based compounds are hardly included. Thus, at the international conferences of the BIC series (e. g., International Conference on Biological Inorganic Chemistry – ICBIC, European and Asian Biological Inorganic Chemistry Conferences – EuroBIC and AsBIC), which are the topical meetings worldwide, contributions from the boron community are extremely rare.[ 5a , 5b ] Furthermore, to date no boron cluster compounds have made it to the clinics, and clinical trials have been very sparse. [3] What is the reason for this? Why is a whole class of often so‐called very promising inorganic compounds, on which scientists have concentrated many efforts for more than two decades, not well integrated in the medicinal inorganic chemistry community?
In this review, we have targeted these questions using a series of examples from the available literature on boron‐based compounds and appropriate comparative studies from medicinal (in)organic chemistry, with a focus on polyhedral molecular clusters of type closo‐C2B10H12 (closo‐carboranes), nido‐[C2B9H12]− (nido‐carborate(−1)) and metal complexes of nido‐[C2B9H11]2− (nido‐carborate(−2), dicarbollide), namely [3‐Ln‐3,1,2‐MC2B9H11] (metallacarboranes, with Ln=ancillary ligands, and M=transition metal; Figure 1).
Figure 1.
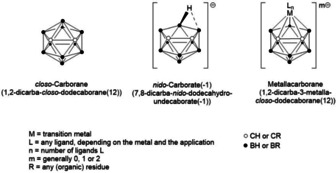
General structure of polyhedral boron cluster compounds discussed in this review. From left to right, closo‐carborane (only one isomer is shown), nido‐carborate(−1) and metallacarborane.
The confinement of boron‐based compounds in medicinal chemistry seems to be attributed to two main factors, which are inevitably correlated. First, in contrast to small organic and metal‐based drugs, hydrophobic (metalla)carboranes self‐assemble as micelle‐forming building blocks in aqueous systems, including those mimicking biological conditions. [6] Despite being a well‐investigated phenomenon, especially for anionic boron cluster compounds (ABCCs) such as the sandwich complex [commo‐3,3′‐Co(C2B9H11)2]− (COSAN),[ 7 , 8 , 9 , 10 ] its implications for the actual biological application of cluster‐containing drug candidates have been overlooked in the vast majority of the studies. Second, investigations by the medicinal boron community have traditionally been focused either on the chemical synthesis and characterisation of the target compounds (chemical identity), or on the biological activity of the drugs per se, but have mostly ignored the “bridge” between chemistry lab and, for example, tumour model, namely, the field of bioanalytics. [11] In drug discovery, bioanalytics focuses on the comprehensive, multi‐spectroscopic and ‐methodologic study of small drug molecules under simulated biological conditions, in terms of composition, temperature, ionic strength, drug concentration and interaction with biomolecules, with the ultimate scope of finding a rationale behind a drug‘s peculiar physicochemical properties and its solution behaviour. [12]
On the other hand, a vast number of organic compounds, many of which are approved drugs on the market, form colloidal drug aggregates (typically, 50–1000 nm) in high‐serum media,[ 13a , 13b , 13c , 13d , 14 ] which are the conditions to be considered for the pharmacological profile, or, in other words, for enabling translation from academic drug design to its final application. [15]
Colloidal drug aggregates have always been a problem in drug development, because they do not conform with routine methods of screening and analytical techniques. [16] Organic drug research makes intensive use of high‐throughput screening (HTS) tools, for “rational” identification of the most promising drug candidates. [17] The classical ones rely on structure–activity relationships (SARs), others on the best docking results, but they suffer from many false positive and negative hits.[ 14 , 18 ] Other newer algorithms search specifically for Pan Assay INterference compoundS (PAINS).[ 19a , 19b ] The Aggregation Advisor predictive tool approach is based on the chemical similarity to known aggregators and physical properties (http://advisor.bkslab.org). [20] It still suffers from false positives and false negatives, but it is a first step to structure the continuously increasing number of colloidal drug aggregates.
These PAINS are nowadays widely recognised as one of the most common causes of analytical and biological artifacts in early drug discovery.[ 21a , 21b ] Accordingly, for example the peer‐reviewed Journal of Medicinal Chemistry requires appropriate screening of suspected PAINS for publication. [22] Limited knowledge about the molecular interactions between small molecules and proteins complicates matters. The aggregation conditions are extremely sensitive to the composition of the medium (e. g., salts, protein nature and concentration), temperature, and additional excipients (e. g., detergents, such as polysorbate 80 or Triton X‐100).
Pioneers in the biophysical, biological and computational research on organic colloidal drug aggregates are without doubt the groups of Molly Shoichet at the University of Toronto (www.shoichetlab.utoronto.ca) and Brian K. Shoichet at the University of California‐San Francisco (UCSF, http://bkslab.org). Since 20 years, they focus on high‐throughput identification (experimentally and computationally) of promiscuous (organic) inhibitors forming aggregates at low micromolar concentrations that cause unspecific inhibition in assays,[ 20 , 23 ] which we believe should become an inspiration also for carborane chemists who want to apply their inorganic compounds in medicine. The term promiscuous inhibitors is now widely found in the literature to classify those compounds which inhibit a protein (often reversibly) via interaction of molecular aggregates (≈108 molecules), rather than the binding of individual molecules to specific protein pockets (Figure 2). [24]
Figure 2.
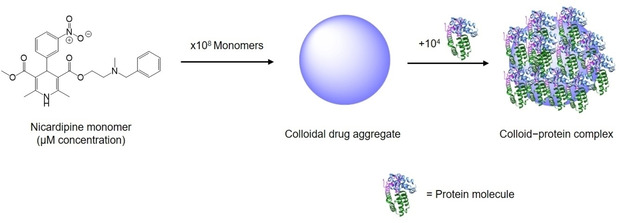
Simplified representation of colloidal drug aggregate and colloid–protein adsorption complex. Nicardipine (sold under the trade name Cardene®) is a calcium channel blocker used to treat high blood pressure and angina. Here, it is chosen as a representative example of a marketed drug that is a known colloidal aggregator. Adapted from ref. [11].
The nature of this small molecule aggregate–protein interaction is not trivial to elucidate and has been approached with a multitude of spectroscopic methods, for example, gel electrophoresis, fluorescence and electron microscopy, and light scattering techniques,[ 14 , 18 ] as well as appropriate formulation strategies to overcome problems such as polydispersity and transient stability in biological media. [25] It is still not fully understood, but it is rather clear that it is a surface adsorption phenomenon, highly dependent on high surface area and, probably, the apolar nature of the colloidal aggregates′ surface, which results in non‐site‐specific macromolecule sequestration. [23]
A rather intriguing feature is that colloidal aggregates seem to adsorb specifically on proteins, but not on other biomolecules such as DNA, and that the strength of adsorption is generally higher with large proteins than with smaller peptides. [25] This phenomenon is reminiscent of the spontaneous adsorption of proteins on nanoparticles (NPs) in a biological environment, known in nanomedicine as protein corona, which determines the biological identity of the nano‐entity and, thus, has a tremendous impact on physicochemical properties, biocompatibility and pharmacology of NPs.[ 26 , 27 ]
Although at first glance colloidal drug aggregates might seem unsuitable for application in medicine, these aggregates can be highly intriguing as delivery systems, because they are self‐assemblies of pure active drug and have nanoscale dimensions.[ 28 , 29 ] The key is to find the appropriate formulation strategy that allows sufficient physical and biological stability of the colloidal aggregates. The Shoichet siblings and co‐workers (2017, 2019) have shown that it is possible to produce stable co‐assemblies of colloidal drugs, such as fulvestrant and pentyloxycarbonyl‐(p‐aminobenzyl)doxazolidinylcarbamate (PPD), with the polymeric excipients polysorbate 80 (UP80) and poly(d,l‐lactide‐co‐2‐methyl‐2‐carboxytrimethylene carbonate)‐graft‐poly(ethylene glycol) (PLAC‐PEG), reaching drug loadings of 75 and 50 wt%, respectively. Such optimised formulations increased in vivo plasma half‐lives of the colloids, in comparison to their respective monomeric forms.[ 28 , 29 ]
Therefore, for carborane medicinal chemists the formation of colloidal aggregates in aqueous solutions should also receive appropriate attention and consideration in view of the final application of the compounds. A plethora of very sophisticated physicochemical and theoretical studies exist on the aggregation behaviour of ABCCs in water. Several explanations have been proposed since the early 2000s, trying to include these compounds in pre‐existing categories of self‐assembling substances,[ 8 , 10 , 30 , 31 , 32 , 33 , 34 ] or, more recently, to propose new terms and new classifications, more appropriate to the peculiar nature of the clusters, for example, (super)chaotropic ions or nano‐ions.[ 9 , 35 , 36 , 37 , 38 , 39 ] In contrast, only a few reports exist which suggest that also non‐ionic icosahedral (metalla)carboranes show a tendency to aggregate into nano‐ and micro‐entities in aqueous solutions.[ 40 , 41 , 42 , 43 , 44 , 45 , 46 ] However, investigations which translate the physicochemical self‐assembling behaviour to the biological concept of colloidal drug aggregates are missing, except our most recent work. [47] Thus, we have shown that neutral boron cluster compounds spontaneously aggregate in buffered solutions and/or in cell culture media. For example, the ruthenacarborane complex closo‐[3‐(η 6‐p‐cymene)‐1,2‐Me2‐3,1,2‐RuC2B9H9] forms polydisperse self‐assemblies in phosphate‐buffered saline (PBS) solutions, within 1 h after dissolution, in concentration as high as 109 particles mL−1, as revealed by nanoparticle tracking analysis (NTA; black trace in Figure 3, left). [48] An analogous behaviour was observed for the ortho‐carborane analogue of the cyclooxygenase (COX) inhibitor indomethacin, namely 1‐(1‐carboxy‐1,2‐dicarba‐closo‐dodecaboranyl)‐5‐methoxy‐2‐methyl‐1H‐indole‐3‐acetic acid methyl ester, known as indoborin, [49] with particle concentration of 108 particles mL−1, shortly after dissolution in PBS (orange trace in Figure 3, left). Furthermore, incubation of human breast adenocarcinoma cells (MCF‐7) with the ruthenacarborane derivative (30 μM, 72 h) revealed the presence of large agglomerates of the compound, which had precipitated from the culture medium (Figure 3, right).
Figure 3.
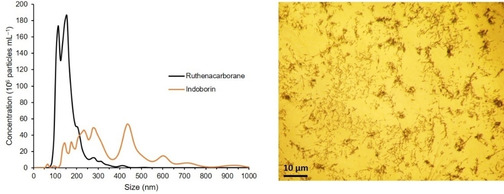
Left: Size distribution of self‐assembled nanoparticles of closo‐[3‐(η6‐p‐cymene)‐1,2‐Me2‐3,1,2‐RuC2B9H9] (denoted Ruthenacarborane) [48] and 1‐(1‐carboxy‐1,2‐dicarba‐closo‐dodecaboranyl)‐5‐methoxy‐2‐methyl‐1H‐indole‐3‐acetic acid methyl ester (denoted Indoborin), [49] in PBS/DMSO mixture, from NTA. Concentration for both compounds: 20 μM. DMSO content: 1.0 vol %, T=25 °C. Average data from five replicates are shown. Standard deviations are for a given concentration in the range of ±3.3–8.9×107 particles mL−1 and for a given size in the range of 2.4–26.9 nm. Right: Inverted‐light microscope image (40× optical zoom) of MCF‐7 cells, after 72 h of incubation with the ruthenacarborane derivative (30 μM). Dark spots are “big” agglomerates (i. e., precipitated aggregates) of the ruthenacarborane derivative. Experimental details of the NTA measurements and the cell culture experiment are given in ref. [53]; NTA measurements for indoborin are unpublished data (experimental details analogous to ref. [53]).
The biological performance of both compounds, the ruthenium complex and indoborin, has not been further investigated mainly due to concerns related to their very low water solubility and chemical stability (for indoborin), which may further hamper medical application, in addition to the self‐assembly behaviour. The nido analogue of indoborin should ensure at least better chemical stability under biological conditions. [50] As for the ruthenium complex, the observation of strong aggregation proclivity prompted further biophysical investigations on the self‐assembly of neutral metallacarboranes, namely ruthena‐ and molybdacarboranes, with the main scope of finding an appropriate formulation strategy to ensure biological stability for in vitro cell assays.[ 47 , 51 , 52 ]
Nonetheless, we just started to scratch the surface of a phenomenon that, for organic colloidal drug aggregates, has been investigated for over two decades. As for the latter, also (metalla)carborane colloidal drug aggregates can be seen as self‐assemblies of pure active drugs with nanometre size. Therefore, the carborane community should also refer to (metalla)carborane clusters in water as nanoparticles per se, and not only when the cluster is grafted onto the surface of metal nanoparticles. In turn, (metalla)carborane medicinal chemistry should form a new platform in the field of nanomedicine.
Today, established nanomedicine platforms are categorised into i) lipid‐based nanocarriers, ii) polymer‐based nanocarriers, iii) drug conjugates, iv) viral nanoparticles and v) inorganic nanoparticles (Figure 4), as elegantly summarised by Wicki et al. in 2015. [54] We suggest to add another platform which cannot be assigned to the existing ones, namely molybda‐ and ruthenacarboranes which provoke self‐assembly with bovine serum albumin (BSA) to form nanoparticles (Figure 4). [47]
Figure 4.
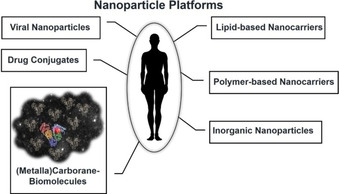
Schematic view of established nanotherapeutic platforms. Inspired by refs. [47, 54].
Inorganic nanoparticles are mostly metal NPs, such as iron oxide and gold NPs, developed for imaging and/or theranostic applications. Despite their promising therapeutic potential, no inorganic NP system has made it to the clinics to date, and only a few NP‐based delivery systems have reached clinical trials.[ 55a , 55b , 56 ] Major reasons are the inherent difficulty in studying the pharmacological profile of the NPs in biological fluids, clearly characterising their biological identity, besides the chemical one, and, for a long time, the lack of solid risk and safety assessment protocols in view of their implementation in vivo. [57]
Thankfully, two large institutions have been founded, the Nanotechnology Charaterization Lab (NCL, https://ncl.cancer.gov) in 2004, and the European Nanomedicine Characterisation Laboratory (EUNCL, http://www.euncl.eu) in 2017, which have developed and established a series of standard operating protocols (SOPs), also named assay cascades,[ 58a , 58b , 58c , 59 , 60 , 61 ] that serve as uniform recommendations for a well‐rounded and comprehensive characterisation of NP‐based therapeutics, with a clear focus on their final application in medicine. The developed step‐by‐step approach consists of four fundamental categories for characterisation of therapeutic NPs (Figure 5): 1) bulk characterisation, mostly dealing with size, size distribution and shape, 2) surface characterisation, in terms of surface‐grafted targeting ligands, PEGylation, zeta potential, 3) chemical characterisation, that is, chemical composition and identity, drug loading and release in formulation, and 4) biological characterisation, including protein corona, sterility and drug release in complex media.[ 62a , 62b , 63 , 64 , 65 , 66 , 67 ]
Figure 5.
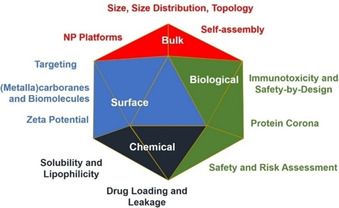
Classification of the step‐by‐step approach to therapeutic NPs comprehensive characterisation, as proposed by NCL and EUNCL.[ 56 , 64 , 67 ]
The goal of this review is thus to strongly advise the medicinal chemistry community to consistently regard (metalla)carboranes and borates as nanomedicine platforms and to arise awareness also of unreliable results, due to a lack of standard operation protocols. However, there are undoubtedly also very good studies including (metalla)carboranes and borates in line with good scientific practise references. We will highlight examples of characterisation experiments of boron cluster and nanosized compounds in view of the recommendations of EUNCL and NCL. As such, the review is organised into four main parts, following the fundamental categories for characterisation of therapeutic NPs (Figure 5), and ends with a recommendation for future directions for boron clusters in nanomedicine.
In each of the four main sections, the most relevant spectroscopic techniques will be briefly discussed, with a focus on the kind of chemical, physical, surface and/or biological information which can be derived when applied to NP‐based systems. The main focus is on the literature of (metalla)carboranes and borates in medicinal chemistry covering the last 10–15 years, with specific focus on contributions presenting experimental data, either in physicochemical or biological terms, which clearly suggest an aggregation behaviour of the clusters in aqueous solutions. Older publications are cited, when relevant. For a more comprehensive overview of all the most recent boron medicinal chemistry literature the reader is referred to refs. [1, 4, 68a,b].
2. Part I: Bulk Characterisation
From a nano‐object point of view, there are a few properties which define an object as nano‐entity, namely small size, theoretical description by quantum chemistry and size‐dependent properties, such as colour, solubility, material strength, electrical conductivity, magnetic behaviour, mobility and chemical and biological reactivity. Another physical nano‐property is the aggregation/agglomeration behaviour. The term nanoparticles (NPs) is used for assemblies of single particles or small particles in the nanometre scale, which can form aggregates, typically through covalent bonds, interactions from sintering or complex entanglements. These aggregates can then assemble to agglomerates, stabilised by Van der Waals forces or simple entanglements. All states are in equilibrium with the preceding state and convert into each other. [69] Other properties to investigate are structure, size, surface properties, porosity and charge.
An important term to introduce in this context is the polydispersity of the nano‐entity, meaning heterogeneity regarding size, shape and mass. Small changes in physicochemical properties might result in dramatic changes in secondary properties (e. g., biocompatibility, toxicity and in vivo behaviour). Therefore, it is desirable to have NPs being as homogeneous and defined as possible to easily predict or follow their changes in biological systems.[ 63 , 67 , 70 ]
Related to that, fundamental issues faced throughout the preparation of nano‐based drugs are the characterisation before and after administration, because of the variability between chemical and biological identity (see Section 5). For this situation, it is difficult to develop one quantitative analytical method.
2.1. Analytical techniques for determination of size, size distribution and topology
There are several techniques that can be used for characterising the size of nanoparticles. Each has its advantages, disadvantages and restrictions regarding the size range that can be accurately detected: 1–1000 nm (electron microscopy, EM; dynamic light scattering, DLS; asymmetric‐flow field‐flow fractionation multiangle light scattering, AF4‐MALS/DLS; size‐exclusion chromatography, SEC‐MALS/DLS), 20–1000 nm (laser diffraction, LD), 40–800 nm (NTA), 50–900 nm (tuneable resistive pulse sensing, TRPS) and 1–60 nm (Taylor dispersion analysis, TDA). [59] The most relevant techniques for the present review will be discussed here briefly (Figure 6). For a detailed description of the discussed and other additional techniques the reader is referred to Clogston et al. (2020). [59]
Figure 6.
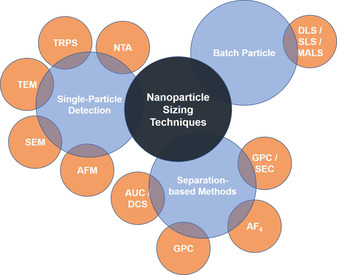
Summary of nanoparticle sizing techniques discussed in this section.
Nanoparticle sizing techniques can be essentially classified into batch particle, single particle and separation‐based methods. The most widespread technique to measure size distribution in batch suspensions is DLS, which detects time‐dependent fluctuations (Brownian motion) of the particles based on their scattering of the incoming laser light (called auto‐correlation function). After fitting of the auto‐correlation function, the NP diffusion coefficients are deduced, from which the hydrodynamic diameter can be determined by using the Stokes‐Einstein equation. [60] The same principles and devices are used for static light scattering (SLS), with the striking difference that the photon detector is moved around the sample. From the radius of gyration, the function of size and surface morphology can be obtained. This technique is usually used as a multiangle light scattering (MALS) setup and in conjunction with a separation step (e. g., AF4, SEC, etc.). [60] Importantly, these techniques assume a spherical shape of the particles and known viscosity of the solution; if these criteria are not satisfied, the results may be unreliable. In addition, major limitations are that for polydisperse samples unreliable data is collected, because the scattered intensity is proportional to the diameter to the power of six, causing large agglomerates to have a much bigger impact than smaller ones, which leads to a biased size distribution. Their use is, however, still justifiable, when sample integrity needs to be checked or the stability of sample formulation verified, under high salt concentrations, in strongly acidic or basic conditions or in the presence of plasma proteins. [59] On the other side, these are very fast measurements with an inexpensive setup and do not require highly specialised personnel.
One of the single‐particle techniques is NTA, a nascent technique for the measurement of colloidal and nanosized suspensions, which was first commercialised in 2006 by NanoSight Ltd. (UK). [71] The analysis principles and instrument setup have been extensively discussed in the literature.[ 72a , 72b , 72c , 72d ] NTA has been used for the study of different kinds of samples, ranging from atmospheric,[ 73a , 73b ] to food [74] and biological samples,[ 75a , 75b , 75c , 75d , 75e ] as well as engineered nanomaterials. [76] Often, the same samples have been analysed with different light‐scattering‐based techniques in parallel to NTA, for example, DLS, EM, atomic force microscopy (AFM) and flow cytometry, to provide experimental evidence of the validity of the former. NTA detects trajectories of each particle driven by Brownian motion over time and calculates the hydrodynamic radius of every detected particle using the Stokes‐Einstein equation. In comparison to DLS, for example, NTA gives number distributions instead of intensity‐weighted size distributions.
The single particle technique TRPS allows complex mixtures of nanoparticles with different size and shape, [77] zeta potentials [78] and concentrations to be analysed, [79] resulting in a high information output, [80] thus making it an emerging tool as a biosensor technique (e.g proteins, DNA, DNA−protein interactions, etc.). The simple setup can be described as two electrodes, separated by a membrane with only one pore, between which an ionic current flow is applied. When analyte particles pass through the pore, the potential drops during the transition event (so‐called “blockage” event), which causes “dips” in the potential vs. time plot. The peaks are characterised by a full width at half maximum (FWHM; particle velocity), a blockade duration (surface charge), a peak magnitude (Δi p; particle volume), peak shape (particle shape) and a peak frequency (events/min; concentration), resulting in the aforementioned information about the NPs. [81]
Other often used techniques are scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The scanning setup works according to the principle of scattered electrons, while TEM is based on transmitted electrons through ultrathin films. The scanning method allows a fast scanning of the whole sample's surface with a lower level of details compared to TEM, but is less restrictive (industrial metals, geological samples, biological specimens etc.) and involves facile sample preparation. [82] TEM shows characteristics such as morphology, internal composition and sub‐structures (as electrons pass through the sample), surface structures in a 2D picture, while SEM offers 3D projection of the sample. Field emission scanning electron microscopy (FE‐SEM) is the higher resolution variant of SEM, which works essentially in the same way, but is equipped with a field emission gun providing extremely focused high‐ and low‐energy electron beams. This allows the investigation of very sensitive specimen. [82] In contrast to TEM, the high‐resolution TEM (HR‐TEM) uses both the transmitted and the scattered beams to create an interference image. As information at atomic scale can be obtained, it is more often used for crystalline materials in materials science. For biological samples, usually the cryo‐TEM technique is employed, because it uses frozen samples, gentle electron beams and sophisticated image processing (to get 3D structures). [83] For all electron microscopy techniques, highly trained personnel and proper sample preparation are needed, and the machines are quite expensive.
Atomic force microscopy is a scanning probe microscopy for imaging surface structures on the nanometre to sub‐nanometre scale. A tip attached to the cantilever spring moves when adhesive interactions with the surface are present. These minor movements are detected by a position‐sensitive photodetector after irradiation of the cantilever through the laser. The AFM as a multifunctional nanotool measures a wide variety of mechanical properties of living systems and correlates them with well‐defined mechanical cues (even pressure, tension, adhesion, friction, elasticity, viscosity and energy dissipation of biological systems). [84]
Techniques based on separation of particles are gel permeation chromatography (GPC), which is also often used to determine molecular weight, purify and assess the physical stability of polymeric (nano)materials in organic solvents. As a subcategory of size‐exclusion chromatography (SEC) techniques, it separates polymeric analytes based on their size (hydrodynamic volume), in contrast to other chromatographic separation techniques which discriminate based on chemical or physical interactions – the bigger the particles, the faster the elution. GPC actually measures the molecular volume and shape function as defined by the intrinsic viscosity. Quantification is achieved by means of a UV/vis, refractive‐index or light‐scattering detector. A potential drawback could be the interaction of the NPs with the stationary phase interfering with the size determination accuracy. An important value obtained by this technique is the polydispersity index (PDI), which is calculated somewhat differently as from DLS, because it is the ratio of the weight‐average molecular weight (M w) and the number‐average molecular weight (M n), which is equal to 1 for uniform (monodisperse) samples. On the other side, the PDI for a peak from DLS is the square of its standard deviation divided by the square of its mean value. Therefore, a perfectly monodisperse sample would have a PDI of 0.0 from DLS. [85]
Another separation technique, asymmetrical‐flow field‐flow fractionation (AF4), reported for the first time already in 1966, [86] is very powerful, because it can be connected online to a large variety of analytical techniques corresponding to the properties of the analytes, e. g. UV/vis (to obtain qualitative information about drug loading of active pharmaceutical ingredients), ICP‐MS (inductively coupled plasma mass spectrometry; to elucidate chemical composition, impurities of inorganic particles or differentiation of shape/topology (spheric AuNPs vs. Au‐nanorods)), or light scattering techniques (to detect small changes in particle size distribution (consistency and stability), to check for NP modification in a biological environment after separation from plasma proteins or to conclude the particle shape from a DLS/MALS ratio or even the molecular weight (MALS+differential refractometer (DRI).[ 87 , 88 ] The apparatus consists of a flat channel with a height of 50–500 μm in which a parabolic laminar channel flow of a carrier liquid carries an injected sample from the inlet to the outlet. An additional crossflow, perpendicular to the laminar flow, locates the lighter particles in the middle of the chamber, which are then transported faster due the laminar flow profile, and in turn, separated from larger/heavier particles. [88] At present, this method is rarely used in nanomedicine (and not at all for carborane‐containing molecules), but its use is highly recommended for preclinical evaluation. [61] Admittedly, there are also some limitations for using this analysis as a routine method, mainly because the complex instrumentation requires trained personnel and the conditions must be adapted for each sample; thus, there are no standardised settings and specific/laborious method optimisations are needed. The possible loss of sample in the channel due to irreversible interactions with the channel surface should be considered as well. [60]
A technique called fluorescence correlation spectroscopy (FCS) is often used to study nanocarriers′ or nanoparticles′ formation, [89] drug loading,[ 90a , 90b ] stability, [91] interactions with plasma proteins,[ 92a , 92b ] and even the (triggered) release approach. [93] Recently, Negwer et al. were able to apply FCS to human blood serum and whole‐blood samples while monitoring the behaviour of drug nanocarriers in this complex medium. [94] This is somewhat remarkable as, in this way, the biological identity of nanoparticles under physiological conditions could be investigated, which nowadays is one of the major challenges in nanomedicine (see Section 5.2). To be best of our knowledge, there is only one research paper about (metalla)carboranes and FCS, [95] even though this technique is widely applied in nanomedicine.
Other interesting methods, which have so far not been reported in publications dealing with carboranes, are centrifugation techniques, such as differential centrifugal sedimentation (DCS) or analytical ultracentrifugation (AUC). Advantages are that no labelling is needed, and biologically relevant conditions can be used without unwanted interactions with surfaces or matrices, etc. Information that can be obtained comprises sedimentation/diffusion coefficients, size and shape of particles, mass and molecular weight, purity, oligomerisation and interaction with binding partners. For more information the reader is referred to the literature.[ 60 , 96a , 96b , 96c ]
The publication of Anderson and co‐workers (2013) shows nicely the differences between the various techniques (here TEM, DLS, TRPS, NTA/PTA, DCS) for both an individual and a mixed sample of monodisperse, submicron (220, 330, and 410 nm – nominal modal size) polystyrene particles. [97] The authors conclude that the particle size distribution (PSD) of complex samples must be investigated thoroughly by several techniques before comments or conclusions on the PSD can be made.
2.2. Nanoparticle platforms
Mainly three types of NP platforms are used for carboranes and metallacarboranes, namely i) polymer/co‐polymer matrices, ii) liposomes, and iii) inorganic NPs. These platforms are typically employed for promoting selective delivery and uptake of boron‐rich compounds (liposomes), or to enhance biophysical stability (polymer/co‐polymer), or as theranostic platform (inorganic NPs). In the following sections, some examples are discussed with a focus on physical aspects (even if they may contain also biological aspects).
2.2.1. Inorganic NPs
Inorganic NPs made of gold, silver or iron oxide are a common NP platform, used mainly because of their physical and chemical properties rather than their biological activities. Gold nanoparticles (AuNPs), for example, exist in a variety of sizes and shapes, and are well‐investigated for biomedical applications in imaging, as drug‐delivery platforms, as well as in photothermal (PTT) and photodynamic (PDT) therapy. Their toxicity in biological systems is highly dependent on the chemical composition of the surface ligands. [98] Be it a gold nanosphere (2–100 nm), nanorod (10–100 nm), silicon (core)‐gold nanoshell (100 nm) or a nanocage (40–50 nm), AuNPs can all be easily modified with a variety of coordinating groups (citrate, sulfide functionalised molecules, PEG, DNA, etc.). Basic properties of gold nanoparticles, such as the so‐called localised surface plasmon resonance effect (LSPR), allow the use of surface‐enhanced Raman spectroscopy (SERS), surface enhanced fluorescence (SEF) and non‐invasive in vivo and in situ detection methods, as well as imaging, PTT, PDT and in vitro diagnostics (IVD). [99] Additionally, radioactivity of synthetic isotopically enriched AuNPs (198Au: t 1/2=2.7 d and 199Au: t 1/2=3.2 d) facilitates radiotherapy and radionuclide imaging (RNI), and high X‐ray absorption coefficients increase the efficiency of radiotherapy sensitisation.[ 99 , 100 ] Furthermore, from a chemical point of view the fabrication procedures are easy to apply (different shapes, sizes, surface covers, etc.), as well as the surface functionalisation (polyvinylpyrrolidone (PVP) and tannic acid, PEG, BSA, or numerous other proteins, peptides, and oligonucleotides), and the concentrations in cells are easy to detect with ICP‐MS and UV/vis spectroscopy. [99] Silver nanoparticles are comparable with AuNPs and share many of the basic properties described above,[ 101a , 101b ] as well as applications, for example, in cancer nanomedicine. [102] Thus, due to this analogy, also carborane‐modified AgNPs were produced, analysed and applied in cell imaging and boron delivery to cancer cells. [103] Examples with carboranes will be discussed in Section 3.3.
There are also magnetic NPs based on Fe3O4 used in hyperthermia anticancer therapy, in which body tissue is exposed to high temperatures damaging or killing cancer cells. [104] In clinical trials hyperthermia has been studied often in combination with radiation therapy and/or chemotherapy. [105] The studies available containing carborane structures are very scarce and relatively new. Tulebayeva et al. (2018) synthesised magnetic Fe3O4‐NPs modified with (3‐aminopropyl)‐trimethoxysilane (APTMS) and capped with a di(ortho‐carborane‐1,2‐dimethyl) borate system and tested their physicochemical properties and stability (using DLS, zeta potential, TEM, FTIR, X‐ray diffraction); however, experimental details are only very poorly described and there are also major language problems. [106] In a follow‐up publication, stability tests in PBS buffer and in vitro cytotoxicity studies on this system were reported. However, the interpretation and conclusion are imprecise and inconclusive, and again the documentation of the experiment and wording have many deficits. [107]
2.2.2. Polymer/co‐polymer matrices
Polymeric and co‐polymeric matrices have been used in combination with metallacarboranes to produce loaded polymeric micelles with defined shape and size distribution, which could either improve the aqueous solubility of the metallacarborane molecules, [108] or stabilise colloidal suspensions of the latter. [109] The most widely used polymers are poly(ethylene oxide) (PEO), poly(methacrylic acid) (PMA) and poly(2‐vinylpyridine) (P2VP), but many others are also used, such as poly(2‐ethyl‐2‐oxazoline) (PEOX), poly(p‐phenylene oxide) (PPO), etc. (see series of publications by Matějíček and co‐workers discussed in the following). A common theme in the research of Matějíček and co‐workers over 10 years is the combination of COSAN with polymers, for example, with poly(ethylene oxide) (PEO), to form a metallacarborane/polymer complex with uniformly dispersed COSAN molecules in the polymer matrix, which is, however, insoluble in aqueous solutions. [109] The interaction between the two components is based on dihydrogen bonding between B−H and C−H in repeating ethylene oxide fragments in one part of the polymer, and interactions with the counterion in a space‐separated second unit (here Na+), verified by solid‐state NMR spectroscopy and wide‐angle X‐ray scattering (WAXS). [110] To increase the solubility in aqueous media, a second, more hydrophilic polymer was incorporated, namely poly(methacrylic acid) (PMA), which does not interact with COSAN molecules per se, but is sensitive towards changes in pH and the manufacturing process. In this way, core(COSAN‐PEO)‐shell(PMA) nanoparticles are spontaneously formed. [109] Fluorophore‐labelled COSAN derivatives were prepared with the PEO‐PMA block copolymer, where a 2 : 1 COSAN/fluorescein conjugate forms more defined rigid and spherical nanoparticles, whereas a 1 : 1 conjugate is less compact and irregular (probably due to higher hydrophilicity of the fluorescein moiety), verified by AFM and DLS. [111] Due to non‐biocompatibility, in subsequent studies the PMA fragment was exchanged with PEOX, which is also able to interact with the COSAN molecule resulting in hybrid gel‐like nanostructures. [112] Their sizes depend strongly on the preparation protocol, but not their composition and constitution. For elucidating this information (morphology, size, size distribution), a large ensemble of complementary techniques was employed, such a LS, AFM, TEM, small‐angle X‐ray scattering (SAXS), isothermal titration calorimetry (ITC) and 1H NMR spectroscopy.
Another study on the same polymer systems shows the versatility and the high loading capacity of the polymer‐NP platform based on the preparation procedure. A star‐shaped [PEO‐PMeOx]4 macromolecule, where PMeOx is poly(2‐methyl‐2‐oxazoline), forms nanoparticles with about 30 compartments and about 80 COSAN molecules in the centre of each compartment (with spacial proximity to PEO). [113] In another study, Matějíček and co‐workers employed a polymeric system of poly(ethylene oxide) (PEO) and two isomers of poly(vinylpyridine) (P2VP and P4VP) to analyse the principles of electrostatically driven self‐assembly (charge‐transfer‐assisted hydrogen bonding, B−H⋅⋅⋅(H+)−N), in contrast to the systems discussed before, where only weak unipolar B−H⋅⋅⋅H−C bonds were at play, using WAXS, advanced solid‐state NMR, and quantum chemical calculations. [114] These systems are called amphidynamic nanocomposites (long‐range molecular order with well‐defined site‐specific dynamics), and these findings and the principles behind could be useful for the design of (metalla)carborane‐loaded NPs.
The choice of a thermoresponsive block polymer system, for example, poly(2‐methyl‐2‐oxazoline)block‐poly(2‐n‐propyl‐2‐oxazoline) (PMeOx‐PPrOx), allows the use of a physical external trigger to release the loading (here COSAN). [115] In all the studies, cations have a crucial role, which was elucidated in a study from 2016 by Matějíček and co‐workers, who prepared a double‐hydrophilic block copolymer, poly(ethylene oxide)‐block‐poly(2‐alkyloxazoline) (PEO‐POX), assembled together with COSAN, which forms B−H⋅⋅⋅H−C dihydrogen interactions leading to compact nanomaterials (star‐like [PEO‐POX]4/M[COSAN] in 3 M solution of MCl, with M=Na+, Li+, and K+). The counterions (Li+, Na+ and K+) drive the affinity of the binding of COSAN to either of the two block copolymers, and thus, determine the compartmentalisation. [116]
The reported studies on polymer‐COSAN systems have been performed very thoughtfully by using orthogonal techniques and give sufficiently detailed experimental data.
2.2.3. Liposomes
Liposomes are used for encapsulation of active substances for use in different therapies. Their lipid bilayer construction consisting of hydrophilic and hydrophobic parts resembles cell membranes, but they often require a surface modification to facilitate cell penetration (see Section 3.2).[ 117 , 118 ] The difficulty here is to gain a satisfactory encapsulation efficiency of the biologically active loading in the lab and a quantitative and selective release at the place of action in biological systems. [118] In most cases, liposomes are thus loaded with molecules, including carborane‐containing ones (see Sections 2.3 and 3.3.3). Recently, Bregadze et al. (2020) reported cholesterol derivatives of closo‐dodecaborane, cobalt and iron bis(dicarbollide), which form liposomes together with hydrogenated (Soy) L‐α‐phosphatidylcholine (HSPC) and 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanol‐amine‐N‐[methoxy(polyethylene glycol)‐2000] (DSPE‐PEG). [119] DLS and TEM experiments were performed to identify the size distribution of the new liposomes and the lipid bilayer structure. One sample with different sizes was presented, however, without showing the size distribution from TEM experiments; unfortunately, no detailed information was given on the DLS measurements. For liposome characterisation, some data (diameter, PDI, zeta potential) was collected before and after lyophilisation. Finally, the encapsulation efficiency (boron and cobalt content) was determined by UV/vis spectroscopy showing moderate to excellent entrapments; however, few experimental details were given. Confirmation of these results with another quantification technique (e. g., ICP‐OES) and a focus on the biological stability, besides the physical stability, would have been beneficial prior to in vitro experiments.
2.3. Self‐assembly
While researchers in medicinal chemistry are certainly aware of the possibility of self‐aggregation of potential new drugs, this effect is not always considered in the characterisation. The same is true for (metalla)carboranes and borates. COSAN is the most investigated metallacarborane in terms of self‐aggregation properties or interaction with biological systems, but as yet, no general consensus was found regarding the behaviour in aqueous solution when used in aqueous biological media.
On its website, the journal Nature defines self‐assembly as: “the process by which an organised structure spontaneously forms from individual components, as a result of specific, local interactions among the components.” [120] For highly hydrophobic substances in aqueous media, we would like to extend this definition to …among the components, but also due to the interactions between the components and their environment, as especially the latter plays an important role in this context.
Figure 7 summarises the most important parameters which play a role in the self‐assembly of carborane‐containing structures and are discussed in the following sections.
Figure 7.
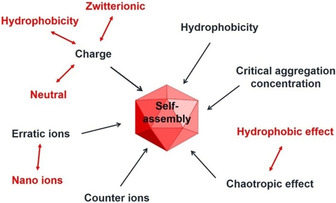
Parameters with the largest impact on self‐assembly.
One of the earliest basic studies on self‐assembly in 2001 and 2005 were conducted by Hawthorne and co‐workers on ortho‐, meta‐ and para‐carboranes mono‐ and di‐C‐substituted with aminoalkyl chains of varying lengths, resulting in amphiphilic structures which spontaneously formed self‐assembled rod‐shaped micro‐ and nanostructures upon sonication in aqueous solutions.[ 40 , 41 ] TEM and optical microscopy showed that the orientation of the side chains (ortho, meta, or para, affect the dipole moment, little effect), the side‐chain length (little effect), the counterion identity (varying effect), the number of the side chains attached, and the hydrophobicity (high impact) have an effect on the self‐assembly. These conclusions run like a golden thread through the whole story of influencing parameters on self‐assembly of carboranes.
One of the early studies on the “Molecular assembly of metallacarboranes in water”, published in 2006 by Matějíček et al., [8] starts with a discussion about why it is important to understand and characterise the self‐assembly behaviour in water of pharmacologically active compounds. Our reason would be, because it definitely has an effect on pharmacokinetics, biological activity and specificity of interactions. A combination of orthogonal techniques for studying size, shape, dispersity, and behaviour of COSAN (light scattering, microscopy, conductometry, gel permeation chromatography) was employed. Each technique was critically discussed, and conclusions were drawn with diligence. An equilibrium size of 115 nm was concluded for the discussed molecules above a certain concentration, as well as the potency for a secondary aggregation upon dilution. [8] However, no comment on the influence of the observed aggregation on the HIV protease inhibition ability of COSAN was included.
In a follow‐up study, [33] they report on closo‐carborane‐, nido‐carborate‐ and COSAN‐conjugated nucleosides, as an interesting platform for selective delivery of boronated agents for BNCT. Inspired by several theoretical and experimental reports on self‐assembling (metalla)carborane compounds, they analysed the influence of size, charge, exoskeletal substitution pattern and type of cluster on the self‐assembly behaviour in water. They critically assessed techniques used, commenting on qualitative versus quantitative information one can infer from the respective methods (e. g., DLS/SLS and AFM). Calculation of partition coefficients (n‐octanol/water, P ow or log P ow) is also treated critically: it is indicated as a useful parameter for predicting self‐assembly, but the reader is invited to use caution, as to not over‐interpret the practical significance of partition coefficient values (see also Section 4.2). In the concluding remarks, a very important aspect that needs to be considered when choosing a lead structure for biological applications is mentioned: hydrophobic and hydrophilic interactions are highly depending on the nature of the clusters (as discussed before). Thus, nucleosides with attached neutral (e. g., closo‐carborane) or bulky charged clusters (COSAN) aggregate spontaneously, but molecules containing smaller and charged nido‐carborate(−1), as well as unmodified nucleosides can be expected as true solutions or oligo‐molecular associates not observable by DLS. Furthermore, zwitterionic species show highly increased agglomeration tendency being sparingly soluble in aqueous solution. [33]
Recently, self‐aggregating ionic boron clusters have been called “erratic ions”, defined as ions of irregular chemical composition and hydration shell, but also based on their atypical solution behaviour compared to classical ions or surfactants. [39] These irregular ions are proposed as nano‐ions, which should be considered as building blocks for nanomedicine and materials science, that is, not treated just as ions, with only the classically associated properties of salting‐in/‐out effects and ion‐pairing for describing their interactions with co‐solutes (e. g., counterions, biological membranes, synthetic water‐soluble polymers). It is stressed that there is an urgent need to go beyond the dichotomy cosmotropic/chaotropic, a model that is too simple to explain solvation and (de)hydration of nano‐ions at a molecular level. For nano‐ions with low charge density and a certain surface activity, long range hydrophobic interactions between hydrophobic surfaces and the structure of water in the vicinity of hydrophobic nano‐ions need to be considered.
In the case of COSAN, of the two characteristics negative charge and high hydrophobicity, the second predominates in most cases in the interaction with the surrounding, especially in water, in which the corresponding solute needs polar/charged functionalities to interact. Molecular dynamics studies on the n‐octanol/water system also predict COSAN to be dissolved completely in the n‐octanol phase rather than in water; [32] the same results were obtained for chloroform [31] and 1,2‐dichloroethane instead of n‐octanol. [34] Interestingly, one feature of H[COSAN] that was only described theoretically is a capability to extract different counterions (such as Na+, K+, Cs+, H3O+ as close contact, or very hydrophilic [Eu(H2O)9]3+ and [UO2(H2O)5]2+ as hydration shell‐separated moieties) from water into the organic phase. This complex theoretical experiment was critically assessed regarding model settings and parameters to avoid computational artefacts (e. g., different solvation models and grids for counterion positions within the two phases and at the interface). [31]
The use of orthogonal techniques such as surface‐tension measurements and nonlinear optics (second harmonic generation, SHG), is a good example for the importance of cross‐validation by at least two independent techniques to prove characteristics of a compound. It could be shown that H[COSAN] adopts, in relation to its long axis, an orientation orthogonal to the air–liquid interface, [35] facilitating the intermolecular formation of B−Hδ−⋅⋅⋅δ+H−C interactions at the interface. An experimental study employing several advanced techniques for size determination showed nicely the complexity of the aggregation states of H[COSAN] in aqueous solutions depending on the concentration. [35] Monolayer vesicles were formed at low concentrations in water (critical aggregation concentration, CACvesicle=0.01 mM), with a radius of approximately 20 nm (at concentrations between 0.02 and 0.69 mM), which was determined by small angle neutron scattering (SANS) and SLS. However, when reaching the critical micelle concentration (CMC), coexistence of vesicles and small micelles (r mic=1.16 nm) could be confirmed by SANS measurements (CMCmic,SANS ≈ 18.6 mM). [30] Cryo‐TEM experiments (above CMCmic) confirmed the sizes of the micelles and vesicles as well as their coexistence. [30] These are great findings with highly elaborated experiments, but the results also imply sombre prospects for the practical application in complex biological systems, especially considering the NaCl titration experiments with a solution containing vesicles (H[COSAN]=3.7 mM in H2O) showing an almost linear increase of the diameter from 80 to 890 nm (by DLS).
1,2‐Dicarba‐closo‐dodecaborane functionalised with β‐d‐galactopyranose and β‐d‐glucopyranose derivatives were mixed with a spin probe (5‐doxyl stearic acid (5‐DSA) nitroxide), to study the interaction with a cationic liposome based on di‐oleoyltrimethylammonium propane (DOTAP) and l‐α‐di‐oleoylphosphatidyl‐ethanolamine (DOPE). [42] The findings (using EPR spectroscopy and DLS) suggested that the carborane‐sugar derivatives and the spin probes are hosted in the liposomal bilayer, without increasing the polydispersity and the hydrodynamic diameter of the liposomes (ca. 125 nm). Concentration‐dependent aggregation was studied (qualitatively) and an attempt was made to quantify the extent of aggregation. We highlight the critical comments on the limits of each technique for interpretation of the results, and the specific statement that self‐assembly must be considered when using these types of compounds in aqueous media. The aggregation properties in solution of a different sugar‐based derivative, namely [1,2‐dicarba‐closo‐dodecaboran‐1‐yl‐methyl](β‐d‐galactopyranosyl)‐(1→4)‐β‐d‐glucopyranoside, were investigated by NMR spectroscopy (1H, 13C relaxation, 1H,1H NOESY/ROESY) and ab initio calculations under both aggregating and non‐aggregating conditions. [121] The found aggregates showed rapid exchange with the bulk and high sensitivity to temperature changes. Interestingly, strong intramolecular hydrogen bonding (CHcarborane⋅⋅⋅Osugar) could be observed in both aggregating and non‐aggregating states, which are, however, not responsible for the aggregation process, but only hydrophobic interactions.
A term often mentioned in the field of carboranes is the chaotropic effect, describing an intrinsic property of (chaotropic) anions to break up the intermolecular water structures and rather assemble with hydrophobic and neutral polar phases, in contrast to the hydrophobic effect which is a distinct assembling motif. [37] Consequently, chaotropic anions can disrupt hydrogen bonding between water molecules and proteins reducing the stability of their native state. [122] This concept is also used in conjunction with host‐guest complexation of carboranes with β‐cyclodextrins (β‐CD)[ 44 , 123 ] or with cucurbit[n]‐urils (CBn, n=5–8). CBn are a series of pumpkin‐shaped glycoluril oligomers that are water‐soluble macrocycles with unique recognition properties. Wang et al. (2018) presented a comprehensive multi‐spectroscopic study on orthogonal self‐assembly, guided by either the chaotropic effect (borates+CBn: exclusion complex) or hydrophobic effect (azobenzene (AZO)+CBn: inclusion complex). [124] The complexation could be followed by 1H NMR spectroscopy (for borate/CBn, or AZO/CBn, but not for borate/AZO/CBn, due to extensive aggregation), morphology changes by TEM and SEM, and changes in hydrodynamic radii via DLS. Remarkably, XRD structures of the exclusion complexes CB7/B12H11OH2− (1:2), CB7/B12Cl12 2‐ (1:1) and CB6/B12H12 2‐ (3:2) could be obtained. This study showed the development of interesting and promising multi‐responsive self‐assembled systems (pH change for exclusion complex, irradiation for inclusion complex), whose size, size distribution and shapes can be controlled. The new insights were used to expand this unique strategy for exploiting orthogonal phenomena (chaotropic/hydrophobic effects) for the ditopic couple dodecaborate anions (exclusion complex) and adamantane (inclusion complex) to create supramolecular self‐assemblies, with a focus on physical characterisation (DLS, TEM, SEM, ssNMR, UV/vis, etc.) and immobilisation of the new systems on stationary phases (i. e., silicon wafer). [125]
In aqueous solution, the aggregation of ionic metallacarboranes is already clearly described, but the question, what happens when interacting macromolecular components are additionally present, remained. Therefore, two model systems, incorporating one or two COSAN moieties and a fluorescein molecule, were used by Uchman et al. (2010) to investigate the aggregation behaviour in aqueous systems. [95] Addition of cyclodextrins or model lipid membranes was tested for controlling aggregation and enhancing solubility. The fluorescent probe was used to study the distribution in model systems using steady‐state and time‐resolved fluorometry, fluorescence correlation spectroscopy (FCS), and fluorescence lifetime imaging (FLIM). It was shown that both compounds can be loaded into phospholipid membranes, which is promising in terms of use of phospholipids as delivery system, but bears the risk that the compounds may get trapped in cellular membranes and do not reach the intended target. Additionally, differences between the studied model compounds in a model system, and “real” compounds (best without fluorescent probe which potentially changes the biological activity of the COSAN structures) in real biological systems were emphasised. [95]
In 2017, Musah and co‐workers reported a neutral meta‐carborane‐containing derivative of the amino acid cysteine (2‐amino‐3‐(1,7‐dicarba‐closo‐dodecaboranyl‐1‐thio)propanoic acid), which is one of the few publications taking self‐assembly of closo‐carborane derivatives into account. [45] Evaporation of the solvent from an aqueous solution of the new substance gave fibrils, which can be converted into florets by resuspension in ethanol followed by evaporation (verified by FE‐SEM). The same chemical composition of the differently shaped nano‐structures was verified by energy dispersive X‐ray spectroscopy (EDS). TEM was also attempted, but the sample preparation seemed to be problematic. DLS gave a size distribution, but since this sizing technique assumes a spherical particle shape, which is, based on FE‐SEM, evidently not the case, the significance is limited. Circular dichroism (CD) spectroscopy experiments indicated the formation of secondary structures at a certain substrate concentration which could be related to the self‐assembly formation. [45]
The topics “self‐assembly” and “medicinal chemistry” are new to the boron community; it is difficult and labour‐intensive to establish new standard protocols for physical characterisation, because the majority of techniques requires more in‐depth expertise in synthetic and biophysical research and in handling of the respective equipment. However, the promising examples presented in the nanomedical literature prove that this is worth the effort. An important and general conclusion is that the strongly hydrophobic character of the carborane dominates the behaviour of compounds or materials in aqueous solution independent of their derivatisation.
3. Part II: Surface Characterisation
When talking about surface characterisation of NPs or colloidal aggregators, we specifically refer to chemical modifications of the surface to impart specific properties, and to surface‐specific properties, that is, not shared by the bulk, which determine the nature and extent of NPs interaction with the surrounding environment. The following section will thus be focused on two main aspects, namely zeta potential, as fundamental parameter in the characterisation of a nano‐surface in a given environment, and targeting strategies, which exploit a particular surface functionalisation pattern (active targeting), or a specific surface property (triggered release). A brief excursus on passive targeting will also be discussed since it is omnipresent in the medicinal chemistry literature of carboranes and metallacarboranes.
3.1. Zeta potential
The so‐called zeta potential (ζ‐potential), which can be determined, for example, with the same apparatus as DLS, [126] is a parameter describing the electrochemical equilibrium at interfaces and is a cornerstone of the theory by Derjaguin, Landau, Vervey, and Overbeek (DLVO theory) for colloidal dispersion stability. Since the values of the electrostatic potential on the imaginary slipping plane of a particle mirror the repulsion between particles or surfaces, the rule is that the higher the zeta potential, the stronger the repulsion and the more stable the system becomes.[ 127 , 128 ] As a consequence, the zeta potential is a system‐dependent property consisting of particles and their surrounding environment, which in turn determines the fate, behaviour and toxicity of NPs in environmental and biological systems. Depending on the surface modification of the particles (e. g., PEGylation) the potential distance from surface plot of the zeta potential differs (bare/hard particle, soft particle with thick charged layer or soft particle with thin charged layer, etc.). [127] Lowry et al. (2016) discussed model experiments consisting of three descriptors, namely the solution (e. g., ionic strength, pH, ionic composition, viscosity), the particle (e. g., concentration, size, composition, charge density, etc.) and the applied current. With methods such as DLS, TRPS or NTA, the so‐called electrophoretic mobility μ (EPM) can be determined in unit of [m2 V−1 s−1], which is the observed electrophoretic velocity v ([m s−1]), divided by the electric field strength E ([V m−1]). Applying a specific theoretical model (e. g., Henry's equation), a zeta potential (V) can be estimated. In the context of the studied system and the newly obtained information on the effective surface charge, predictions can be made on the interaction with surfaces or forecast of transport, transformation, toxicity or adsorbed biomolecules. [127]
When performing a zeta potential experiment on NPs with varying pH values, a parameter similar to the isoelectric point (pI) for proteins can be determined, which can give information on solubility issues,[ 129a , 129b ] programmed size changes leading to drug release, NP retention at tumour sites, easier penetration into tumours and escape mechanisms from endosomes and lysosomes. [130] There are several studies about carboranes using the zeta potential measurement,[ 131 , 132 ] but unfortunately often without considering the environment of the particles and without giving sufficient meta‐data for measurement conditions and the model used to convert electrophoretic mobility measurements into zeta potentials, which is required for using the zeta potential as an explanatory variable. [127]
3.2. Key principles of the targeting approach
Following Ehrlich's perspective of “magic bullets” in chemotherapy, three major physicochemical targeting strategies are used in nanomedicine. The oldest targeting principle is the passive targeting, which works according to the principle of alleviated perfusion. Mainly, first‐generation nanomedicine drugs use this way to diffuse into cancer cells (or in direct vicinity of the cells) through the leaky vasculature of the endothelial cells. Formulations like this typically improve the pharmacokinetics of the drugs, as shown for the well‐known cancer chemotherapeutics doxorubicin formulated in a PEGylated liposome (sold as Doxil® (US) or Caelyx® (Canada)). [133] A 25‐fold increased concentration of doxorubicin was found at the tumour site, and additionally, significantly improved tumour growth inhibition and overall therapeutic efficiency compared to the free drug were observed, while having only minor and temporary systemic toxic effects. [133] The same strategy was used for sodium mercaptoundecahydrododecaborate (BSH) back in 1996, [134] when BSH was encapsulated into PEGylated liposomes which increased the amount of BSH in circulation after 24 h (7 % for conventional liposomes, 19 % for PEGylated liposomes). At the same time, accumulation in liver and spleen was comparable, but the blood/reticuloendothelial system (RES) ratio was higher for the PEGylated liposomes indicating a higher amount of BSH in circulation, which however does not necessarily imply enhanced tumour uptake.
The passive targeting is confronted with some barriers. Abnormal tumour vascularity is one of cancer's hallmarks which causes heterogeneity of tumour blood flow and at the same time poor and heterogeneous perfusion. Further consequences are elevated interstitial fluid pressure (IFP) from constant extravasation of fluid, which, in turn, generates hypoxic and acidic intra‐tumoural conditions. The IFP limits the convection of nanosized drugs into the tumour tissue, in fact, even promotes passive diffusion out of the tumour. Furthermore, there is solid stress induced by tumour growth causing in turn cell compression, cell invasion, changed gene expression, apoptosis and cell‐related extracellular matrix production and organisation. [135]
An often discussed effect, especially in carborane medicinal chemistry, is the enhanced permeability and retention (EPR) effect, which describes a concept, in which a drug should preferentially accumulate within tumours through invasion allowed by leaky vasculature (gaps of ca. 0.1 to 3 μm) and poor lymphatic drainage.[ 136 , 137a , 137b ] On average, it only gives a twofold increase of nanodrug delivery in comparison to critical normal organs, namely liver, kidney or lungs. Thus, more possible (severe) side effects must be considered. However, there are possibilities to improve the efficiency of the EPR effect, which basically imply exploitation of cooperative effects from dual therapies or combination of different therapeutic agents. For example, a cytostatic/cytotoxic drug can be co‐administered with blood flow modulators (vasoconstrictors or vasodilators, e. g., nitric oxide, prostaglandins, etc.) or with growth factors to act directly on tumour vasculature and stroma. Combination with hyperthermia or sonoporation can also enhance vascular permeability, or an anti‐angiogenic treatment can help normalise the vasculature. Another option is to assist chemotherapeutic treatment with other apoptosis‐inducing techniques, such as radiation therapy, photodynamic, photothermal, radioimmunotherapy, or NIR‐photoimmunotherapy. [136] Nevertheless, it should be clear that the EPR effect is not the holy grail in nanomedicine; even though it is often used as an argument for carboranes to be of potential use in medicine, it is far from the aimed selectivity a drug should have to be employed as personalised medicine in future.
On the other hand, active targeting is a more elegant and effective way to treat diseases like cancer, but also more difficult regarding the development procedures in terms of synthesis, tuning the biophysical properties and investigation of the interaction in biological systems. The working principle is based on a specific target (biomolecule) being unique for the respective target cells, which is addressed with a vector (e. g., on the surface of a NP) in a lock‐and‐key pair approach to ensure selectivity. In principle, full internalisation of the nanocarriers is not needed, because the target receptor could be also present on the endothelial cells. Thus, either a depot is formed in the vicinity of a tumour, which slowly releases the loading, or recognition of the NPs by endothelial cells initiates release of small molecules, such as doxorubicin, which could passively diffuse through cell membranes. Some platforms, such as antibody–drug conjugates, even directly target the extracellular matrix.
For the already mentioned BSH‐loaded liposomes, the fundamental issue of selective or preferential accumulation in tumours was targeted using transferrin (TF)‐conjugated PEG liposomes, exploiting the TF receptor recognition and subsequent endocytosis. [138] The EPR effect was made responsible for the (passive) targeting. During the preparation process, the encapsulation efficiency of BSH into the PEG‐liposomes was determined to be only about 6–8 % and the boron content as 26–30 μg per μmol lipid. The focus of this study was only on biological aspects; therefore, no stability study and formulation protocol or self‐assembling properties of carboranes were investigated.
Very sophisticated is the triggered release approach. Suitable drugs are positioned in or in the vicinity of a tumour cell, followed by an internal pathophysiological or chemical stimulus such as changes in pH, redox potential, ionic strength, shear stress or presence of enzymes. Also, external (physical) triggers can be used, like temperature, light, ultrasound, magnetic force or electric fields. [54]
Important factors to be considered in design strategies are that the vector must bind to target cells (not healthy cells) and the ligand should be stable enough avoiding premature release and degradation. In addition, the density of targeting ligands on the surface of a nanoparticle platform must be optimised to avoid excessive interaction with serum proteins or the immune system, as well as to reach a certain targeting efficiency.
When performing inhibition assays to show the performance of an inhibitor, it is often observed that many colloidal aggregates exhibit strong unspecific protein adsorption on their surface. In the case of enzymes often there is a loss of enzymatic activity connected to that phenomenon, so random inhibition results are obtained. [139] This observation is important especially for hydrophobic carborane‐based compounds to avoid obtaining wrong results.
3.3. Examples of surface‐modified NP platforms
3.3.1. Inorganic NPs
For the surface modification of nanoparticle platforms (Figure 8), metal nanoparticles like silver NPs (AgNPs) have been modified with 45 % 1‐thiol‐1,2‐dicarba‐closo‐dodecaborane (mercaptocarborane, CBT) as boron source for BNCT, 10 % thiosuccinimidyl propionate (TSP) with molecules attached that target extracellular domains, such as the anti‐EGFR AB (epidermal growth factor receptor antibody), as well as 45 % of a passivating low molecular weight compound, (1‐mercaptoundec‐11‐yl)tri(ethylene glycol) (EG3SH), to saturate the remaining surface with a water‐soluble and water‐stable component and at the same time separate the CBT units. [103] Bright‐field optical images and high‐resolution SEM as well as SERS microscopy were used to characterise the labelled nanoparticles inside cells. In addition, optical spectroscopy helped to visualise the NPs due to LSPR of the silver atoms. With this approach, a boron‐loading of about 4.5×108 atoms, calculated as 10 boron atoms per carborane molecule, 9000 carborane molecules per AgNP and 5000 AgNP per cell, was achieved. [103]
Figure 8.
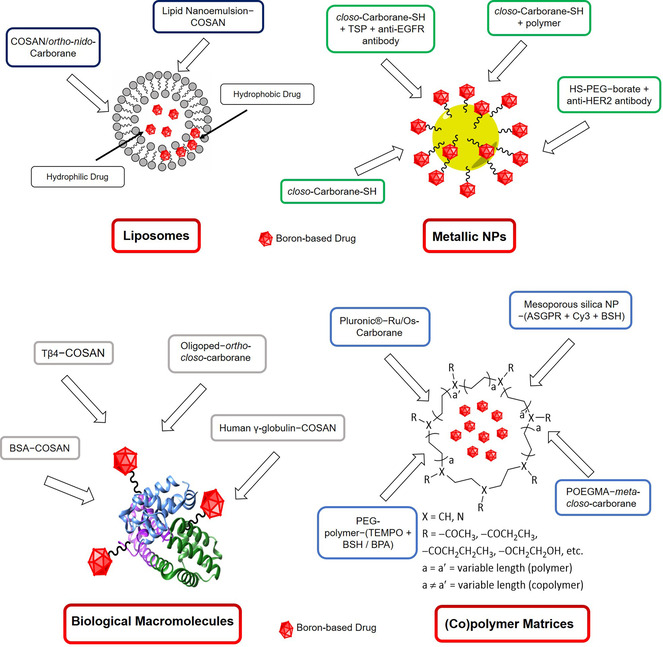
Overview of the discussed specific type of carrier systems (liposomes, metallic NPs, biological macromolecules and (co)polymer matrices) and their surface modifications.
Brust and co‐workers (2017) used the simple mercaptocarborane to cap small gold nanoparticles (2–3 nm), which can be used as artificial selective ion transporters (for Na+ and K+, but not Mg2+) across phospholipid membranes. [131] This study combined several techniques for orthogonal characterisation, such as voltage‐dependent fluorescence spectroscopy, potentiometric/potential step experiments, UV/vis, HR‐TEM, cryo‐TEM, zeta potential, DLS, mass spectrometry, and attenuated total reflection (ATR) infrared and NMR spectroscopy. Self‐assembling properties of carboranes were not discussed because AuNPs are used as a platform. Furthermore, in terms of a targeting principle, this would be assigned to passive targeting.
Baše et al. (2005) investigated a monolayer of mercaptocarborane derivatives on the densely covered surface of gold NPs. It was found that the mercaptocarborane molecules are also incorporated into the gold NPs. Interestingly, after thermal treatment of the loaded NPs, 1,2‐C2B10H10 fragments are released leaving behind the sulfur atoms on the gold surface. [140] These AuNPs loaded with mercaptocarborane are only soluble in organic solvents, but not in water. AuNPs coupled with 9‐SH‐ortho‐carborane or 9,12‐(SH)2‐ortho‐carborane will not solve the issue of water insolubility; therefore, another modification, namely a poly(ethylene oxide)‐b‐poly(caprolactone) diblock‐copolymer (PEO‐b‐PCL), was investigated by Ciani et al. (2013). [141] The step‐wise development of water‐soluble mercaptocarborane‐modified AuNPs was explained and confirmed by TEM data and microscopic observations during biological evaluation. It was intended to produce NPs that passively diffuse into the cell plasma of osteosarcoma cells (UMR‐106). However, having two thiol groups on the carborane did not change the properties of the nanoparticles regarding the size and boron uptake by the cell (13.5 ppm and 5.75 ppm for 9‐SH‐ortho‐carborane@AuNPs and 9,12‐(SH)2‐ortho‐carborane@AuNPs, respectively). While size information from DLS is discussed in much detail, TEM data are only briefly discussed.
Six years later, Kuo and co‐workers (2019) took up the knowledge from previous studies and used standardised AuNPs (20 nm diameter), covered them with thiolated PEG, azides (−N3, for further 123I labelling via copper(I)‐mediated click reaction), and dodecaborate (B) derivatives. [142] Additionally, the NPs were equipped with a self‐developed anti‐HER2 (human epidermal growth factor receptor 2) immunoglobulin G antibody (61 IgG) for receptor‐mediated endocytosis (trastuzumab was used as anti‐HER2 antibody reference). The biophysical characterisation of the NPs was kept to a minimum (only DLS), with a clear focus on the imaging (single‐photon emission computed tomography, SPECT/computed tomography, CT) in N87 gastric xenografts in mice and the quantification of the boron content in the tumour with ICP‐MS. The targeting approach worked out, because most of the 123I‐B‐AuNPs were detected in the liver, but in the case of the 123I‐61(IgG)‐B‐AuNPs, the largest amount of NPs was found in the tumour 12 h after injection.
More PEGylated AuNPs with star‐shaped and dendritic surface modifications have been reported, using click chemistry methods. This synthetic method provided bifunctional AuNPs with narrow polydispersity and without any hints of aggregation of AuNPs. [143] The publication is only focused on synthetic methods and physicochemical characterisation (DLS, TEM, infrared, NMR); no stability tests for intended biological application were conducted.
3.3.2. Polymer and co‐polymer matrices
A reasonable approach towards finding a functioning delivery system is the use of commercially available micelle‐forming symmetric triblock polymer Pluronic® P123, which can encapsulate highly hydrophobic exo‐coordinated [M(p‐cymene)(1,2‐dicarba‐closo‐dodecarborane‐1,2‐dithiolate)] (with M=Ru, Os; RuCb/OsCb). [108] However, this led to lower cytotoxicity against an ovarian cancer cell line (A2780: RuCb 0.17±0.02/RuCb@P123 6.69±0.33 μM; OsCb 2.50±0.09/OsCb@P123 117.50±0.18 μM), and against healthy lung fibroblasts (MRC5; RuCb 0.31±0.03/RuCb@P123 51.6±0.9 μM; OsCb n.d./OsCb@P123 n.d). Interestingly, the diameter of the micelles decreased upon encapsulation of the highly hydrophobic metallacarboranes, most likely due to expulsion of water molecules from the micelle, unlike for more polar molecules whose encapsulation led to an increase of size for the Pluronic® P123 system. [144] However, strangely, DLS and cryo‐TEM gave total diameters which were equivalent to only the core diameter of the core‐shell particles, but only half of the total diameter determined by SAXS. This publication competently highlights advantages and disadvantages of the concept of the use of micelles in drug delivery. The selectivity between malignant and benign cells can be enhanced, but often at the expense of cytotoxicity. The self‐assembling behaviour of metallacarboranes was, however, not addressed and other applications of the new formulated drugs were not considered, because cytotoxicity and accumulation for BNCT are in principle of opposite nature. [108]
Lai et al. (2013) prepared a target‐specific delivery system for BNCT based on mesoporous silica NPs (MSN), surface‐modified with a Cy3 fluorescent dye and cell‐targeting trivalent galactosyl ligands, which interact with the asialoglycoprotein receptor (ASGPR) being present on the surface of hepatic cancer cells (HepG2), enhance water dispersibility and inhibit leakage of ortho‐carborane loading. [145] The drug loading was relatively high (60 wt% boron atoms per MSN), and the drug delivery efficiency even surpassed the one of routinely used sodium BSH under the tested conditions. The MSN could be traced in the cells and a potential mechanism was proposed, which is uptake through endosomes, transport via lysosomes, release into cytoplasm and diffusion over the whole cell. Also, in this case the self‐assembling properties of carborane‐based molecules were not investigated, but rather a known nanosized system was used.
Yan and co‐workers (2018) used the same cancer type (HepG2), same target (ASGPR, with d‐galactose as targeting moiety), same therapeutic approach (BNCT), but a different NP platform (amphiphilic copolymer), incorporating a cyanine dye for NIR imaging. [146] The meta‐carborane (mCb) species was covalently attached to a self‐assembling copolymer (acryloyl‐d‐galactose pentaacetate, POGal‐PmCb‐PCL), which increased the polydispersity index slightly (from 1.18 to 1.27, based on GPC measurements). For determination of size and polydispersity, DLS, TEM and GPC were used giving concurrent results. For the in vitro cell imaging no delivery efficiency was experimentally determined, but instead, the respective feeds were taken as fully resorbed. In conclusion, the chemical identity of the NPs was studied, and first in vitro cytotoxicity and imaging studies were reported, but the biological identity of the NPs and the delivery efficiency remained unclear. The publication is closely related to previous work by the same group, in which a different polymer backbone, a longer linker between meta‐carborane and polymer backbone, and no sugar unit was used, but still the same dye (cyanine, Cy3; POEGMA‐PmCbA‐PCL‐Cy). However, the basic idea and the results were relatively similar. [147] As this group is focused on functional polymer nanocarriers, they have established their general platform (poly‐oligo(ethylene glycol)methacrylate, POEGMA), which is extended with the required functionalities. In another study they incorporated a pH sensitive unit, which released the loading (here doxorubicin, DOX) at pH<6.5 and was stable at pH>7.0. These changes in morphology and constitution could be followed by DLS, TEM and zeta potential measurements. [148]
With their study on nanoparticles made of a PEGylated anionic block copolymer (PEG‐b‐poly[(closo‐dodecaboranyl)‐thiomethylstyrene] (PEG‐b‐PMBSH) and a PEGylated cationic block copolymer (nitroxide radical‐containing cationic block copolymer PEG‐b‐poly[4‐(2,2,6,6‐tetramethylpiperidine‐N‐oxyl)aminomethylstyrene] (PEG‐b‐PMNT), Nagasaki and co‐workers (2016) aimed at reducing the side effect of an increase of ROS (resulting from γ radiation) during BNCT treatment. [149] New insight for the suitable design of these polymer‐based NPs was obtained. For example, TEMPO (2,2,6,6‐tetramethylpiperidine‐N‐oxyl) containing NPs could effectively lower the amount of ROS and the level of leucocytes, while both BNCT agents, BPA and BSH, increased the amount of ROS compared to the control. Furthermore, the covalent binding of BSH to the polymer prevented premature leakage of boron in blood circulation. Even though the uptake selectivity for PEGylated liposomes was higher for tumour cells (compared to healthy cells), leading to a 3.3‐fold increase of the uptake for the boron‐containing NPs relative to BPA, the absolute amount of boron delivered to tumour cells was higher for BPA in vitro. Nonetheless, benefits in in vivo studies can be expected, as the relatively long retention in the tumour tissue (5.5 % of the injected dose per gram (ID g−1) tissue at 48 h (and 72 h)) after administration of the NPs is high compared to BSH (<0.1 % ID g−1 was left in the blood circulation after 24 h). It should be mentioned that the presentation of the DLS and TEM (meta)data does not allow a meaningful comparison of both data sets.
3.3.3. Liposomes
Nakamura, Leśnikowski and co‐workers (2013) coupled cholesterol to nido‐ortho‐carborane and chromium, iron, or cobalt bis‐(dicarbollides) and incorporated these conjugates in a liposome membrane. [150] As it was speculated that the cobalt‐containing unit is “encrusted” on the surface of the liposome, we have included this paper in the section of this article dealing with surface modifications. The NPs were used as a boron delivery system, and in vivo biodistribution studies in colorectal carcinoma‐bearing mice (colon‐26) were performed. It was found that liposomes with a high content of cholesterol‐metallacarborane conjugates (1 : 1 molar ratio) induced liposome aggregation, although the metallacarborane effect on system aggregation is only briefly recalled in the publication. DLS measurements showed a polydisperse size distribution, indicating a heterogeneous sample mixture already before injection into mice which hampers the predictability of the fate of the NPs in vivo. Contrary to expectations, most of the boron was taken up by the RES system (324 ppm in spleen), presumably because of the instability of the liposomes in blood. This assumption was, however, not tested under simulated condition before starting in vivo studies, or at least not documented. However, a boron content of 43 ppm was found in the tumour after 24 h (42 ppm after 36 h, and 3–4 ppm in blood), which satisfies the theoretical requirements for effective BNCT (boron concentration in tumour >20 ppm and tumour/blood ratio >3). [151] Besides quantification of the boron content, the metal content was also determined in the same tissues, however, only for the cobalt‐containing derivative and not for the iron or chromium analogues. Furthermore, the nido‐ortho‐carborane derivative is only mentioned in the synthetic part.
Navascuez et al. (2020) used nanoemulsions (NEs) of the ω‐3 fatty acid DHA (docosahexaenoic acid) in water stabilised by COSAN molecules on the oil‐water interface for application as nanocarrier system for poorly water‐soluble drugs (here: [18F]‐16α‐fluoroestradiol). [152] Different sizing techniques were used (cryo‐EM, LS and DLS) which gave diverging results for the NEs (39±27 nm, 270 nm and 170 nm (PDI=0.18), respectively). The differences were explained with a high PDI, which is, however, in terms of liposomes (<0.3) [153] and nanoemulsions (<0.22) [154] still acceptable, and thus, probably not the whole truth. The stability of the COSAN‐NEs with water is remarkable, but it is probably very problematic that contact with salt‐containing solutions (here phosphate buffered saline, PBS) causes quick separation of the nanoemulsion into oil and aqueous phase. One conclusion was also that the stabiliser (COSAN) and the loaded drug are rapidly cleared from the lungs, which is, however, debatable.
3.4. (Metalla)carboranes and biomolecules
COSAN derivatives are by far the most widely studied metallacarboranes, and, surprisingly, they seem to work successfully in almost every kind of application. COSAN and its derivatives can stabilise nanoemulsions, [152] are effective antimicrobial and antibiofilm agents, [155] interact with DNA, [156] are potent and highly selective inhibitors of tumour‐specific carbonic anhydrase IX [157] and form robust shields on proteins. [158] They are not only potent in medical applications, but also capable of performing photo‐redox catalysis in water, [159] tune the properties of conducting organic polymers [160] or selectively extract lanthanide and actinide cations from solutions. [161]
However, some interpretations and predictions about COSAN in general should be treated with caution. For example, some authors[ 162 , 163 ] attest COSAN a good solubility in water, assuming solution by formation of a solvent shell around each individual molecule (referring to a study by Plešek et al. (1984) of <10−3 mol L−1 for most of the salts, which is about 324–500 mg L−1). [164] A similar solubility in water was also determined by Navascuez et al. (2020; ca. 500 mg L−1), which is a low to moderate water solubility according to the solubility scale of the U.S. National Pesticide Information Center (NPIC). [165] In contrast, several studies showed that COSAN aggregates in aqueous solutions. [166]
It was investigated if COSAN has the capability to enhance the activity of therapeutic peptides, specifically thymosin β4 (Tβ4), a peptide containing 43 amino acids, chosen by the authors based on its known biological functions in promoting tissue healing and regeneration. Due to the assumed binding of Tβ4‐COSAN derivatives to human serum albumin (HSA), the half‐life in blood circulation of the therapeutic peptide is potentially prolonged. [162] Binding constants with HSA were determined based on single steady‐state fluorescence experiments (Trp, λex=280 nm and λem=354 nm); however, experiments at three different temperatures or/and displacement experiments are needed for determination of the quenching mechanisms, as previously conducted and reported by the authors for the COSAN molecule alone, [163] since the system and the mechanism changed after derivatisation. The dissociation constants (KD) were determined by surface plasmon resonance (SPR) measurements under dynamic conditions in a CM5 chip setup, and the binding constants (Kb) were obtained by fluorescence quenching measurements. The discussion of the obtained data was done confidently and explanations for differences were given. While COSAN was involved in this study, the possibility of formation of nanoparticles with the peptide or HSA was not investigated.
Back in 1974, Lipscomb and co‐workers were the first to study the interaction of ionic boron hydride clusters with proteins (human γ‐globulin and bovine serum albumin). [167] Different salts of the different icosahedral carboranes or boron hydrides induced agglomerisation or precipitation of the proteins to a varying extent (0−100 %). This early publication already documented the aggregating interactions of carboranes with proteins. The idea of using this concept for applications in BNCT was highlighted in the title but was still in the far future at that time.
Very recently, Kaniowski et al. (2020) presented an automated synthesis of so‐called oligopeds, which are short DNA adapters (5’‐d(TTT CTT TTC CTC CAG AGC CCGA)‐3’ and the antisense sequence 5d’‐(TCG GGC TCT GGA GGA AAA GAAA)‐3’) connected to boron clusters (9,12‐bis‐functionalised 1,2‐dicarba‐closo‐dodecaborane). [46] When both corresponding 9,12‐bis‐functionalised carboranes (each carborane is functionalised twice with the same sequence of DNA 22‐mers) were mixed (1 : 1), the formed nanohybrids adopted torus‐like shapes, which were verified by AFM and cryo‐TEM. An MTT assay showed that the nanostructure does not interfere with the metabolic activity of HeLa cells. The new oligoped system seemed to be less effective in expression silencing than one of the non‐modified DNA 22‐mer under the tested conditions in breast adenocarcinoma (MCF‐7), squamous carcinoma (A431) and cervical carcinoma (HeLa); a prolongation of the incubation time (from 48 to 72 h), however, increased silencing ability. It was assumed that this effect is due to higher stability of the nanostructures. Unfortunately, it was not discussed whether the DNA 22‐mer without carboranes also forms nanostructures; this would have shown the impact of the carboranes on the biomolecules.
As carboranes will interact with biomolecules when used in medical applications, it is important to study and understand this phenomenon. On the other hand, synthetic design allows to influence when carboranes meet specific biomolecules, how they meet and what the results of the meetings are.
4. Part III: Chemical Characterisation
From a chemical perspective, nanomedicine means first of all synthetic accessibility of the target, namely nanosized potential therapeutics, which goes hand in hand with its structural characterisation (composition, binding motifs, spatial arrangement of functional groups), chemical stability (loading capacity, leakage, size evolution, etc.) and aqueous solubility properties.
NP drug design strategies mostly deal with targeted stabilisation and functionalisation of a metallic NP, such as gold or silver NPs, or with encapsulation of an organic drug in a polymer matrix or a liposome, or with conjugation of an active drug to a biological macromolecule (transport protein, antibody, virus). [54] Structural characterisation of the final nano‐product is typically based on a combination of techniques, for example NMR spectroscopy, AFM, SEM, TEM, SERS, when a metallic core is present, HPLC and zeta potential. A detailed discussion of synthetic design strategies for NP‐based therapeutics is beyond the scope of this review; therefore, the reader is referred to the reviews by Chugh et al. (2018), [102] Liang et al. (2020), [168] Yang (2016), [169] and Arranja, Pathak et al. (2017), [170] for general synthetic approaches in nanomedicinal chemistry. For nano‐encapsulation strategies for (metalla)carboranes and borates, numerous publications by Matějíček (polymeric and co‐polymeric matrices),[ 110 , 111 , 113 ] Nakamura (liposomes),[ 171 , 172 ] Hosmane, [143] Viñas and Teixidor (metallic NPs)[ 173 , 174 ] and co‐workers, among others, can be recommended. A combination of techniques for structural characterisation of COSAN‐loaded co‐polymer matrices was reported by Ďord′ovič et al. (2013). [112] In the following sections, we will focus on the chemical stability and the aqueous solubility of NP‐based boron‐rich drugs.
4.1. Chemical stability: Drug loading and leakage
Loading of a biologically active drug into a nanosized carrier system can be achieved covalently or noncovalently. The first approach, used also for grafting a molecule onto the surface of a carrier system or a macromolecular vector, requires appropriate functional groups on both components (e. g., azide/alkyne, thiol/maleimide, amine/carboxylic acid, etc.). The second one requires a good understanding of intermolecular interactions (hydrophobic/hydrophilic, hydrogen and dihydrogen bonding, ion pairs, etc.), to be able to engineer a structurally stable system.
The boron medicinal chemistry literature is rich of examples of elaborated and well‐established synthetic approaches to boron‐loaded NP platforms and biological macro‐vectors,[ 108 , 109 , 110 , 112 , 113 , 138 , 150 , 175 , 176 ] which are accompanied by experiments to determine the loading efficiency and investigate drug release kinetics, at least as preliminary evaluation under simulated biological conditions, especially in the latest works.[ 107 , 119 , 149 , 177 , 178 ]
Loading efficiency is generally investigated using a combination of LS techniques (SLS, DLS), calorimetry (mostly, ITC) and 1H NMR spectroscopy, particularly in the case of (co)polymeric matrices as carrier systems (Figure 9).[ 110 , 111 , 112 , 113 , 116 ] These techniques can give direct access to nano‐object formation mechanisms, preferential binding sites and motifs, which indirectly give (semi‐)quantitative information on, for example, maximum loading capacity and/or loading–carrier ratios, often expressed as segment/probe ratios (see, e. g., ref. [111]).
Figure 9.
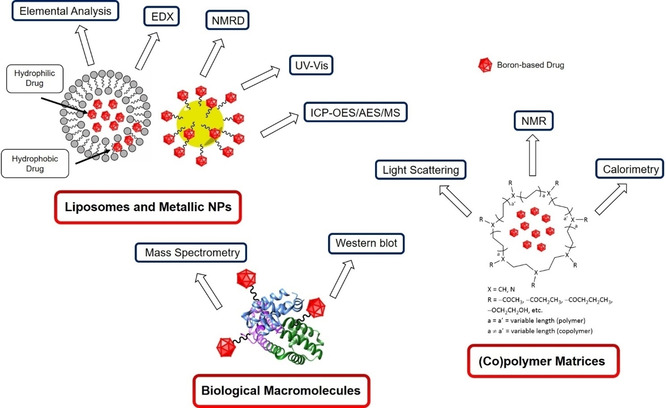
Schematic view of the main spectroscopic techniques used to study loading efficiency, based on the specific type of carrier system (liposomes, metallic NPs, biological macromolecules and (co)polymer matrices).
Furthermore, loading capacity and binding sites are also determined using mass spectrometry methods or Western blot analyses, often in combination with chromatographic or electrophoretic separations, specifically for peptide, protein or enzyme conjugates.[ 162 , 176 , 179 , 180 , 181 ] A rather atypical example is one of the latest works by Ventura, Teixidor and co‐workers (2019) on the interaction between BSA and Na[COSAN]. [158] They used DLS, zeta potential and far‐UV circular dichroism (CD) spectroscopy to infer saturation stoichiometry and co‐assembly motifs of BSA–COSAN nanostructures, concluding that COSAN molecules, when used in large excess (100×) over BSA, can form a sort of protective crown around the BSA molecule, shielding it from thermal and light‐induced denaturation and acetylation. In conclusion, the authors proposed the BSA–COSAN nano‐entity as stable inorganic self‐assembling system that can be used to modulate the biological functions of encapsulated proteins. However, in real biological systems, there will always be a large excess of serum (and other) proteins with respect to COSAN concentration. Furthermore, since COSAN forms colloidal suspensions in aqueous solutions, in a biological system it will most likely have a different biological identity as its in vitro chemical one, which will have a tremendous influence on the overall biological performance (see Section 5 below).
Finally, in the case of liposomes and metallic nanocarriers (Au‐ and AgNPs, magnetic Fe3O4‐NPs), boron loading is typically measured by 1H nuclear magnetic resonance dispersion (NMRD), provided that a suitable imaging probe such as gadolinium(III)[ 123 , 175 ] is present, inductively coupled plasma atomic or optical emission spectroscopy (ICP‐AES/OES) and/or mass spectrometry (ICP‐MS), microwave‐induced plasma mass spectrometry (MIP‐MS), elemental analysis, energy‐dispersive X‐ray spectroscopy (EDX), and UV/vis spectroscopy.[ 107 , 119 , 138 , 150 , 174 , 182 , 183 , 184 ]
One highlight among the synthetic approaches to boron‐rich drug–carrier systems is the work by Sumitani et al. (2012), which uses a “synthetic chemist” approach (covalent vs. noncovalent bonding) to tackle an effective problem of applicability for AB‐type amphiphilic block copolymers, that is, drug leakage during blood circulation. [185] Leakage of covalently bonded 1‐(4‐vinylbenzyl)‐closo‐ortho‐carborane from core‐polymerised acetalated and methacryloylated poly(ethylene glycol)‐block‐poly‐(lactide) (acetal‐PEG‐b‐PLA‐MA) micelles in 10 wt% FBS solution was significantly suppressed, compared to analogous non‐crosslinked micelles, as determined by ICP‐AES. ICP‐AES or ICP‐MS are often used to quantify the amount of boron released from a carrier system, under specific conditions, as descriptor of drug leakage, sometimes coupled with a membrane or micropore dialysis pre‐step.[ 43 , 178 , 186 ] UV/vis spectroscopy can also be used, [109] provided that a chromophore is present and accurate scattering correction to the measured absorbance is used. Olusanya et al. (2019) recently used zeta potential and fluorescence spectroscopy to assess constitutional stability (zeta potential) of dipalmitoyl‐phosphatidylcholine/1,2‐distearol‐sn‐glycerol‐3‐phosphocholine (DPPC/DSPC) liposomal carriers, and loading stability via retention experiments of calcein (fluorescent dye) in PBS and human serum solutions. [187]
The chemical stability descriptors (drug loading and leakage) discussed here are solid and well‐established parameters in the boron chemistry medicinal community. The term nanomedicine is typically only used for engineered drug carriers connected with boron‐based small molecules, neglecting many other boron‐based drugs, when applied without a carrier. One prominent example is the COSAN molecule, a known surfactant, which can spontaneously form NPs in water (see Section 2.3), but when it is biologically evaluated without carrier, it is only sporadically defined as nanosized system per se. The result are inconsistencies in overall drug design and testing protocols throughout the literature (see Section 5), referring to nanomedicine and organic medicinal chemistry without a clear rationale. One striking example are the porphyrin–COSAN conjugates, which are frequently designated as promising dual agents for BNCT and photodynamic therapy (PDT). Porphyrins tend already per se to form self‐assemblies via stacking mechanisms, [188] and their aggregation properties in water are often evident during photo‐ and physicochemical investigations.[ 189a , 189b , 189c ] However, the associated implications for their biological end‐use application are rarely considered.
Inspired by the works of the Shoichet groups on colloidal aggregators in medicine, [23] we therefore suggest to extend the concept of chemical stability to a broader spectrum of parameters, including properties such as self‐assembly stability and evolution over time in simulated biological milieus, as shown also in recent studies in our labs. [47]
4.2. Solubility and lipophilicity
For a nanoparticle, solubility is typically described as monomer concentration in solution (suspension) in equilibrium with its agglomerated form (precipitate). [190] Specific surface grafting patterns are used as stabilising groups to prevent self‐aggregation and ‐agglomeration of nanoparticles in aqueous solution, e. g. glutathione or PEG fragments for AuNPs, [191] because for the successful application in real biological systems, the nanotherapeutics need to be available as stable suspensions (bioavailability).
A series of three papers by Rak et al. (2010, 2011, 2013) on the de‐aggregation and solubilisation of COSAN and its derivatives illustrates this solubility/suspension stability concept for self‐aggregating, and thus nanoparticle‐forming, boron cluster molecules.[ 166 , 192 , 193 ] The general idea here was to use a biocompatible excipient to hamper self‐aggregation using host–guest chemistry principles, in other words, to stabilise the monomeric form of the drug, with monomer being a single molecule (Figure 10). This is analogous to our recent approach of using the BSA protein to stabilise water suspensions of self‐aggregating molybda‐ and ruthenacarborane complexes as monomers by complexation with one molecule of the protein, protected by a layer (or crown) of non‐complexed protein molecules, to ensure applicability under high serum conditions. [47]
Figure 10.
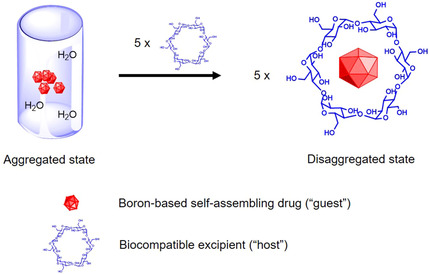
Schematic representation of the host–guest approach to stabilise boron‐based compounds in aqueous solution as monomers (i. e., single molecules). Here, α‐cyclodextrin is shown (right) as example of a biocompatible host. For simplicity, the aggregated state (left) is represented by five molecules of a boron‐containing compound.
Rak et al. (2010) investigated different excipients, mainly derivatives of β‐cyclodextrin, surfactants, copolymers and HSA, and studied the solubilisation properties by UV/vis spectroscopy, as ratio of absorbance of metallacarborane with and without excipient, from which only qualitative conclusions on relative excipient effects could be drawn, within the series of tested systems. [192] Quasi‐elastic light scattering (QELS) was then used for the best metallacarborane‐excipient combinations to determine radius and concentration of self‐assemblies of COSAN derivatives alone, excipients alone and mixtures thereof. It was observed that only heptakis(2,6‐di‐O‐methyl)‐β‐cyclodextrin (DIMEB) was able to completely suppress self‐aggregation of the metallacarborane molecules. [193]
Finally, the lipophilicity of the metallacarboranes was determined under different conditions, namely in pure water, saline solution (0.9 wt% NaCl) and saline solution in presence of HSA (50 g L−1). [166] The typical descriptor of lipophilicity in medicinal organic chemistry is the n‐octanol/water partition coefficient (P ow or log P ow), measured with the shake‐flask method, which often shows good correlation with water solubility and can be used to predict (or explain) binding affinities with biological substrates. [194] However, for COSAN and its derivatives, not only poor correlations between P ow and water solubility were found, but also between P ow and IC50 values or mechanism of action for HIV protease inhibition activities, expressed as ligand lipophilicity efficiency (LLE=−log IC50 – log P ow). This could be likely due to two reasons: either the classical spectrophotometric determination of P ow is not appropriate for this type of cluster molecules and other measuring techniques should be evaluated, or lipophilicity might not be the only property used to describe the biological interaction between the self‐aggregating cluster molecule and its intended substrate. In fact, when we look at nanoparticles, the interpretation of P ow values is typically discussed depending on many other parameters, such as surface charge, functionalisation and composition, dispersity, ionic strength of the solvent and pH, as a kind of structure–nano‐entity chemical relationship. [195] It seems thus, once again, that approaches from nanomedicinal chemistry should be implemented in the medicinal chemistry of boron clusters, as we observed before for the chemical stability (see Section 4.1).
For the specific topic of solubility and lipophilicity of boron cluster compounds in medicine, there is still a lot to be done ‐ the cited series of papers by Rak et al. is only an isolated example of a systematic study on the topic, with some “spin‐off” studies published afterwards (see for example ref. [163]). We thus encourage the medicinal boron community to move towards nanomedicinal chemistry approaches and to reconsider the concepts of chemical stability, solubility and lipophilicity for the particular class of inorganic boron cluster compounds.
5. Part IV: Biological Characterisation
To assess the biological performance of NP‐based or colloidal therapeutics, two main aspects need to be taken into account, namely i) safety and risk assessment, and ii) identity and evolution profile of the protein corona.[ 56 , 57 , 196 , 197 ] The first aspect will be described in the following paragraph, in terms of immune toxicity and “safety‐by‐design”. The next paragraph will deal with the protein corona topic, which, in our opinion, could, and should, also be considered in the current and future research on (metalla)carboranes in medicinal chemistry. Note that we use the term biological performance to describe the biological activity of the nano‐entity in its most comprehensive form. This includes the biochemical activity at the target‘s sites or interaction with the target, distribution in tissues, blood circulation time, clearance pathways and immunological response, as all of these elements together determine the actual applicability of a drug in real biological systems, and ultimately define whether a compound is a promising drug candidate or not.[ 66 , 198 ]
5.1. Safety and risk assessment
Safety and risk assessment of NP‐based or colloidal therapeutics usually becomes important at a relatively late stage of the drug‐development process, namely when it is necessary to assess large scale manufacturing and in‐patients application.[ 56 , 65 , 199 , 200 ] Nonetheless, preliminary studies towards risk assessment should as well be performed at earlier stages of the drug development process to discover failures early, for example, by in vitro oxidative stress assays in nontarget compartments and immunogenicity assays, to avoid or minimise waste of resources on studying nano‐entities with highly unfavourable toxicity profiles. [70]
Such an assessment starts with a thorough physicochemical characterisation, continues with investigations of the biological identity (see Section 5.2), followed by evaluation of the toxicity of the nano‐entity (nanotoxicity), which depends on the route of administration (systemic, oral, nasal, gastrointestinal, etc.), size, shape and dispersity of the particles, biochemical and immunogenic response on‐ and off‐target in the human organism (Figure 11), and ultimately ends with the reproducibility of the therapeutic efficiency.[ 63 , 70 ]
Figure 11.
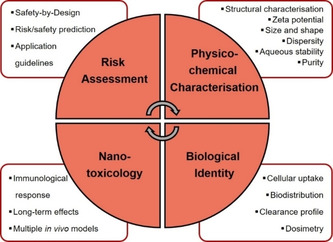
Schematic view of the safety and risk assessment strategy in nanomedicine. Adapted from ref. [191].
5.1.1. Immunotoxicity
In line with other chemotherapeutic treatments, also nanomedicines have been shown to either promote or suppress immune response in both in vivo animal models and in humans, with activation of the immune response being the most frequently observed effect.[ 65 , 201 ] Immune response to administered NPs follows mostly two possible pathways, often occurring in parallel: activation of innate immune response (e. g., via macrophage‐mediated phagocytosis, uptake in liver and spleen tissue, or complement activation) and of adaptive immune response. [198] Complement‐activated pseudo‐allergic reactions and adverse drug reactions (ADRs, e. g., hypersensitivity/allergic reactions) are the most recurrent side‐effects encountered in vivo after NPs′ administration, often reported as common side‐effects of liposomal and polymeric drug formulations, which might impair their therapeutic applicability.[ 198 , 202a , 202b ] Liposomal and polymeric formulations deserve specific attention, when translated to the application of (metalla)carboranes in medicine, since the use of liposomes and polymers (or co‐polymers) as nanosized drug delivery platforms is widely spread in the (metalla)carboranes literature, especially for applications in boron neutron capture therapy (BNCT), often justified by improved target selectivity, solubility properties and/or accumulation profiles.[ 43 , 108 , 119 , 138 , 146 , 149 , 150 , 178 , 185 , 186 ] It is particularly striking that the term “toxicity” is practically exclusively used to indicate the ability of the (metalla)carborane‐based drug, with or without carrier, to promote cell death in the tumour cell line or tissue examined (acute toxicity), and is solely evaluated in terms of half‐maximal inhibitory concentration (IC50). A handful of examples exist, where IC50 values were calculated for (metalla)carborane and borate compounds against model cell cultures for immune response, based on simple in vitro colorimetric assays, which will be briefly discussed below.[ 51 , 52 , 203a , 203b ] But to the best of our knowledge, no specific investigations on the immune response upon (metalla)carborane‐based treatment, with or without encapsulation in a carrier system, have been reported.
Passing from in vitro to in vivo conditions, there is a plethora of studies focused on the evaluation of the potential therapeutic efficiency for BNCT applications of several boron‐rich compounds, either carrying lipophilic probes for specific target recognition (e. g., cholesterol or fatty acids), [123] or encapsulated in liposomal or polymeric matrices,[ 138 , 142 , 149 , 150 , 178 , 185 , 204 ] or grafted onto the surface of inorganic nanoparticles (e. g., magnetic iron oxide, silver or gold NPs),[ 174 , 205 , 206 ] where factors such as toxicity, biosecurity, safety and organism tolerance become important.
By far, the predominant focus of these studies is on reaching the required amount of 10B inside the target cells, for ensuring therapeutic efficiency of the treatment (10–30 μg 10B per gram tumour mass, as typically accepted), [3] while guaranteeing an acceptable systemic biodistribution profile. The amount of 10B uptake is usually evaluated measuring the 10B content (in ppm) in cells, cell compartments or tissues after selected exposure times and within relevant concentration ranges after digestion. Inductively coupled plasma atomic or optical emission spectroscopy (ICP‐AES, ICP‐OES),[ 132 , 142 , 145 , 149 , 150 , 174 , 175 , 176 , 180 ] or energy dispersive spectroscopy (EDS) [205] have been established as common spectroscopic tools by many groups for in vivo and in vitro analyses. When the boron‐based drug contains a suitable imaging probe, boron content in target and non‐target tissues can be also indirectly quantified, measuring the probe signal with the appropriate spectroscopic technique, provided that accurate calibration of the probe‘s signal intensity is possible. Some examples are gold nanoclusters decorated with [7‐NH2(CH2)3S‐7,8‐nido‐C2B9H11]– reported by Yan, Wang and co‐workers (2017) [205] for real‐time fluorescent imaging of HeLa xenograft tumours in mice, gold NPs functionalised with radioiodinated anti‐HER2 antibodies, PEG6000 and dodecaborate clusters by Wu, Kuo and co‐workers (2019) [142] for single‐photon emission computed tomography (SPECT)/computed tomography (CT) imaging (see also Section 3.3.1), or the magnetic resonance imaging (MRI)/BNCT dual probe by Aime and co‐workers (2011), [123] consisting of an ortho‐carborane derivative labelled with a gadolinium(III)‐DOTAMA probe (DOTAMA=1,4,7,10‐tetraazacyclododecane monoamide), for MR imaging of melanoma cells (B16) in C57BL/6 mice (Figure 12).
Figure 12.
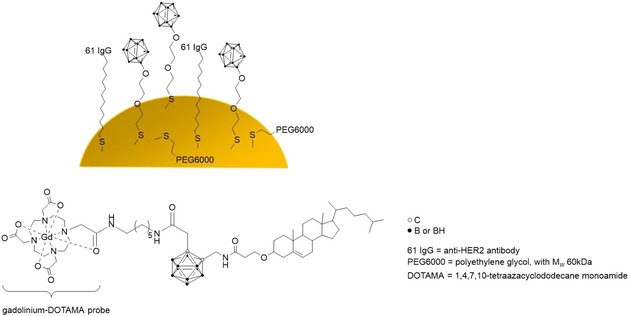
Imaging probes by Wu, Kuo (top) [135] and Aime (bottom) and co‐workers. [116]
Many more examples of such dual probes exist in the literature,[ 103 , 132 , 145 , 146 , 147 , 175 , 207 ] but in this chapter only selected studies dealing with in vivo experiments are highlighted. Even though, strictly technically speaking, the rationally designed compounds do exhibit promising properties, major conclusions on their therapeutic efficiency for BNCT applications in vivo are often drawn using as sole descriptor the amount of boron uptake in the tumour at specific points of time, relative to other nontarget organs,[ 150 , 204 , 205 ] which might be oversimplified. In recent years, more studies have been published, where also short‐term effects of neutron irradiation exposure have been evaluated, after injection of the BNCT agent, in terms of tumour size, which at least provides an additional evaluation parameter for the on‐target therapeutic effect.[ 123 , 138 , 176 , 185 ] For in vivo toxicity of the BNCT agent, body weight loss is the only descriptor used, accompanied sometimes by brief comments on general tolerance of the treatment, based on animal behaviour observations.[ 43 , 174 , 185 ] No specific investigations on the in vivo immune response to such treatments have been published so far, which would be required to draw conclusions on the therapeutic efficiency of a BNCT agent in vivo. An exception is the previously mentioned study by Nagasaki and co‐workers (see Section 3.3.2) on boron‐loaded redox‐active NPs with ROS scavenging properties, where a hematologic analysis was performed to check the leukocyte levels, as one of the possible markers of inflammation. [149]
While in‐depth investigations into on‐ and off‐target biological performance and safety are costly, both in terms of money and time, a certain diligence concerning conclusions about safety and systemic toxicity of (metalla)carborane‐ and borate‐based drugs is advised, because there is substantial lack of mechanistic insights on the comprehensive effects on the immune response, both in vitro and in vivo. In 2009, Aillon et al. criticised the traditionally performed tests to assess immune response to NP‐based treatments as sole biomarkers for tolerance and safety, namely histological tests in vitro, studies on overall animal behaviour and weight loss in vivo, [70] and suggested implementation of a series of biomarkers for inflammation, oxidative stress and cell viability. Today, there are still very few in vitro immunotoxicity assays which are able to give an accurate picture of the immune response to nanomedicines. Some progress has been made in that more orthogonal tests are run in parallel and are used by many research groups. These include methods like complement activation assays, secretion of proinflammatory cytokines, reverse transcription polymerase chain reaction, lymphocytes′ activation, enzyme‐linked immunosorbent assay, T cell‐dependent antibody response and, most recently, human‐based skin explant assays for assessment of adverse immune reactions to chemicals and small molecule drugs.[ 198 , 208 ]
Although nanomedicine and nanotoxicology still face unresolved challenges with regard to safety and risk assessment of immune responses, the medicinal chemistry of metallacarboranes, carboranes and borates is yet one step behind, probably comparable with the situation criticised by Aillon et al. about ten years ago (see above), with many inconsistencies and, in some cases, outdated approaches to safety assessment protocols.
5.1.2. Safety‐by‐design
The extensive work of Matějíček, Uchman and co‐workers, already extensively discussed in Section 2 from a physicochemical perspective, is addressed again here from a different viewpoint, for discussing the safety‐by‐design concept.
Reproducibility of the therapeutic effect raises great concerns for the end‐use application of nanomedicines, because today there still is a fundamental lack of clear and uniform directives and specifications from the responsible regulatory agencies (e. g., the American Food and Drug Administration, FDA, and European Medicines Agency, EMA).[ 57 , 197 , 209 ] The result is a plethora of different risk assessment methods and strategies, difficult to compare to each other and sometimes scarcely reproducible. Accurate determination of size, shape and dispersity of a formulation, as well as their evolution over time in several biological fluids, is of utmost importance for properly evaluating therapeutic efficiency and nanotoxicity in vivo, [64] as well as reproducibility of the treatment, [197] and is often a strong limiting factor in nanomedicine: in this context, we use the term safety‐by‐design. It is strongly recommended that several orthogonal techniques are used in parallel to get a comprehensive picture of size, shape and dispersity, since each technique has its drawbacks (see Section 2.1). [210] Following, selected examples are presented, where the safety‐by‐design issue (i. e., shape, size, dispersity and stability) is appropriately taken into account, or, conversely, not.
Based on their long‐term knowledge of the physicochemical properties and dynamics of the self‐assembly of anionic boron cluster compounds (ABCCs) in water solutions (see Section 2.3), Matějíček, Uchman and co‐workers have developed nano‐hybrid composites of block co‐polymer matrices and COSAN or closo‐dodecaborate molecules.[ 110 , 112 , 114 , 211 ] These investigations are one of the rare examples of continuity and consistency in the medicinal chemistry of metallacarboranes and borates by the same group, both in strictly technical (e. g., experimental setup) and methodological (e. g., orthogonal approaches to answer a single research question) terms. Similarly consistent approaches in medicinal organic chemistry are those of the Shoichet siblings′ groups, as discussed in the introduction. On the other hand, many reports on various biological activities of similar systems (i. e., COSAN derivatives and borates) lack a comparable consistency of approaches, particularly with regard to the colloidal behaviour of ABCCs in water, but also in studies on neutral icosahedral carboranes and metallacarboranes.
In 2019, Matějíček's group reported nanocomposites with core‐shell structure, resulting from an optimised co‐assembly procedure of glucose‐functionalised electroneutral hydrophilic poly(ethylene oxide) (glc‐PEO‐glc) and fluorescently labelled COSAN molecules, for visualisation of nanocomposite uptake in vitro in HeLa cell cultures. [211] The in vitro tests consisted of colorimetric cell viability assays, and confocal fluorescence microscopy measurements for cell internalisation and distribution. The main focus was on the investigation of the morphology, shape, and relative solution stability of the nanocomposites in this particular formulation, using a combination of techniques, even though the authors have studied analogous systems for already a decade.[ 109 , 110 , 111 , 112 , 113 , 114 , 116 ] The molecular picture of the nanocomposites, that is, glc‐PEO‐glc/COSAN unilamellar membranes, fused into layered nanostructures, is concluded with diligence, and unclear aspects are clearly stated. Here, the evaluation of the practical (medicinal) applicability of these materials can be comprehensively rationalised and justified on the basis of year‐long consolidated physical investigations of each component of the nanocomposite (COSAN, COSAN–fluorophore conjugate, PEO fragment).[ 8 , 109 , 110 , 111 , 212 ]
Viñas and co‐workers have studied the physicochemical behaviour of self‐assembling COSAN molecules in water for a long time[ 10 , 30 , 35 , 213a , 213b ] and have now extended this also to DNA–Na[COSAN] nano‐hybrids. Their article (2018) on DNA–Na[COSAN] interactions and biodistribution of Na[COSAN] in normal healthy mice is a multi‐spectroscopic investigation (UV/vis spectroscopy, circular dichroism, electrochemistry, cryo‐TEM and light scattering), which attempts to cover several fundamental questions of the biological fate of COSAN molecules. [156] For a biodistribution study in vivo, the boron content was measured via ICP‐OES in the organs of normal healthy mice after a single injection dose at 30 and 60 min post‐injection. These measurements revealed that Na[COSAN] molecules tend to preferentially accumulate in the lungs over time. This is probably due to the fact that Na[COSAN] forms aggregates with blood proteins, which in turn tend to stick inside the pulmonary capillaries, and that such a distribution within the pulmonary tissue is size‐dependent. Although 11B NMR experiments in cell culture medium with and without foetal bovine serum (FBS) have been previously used to postulate the existence of Na[COSAN]–serum proteins aggregates, considering all the possible factors affecting physical and biological identity of a colloidal system,[ 23 , 63 , 64 , 70 ] such as Na[COSAN], additional investigations would be indicated to confirm similar identity and size of Na[COSAN]–protein aggregates in vitro and in vivo. The interpretation of the binding mode of [COSAN]– with DNA as intercalation or electrostatic interaction, depending on ionic strength of the medium, is somewhat unusual as DNA is highly negatively charged (phosphodiester backbone), and the majority of intercalator molecules are expectedly positively charged. [214]
In this regard, an interesting and promising perspective emerged from the latest study by Llop and co‐workers (2019) on radioiodinated (124I) gold NPs decorated with COSAN molecules for combined BNCT/PET therapy. [132] The in vivo biodistribution of size‐ and shape‐tuned NPs (spherical shape with a hydrodynamic diameter of 37.8±0.5 nm, core diameter of 19.2±1.4 nm and zeta potential of −18.0±0.7 mV at pH 7) was investigated in a xenograft mouse model of human fibrosarcoma (HT1080 cells) and their relative amounts in the different tissues were quantified using the PET signal of the probe. The accumulation profile, that is, concentration in liver, spleen and lungs, was as expected based on shape and size considerations (spherical NPs, with ca. 38 nm diameter), corroborating the validity of the rational design of the nanoparticles. In addition, ex vivo studies were performed on liver tissue, using ion beam microscopy (IBM) and confocal Raman microspectroscopy (CRM), to confirm NPs accumulation in the liver and analyse their subcellular distribution. It was also suggested that another form‐specific type of AuNPs, i. e. rod‐like, could result in a better tumour load. The authors concluded that it is rather difficult to discuss and evaluate these findings, specifically in terms of tumour accumulation profile, in comparison with other studies from the literature on similar boron carrier systems,[ 142 , 183 , 215 ] because the different studies in the literature use very heterogeneous experimental setups and tumour models.
Another significant example from the Matějíček group is the already mentioned latest work on nanocomposites of PEOn‐block‐PGEAm with closo‐dodecaborate. [177] Besides the in vitro cell viability tests discussed above, three types of nano‐hybrids with different shapes, namely worm‐like (BWN), rod‐like (BRN) and spherical (BSN), were prepared and the correlation between cellular uptake and morphology of the nanocomposites was studied using laser scanning confocal microscopy (LSCM) and flow cytometry. It was shown that the shape of the particles has a profound influence on the efficiency of internalisation: the rod‐shaped ones showed significantly higher values than the others. For understanding the uptake mechanism, more in‐depth studies would be required. This is a rare example in the medicinal chemistry of borates where the influence of the shape of nanoparticles is used as descriptor of a specific biological performance. This highly interesting approach should certainly be more widely used within the nanomedicine community.
Looking at in vivo studies on closo‐dodecaborates for BNCT applications, the group of Nakamura at the Tokyo Institute of Technology in Japan has been studying such systems for quite some time now, especially in terms of tumour uptake studies (experimental setup and quantification of boron accumulation), biodistribution and irradiation protocols, making them specialists in the field.[ 171 , 172 , 180 , 183 , 184 , 216 ] They often make use of liposomes as drug carriers, as general means for delivering sufficient amounts of 10B to the tumour of interest, with the main focus on the biological characterisation of the boron agent–carrier system. [183]
One of their latest publications (2019) reports the design of a pteroyl‐closo‐dodecaborate conjugate (PBC) and the evaluation of its cellular uptake in vitro in glioma F98 cells, accumulation profile and therapeutic efficiency for BNCT in glioma‐bearing rat tumour model, employing a convection‐enhanced delivery (CED) administration route. [216] Although the PBC was designed to specifically interact with folate receptors, usually overexpressed in several tumour tissues, to promote selectivity of drug–tissue interaction, no immunohistochemical analyses were reported. One very positive point to highlight in the study is that p‐boronophenylalanine (BPA), the only clinically approved BNCT agent together with BSH, is used as reference substance for BNCT performance of PBC. A second very interesting point is the strategy of correlating in vitro and in vivo relative performance of PBC and BPA agents, based on respective observations of 10B accumulation. In vitro, BPA showed significantly higher intracellular accumulation than PBC, while in vivo, intra‐tumoral boron concentration of CED‐administered PBC was higher than for i. v.‐injected BPA (64.6 ± 29.6 vs. 19.9±1.0 μg 10B per gram tumour mass), whereas median survival time (MST) after irradiation was the same for both agents. A tentative explanation of this difference between in vitro and in vivo is given using the effective boron concentration parameter as descriptor, suggesting a strong influence of two competing factors, namely intra‐tumoral and intracellular distribution. In the light of the well‐known colloidal aggregator properties of closo‐dodecaborates in water,[ 6 , 7 , 38 , 124 ] these conclusions point to size and shape of self‐assemblies of dodecaborates as determining factor in biological milieus (see also the higher variance of measured 10B content when PBC is used, with respect to BPA), which in future should be systematically investigated together with in vivo studies to get a better understanding of the underlying physicochemical factors.
Implications in medicine of the self‐assembly behaviour are not only largely neglected or overlooked for ABCCs but also for neutral carboranes and metallacarboranes. For example, in publications by Wu et al. (2012) on Cd/Te quantum dots decorated with ortho‐carboranyl carboxylic acid, [217] by Alberti et al. (2014) on investigations of in vitro cellular uptake for an ortho‐carborane derivative labelled with a gadolinium(III)‐DOTAMA probe, [175] or by Goswani et al. (2015) on an ortho‐carboranylamide labelled with a gadolinium(III)‐DOTA probe (DOTA=1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid), [207] extensive characterisation of the self‐assembly behaviour of neutral cluster compounds in water, in analogy to the studies on ABCCs, might have helped to evaluate their biological performance.
One last remark within the safety and risk assessment aspect concerns the sterility of the NP formulation. Manufacturing NPs void of endotoxins, microbial and viral contaminations is also of primary importance to ensure low nanotoxicity in vivo, along with the therapeutic efficacy of the formulation per se. [65] Sterility can become an issue on large scale batch manufacturing of an NP‐based or colloidal drug, as in the case of Doxil®, a liposomal formulation of the anti‐neoplastic drug doxorubicin. The production was suspended in 2011 due to sterility problems, resulting in massive drug shortage for patients. [54] We are aware of only one report in the carborane literature, where the sterility aspect was mentioned, although not thoroughly investigated. Oleshkevich et al. (2019) simply mentioned the use of sterilised iron(III) magnetic nanoparticles (MNP), grafted with meta‐carboranylphosphate, for performing in vitro uptake studies with brain endothelial (hCMEC/D3) and glioblastoma (A172) cell lines. [174] To the best of our knowledge, no reports on sterility of (metalla)carborane‐based NPs or colloidal formulations are found in the literature.
5.2. Protein corona
Investigations on protein coatings of nano‐entities in biological fluids began already in the late 1970s, [218] but intensive research on the topic started only in 2007, when Dawson and co‐workers first coined the term protein corona. [219] The latter is described as “the spontaneous adsorption of biomolecules on the surface of nanoparticles in a biological environment”, [26] for example, proteins and lipids, among others (Figure 13). This process is driven by a series of attractive intermolecular forces, often acting in parallel: electrostatic and hydrophobic forces, van der Waals, hydrogen bonding and disulfide interactions. [220] The evolution of the protein corona over time is usually described in terms of the Vroman effect: the smaller the size, the higher the diffusion rates and the concentration, the faster a protein will adsorb on a NP surface. Gradually, this first layer of proteins will be replaced by other proteins, which have higher affinity (higher binding constants) for the specific NP surface. [221] Serum albumin and globulins belong to the first group of proteins, whereas apolipoproteins are examples of proteins with higher binding affinity to hydrophobic surfaces. The particular identity of a protein corona depends mainly on four factors: i) physicochemical characteristics of the NP system (shape, size, composition), ii) chemical properties of the medium (pH, ionic strength, temperature), iii) composition of the biological milieu (nucleic acids, proteins, peptides, etc.), and iv) administration route. The latter determines which fluids and biological compartments the NPs come into contact with and in which order, leading in some cases to multiple parallel displacement equilibria, which partially alter the corona composition. The protein corona can thus also be seen as a memory of the biological fate of an NP system, if one can identify all its components. [222]
Figure 13.
Schematic overview of selected types of nano‐object–protein corona interactions, as well as interactions with cell membranes and nucleic acids. Adapted from ref. [215].
In their recent review article, Sudheesh and co‐workers (2017) questioned whether the protein corona on nanoparticles might be the missing link between in vitro and in vivo correlations, [26] and emphasised the need for a better understanding of the interactions at the bio–nano interface, although this is an extremely challenging task. Synthetic identity of a nanoparticle is significantly different from its biological identity, but it is the latter that ultimately determines its performance as nanomedicine, in terms of biodistribution, uptake and clearance pathways, as well as on‐target activity and immune response. It is thus of utmost importance to consider the implications of protein coronas carefully and diligently, when working with NP‐based drugs or colloidal aggregates for application in medicine. As until today, the implications of protein coronas on the biological performance of (metalla)carboranes and borates as colloidal aggregators have hardly been studied, but some publications on metallacarborane–protein conjugates deal with adsorbed protein phaenomena (see, e. g., refs. [158] and [179]). The following sections will thus highlight selected effects of protein coronas on immune toxicity, active targeting, and cellular uptake and clearance pathways, and appropriate examples from the (metalla)carboranes literature will be briefly discussed.
5.2.1. Protein corona influence on immune toxicity
The presence and specific type of protein corona plays a crucial role in the immune response to a nanoparticle‐ or colloidal aggregator‐based treatment. Physicochemical properties of the adsorbed protein layer (shape, size, charge, rigidity, hydrophobicity, etc.) in fact strongly affect complement activation, in terms of immunogenic reaction pathways and clearance from blood circulation. Moreover, specific proteins can promote recognition by macrophages or by scavenging receptors on Kupffer cells in the liver, thus modulating their phagocytosis. [64]
From the point of view of the end‐use application, one particularly undesired effect is the accumulation of the nano‐entity in the spleen, because there is a high chance of occurrence of immunogenic reactions, exemplified by the accelerated blood clearance (ABC) effect. [223] Briefly, direct interaction of the nano‐drug with lymphocytic B cells in the spleen induces nano‐drug‐specific antibody production, which, once translated into blood circulation, will recognise the nano‐drug, sequester it and transport it directly to the liver for clearance.
The literature on (metalla)carboranes and borates is full of examples of in vivo biodistribution studies, where high boron accumulation in spleen and liver is observed.[ 123 , 178 , 183 , 184 , 185 , 204 ] However, this accumulation behaviour is typically only descriptively discussed, whereas the outcome of a biodistribution study is critically evaluated solely in terms of tumour‐to‐blood or tumour‐to‐tissue boron ratio. If the latter are above a certain threshold value, then the biodistribution profile is evaluated as positive and promising. Also, in vivo studies are systematically conducted following a single injection protocol, which is not recommended in nanomedicine, since it prevents the evaluation of possible occurrence of the ABC effect due to immunogenic response. [64] An exception is the work by Nigg, Hawthorne and co‐workers (2013, 2017) on lecithin/cholesterol liposomes loaded with lipophilic Na3[1‐(2′‐B10H9)‐2‐NH3B10H8] and amphiphilic K[nido‐7‐CH3(CH2)15‐7,8‐C2B9H11], tested as BNCT agents in murine EMT6 mammary adenocarcinoma and CT26 colon carcinoma.[ 224a , 224b ] A double‐injection protocol was developed to be able to reach an acceptable 10B concentration in the tumour and a satisfactory tumour/blood ratio, which allowed time‐resolved neutron irradiation studies to be performed. Immune response to the treatment was, however, not investigated, albeit a high accumulation in the spleen was observed for the case of CT26 tumours.
Liposomes also raise some concerns in nanomedicine, because their size shrinkage upon contact with plasma proteins can affect both the blood clearance via modulation of the immune response and the tumour accumulation profile. [220] Protein corona effects on liposomal formulations should thus be properly investigated in the medicinal boron chemistry community, since they have been exploited for 25 years already as selective drug carriers for BNCT agents.[ 119 , 134 , 138 , 150 , 172 , 175 , 183 , 184 ]
5.2.2. Protein corona influence on active targeting
As discussed before (Section 3.2), active targeting is often used to confer high selectivity for a specific receptor, which implies a specific and often laborious chemical modification of the NP surface. This approach can however be strongly affected by formation of protein coronas, up to the point where it is almost fully invalidated, because the protein coating on the surface of the NP or colloidal aggregate (bio‐nano interface) shields the carefully designed vector from its intended biological target. [220] As a result such nano‐entities often show frequent‐hitter behaviours, seemingly being able to modulate the activity of a multitude of biological targets, such as enzymes, as already discussed (see Section 1), independent of the specific surface modification. This is reminiscent of the broad spectrum of biological activities that are attributed to COSAN and its derivatives throughout the literature.
There are several possible approaches to circumvent this issue, [27] such as designing “adsorption‐proof” NP platforms, or exploiting the protein corona phenomenon itself for targeting and stabilisation, like Ganesh et al. (2017). [29] PEGylation is one of the most common ways to minimise protein adsorption on a nano‐entity, because, among other reasons, it increases the hydrophilicity of the interface. [225] It is a well implemented approach for drug carriers of boron cluster compounds, usually justified in terms of increased aqueous solubility, especially when AuNPs or micellar systems (liposomes, co‐polymer matrices, etc.) are considered.[ 43 , 44 , 119 , 131 , 132 , 138 , 142 , 149 , 150 , 175 , 185 , 204 ] Their stability is typically assessed by UV/vis spectroscopy, gel permeation chromatography, powder XRD or HPLC, in terms of leakage of the loading and/or decomposition of the carrier itself, in physiological buffer or, rarely, cell culture media. However, possible destabilising interactions with blood proteins (serum albumin and immunoglobulins) and cell membranes, [226] as well as characterisation of the adsorbed protein layers under high serum conditions, should also be performed to be able to draw conclusions on the effective performance (stability and real targeting capabilities) of the carrier system.
Worth highlighting is that some studies report on the evaluation of in vitro biological activity, usually in cell cultures, under serum‐free and serum‐containing conditions. This is an appropriate indirect way of preliminary assessing if the chosen vector gives the expected biological response and if and to what extent the biological activity of the whole carrier system is influenced by different types of protein coronas. An example are ferrocene‐substituted dithio‐ortho‐carboranes, developed by Wu et al. (2011), which showed significantly lower cytotoxicity against human hepatocellular carcinoma cell line (SMMC‐7721) under serum‐containing conditions with respect to serum‐free conditions. [227] Another example is the maleimide‐functionalised closo‐dodecaborate–BSA conjugate by Kikuchi et al. (2016), whose uptake by colon‐26 cells was arrested in the presence of 10 wt% foetal bovine serum (FBS), in contrast to BSH uptake, which was effective only with FBS in the culture medium. [176] Modulation of the in vitro biological activity due to a specifically stabilised protein‐aggregator colloidal suspension was also observed in our recent work on the stabilisation of spontaneous self‐assemblies of molybdacarboranes, [47] which turned out to be very important for the appropriate formulation of such metallacarborane complexes carrying a vector for oestrogen receptor recognition. [51]
There are thus already approaches which, indirectly, give a preliminary indication of the effect of protein coronas on the biological activity of a targeted boron cluster‐containing drug or carrier system, but it should be emphasised that these investigations need to be performed on a regular basis for a better understanding of the bio–nano interface of boron cluster compounds in medicine.
5.2.3. Protein corona influence on cellular uptake and blood clearance
The specific nature of the bio‐nano interface has a profound influence on cellular uptake pathways and blood circulation time. [27] For example, the former can be receptor‐mediated and thus modulated depending on the specific types of proteins adsorbed on the NP surface. Then the evaluation of cellular uptake routes becomes very important, in particular for those cases where either the monomer (i. e., carborane‐containing molecule) or the carrier system is designed to interact with receptors on the surface of cellular membranes.
For example, a vector for recognition of the transmembrane epidermal growth factor receptor (EGFR) was used by Couto et al. (2017, 2018) to target tumours which overexpress such receptors, like gliomas and glioblastomas, and promote selectivity.[ 228a , 228b ] In vitro enzyme inhibition assays were performed on a series of closo‐carborane derivatives, showing IC50 values in the low to high nanomolar range; however, it is not clear whether cellular uptake was EGFR‐dependent, due to lack of direct investigations on cellular uptake pathways. On the other hand, Maruyama et al. (2004) reported on liposome–cell association assays for investigating the ability of transferrin‐functionalised PEGylated liposomes to mediate cellular uptake by binding to transferrin receptors. [138] This approach allowed a clear conclusion regarding a mechanism of receptor‐mediated endocytosis, at least in vitro, in cell culture medium (RPMI) and with 10 wt% FBS.
In those cases where no specific vector for receptor recognition is used, it would be helpful if major conclusions on uptake mechanisms or uptake itself would be supported by convincing experimental evidence and presented in detail in the experimental section of a publication, otherwise it is rather difficult to follow the conclusions drawn and, sometimes, even the experiments performed.
Concerning the blood circulation time, in nanomedicine there is nowadays a rather established classification of adsorbed proteins into opsonins and dysopsonins, according to whether they shorten (opsonins) or prolong (dysopsonins) blood clearance of the nano‐entity. [27] Opsonins are for example complement proteins, fibronectin and immunoglobulins, dysopsonins are serum albumin and apolipoproteins. Precise characterisation of the nature and the displacement equilibria of protein coronas on nano‐entities is thus very important also in this case, in order to provide an accurate blood clearance profiling.
Persistence of boron compounds in blood is widely present in in vivo studies of new potential BNCT agents, typically evaluated as time‐resolved 10B concentration in blood at specific points in time after injection and/or neutron irradiation, and often compared to corresponding concentrations in other tissues and organs. However, as for biodistribution studies (see Section 5.2), blood clearance profiles have rarely received proper attention, but can have tremendous impact on the observed biodistribution. Understanding which kind of proteins adsorb on a specific drug carrier and their kinetic adsorption profile might shed some light on, for example, why certain agents accumulate preferentially in the liver, others in the spleen or in the lungs, and so on.
In conclusion, the implementation of boron clusters, either borates, carboranes or metallacarboranes, in medicine is everything but trivial. There are excellent teams of scientists worldwide, who have been working on the development of boron‐containing pharmaceuticals for over two decades, mainly either as chemists or as biologists, demonstrating a plethora of competencies. However, at the level of comprehensive biological performances, the implementation of approaches and techniques from the “traditional” nano‐world into the medicinal chemistry of boron cluster compounds is still in the early stages. With this review, we therefore want to motivate the community to develop interdisciplinary tailored approaches for a better understanding of boron‐containing molecules in nanomedicine.
6. What Is Known To Be True and What Is Believed To Be True?
Diverse classes of boron cluster‐containing systems have been investigated in the last 20 years for potential application as therapeutics. These include, but are not limited to, icosahedral closo‐carboranes, nido‐carborates(−1), metallacarboranes and borates, as small molecule conjugates with organic moieties or biological macromolecules, or as nanosized drug–carrier systems (polymeric matrices, liposomes, metallic and magnetic NPs), for which today synthetic strategies, chemical (structural) and physicochemical characterisation approaches are widely established. Very often, a list of advantages of boron clusters over classical organic molecules in medicine is given, including metabolic and/or enzymatic stability, low systemic toxicity, high lipophilicity, superior structural fine‐tuning possibilities, etc. These are generally well‐accepted points, in other words, they are believed to be true. However, such statements are rarely critically approached, or even studied for a specific boron‐based drug under investigation. What is known to be true is that the chemistry of carboranes, metallacarboranes and borates is mainly the domain of chemists with a primary focus on their synthesis and chemical/physical characterisation. As a result, the bridge between the chemistry laboratory and “real” applications in living systems is not yet well established, and the translation of boron‐based medicinal chemistry into nanomedicine topics must be encouraged. “The time has come, the Walrus said, to talk of many things” [Lewis Carroll]: The boron chemistry community should be encouraged to cross borders and become truly interdisciplinary.
“What we observe is not nature itself, but nature exposed to our method of questioning.” [Werner Heisenberg]
With respect to good scientific practice (GSP), we would like to give the following citations from a perspective by Hofmann‐Amtenbrink et al. (2015) in the area of nanomedicine, [199] one subchapter of which inspired the title of this concluding section: “current scientific outcomes are often not published in ways that readily allow meta‐studies to help establish more general relationships between particle properties and observed in vitro cell behaviors”, “published results often do not contain information sufficient to allow experimental conduct to compare results in a consistent way”, “properties of nanoparticles responsible for possible toxic effects […] should be clearly analyzed and technically communicated in publications, be it a supplement or on designated Web page repository, to enable comparison”, “this information gap caused by the absence of guidelines and peer review expectations for proper descriptions of nanomaterials and methods in most publications makes continued funding of nanoparticle research (including toxicology) inefficient and may even confound the appropriate future directions of such research in both academia and industry”, “a repository for negative results is also valuable to avoid repetition”.
Basically, Hofmann‐Amtenbrink et al. were telling us that uniform regulations for the application of nanomaterials in medicine are needed, and must be tightly connected with accurate and critical experimental procedures. This is even more true for self‐aggregating (metalla)carboranes and borates. Therefore, we recommend consulting the websites of the European Nanomedicine Characterisation Laboratory (EUNCL), launched in July 2015 as part of the European Union's Horizon 2020 research and innovation programme, and the US National Cancer Institute Nanotechnology Characterization Laboratory (NCI‐NCL) and the Nanomedicine – European Technology Platform and their affiliated partners. The EUNCL for example does not only offer a trans‐disciplinary testing infrastructure, but also wide access to their state‐of‐the‐art characterisation platforms. Their self‐chosen tasks are four‐step knowledge sharing, open access standard operation protocols (SOPs) and lab protocols, offering a platform for educating the community, and free of charge and unbiased access to their state‐of‐the‐art characterisation service. [60] SOPs align research with regulatory needs and guarantee overall applicability and suitability. SOPs and quality control seem to be a boring subject for researchers, but scientists can only contribute to the progress of the scientific community by producing quality data.
Abbreviations
- 5‐DSA
5‐doxyl stearic acid
- ABC
accelerated blood clearance (effect)
- ABCC
anionic boron cluster compound
- AF4‐MALS
asymmetric‐flow field‐flow fractionation multiangle light scattering
- AFM
atomic force microscopy
- anti‐EGFR AB
epidermal growth factor receptor antibody
- APTMS
(3‐aminopropyl)trimethoxysilane
- ATR
attenuated total reflection (infrared spectroscopy)
- AUC
analytical ultracentrifugation
- AgNP
silver nanoparticle
- AuNP
gold nanoparticle
- AZO
azobenzene
- BIC
biological inorganic chemistry
- BNCT
boron neutron capture therapy
- BPA
4‐dihydroxyboryl‐l‐phenylalanine
- BRN
rod‐like boron‐rich particles
- BSA
bovine serum albumin
- BSH
sodium mercaptoundecahydrododecaborate
- BSN
spherical boron‐rich particles
- BWN
worm‐like boron‐rich particles
- CAC
critical aggregation concentration
- CBn
cucurbit[n]urils
- CBT
1‐thiol‐1,2‐dicarba‐closo‐dodecaborane (mercaptocarborane)
- CD
circular dichroism
- β‐CD
β‐cyclodextrin
- CED
convection‐enhanced delivery
- CMC
critical micelle concentration
- COSAN
cobaltabis(dicarbollide) [3,3′‐Co(1,2‐C2B9H11)2]−
- COX
cyclooxygenase
- CRM
confocal Raman microscopy
- CT
computed tomography
- DCS
differential centrifugal sedimentation
- DHA
docosahexaenoic acid
- DIMEB
heptakis(2,6‐di‐O‐methyl)‐β‐cyclodextrin
- DNA
deoxyribonucleic acid
- DLS
dynamic light scattering
- DLVO
Derjaguin, Landau, Vervey, and Overbeek (theory)
- DOPE
l‐α‐di‐oleoylphosphatidylethanolamine
- DOTA
1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid
- DOTAMA
1,4,7,10‐tetraazacyclododecane monoamide
- DOTAP
di‐oleoyltrimethylammonium propane
- DOX
doxorubicin
- DPPC
dipalmitoyl‐phosphatidylcholine
- DRI
differential refractometer
- DSPC
1,2‐distearol‐sn‐glycerol‐3‐phosphocholine
- DSPE‐PEG
1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanol‐amine‐N‐[methoxy(polyethylene glycol)‐2000]
- EDS or EDX
energy‐dispersive X‐ray spectroscopy
- EG3SH
(1‐mercaptoundec‐11‐yl)tri(ethylene glycol)
- EGFR
epidermal growth factor receptor
- EM
electron microscopy
- EPR effect
enhanced permeability and retention effect
- EPR
electron paramagnetic resonance (spectroscopy)
- EUNCL
European Nanomedicine Characterisation Laboratory
- FBS
foetal bovine serum
- FCS
fluorescence correlation spectroscopy
- FE‐SEM
field emission scanning electron microscopy
- FLIM
fluorescence lifetime imaging
- FWHM
full width at half maximum
- glc‐PEO‐glc
glucose‐functionalised poly(ethylene oxide)
- GPC
gel permeation chromatography
- HepG2
hepatic cancer cells
- HER2
human epidermal growth factor receptor 2
- HIV
human immunodeficiency virus
- HPLC
high‐performance liquid chromatography
- HR‐TEM
high‐resolution transmission electron microscopy
- HSA
human serum albumin
- HSPC
hydrogenated (Soy), l‐α‐phosphatidylcholine
- HTS
high‐throughput screening
- IBM
ion beam microscopy
- IC50
half maximal inhibitory concentration
- ICP‐AES/OES
inductively coupled plasma atomic or optical emission spectroscopy
- ICP‐MS
inductively coupled plasma mass spectrometry
- ID
injected dose
- IFP
interstitial fluid pressure
- IgG
immunoglobulin G
- ITC
isothermal titration calorimetry
- IVD
in vitro diagnostics
- LD
laser diffraction
- LLE
ligand lipophilicity efficiency
- LSCM
laser scanning confocal microscopy
- LSPR
localised surface plasmon resonance
- MALS
multiangle light scattering
- MIP‐MS
microwave‐induced plasma mass spectrometry
- MR(I)
magnetic resonance (imaging)
- MSN
mesoporous silica NPs
- MST
median survival time
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- Mw
molecular weight
- NCL
Nanotechnology Characterization Lab
- NE
nanoemulsion
- NMRD
nuclear magnetic resonance dispersion
- NOESY
nuclear Overhauser enhancement spectroscopy
- NP
nanoparticle
- NTA/PTA
nanoparticle/particle tracking analysis
- P2VP
poly(2‐vinylpyridine)
- PAINS
pan assay interference compounds
- PBC
pteroyl‐closo‐dodecaborate conjugate
- PBS
phosphate‐buffered saline
- PDI
polydispersity index
- PDT
photodynamic therapy
- PEG
poly(ethylene glycol)
- PEO
poly(ethylene oxide)
- PEOn‐block‐PGEAm
poly(ethylene oxide)‐block‐poly(2‐(N,N,N’,N’‐tetramethyl guanidium)ethyl acrylate)
- PEO‐PMA
poly(ethylene oxide) poly(methacrylic acid) block polymer
- PEOX
poly(2‐ethyl‐2‐oxazoline)
- PET
positron emission tomography
- PLAC‐PEG
poly(d,l‐lactide‐co‐2‐methyl‐2‐carboxytrimethylene carbonate)‐graft‐poly(ethylene glycol)
- PMA
poly(methacrylic acid)
- PMBSH
poly[(closo‐dodecaboranyl)thiomethylstyrene]
- PMeOx
poly(2‐methyl‐2‐oxazoline)
- PMeOx‐PPrOx
poly(2‐methyl‐2‐oxazoline)block‐poly(2‐n‐propyl‐2‐oxazoline)
- PMNT
poly[4‐(2,2,6,6‐tetramethylpiperidine‐N‐oxyl)aminomethylstyrene]
- Pow or log Pow
n‐octanol/water partition coefficient
- PPD
pentyloxycarbonyl‐(p‐aminobenzyl)doxazolidinylcarbamate
- PPO
poly(p‐phenylene oxide)
- PTT
photothermal therapy
- PVP
polyvinylpyrrolidone
- QELS
quasi‐elastic light scattering
- RES
reticuloendothelial system
- RNA
ribonucleic acid
- RNI
radionuclide imaging
- ROESY
rotating frame Overhauser enhancement spectroscopy
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute
- SANS
small‐angle neutron scattering
- SAR
structure‐activity relationship
- SAXS
small‐angle X‐ray scattering
- SEC
size‐exclusion chromatography
- SEM
scanning electron microscopy
- SERS
surface‐enhanced Raman scattering/spectroscopy
- SHG
second harmonic generation
- SLS
static light scattering
- SOP
standard operation protocol
- SPECT
single‐photon emission computed tomography
- TDA
Taylor dispersion analysis
- TEM
transmission electron microscopy
- TEMPO
2,2,6,6‐tetramethylpiperidine‐N‐oxyl
- TRPS
tuneable resistive pulse sensing
- TSP
thiosuccinimidyl propionate
- Tβ4
thymosin β4
- UMR‐106
osteosarcoma cell line
- UP80
polysorbate 80
- WAXS
wide‐angle X‐ray scattering
- XRD
X‐ray diffraction.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Marta Gozzi received her BSc degree from the University of Bologna (Italy). She joined the Erasmus Mundus program Advanced Spectroscopy in Chemistry and received a double MSc degree (University of Lille1 and Leipzig University). She carried out her doctoral studies at Leipzig University (with Prof. E. Hey‐Hawkins), receiving her degree in 2019. Her research interests are in the fields of bioorganometallic chemistry, particularly metallacarborane complexes for application in medicine, and of analytical chemistry. She contributed chapters to Boron‐Based Compounds: Potential and Emerging Applications in Medicine, and the Handbook of Boron Chemistry in Organometallics, Catalysis, Material Science and Medicine.

Biographical Information
Benedikt Schwarze received his BSc and MSc at Leipzig University, with a semester at Monash University (Melbourne, Australia). He conducted his doctoral studies at Leipzig University (with Prof. E. Hey‐Hawkins), receiving his degree in 2019 on bioorganometallic chemistry using metallacarborane complexes for application in medicine. He contributed chapters to Boron‐Based Compounds: Potential and Emerging Applications in Medicine, and the Handbook of Boron Chemistry in Organometallics, Catalysis, Material Science and Medicine. Since January 2020, he has been a postdoctoral researcher at the Institute of Medicinal Physics and Biophysics at Leipzig University (with Prof. D. Huster) working on interdisciplinary projects between biophysics, biochemistry, and chemistry.

Biographical Information
Evamarie Hey‐Hawkins has been Professor of Inorganic Chemistry at Leipzig University, Germany, since 1993. She received her diploma (1982) and doctoral degree (1983) at Philipps University Marburg, Germany. After stays in the UK (1984/85) and Australia (1985/87), she completed her habilitation in Marburg (1988). Her scientific interests are organophosphorus chemistry, biologically active boron compounds, and heterometallic transition metal complexes with applications in catalysis and materials science. She has published over 530 papers, edited the book Boron‐Based Compounds: Potential and Emerging Applications in Medicine (with Clara Viñas Teixidor), is a member of the European Academy of Sciences, and has received several prestigious awards.

Acknowledgements
Support from the Deutsche Forschungsgemeinschaft (DFG; HE 1376/38‐1) is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
M. Gozzi, B. Schwarze, E. Hey-Hawkins, ChemMedChem 2021, 16, 1533.
Some minor language corrections were implemented under Section 3.4. (Metalla)carboranes and biomolecules after publication of this article in Early View on 19.March 2021, but prior to final publication in issue 10.
References
- 1.E. Hey-Hawkins, C. V. Teixidor (Eds.) Boron-Based Compounds, John Wiley & Sons, Ltd, Chichester, UK, 2018.
- 2.
- 2a. Leśnikowski Z. J., J. Med. Chem. 2016, 59, 7738–7758; [DOI] [PubMed] [Google Scholar]
- 2b. Stockmann P., Gozzi M., Kuhnert R., Sárosi M. B., Hey-Hawkins E., Chem. Soc. Rev. 2019, 48, 3497–3512; [DOI] [PubMed] [Google Scholar]
- 2c. Goszczyński T. M., Fink K., Boratyński J., Exp. Opin. Biol. Ther. 2018, 18, 205–213. [DOI] [PubMed] [Google Scholar]
- 3. Barth R. F., Mi P., Yang W., Can. Commun. 2018, 38, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zargham E. O., Mason C. A., Lee M. W. Jr., Int. J. Cancer Clin. Res. 2019, 6, 110–114. [Google Scholar]
- 5.
- 5a. https://www.birmingham.ac.uk/facilities/mds-cpd/conferences/eurobic/index.aspx;
- 5b. https://www.chem.uzh.ch/en/icbic19.html.
- 6. Fernandez-Alvarez R., Ďorďovič V., Uchman M., Matějíček P., Langmuir 2018, 34, 3541–3554. [DOI] [PubMed] [Google Scholar]
- 7. Ďorďovič V., Tošner Z., Uchman M., Zhigunov A., Reza M., Ruokolainen J., Pramanik G., Cígler P., Kalíková K., Gradzielski M., Matějíček P., Langmuir 2016, 32, 6713–6722. [DOI] [PubMed] [Google Scholar]
- 8. Matejícek P., Cígler P., Procházka K., Král V., Langmuir 2006, 22, 575–581. [DOI] [PubMed] [Google Scholar]
- 9. Uchman M., Ďorďovič V., Tošner Z., Matějíček P., Angew. Chem. Int. Ed. 2015, 54, 14113–14117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14319–14323. [Google Scholar]
- 10. Viñas C., Tarrés M., González-Cardoso P., Farràs P., Bauduin P., Teixidor F., Dalton Trans. 2014, 43, 5062–5068. [DOI] [PubMed] [Google Scholar]
- 11.F. Lottspeich, J. Engels, Bioanalytics: Analytical Methods and Concepts in Biochemistry and Molecular Biology, Wiley, 2018.
- 12. Hill H., Bioanalysis 2011, 3, 2155–2158. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Coan K. E. D., Shoichet B. K., J. Am. Chem. Soc. 2008, 130, 9606–9612; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Lucas A. T., Herity L. B., Kornblum Z. A., Madden A. J., Gabizon A., Kabanov A. V., Ajamie R. T., Bender D. M., Kulanthaivel P., Sanchez-Felix M. V., Havel H. A., Zamboni W. C., Int. J. Pharm. 2017, 526, 443–454; [DOI] [PubMed] [Google Scholar]
- 13c. Owen S. C., Doak A. K., Wassam P., Shoichet M. S., Shoichet B. K., ACS Chem. Biol. 2012, 7, 1429–1435; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Shoichet B. K., Drug Discovery Today 2006, 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGovern S. L., Caselli E., Grigorieff N., Shoichet B. K., J. Med. Chem. 2002, 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- 15. Pharmaceutical Medicine and Translational Clinical Research (Eds.: D. Vohora, G. Singh), Elsevier, London, 2018.
- 16. Jadhav A., Ferreira R. S., Klumpp C., Mott B. T., Austin C. P., Inglese J., Thomas C. J., Maloney D. J., Shoichet B. K., Simeonov A., J. Med. Chem. 2010, 53, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szymański P., Markowicz M., Mikiciuk-Olasik E., Int. J. Mol. Sci. 2012, 13, 427–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGovern S. L., Helfand B. T., Feng B., Shoichet B. K., J. Med. Chem. 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Dahlin J. L., Walters M. A., Assay Drug Dev. Technol. 2016, 14, 168–174; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Matlock M. K., Hughes T. B., Dahlin J. L., Swamidass S. J., J. Chem. Inf. Model. 2018, 58, 1483–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irwin J. J., Duan D., Torosyan H., Doak A. K., Ziebart K. T., Sterling T., Tumanian G., Shoichet B. K., J. Med. Chem. 2015, 58, 7076–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Coan K. E. D., Maltby D. A., Burlingame A. L., Shoichet B. K., J. Med. Chem. 2009, 52, 2067–2075; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Yang Z.-Y., He J.-H., Lu A.-P., Hou T.-J., Cao D.-S., Drug Discovery Today 2020, 25, 657–667. [DOI] [PubMed] [Google Scholar]
- 22. Aldrich C., Bertozzi C., Georg G. I., Kiessling L., Lindsley C., Liotta D., Merz K. M., Schepartz A., Wang S., J. Med. Chem. 2017, 60, 2165–2168. [DOI] [PubMed] [Google Scholar]
- 23. Ganesh A. N., Donders E. N., Shoichet B. K., Shoichet M. S., Nano Today 2018, 19, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryan A. J., Gray N. M., Lowe P. N., Chung C.-w., J. Med. Chem. 2003, 46, 3448–3451. [DOI] [PubMed] [Google Scholar]
- 25. Duan D., Torosyan H., Elnatan D., McLaughlin C. K., Logie J., Shoichet M. S., Agard D. A., Shoichet B. K., ACS Chem. Biol. 2017, 12, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jain P., Pawar R. S., Pandey R. S., Madan J., Pawar S., Lakshmi P. K., Sudheesh M. S., Bioptechnol. Adv. 2017, 35, 889–904. [DOI] [PubMed] [Google Scholar]
- 27. Cai R., Chen C., Adv. Mater. 2018, 31, 1805740–1805753. [Google Scholar]
- 28. Ganesh A. N., Aman A., Logie J., Barthel B. L., Cogan P., Al-Awar R., Koch T. H., Shoichet B. K., Shoichet M. S., ACS Chem. Biol. 2019, 14, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ganesh A. N., Logie J., McLaughlin C. K., Barthel B. L., Koch T. H., Shoichet B. K., Shoichet M. S., Mol. Pharmaceutics 2017, 14, 1852–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bauduin P., Prevost S., Farràs P., Teixidor F., Diat O., Zemb T., Angew. Chem. Int. Ed. 2011, 50, 5298–5300; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5410–5412. [Google Scholar]
- 31. Chevrot G., Schurhammer R., Wipff G., J. Phys. Chem. B 2006, 110, 9488–9498. [DOI] [PubMed] [Google Scholar]
- 32. Chevrot G., Schurhammer R., Wipff G., Phys. Chem. Chem. Phys. 2007, 9, 1991–2003. [DOI] [PubMed] [Google Scholar]
- 33. Matejícek P., Cígler P., Olejniczak A. B., Andrysiak A., Wojtczak B., Prochazka K., Lesnikowski Z. J., Langmuir 2008, 24, 2625–2630. [DOI] [PubMed] [Google Scholar]
- 34. Popov A., Borisova T., J. Colloid Interface Sci. 2001, 236, 20–27. [DOI] [PubMed] [Google Scholar]
- 35. Gassin P.-M., Girard L., Martin-Gassin G., Brusselle D., Jonchère A., Diat O., Viñas C., Teixidor F., Bauduin P., Langmuir 2015, 31, 2297–2303. [DOI] [PubMed] [Google Scholar]
- 36. Hohenschutz M., Grillo I., Diat O., Bauduin P., Angew. Chem. Int. Ed. 2020, 59, 8084–8088. [DOI] [PubMed] [Google Scholar]
- 37. Assaf K. I., Nau W. M., Angew. Chem. Int. Ed. 2018, 57, 13968–13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karki K., Gabel D., Roccatano D., Inorg. Chem. 2012, 51, 4894–4896. [DOI] [PubMed] [Google Scholar]
- 39. Matějíček P., Curr. Opin. Colloid Interface Sci. 2020, 45, 97–107. [Google Scholar]
- 40. Maderna A., Herzog A., Knobler C. B., Hawthorne M. F., J. Am. Chem. Soc. 2001, 123, 10423–10424. [DOI] [PubMed] [Google Scholar]
- 41. Ma L., Hamdi J., Huang J., Hawthorne M. F., Inorg. Chem. 2005, 44, 7249–7258. [DOI] [PubMed] [Google Scholar]
- 42. Morandi S., Ristori S., Berti D., Panza L., Becciolini A., Martini G., Biochim. Biophys. Acta 2004, 1664, 53–63. [DOI] [PubMed] [Google Scholar]
- 43. Xiong H., Zhou D., Qi Y., Zhang Z., Xie Z., Chen X., Jing X., Meng F., Huang Y., Biomacromolecules 2015, 16, 3980–3988. [DOI] [PubMed] [Google Scholar]
- 44. Xiong H., Zhou D., Zheng X., Qi Y., Wang Y., Jing X., Huang Y., Chem. Commun. 2017, 53, 3422–3425. [DOI] [PubMed] [Google Scholar]
- 45. He T., Misuraca J. C., Musah R. A., Sci. Rep. 2017, 7, 16995–17005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaniowski D., Ebenryter-Olbinska K., Kulik K., Janczak S., Maciaszek A., Bednarska-Szczepaniak K., Nawrot B., Lesnikowski Z., Nanoscale 2020, 12, 103–114. [DOI] [PubMed] [Google Scholar]
- 47. Schwarze B., Gozzi M., Zilberfain C., Rüdiger J., Birkemeyer C., Estrela-Lopis I., Hey-Hawkins E., J. Nanopart. Res. 2020, 22, 2165–2187. [Google Scholar]
- 48. Gozzi M., Schwarze B., Coburger P., Hey-Hawkins E., Inorganics 2019, 7, 91–104. [Google Scholar]
- 49. Scholz M., Blobaum A. L., Marnett L. J., Hey-Hawkins E., Bioorg. Med. Chem. 2012, 20, 4830–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neumann W., Xu S., Sárosi M. B., Scholz M. S., Crews B. C., Ghebreselasie K., Banerjee S., Marnett L. J., Hey-Hawkins E., ChemMedChem 2016, 11, 175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwarze B., Jelača S., Welcke L., Maksimović-Ivanić D., Mijatović S., Hey-Hawkins E., ChemMedChem 2019, 14, 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gozzi M., Murganic B., Drača D., Popp J., Coburger P., Maksimović-Ivanić D., Mijatović S., Hey-Hawkins E., ChemMedChem 2019, 14, 2061–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.M. Gozzi, Ruthenacarborane Complexes as Building Blocks for the Design of Novel Anti-Tumour Agents, 2019, Doctoral Thesis, Leipzig University, Leipzig.
- 54. Wicki A., Witzigmann D., Balasubramanian V., Huwyler J., J. Controlled Release 2015, 200, 138–157. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Duncan R., Gaspar R., Mol. Pharm. 2011, 8, 2101–2141; [DOI] [PubMed] [Google Scholar]
- 55b. Gaspar R., Duncan R., Adv. Drug Delivery Rev. 2009, 61, 1220–1231. [DOI] [PubMed] [Google Scholar]
- 56. Boverhof D. R., David R. M., Anal. Bioanal. Chem. 2010, 396, 953–961. [DOI] [PubMed] [Google Scholar]
- 57. Siegrist S., Cörek E., Detampel P., Sandström J., Wick P., Huwyler J., Nanotoxicology 2019, 13, 73–99. [DOI] [PubMed] [Google Scholar]
- 58.
- 58a. Gioria S., Caputo F., Urbán P., Maguire C. M., Bremer-Hoffmann S., Prina-Mello A., Calzolai L., Mehn D., Nanomedicine 2018, 13, 539–554; [DOI] [PubMed] [Google Scholar]
- 58b. Crist R. M., Grossman J. H., Patri A. K., Stern S. T., Dobrovolskaia M. A., Adiseshaiah P. P., Clogston J. D., McNeil S. E., Integr. Biol. 2013, 5, 66–73; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58c. Halamoda-Kenzaoui B., Holzwarth U., Roebben G., Bogni A., Bremer-Hoffmann S., WIREs Nanomed. Nanobiotechnol. 2019, 11, 1531–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clogston J. D., Hackley V. A., Prina-Mello A., Puri S., Sonzini S., Soo P. L., Pharm. Res. 2020, 37, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Caputo F., Clogston J., Calzolai L., Rösslein M., Prina-Mello A., J. Controlled Release 2019, 299, 31–43. [DOI] [PubMed] [Google Scholar]
- 61. Caputo F., Arnould A., Bacia M., Ling W. L., Rustique E., Texier I., Mello A. P., Couffin A.-C., Mol. Pharm. 2019, 16, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.
- 62a. Allen C., J. Controlled Release 2019, 315, 214–215; [DOI] [PubMed] [Google Scholar]
- 62b. D′Mello S. R., Cruz C. N., Chen M.-L., Kapoor M., Lee S. L., Tyner K. M., Nat. Nanotechnol. 2017, 12, 523–529. [DOI] [PubMed] [Google Scholar]
- 63. Baer D. R., Front. Chem. 2018, 6, 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bertrand N., Leroux J.-C., J. Controlled Release 2012, 161, 152–163. [DOI] [PubMed] [Google Scholar]
- 65. Dobrovolskaia M. A., McNeil S. E., Nat. Nanotechnol. 2007, 2, 469–478. [DOI] [PubMed] [Google Scholar]
- 66. Lammers T., Kiessling F., Hennink W. E., Storm G., J. Controlled Release 2012, 161, 175–187. [DOI] [PubMed] [Google Scholar]
- 67. Nel A. E., Mädler L., Velegol D., Xia T., Hoek E. M. V., Somasundaran P., Klaessig F., Castranova V., Thompson M., Nat. Mater. 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a.“Carboranes in Medicine”, R. N. Grimes, Carboranes 3rd ed., Academic Press, Amsterdam, 2016, Chapter 16;
- 68b. Barth R. F., Grecula J. C., Appl. Radiat. Isot. 2020, 160, 109029–109034. [DOI] [PubMed] [Google Scholar]
- 69.International Organization for Standardization, Nanotechnologies: Vocabulary. Part 2: Nano-objects, 07.120, 2015, Geneva.
- 70. Aillon K. L., Xie Y., El-Gendy N., Berkland C. J., Forrest M. L., Adv. Drug Delivery Rev. 2009, 61, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.B. Carr, M. Wright, Nanoparticle Tracking Analysis. A Review of Applications and Usage in the Analysis of Exosomes and Microvesicles, Malvern Panalytical Ltd., 2012.
- 72.
- 72a. Nanoscale Material Characterization: A Review of the Use of Nanoparticle Tracking Analysis, Malvern Instruments Ltd;
- 72b. Walker J. G., Meas. Sci. Technol. 2012, 23, 65605–65612; [Google Scholar]
- 72c. Gallego-Urrea J. A., Tuoriniemi J., Hassellöv M., TrAC Trends Anal. Chem. 2011, 30, 473–483; [Google Scholar]
- 72d. Filipe V., Hawe A., Jiskoot W., Pharm. Res. 2010, 27, 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.
- 73a. Luo P., Roca A., Tiede K., Privett K., Jiang J., Pinkstone J., Ma G., Veinot J., Boxall A., J. Environ. Sci. 2018, 64, 62–71; [DOI] [PubMed] [Google Scholar]
- 73b. Sanchís J., Bosch-Orea C., Farré M., Barceló D., Anal. Bioanal. Chem. 2015, 407, 4261–4275. [DOI] [PubMed] [Google Scholar]
- 74. Jarzębski M., Bellich B., Białopiotrowicz T., Śliwa T., Kościński J., Cesàro A., Food Hydrocolloids 2017, 68, 90–101. [Google Scholar]
- 75.
- 75a. van der Pol E., Coumans F. A. W., Grootemaat A. E., Gardiner C., Sargent I. L., Harrison P., Sturk A., van Leeuwen T. G., Nieuwland R., J. Thromb. Haemostasis 2014, 12, 1182–1192; [DOI] [PubMed] [Google Scholar]
- 75b. Chen X.-X., Cheng B., Yang Y.-X., Cao A., Liu J.-H., Du L.-J., Liu Y., Zhao Y., Wang H., Small 2013, 9, 1765–1774; [DOI] [PubMed] [Google Scholar]
- 75c. Gercel-Taylor C., Atay S., Tullis R. H., Kesimer M., Taylor D. D., Anal. Biochem. 2012, 428, 44–53; [DOI] [PubMed] [Google Scholar]
- 75d. Soo C. Y., Song Y., Zheng Y., Campbell E. C., Riches A. C., Gunn-Moore F., Powis S. J., Immunology 2012, 136, 192–197; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75e. Sokolova V., Ludwig A.-K., Hornung S., Rotan O., Horn P. A., Epple M., Giebel B., Colloids Surf. B 2011, 87, 146–150. [DOI] [PubMed] [Google Scholar]
- 76. Mehrabi K., Nowack B., Arroyo Rojas Dasilva Y., Mitrano D. M., Environ. Sci. Technol. 2017, 51, 5611–5621. [DOI] [PubMed] [Google Scholar]
- 77. Vogel R., Willmott G., Kozak D., Roberts G. S., Anderson W., Groenewegen L., Glossop B., Barnett A., Turner A., Trau M., Anal. Chem. 2011, 83, 3499–3506. [DOI] [PubMed] [Google Scholar]
- 78. Vogel R., Anderson W., Eldridge J., Glossop B., Willmott G., Anal. Chem. 2012, 84, 3125–3131. [DOI] [PubMed] [Google Scholar]
- 79. Willmott G. R., Vogel R., Yu S. S. C., Groenewegen L. G., Roberts G. S., Kozak D., Anderson W., Trau M., J. Phys. Condens. Matter 2010, 22, 454116–454126. [DOI] [PubMed] [Google Scholar]
- 80. Blundell E. L. C. J., Mayne L. J., Billinge E. R., Platt M., Anal. Methods 2015, 7, 7055–7066. [Google Scholar]
- 81. Billinge E. R., Platt M., Biosens. Bioelectron. 2015, 68, 741–748. [DOI] [PubMed] [Google Scholar]
- 82. Electron Microscopy and Analysis (Eds.: P. J. Goodhew, J. Humphreys), CRC Press, Boca Raton, 2000.
- 83. Callaway E., Nature 2020, 578, 201. [DOI] [PubMed] [Google Scholar]
- 84. Krieg M., Fläschner G., Alsteens D., Gaub B. M., Roos W. H., Wuite G. J. L., Gaub H. E., Gerber C., Dufrêne Y. F., Müller D. J., Nat. Rev. Phys. 2019, 1, 41–57. [Google Scholar]
- 85. Polydispersity: What Does It Mean for DLS and Chromatography?, U. Nobbmann, https://www.materials-talks.com/blog/2017/10/23/polydispersity-what-does-it-mean-for-dls-and-chromatography/, 2017.
- 86. Giddings J. C., Sep. Sci. 1966, 1, 123–125. [Google Scholar]
- 87. López-Sanz S., Fariñas N. R., Zougagh M., Martín-Doimeadios R. d. C. R., Ríos Á., J. Anal. At. Spectrom. 2020, 35, 1530–1536. [Google Scholar]
- 88. Zhang H., Lyden D., Nat. Protoc. 2019, 14, 1027–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hemmelmann M., Kurzbach D., Koynov K., Hinderberger D., Zentel R., Biomacromolecules 2012, 13, 4065–4074. [DOI] [PubMed] [Google Scholar]
- 90.
- 90a. Nuhn L., Hirsch M., Krieg B., Koynov K., Fischer K., Schmidt M., Helm M., Zentel R., ACS Nano 2012, 6, 2198–2214; [DOI] [PubMed] [Google Scholar]
- 90b. Rigler P., Meier W., J. Am. Chem. Soc. 2006, 128, 367–373. [DOI] [PubMed] [Google Scholar]
- 91. Dakwar G. R., Zagato E., Delanghe J., Hobel S., Aigner A., Denys H., Braeckmans K., Ceelen W., de Smedt S. C., Remaut K., Acta Biomater. 2014, 10, 2965–2975. [DOI] [PubMed] [Google Scholar]
- 92.
- 92a. Milani S., Bombelli F. B., Pitek A. S., Dawson K. A., Rädler J., ACS Nano 2012, 6, 2532–2541; [DOI] [PubMed] [Google Scholar]
- 92b. Pelaz B., del Pino P., Maffre P., Hartmann R., Gallego M., Rivera-Fernández S., de La Fuente J. M., Nienhaus G. U., Parak W. J., ACS Nano 2015, 9, 6996–7008. [DOI] [PubMed] [Google Scholar]
- 93. Mittag J. J., Kneidl B., Preiβ T., Hossann M., Winter G., Wuttke S., Engelke H., Rädler J. O., Eur. J. Pharm. Biopharm. 2017, 119, 215–223. [DOI] [PubMed] [Google Scholar]
- 94. Negwer I., Best A., Schinnerer M., Schäfer O., Capeloa L., Wagner M., Schmidt M., Mailänder V., Helm M., Barz M., Butt H.-J., Koynov K., Nat. Commun. 2018, 9, 5306–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Uchman M., Jurkiewicz P., Cígler P., Grüner B., Hof M., Procházka K., Matejícek P., Langmuir 2010, 26, 6268–6275. [DOI] [PubMed] [Google Scholar]
- 96.
- 96a. Liu J., Andya J. D., Shire S. J., AAPS J. 2006, 8, 580–589; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96b. Davidson A. M., Brust M., Cooper D. L., Volk M., Anal. Chem. 2017, 89, 6807–6814; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96c. Lebowitz J., Lewis M. S., Schuck P., Protein Sci. 2002, 11, 2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Anderson W., Kozak D., Coleman V. A., Jämting Å. K., Trau M., J. Colloid Interface Sci. 2013, 405, 322–330. [DOI] [PubMed] [Google Scholar]
- 98. Giljohann D. A., Seferos D. S., Daniel W. L., Massich M. D., Patel P. C., Mirkin C. A., Angew. Chem. Int. Ed. 2010, 49, 3280–3294; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3352–3366. [Google Scholar]
- 99. Bai X., Wang Y., Song Z., Feng Y., Chen Y., Zhang D., Feng L., Int. J. Mol. Sci. 2020, 21, 2480–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mesbahi A., Rep. Pract. Oncol. Radiother. 2010, 15, 176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.
- 101a. Solati E., Dorranian D., J. Cluster Sci. 2015, 26, 727–742; [Google Scholar]
- 101b. Carlini L., Fasolato C., Postorino P., Fratoddi I., Venditti I., Testa G., Battocchio C., Colloids Surf. A 2017, 532, 183–188. [Google Scholar]
- 102. Chugh H., Sood D., Chandra I., Tomar V., Dhawan G., Chandra R., Artif. Cells, Nanomed., Biotechnol. 2018, 46, 1210–1220. [DOI] [PubMed] [Google Scholar]
- 103. Kennedy D. C., Duguay D. R., Tay L.-L., Richeson D. S., Pezacki J. P., Chem. Commun. 2009, 6750–6752. [DOI] [PubMed] [Google Scholar]
- 104. Wu A., Ou P., Zeng L., NANO 2010, 5, 245–270. [Google Scholar]
- 105. van der Zee J., Ann. Oncol. 2002, 13, 1173–1184. [DOI] [PubMed] [Google Scholar]
- 106. Tulebayeva D. Z., Kozlovskiy A. L., Korolkov I. V., Gorin Y. G., Kazantsev A. V., Abylgazina L., Shumskaya E. E., Kaniukov E. Y., Zdorovets M. V., Mater. Res. Express 2018, 5, 105011–105016. [Google Scholar]
- 107. Tishkevich D. I., Korolkov I. V., Kozlovskiy A. L., Anisovich M., Vinnik D. A., Ermekova A. E., Vorobjova A. I., Shumskaya E. E., Zubar T. I., Trukhanov S. V., Zdorovets M. V., Trukhanov A. V., J. Alloys Compd. 2019, 797, 573–581. [Google Scholar]
- 108. Barry N. P. E., Pitto-Barry A., Romero-Canelón I., Tran J., Soldevila-Barreda J. J., Hands-Portman I., Smith C. J., Kirby N., Dove A. P., O′Reilly R. K., Sadler P. J., Faraday Discuss. 2014, 175, 229–240. [DOI] [PubMed] [Google Scholar]
- 109. Matějíček P., Zedník J., Ušelová K., Pleštil J., Fanfrlík J., Nykänen A., Ruokolainen J., Hobza P., Procházka K., Macromolecules 2009, 42, 4829–4837. [Google Scholar]
- 110. Matějíček P., Brus J., Jigounov A., Pleštil J., Uchman M., Procházka K., Gradzielski M., Macromolecules 2011, 44, 3847–3855. [Google Scholar]
- 111. Uchman M., Cígler P., Grüner B., Procházka K., Matejícek P., J. Colloid Interface Sci. 2010, 348, 129–136. [DOI] [PubMed] [Google Scholar]
- 112. Ďorďovič V., Uchman M., Procházka K., Zhigunov A., Pleštil J., Nykänen A., Ruokolainen J., Matějíček P., Macromolecules 2013, 46, 6881–6890. [Google Scholar]
- 113. Ďorďovič V., Uchman M., Zhigunov A., Nykänen A., Ruokolainen J., Matějíček P., ACS Macro Lett. 2014, 3, 1151–1155. [DOI] [PubMed] [Google Scholar]
- 114. Brus J., Zhigunov A., Czernek J., Kobera L., Uchman M., Matějíček P., Macromolecules 2014, 47, 6343–6354. [Google Scholar]
- 115. Ďorďovič V., Verbraeken B., Hogenboom R., Kereïche S., Matějíček P., Uchman M., Chem. Asian J. 2018, 13, 838–845. [DOI] [PubMed] [Google Scholar]
- 116. Ďorďovič V., Uchman M., Reza M., Ruokolainen J., Zhigunov A., Ivankov O. I., Matějíček P., RSC Adv. 2016, 6, 9884–9892. [Google Scholar]
- 117. Allen T. M., Cullis P. R., Adv. Drug Delivery Rev. 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- 118.“Calcein as a Tool in Liposome Methodology”, Liposome Technology, Vol. 2 (Ed. G. Gregoriadis), CRC Press, 2018, Chapter 12.
- 119. Bregadze V. I., Sivaev I. B., Semioshkin A., Shmal′ko A. V., Kosenko I. D., Lebedeva K. V., Mandal S., Sreejyothi P., Dubey R. D., Sarkar A., Shen Z., Wu A., Hosmane N. S., Chem. Eur. J. 2020, 26, 13832–13841. [DOI] [PubMed] [Google Scholar]
- 120. Nature Research, “Self-assembly”, https://www.nature.com/subjects/self-assembly.
- 121. Bonechi C., Ristori S., Martini S., Panza L., Martini G., Rossi C., Donati A., Biophys. Chem. 2007, 125, 320–327. [DOI] [PubMed] [Google Scholar]
- 122. Salvi G., de Los Rios P., Vendruscolo M., Proteins 2005, 61, 492–499. [DOI] [PubMed] [Google Scholar]
- 123. Geninatti-Crich S., Alberti D., Szabo I., Deagostino A., Toppino A., Barge A., Ballarini F., Bortolussi S., Bruschi P., Protti N., Stella S., Altieri S., Venturello P., Aime S., Chem. Eur. J. 2011, 17, 8479–8486. [DOI] [PubMed] [Google Scholar]
- 124. Wang W., Wang X., Cao J., Liu J., Qi B., Zhou X., Zhang S., Gabel D., Nau W. M., Assaf K. I., Zhang H., Chem. Commun. 2018, 54, 2098–2101. [DOI] [PubMed] [Google Scholar]
- 125. Wang W., Wang X., Xiang C., Zhou X., Gabel D., Nau W. M., Assaf K. I., Zhang H., ChemNanoMat 2019, 5, 124–129. [Google Scholar]
- 126. Skoglund S., Hedberg J., Yunda E., Godymchuk A., Blomberg E., Odnevall Wallinder I., PLoS One 2017, 12, 181735–181753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lowry G. V., Hill R. J., Harper S., Rawle A. F., Hendren C. O., Klaessig F., Nobbmann U., Sayre P., Rumble J., Environ. Sci.: Nano 2016, 3, 953–965. [Google Scholar]
- 128. Hotze E. M., Louie S. M., Lin S., Wiesner M. R., Lowry G. V., Environ. Chem. 2014, 11, 257–267. [Google Scholar]
- 129.
- 129a. Zhang H., Chen B., Banfield J. F., J. Phys. Chem. C 2010, 114, 14876–14884; [Google Scholar]
- 129b. Fernando I., Zhou Y., Chemosphere 2019, 216, 297–305. [DOI] [PubMed] [Google Scholar]
- 130. Wu W., Luo L., Wang Y., Wu Q., Dai H.-B., Li J.-S., Durkan C., Wang N., Wang G.-X., Theranostics 2018, 8, 3038–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Grzelczak M. P., Danks S. P., Klipp R. C., Belic D., Zaulet A., Kunstmann-Olsen C., Bradley D. F., Tsukuda T., Viñas C., Teixidor F., Abramson J. J., Brust M., ACS Nano 2017, 11, 12492–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pulagam K. R., Gona K. B., Gómez-Vallejo V., Meijer J., Zilberfain C., Estrela-Lopis I., Baz Z., Cossío U., Llop J., Molecules 2019, 24, 3609–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Vaage J., Barberá-Guillem E., Abra R., Huang A., Working P., Cancer 1994, 73, 1478–1484. [DOI] [PubMed] [Google Scholar]
- 134. Mehta S. C., Lai J. C., Lu D. R., J. Microencapsulation 1996, 13, 269–279. [DOI] [PubMed] [Google Scholar]
- 135. Nakamura Y., Mochida A., Choyke P. L., Kobayashi H., Bioconjugate Chem. 2016, 27, 2225–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Danquah M. K., Zhang X. A., Mahato R. I., Adv. Drug Delivery Rev. 2011, 63, 623–639. [DOI] [PubMed] [Google Scholar]
- 137.
- 137a. Matsumura Y., Maeda H., Cancer Res. 1986, 46, 6387–6392; [PubMed] [Google Scholar]
- 137b. Maeda H., Matsumura Y., Crit. Rev. Ther. Drug Carrier Syst. 1989, 6, 193–210. [PubMed] [Google Scholar]
- 138. Maruyama K., Ishida O., Kasaoka S., Takizawa T., Utoguchi N., Shinohara A., Chiba M., Kobayashi H., Eriguchi M., Yanagie H., J. Controlled Release 2004, 98, 195–207. [DOI] [PubMed] [Google Scholar]
- 139. Duan D., Doak A. K., Nedyalkova L., Shoichet B. K., ACS Chem. Biol. 2015, 10, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Base T., Bastl Z., Plzák Z., Grygar T., Plesek J., Carr M. J., Malina V., Subrt J., Bohácek J., Vecerníková E., Kríz O., Langmuir 2005, 21, 7776–7785. [DOI] [PubMed] [Google Scholar]
- 141. Ciani L., Bortolussi S., Postuma I., Cansolino L., Ferrari C., Panza L., Altieri S., Ristori S., Int. J. Pharm. 2013, 458, 340–346. [DOI] [PubMed] [Google Scholar]
- 142. Wu C.-Y., Lin J.-J., Chang W.-Y., Hsieh C.-Y., Wu C.-C., Chen H.-S., Hsu H.-J., Yang A.-S., Hsu M.-H., Kuo W.-Y., Colloids Surf. B 2019, 183, 110387–110394. [DOI] [PubMed] [Google Scholar]
- 143. Li N., Zhao P., Salmon L., Ruiz J., Zabawa M., Hosmane N. S., Astruc D., Inorg. Chem. 2013, 52, 11146–11155. [DOI] [PubMed] [Google Scholar]
- 144. Parmar A., Aswal V. K., Bahadur P., Spectrochim. Acta Part A 2012, 97, 137–143. [DOI] [PubMed] [Google Scholar]
- 145. Lai C.-H., Lai N.-C., Chuang Y.-J., Chou F.-I., Yang C.-M., Lin C.-C., Nanoscale 2013, 5, 9412–9418. [DOI] [PubMed] [Google Scholar]
- 146. Ruan Z., Yuan P., Liu L., Xing T., Yan L., Int. J. Polym. Mater. 2018, 67, 720–726. [Google Scholar]
- 147. Ruan Z., Liu L., Fu L., Xing T., Yan L., Polym. Chem. 2016, 7, 4411–4418. [Google Scholar]
- 148. Ruan Z., Yuan P., Jing T., Xing T., Yan L., Macromol. Res. 2018, 26, 270–277. [Google Scholar]
- 149. Gao Z., Horiguchi Y., Nakai K., Matsumura A., Suzuki M., Ono K., Nagasaki Y., Biomaterials 2016, 104, 201–212. [DOI] [PubMed] [Google Scholar]
- 150. Białek-Pietras M., Olejniczak A. B., Tachikawa S., Nakamura H., Leśnikowski Z. J., Bioorg. Med. Chem. 2013, 21, 1136–1142. [DOI] [PubMed] [Google Scholar]
- 151. Barth R. F., Coderre J. A., Vicente M. G. H., Blue T. E., Clin. Cancer Res. 2005, 11, 3987–4002. [DOI] [PubMed] [Google Scholar]
- 152. Navascuez M., Dupin D., Grande H.-J., Gómez-Vallejo V., Loinaz I., Cossío U., Llop J., Chem. Commun. 2020, 56, 8972–8975. [DOI] [PubMed] [Google Scholar]
- 153. Danaei M., Dehghankhold M., Ataei S., Hasanzadeh Davarani F., Javanmard R., Dokhani A., Khorasani S., Mozafari M. R., Pharmaceuticals 2018, 10, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Che Marzuki N. H., Wahab R. A., Abdul Hamid M., Biotechnol. Biotechnol. Equip. 2019, 33, 779–797. [Google Scholar]
- 155. Vaňková E., Lokočová K., Maťátková O., Křížová I., Masák J., Grüner B., Kaule P., Čermák J., Šícha V., J. Organomet. Chem. 2019, 899, 120891–120902. [Google Scholar]
- 156. Fuentes I., García-Mendiola T., Sato S., Pita M., Nakamura H., Lorenzo E., Teixidor F., Marques F., Viñas C., Chem. Eur. J. 2018, 24, 17239–17254. [DOI] [PubMed] [Google Scholar]
- 157. Grüner B., Kugler M., El Anwar S., Holub J., Nekvinda J., Bavol D., Růžičková Z., Pospíšilová K., Fábry M., Král V., Brynda J., Řezáčová P., ChemPlusChem 2020, 85, 1–13. [DOI] [PubMed] [Google Scholar]
- 158. Fuentes I., Pujols J., Viñas C., Ventura S., Teixidor F., Chem. Eur. J. 2019, 25, 12820–12829. [DOI] [PubMed] [Google Scholar]
- 159. Guerrero I., Kelemen Z., Viñas C., Romero I., Teixidor F., Chem. Eur. J. 2020, 26, 5027–5036. [DOI] [PubMed] [Google Scholar]
- 160. Fuentes I., Mostazo-López M. J., Kelemen Z., Compañ V., Andrio A., Morallón E., Cazorla-Amorós D., Viñas C., Teixidor F., Chem. Eur. J. 2019, 25, 14254. [DOI] [PubMed] [Google Scholar]
- 161. Grüner B., Plešek J., Báča J., Císařová I., Dozol J. F., Rouquette H., Viňas C., Selucký P., Rais J., New J. Chem. 2002, 26, 1519–1527. [Google Scholar]
- 162. Fink K., Boratyński J., Paprocka M., Goszczyński T. M., Ann. N. Y. Acad. Sci. 2019, 1457, 128–141. [DOI] [PubMed] [Google Scholar]
- 163. Goszczyński T. M., Fink K., Kowalski K., Leśnikowski Z. J., Boratyński J., Sci. Rep. 2017, 7, 9800–9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Plešek J., Baše K., Mareš F., Hanousek F., Štíbr B., Heřmánek S., Collect. Czech. Chem. Commun. 1984, 49, 2776–2789. [Google Scholar]
- 165.“Water Solubility”, U. S. National Pesticide Information Center (NPIC), http://npic.orst.edu/envir/watersol.html, 2016.
- 166. Rak J., Dejlová B., Lampová H., Kaplánek R., Matějíček P., Cígler P., Král V., Mol. Pharm. 2013, 10, 1751–1759. [DOI] [PubMed] [Google Scholar]
- 167. Tolpin E. I., Wong H. S., Lipscomb W. N., J. Med. Chem. 1974, 17, 792–796. [DOI] [PubMed] [Google Scholar]
- 168. Liang K., Chen H., WIREs Nanomed. Nanobiotechnol. 2020, 1616–1640. [DOI] [PubMed] [Google Scholar]
- 169. Yang H., Nanomedicine 2016, 12, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Arranja A. G., Pathak V., Lammers T., Shi Y., Pharmacol. Res. 2017, 115, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Nakamura H., Future Med. Chem. 2013, 5, 715–730. [DOI] [PubMed] [Google Scholar]
- 172. Nakamura H., Methods Enzymol. 2009, 465, 179–208. [DOI] [PubMed] [Google Scholar]
- 173. Cioran A. M., Teixidor F., Krpetić Ž., Brust M., Viñas C., Dalton Trans. 2014, 43, 5054–5061. [DOI] [PubMed] [Google Scholar]
- 174. Oleshkevich E., Morancho A., Saha A., Galenkamp K. M. O., Grayston A., Crich S. G., Alberti D., Protti N., Comella J. X., Teixidor F., Rosell A., Viñas C., Nanomedicine 2019, 20, 101986–101995. [DOI] [PubMed] [Google Scholar]
- 175. Alberti D., Toppino A., Geninatti Crich S., Meraldi C., Prandi C., Protti N., Bortolussi S., Altieri S., Aime S., Deagostino A., Org. Biomol. Chem. 2014, 12, 2457–2467. [DOI] [PubMed] [Google Scholar]
- 176. Kikuchi S., Kanoh D., Sato S., Sakurai Y., Suzuki M., Nakamura H., J. Controlled Release 2016, 237, 160–167. [DOI] [PubMed] [Google Scholar]
- 177. Li J., Janoušková O., Fernandez-Alvarez R., Mesíková S., Tošner Z., Kereïche S., Uchman M., Matejicek P., Chem. Eur. J. 2020, 26, 14283–14289. [DOI] [PubMed] [Google Scholar]
- 178. Takeuchi I., Nomura K., Makino K., Colloids Surf. B 2017, 159, 360–365. [DOI] [PubMed] [Google Scholar]
- 179. Kowalski K., Goszczyński T., Leśnikowski Z. J., Boratyński J., ChemBioChem 2015, 16, 424–431. [DOI] [PubMed] [Google Scholar]
- 180. Kikuchi S., Sato S., Nakamura H., Appl. Radiat. Isot. 2020, 157, 109011–109014. [DOI] [PubMed] [Google Scholar]
- 181. Nakamura H., Kikuchi S., Kawai K., Ishii S., Sato S., Pure Appl. Chem. 2018, 90, 745–753. [Google Scholar]
- 182. Ay A. N., Akar H., Zaulet A., Viňas C., Teixidor F., Zumreoglu-Karan B., Dalton Trans. 2017, 46, 3303–3310. [DOI] [PubMed] [Google Scholar]
- 183. Koganei H., Ueno M., Tachikawa S., Tasaki L., Ban H. S., Suzuki M., Shiraishi K., Kawano K., Yokoyama M., Maitani Y., Ono K., Nakamura H., Bioconjugate Chem. 2013, 24, 124–132. [DOI] [PubMed] [Google Scholar]
- 184. Ueno M., Ban H. S., Nakai K., Inomata R., Kaneda Y., Matsumura A., Nakamura H., Bioorg. Med. Chem. 2010, 18, 3059–3065. [DOI] [PubMed] [Google Scholar]
- 185. Sumitani S., Oishi M., Yaguchi T., Murotani H., Horiguchi Y., Suzuki M., Ono K., Yanagie H., Nagasaki Y., Biomaterials 2012, 33, 3568–3577. [DOI] [PubMed] [Google Scholar]
- 186. Takeuchi I., Ariyama M., Makino K., J. Oleo Sci. 2019, 68, 361–368. [DOI] [PubMed] [Google Scholar]
- 187. Olusanya T. O. B., Calabrese G., Fatouros D. G., Tsibouklis J., Smith J. R., Biophys. Chem. 2019, 247, 25–33. [DOI] [PubMed] [Google Scholar]
- 188. Pasternack R. F., Bustamante C., Collings P. J., Giannetto A., Gibbs E. J., J. Am. Chem. Soc. 1993, 115, 5393–5399. [Google Scholar]
- 189.
- 189a. Hao E., Sibrian-Vazquez M., Serem W., Garno J. C., Fronczek F. R., Vicente M. G. H., Eur. J. Chem. 2007, 13, 9035–9042; [DOI] [PubMed] [Google Scholar]
- 189b. Kubat P., Lang K., Cígler P., Kozísek M., Matejícek P., Janda P., Zelinger Z., Prochazka K., Kral V., J. Phys. Chem. B 2007, 111, 4539–4546; [DOI] [PubMed] [Google Scholar]
- 189c. Olshevskaya V. A., Savchenko A. N., Golovina G. V., Lazarev V. V., Kononova E. G., Petrovskii P. V., Kalinin V. N., Shtil’ A. A., Kuz'min V. A., Dokl. Chem. 2010, 435, 328–333. [Google Scholar]
- 190.J. M. Prausnitz, R. N. Lichtenthaler, E. Gomes de Azevedo, Molecular Thermodynamics of Fluid-Phase Equilibria, 3rd ed., Prentice Hali PTR, Upper Saddle River, 1999.
- 191. Dreaden E. C., Alkilany A. M., Huang X., Murphy C. J., El-Sayed M. A., Chem. Soc. Rev. 2012, 41, 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Rak J., Kaplánek R., Král V., Bioorg. Med. Chem. Lett. 2010, 20, 1045–1048. [DOI] [PubMed] [Google Scholar]
- 193. Rak J., Jakubek M., Kaplánek R., Matějíček P., Král V., Eur. J. Med. Chem. 2011, 46, 1140–1146. [DOI] [PubMed] [Google Scholar]
- 194.B. Testa, P.-A. Carrupt, P. Gaillard, R.-S. Tsai in Methods and Principles in Medicinal Chemistry (Eds.: V. Pliška, B. Testa, H. van de Waterbeemd), Wiley-VCH, Weinheim, 1996.
- 195. Hristovski K. D., Westerhoff P. K., Posner J. D., J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 2011, 46, 636–647. [DOI] [PubMed] [Google Scholar]
- 196. Powers K. W., Brown S. C., Krishna V. B., Wasdo S. C., Moudgil B. M., Roberts S. M., Toxicol. Sci. 2006, 90, 296–303. [DOI] [PubMed] [Google Scholar]
- 197. Wu L.-P., Wang D., Li Z., Mater. Sci. Eng. C 2020, 106, 110302–110308. [DOI] [PubMed] [Google Scholar]
- 198. Dickinson A. M., Godden J. M., Lanovyk K., Ahmed S. S., Appl. In Vitro Toxicol. 2019, 5, 114–122. [Google Scholar]
- 199. Hofmann-Amtenbrink M., Grainger D. W., Hofmann H., Nanomedicine 2015, 11, 1689–1694. [DOI] [PubMed] [Google Scholar]
- 200. Oksel C., Ma C. Y., Liu J. J., Wilkins T., Wang X. Z., Particuology 2015, 21, 1–19. [Google Scholar]
- 201. Fischer H. C., Chan W. C. W., Curr. Opin. Biotechnol. 2007, 18, 565–571. [DOI] [PubMed] [Google Scholar]
- 202.
- 202a. Moghimi S. M., Andersen A. J., Hashemi S. H., Lettiero B., Ahmadvand D., Hunter A. C., Andresen T. L., Hamad I., Szebeni J., J. Controlled Release 2010, 146, 175–181; [DOI] [PubMed] [Google Scholar]
- 202b. Patra J. K., Das G., Fraceto L. F., Campos E. V. R., Rodriguez-Torres M. D. P., Acosta-Torres L. S., Diaz-Torres L. A., Grillo R., Swamy M. K., Sharma S., Habtemariam S., Shin H.-S., J. Nanobiotechnol. 2018, 16, 71–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.
- 203a. Buzharevski A., Paskas S., Sárosi M.-B., Laube M., Lönnecke P., Neumann W., Mijatovic S., Maksimovic-Ivanic D., Pietzsch J., Hey-Hawkins E., ChemMedChem 2019, 14, 315–321; [DOI] [PubMed] [Google Scholar]
- 203b. Buzharevski A., Paskaš S., Sárosi M.-B., Laube M., Lönnecke P., Neumann W., Murganić B., Mijatović S., Maksimović-Ivanić D., Pietzsch J., Hey-Hawkins E., Sci. Rep. 2020, 10, 4827–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Kawasaki R., Sasaki Y., Akiyoshi K., Biochem. Biophys. Res. Commun. 2017, 483, 147–152. [DOI] [PubMed] [Google Scholar]
- 205. Wang J., Chen L., Ye J., Li Z., Jiang H., Yan H., Stogniy M. Y., Sivaev I. B., Bregadze V. I., Wang X., Biomacromolecules 2017, 18, 1466–1472. [DOI] [PubMed] [Google Scholar]
- 206. Wang C., Sang H., Wang Y., Zhu F., Hu X., Wang X., Wang X., Li Y., Cheng Y., Nano Lett. 2018, 18, 7045–7051. [DOI] [PubMed] [Google Scholar]
- 207. Goswami L. N., Cai Q., Ma L., Jalisatgi S. S., Hawthorne M. F., Org. Biomol. Chem. 2015, 13, 8912–8918. [DOI] [PubMed] [Google Scholar]
- 208. Drasler B., Sayre P., Steinhäuser K. G., Petri-Fink A., Rothen-Rutishauser B., NanoImpact 2017, 8, 99–116. [Google Scholar]
- 209. Stefaniak A. B., Hackley V. A., Roebben G., Ehara K., Hankin S., Postek M. T., Lynch I., Fu W.-E., Linsinger T. P. J., Thünemann A. F., Nanotoxicology 2013, 7, 1325–1337. [DOI] [PubMed] [Google Scholar]
- 210. Tinkle S., McNeil S. E., Mühlebach S., Bawa R., Borchard G., Barenholz Y. C., Tamarkin L., Desai N., Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [DOI] [PubMed] [Google Scholar]
- 211. Vrbata D., Ďorďovič V., Seitsonen J., Ruokolainen J., Janoušková O., Uchman M., Matějíček P., Chem. Commun. 2019, 55, 2900–2903. [DOI] [PubMed] [Google Scholar]
- 212. Fernandez-Alvarez R., Medoš Ž., Tošner Z., Zhigunov A., Uchman M., Hervø-Hansen S., Lund M., Bešter-Rogač M., Matějíček P., Langmuir 2018, 34, 14448–14457. [DOI] [PubMed] [Google Scholar]
- 213.
- 213a. Malaspina D. C., Viñas C., Teixidor F., Faraudo J., Angew. Chem. Int. Ed. 2020, 59, 3088–3092; [DOI] [PubMed] [Google Scholar]
- 213b. Tarrés M., Viñas C., González-Cardoso P., Hänninen M. M., Sillanpää R., Dorďovič V., Uchman M., Teixidor F., Matějíček P., Chem. Eur. J. 2014, 20, 6786–6794. [DOI] [PubMed] [Google Scholar]
- 214. Hannon M. J., Chem. Soc. Rev. 2007, 36, 280–295. [DOI] [PubMed] [Google Scholar]
- 215. Yanagie H., Maruyama K., Takizawa T., Ishida O., Ogura K., Matsumoto T., Sakurai Y., Kobayashi T., Shinohara A., Rant J., Skvarc J., Ilic R., Kuhne G., Chiba M., Furuya Y., Sugiyama H., Hisa T., Ono K., Kobayashi H., Eriguchi M., Biomed. Pharmacother. 2006, 60, 43–50. [DOI] [PubMed] [Google Scholar]
- 216. Kanemitsu T., Kawabata S., Fukumura M., Futamura G., Hiramatsu R., Nonoguchi N., Nakagawa F., Takata T., Tanaka H., Suzuki M., Masunaga S.-I., Ono K., Miyatake S.-I., Nakamura H., Kuroiwa T., Radiat. Environ. Biophys. 2019, 58, 59–67. [DOI] [PubMed] [Google Scholar]
- 217. Wu C., Shi L., Li Q., Jiang H., Selke M., Yan H., Wang X., Nanomedicine Nanotechnology, Biol. Med. 2012, 8, 860–869. [DOI] [PubMed] [Google Scholar]
- 218. Tyrrell D. A., Richardson V. J., Ryman B. E., Biochim. Biophys. Acta 1977, 497, 469–480. [DOI] [PubMed] [Google Scholar]
- 219. Cedervall T., Lynch I., Lindman S., Berggård T., Thulin E., Nilsson H., Dawson K. A., Linse S., Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Berrecoso G., Crecente-Campo J., Alonso M. J., Drug Delivery Transl. Res. 2020, 10, 730–750. [DOI] [PubMed] [Google Scholar]
- 221. Casals E., Pfaller T., Duschl A., Oostingh G. J., Puntes V., ACS Nano 2010, 4, 3623–3632. [DOI] [PubMed] [Google Scholar]
- 222. Pulido-Reyes G., Leganes F., Fernández-Piñas F., Rosal R., Environ. Toxicol. Chem. 2017, 36, 3181–3193. [DOI] [PubMed] [Google Scholar]
- 223. Ishihara T., Takeda M., Sakamoto H., Kimoto A., Kobayashi C., Takasaki N., Yuki K., Tanaka K.-i., Takenaga M., Igarashi R., Maeda T., Yamakawa N., Okamoto Y., Otsuka M., Ishida T., Kiwada H., Mizushima Y., Mizushima T., Pharm. Res. 2009, 26, 2270–2279. [DOI] [PubMed] [Google Scholar]
- 224.
- 224a. Kueffer P. J., Maitz C. A., Khan A. A., Schuster S. A., Shlyakhtina N. I., Jalisatgi S. S., Brockman J. D., Nigg D. W., Hawthorne M. F., Proc. Natl. Acad. Sci. USA 2013, 110, 6512–6517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224b. Maitz C. A., Khan A. A., Kueffer P. J., Brockman J. D., Dixson J., Jalisatgi S. S., Nigg D. W., Everett T. A., Hawthorne M. F., Transl. Oncol. 2017, 10, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Kettiger H., Schipanski A., Wick P., Huwyler J., Int. J. Nanomed. 2013, 8, 3255–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Chen H., Kim S., He W., Wang H., Low P. S., Park K., Cheng J.-X., Langmuir 2008, 24, 5213–5217. [DOI] [PubMed] [Google Scholar]
- 227. Wu C., Ye H., Bai W., Li Q., Guo D., Lv G., Yan H., Wang X., Bioconjugate Chem. 2011, 22, 16–25. [DOI] [PubMed] [Google Scholar]
- 228.
- 228a. Couto M., Mastandrea I., Cabrera M., Cabral P., Teixidor F., Cerecetto H., Viñas C., Chem. Eur. J. 2017, 23, 9233–9238; [DOI] [PubMed] [Google Scholar]
- 228b. Couto M., García M. F., Alamón C., Cabrera M., Cabral P., Merlino A., Teixidor F., Cerecetto H., Viñas C., Chem. Eur. J. 2018, 24, 3122–3126. [DOI] [PubMed] [Google Scholar]