

Abstract
The first cationic titanium catalyst system for the intermolecular hydroaminoalkylation of alkenes with various tertiary alkylamines is presented. Corresponding reactions which involve the addition of the α‐C−H bond of a tertiary amine across the C−C double bond of an alkene take place at temperatures close to room temperature with excellent regioselectivity to deliver the branched products exclusively. Interestingly, for selected amines, α‐C−H bond activation occurs not only at N‐methyl but also at N‐methylene groups.
Keywords: alkenes, C−H activation, hydroaminoalkylation, tertiary amines, titanium
Tertiary amines can be used as substrates for the highly regioselective intermolecular hydroaminoalkylation of alkenes in the presence of a cationic titanium catalyst generated in situ from tetrabenzyltitanium and Ph3C[B(C6F5)4]. While in most cases, the involved C−H bond activation reaction occurs at N‐methyl groups, N‐methylazepane and N‐methylpyrrolidine also undergo alkylation at the methylene group in position α to the nitrogen atom.
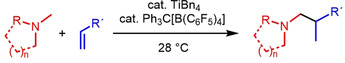
Tertiary amines are important structural motifs in natural products (e.g. alkaloids) and are indispensable for the development of agrochemicals or pharmaceuticals. [1] For example, more than 15 % of the 200 top selling small molecule drugs in 2018 contain a tertiary amine moiety. [1b] An attractive synthetic approach for the synthesis of various amines that has raised a lot of attention in recent years is the hydroaminoalkylation of alkenes which allows the 100 % atom economic addition of the α‐C−H bond of simple amines across the C−C double bond of alkenes (Scheme 1). [2] Corresponding addition reactions can be achieved with late transition metal catalysts, [3] following a photo‐catalytic approach, [4] or most efficiently with early transition metal catalysts.[ 5 , 6 , 7 ] In the latter case, neutral group 4 [5] and 5 [6] metal catalysts have extensively been used for a plethora of successful hydroaminoalkylation reactions of alkenes with primary or secondary amines (Scheme 1 a) but unfortunately, tertiary amines do not react successfully with alkenes in the presence of these catalysts. This lack of reactivity must be regarded as a severe restriction to the use of hydroaminoalkylation reactions, because it prohibits the use of simple tertiary amines as starting materials for the synthesis of more sophisticated tertiary amine products. Although a few late transition metal‐catalyzed hydroaminoalkylation reactions with tertiary amines have been reported,[ 3 , 4 ] in these cases, the amine must contain an additional metal‐binding directing group. However, alkene hydroaminoalkylation with tertiary amine substrates that do not contain an extra directing group could already be achieved in the presence of cationic scandium catalysts (Scheme 1 b). [7] Unfortunately, scandium must be regarded as a highly expensive metal [8] and the catalysts, which need to be synthesized by laborious processes, are only active at elevated temperatures of 70–120 °C.[ 7a , 7c , 7e ] Based on our experiences in the development of titanium‐catalyzed hydroaminoalkylation reactions of alkenes, allenes, [9] and alkynes, [10] and the fact that non‐toxic titanium is the second most abundant transition metal in the earth's crust, [11] we recently started a project to identify an alternative, less expensive, titanium‐based catalyst system for the intermolecular hydroaminoalkylation of alkenes with simple tertiary amines (Scheme 1 c).
Scheme 1.
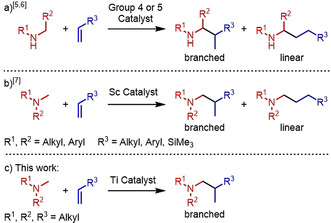
Intermolecular hydroaminoalkylation of alkenes with secondary or tertiary N‐alkylamines in the presence of group 3, 4 or 5 catalysts.
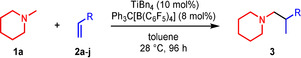
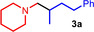
While in the case of titanium‐catalyzed hydroaminoalkylation reactions of unsaturated substrates with primary or secondary amines neutral titanaaziridines formed from a neutral titanium (IV) precursor and the amine substrate are accepted to be the catalytically active species, [12] corresponding scandium‐catalyzed reactions of tertiary amines involve the formation of cationic metallaaziridine intermediates (Scheme 2, M=Sc, n=1).[ 7b , 7d ] Because analogous cationic titanaaziridines (Scheme 2, M=Ti, n=2) should be formed from tertiary amines and cationic titanium (IV) complexes, we initially investigated whether mixtures of the well‐established hydroaminoalkylation catalysts TiBn4 [5b] or Ind2TiMe [5c] (Ind=η5‐indenyl) with Ph3C[B(C6F5)4] catalyze the hydroaminoalkylation of alkenes with N‐methylpiperidine (1 a) as an example of a typical tertiary aliphatic amine (Table 1). The Lewis acid Ph3C[B(C6F5)4] is known to induce benzyl group abstraction from TiBn4. [13] While initial experiments at elevated temperatures (105–160 °C) which are typically required for Ind2TiMe2‐ or TiBn4‐catalyzed hydroaminoalkylation reactions with primary or secondary amines did not lead to good results, we were delighted to see that 4‐phenylbutene (2 a) and N‐methylpiperidine (1 a) smoothly undergo the desired hydroaminoalkylation reaction at temperatures close to room temperature in the presence of 10 mol % TiBn4 and 8 mol % Ph3C[B(C6F5)4]. [14] Best results were obtained when the starting materials, the catalyst, and toluene were simply mixed in a vial and stirred on a stirring plate for 96 h. [15] Although the stirring plate was not purposely heated, this simple experimental setup led to a reaction temperature of 28 °C inside the vial. GC analysis of the crude reaction mixture revealed that the branched hydroaminoalkylation product 3 a was formed exclusively and finally, it was possible to isolate 3 a in 99 % yield (Table 1, entry 1).
Scheme 2.
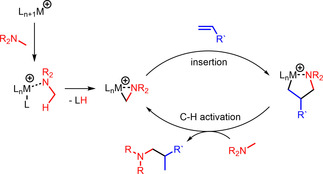
Simplified catalytic cycle of the hydroaminoalkylation of alkenes with tertiary amines which includes a cationic metallaaziridine as the catalytically active species (M=Sc, n=1 or M=Ti, n=2). [7]
Table 1.
Scope of the titanium‐catalyzed hydroaminoalkylation of various alkenes with N‐methylpiperidine (1 a).[a]
|
Entry |
Alkene |
Product |
Yield [%][b] |
|---|---|---|---|
|
1 |
|
|
99 |
|
2 |
|
|
31 |
|
3[c] |
|
|
77 |
|
4 |
|
|
92 |
|
5 |
|
|
66 |
|
6[d] |
|
|
51 |
|
7 |
|
|
64 |
|
8 |
|
|
96 |
|
9 |
|
|
54 |
|
10 |
|
|
55 |
|
11 |
|
|
84 |
|
12 |
|
|
16 |
|
13[e] |
|
|
24 |

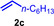
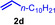
[a] Reaction conditions: N‐methylpiperidine (1 a, 1.00 mmol), alkene (1.50 mmol), TiBn4 (10 mol %), Ph3C[B(C6F5)4] (8 mol %), toluene (5 mL), 28 °C, 96 h, 25 mL vial. [b] Isolated yield. [c] 18 h. [d] Reaction conditions: 1 a (2.00 mmol), alkene (3.00 mmol), TiBn4 (5 mol %), Ph3C[B(C6F5)4] (4 mol %), toluene (5 mL), 28 °C, 18 h, 25 mL vial. [e] 72 h.
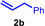
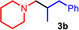
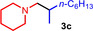
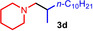
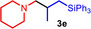
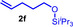
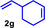
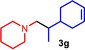
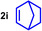
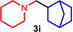
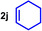
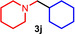
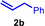
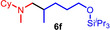
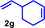
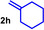
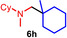
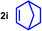
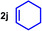
As can be seen from Table 1, a wide variety of additional alkenes (2 b–2 j) could also be reacted successfully with N‐methylpiperidine (1 a) under identical conditions. In the case of monosubstituted terminal alkenes, the corresponding branched hydroaminoalkylation products were always formed exclusively and as a result, the tertiary amines 3 a–3 g could be isolated in good to excellent yields of 54–99 % (Table 1, entries 1, 3–5, and 7–9). When allylbenzene (2 b) was used as a substrate, decomposition of product 3 b was observed under the reaction conditions and as a result, the modest yield of 31 % in which 3 b was isolated after 96 h (Table 1, entry 2) could be improved to 77 % by decreasing the reaction time to 18 h (Table 1, entry 3). With regard to the high oxophilicity of cationic titanium catalysts it is remarkable that the catalyst not only tolerates a triphenylsilyl substituent (Table 1, entry 7) but also the presence of a sterically demanding triisopropylsilyl ether (Table 1, entry 8) which offers a promising possibility for further functionalization. The fact that 4‐vinylcyclohexene 2 g undergoes selective hydroaminoalkylation at the stericaly less hindered vinyl group to give 3 g as the product (Table 1, entry 9) clearly demonstrates that internal alkenes are less reactive than terminal alkenes. Nevertheless, it was possible to convert norbornene (2 i) into the expected hydroaminoalkylation product 3 i in 84 % yield (Table 1, entry 11). The assumption that ring strain plays an important role for reactions of internal alkenes is strongly underlined by the reaction of cyclohexene (2 j) which delivered the expected product 3 j only in poor yields (Table 1, entries 12 and 13). Interestingly, 1,1‐disubstitution of the alkene is better tolerated and as a consequence, methylenecyclohexane (2 h) regioselectively reacted to the corresponding branched amine 3 h in 55 % yield (Table 1, entry 10). In contrast, styrenes could not successfully be used because these substrates underwent fast polymerization. To further demonstrate the efficiency of the new catalyst system, we additionally investigated the reaction of 1‐dodecene (2 d) with 1 a as a typical example in more detail. We tried to reduce the catalyst loading, to minimize the reaction time, and to run the reaction on a multigram scale. During these experiments, it turned out that after stirring of the reaction mixture in a vial at 28 °C for 18 h in the presence of 5 mol % TiBn4 and 4 mol % Ph3C[B(C6F5)4], the hydroaminoalkylation product 3 d is still formed in 51 % yield (Table 1, entry 6). Then an even better yield of 60 % could be obtained from a corresponding experiment which was performed on a 20 mmol scale in a Schlenk tube at a slightly elevated temperature of 35 °C (Scheme 3). In this case, 3.2 g of product 3 d were isolated.
Scheme 3.
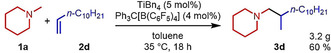
Multigram scale hydroaminoalkylation of 1‐dodecene (2 d) with N‐methylpiperidine (1 a).
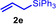
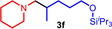
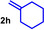
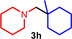
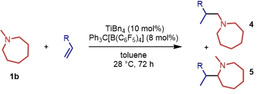
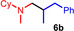
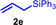
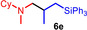
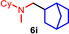
During a subsequent study of the behavior of the closely related tertiary amines N‐methylazepane (1 b) and N‐methylpyrrolidine (1 c) we then recognized that under the typical reaction conditions, these substrates are always converted into two hydroaminoalkylation products. For example, α‐alkylation of N‐methylazepane (1 b) with 4‐phenylbutene (2 a) either took place at the N‐methyl substituent or at the α‐CH2‐group of the seven‐membered ring. As a result, the corresponding products 4 a and 5 a were isolated in 22 % and 37 % yield, respectively (Table 2, entry 1). While in this case, α‐alkylation preferentially takes place at the methylene group of 1 b which leads to a selectivity of 62:38 in favor of 5 a, a corresponding reaction with the sterically more damanding alkene allyltriphenylsilane (2 e) slightly favors alkylation at the sterically less hindered N‐methyl substituent. The corresponding products 4 b and 5 b could be isolated in 35 % and 29 % yield, respectively. After hydrochloride formation, we were able to unambiguously determine the structure of 5 b⋅HCl by single crystal X‐ray diffraction (Figure 1). [16] Additional hydroaminoalkylation reactions performed with N‐methylpyrrolidine (1 c) and various alkenes (2 a–d) then revealed that 1 c behaves similar but unfortunately, we were not able to seperate the two hydroaminoalkylation products which were formed in all theses reactions. However, corresponding product mixtures could be isolated in yields of 18–36 % and GC‐analyses showed that again alkylation at the α‐CH2‐group of the heterocyclic ring is slightly favored. [15] In this context, it should be noted that hydroaminoalkylation reactions of alkenes in which α‐CH2‐alkylation of an amine is preferred over a competing α‐CH3‐alkylation are extremely rare. To the best of our knowledge, not a single example of a corresponding scandium‐catalyzed process of a tertiary amine is known and in the case of group 4 or 5 metal‐catalyzed reactions of secondary amines, only additionally activated amines like N‐methylbenzylamine selectively react at the methylene group.[ 5d , 6c ]
Table 2.
Titanium‐catalyzed hydroaminoalkylation of selected alkenes with N‐methylazepane (1 b).[a]
|
Entry |
Alkene |
R |
Sel. 4/5 [b] |
Yield 4 [%][c] |
Yield 5 [%][c] |
|---|---|---|---|---|---|
|
1 |
2 a |
(CH2)2Ph |
38:62 |
22 (4 a) |
37 (5 a) |
|
2 |
2 e |
CH2SiPh3 |
54:46 |
35 (4 b) |
29 (5 b) |
[a] Reaction conditions: N‐methylazepane (1 b, 1.00 mmol), alkene (1.50 mmol), TiBn4 (10 mol %), Ph3C[B(C6F5)4] (8 mol %), toluene (5 mL), 28 °C, 72 h, 25 mL vial. [b] Selectivity was determined by GC analysis prior to flash chromatography. [c] Isolated yield.
Figure 1.
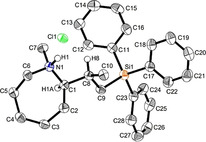
Ellipsoid representation of the molecular structure of 5 b⋅HCl. Hydrogen atoms are omitted for clarity (exception H1, H1A and H8). Ellipsoids are drawn at the 50 % probability level. [16]
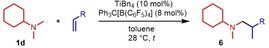
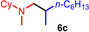
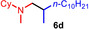
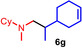
Next we turned our attention towards reactions of non‐heterocyclic tertiary amines and we found that for example, N,N‐dimethylcyclohexylamine (1 d) easily reacts with various alkenes (2 a–2 j) to give the desired hydroaminoalkylation products 6 a–6 j in modest to good yields between 44 % and 81 % (Table 3). Unfortunately, during our entire study, TLC‐visualization of the tertiary amines which was essential for the final chromatographic purification of the products was extremly difficult to achieve. It was just sufficiently possible with a H2PtCl6/KI solution [15] and the modest sensitivity of the detection might be responsible for some of the modest yields. However, as observed before, N,N‐dimethylcyclohexylamine (1 d) and terminal alkenes were selectively converted into the branched hydroaminoalkylation products and internal alkenes gave lower yields than terminal alkenes. Overall, the reactions of 1 d were slightly faster than those of N‐methylpiperidine (1 a) which led to reduced reaction times between 24 h and 72 h.
Table 3.
Scope of the titanium‐catalyzed hydroaminoalkylation of various alkenes with N,N‐dimethylcyclohexylamine (1 d).[a]
|
Entry |
Alkene |
Product |
t [h] |
Yield [%][b] |
|---|---|---|---|---|
|
1 |
|
|
72 |
79 |
|
2 |
|
|
72 |
59 |
|
3 |
|
|
72 |
81 |
|
4 |
|
|
72 |
66 |
|
5 |
|
|
24 |
59 |
|
6 |
|
|
24 |
64 |
|
7 |
|
|
72 |
55 |
|
8 |
|
|
42 |
77 |
|
9 |
|
|
48 |
55 |
|
10 |
|
|
72 |
44 |

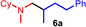
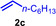
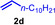

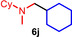
[a] Reaction conditions: N,N‐dimethylcyclohexylamine (1 d, 1.00 mmol), alkene (1.50 mmol), TiBn4 (10 mol %), Ph3C[B(C6F5)4] (8 mol %), toluene (5 mL), 28 °C, t, 25 mL vial. [b] Isolated yield.
While corresponding reactions of sterically demanding amines like N,N‐dicyclohexylmethylamine failed, it was also possible to convert N,N‐dimethylbenzylamine (1 e), N,N‐dimethylethylamine (1 f), and N,N‐dimethylisopropylamine (1 g) into selected hydroaminoalkylation products (Scheme 4). Interestingly, alkylation of N,N‐dimethylbenzylamine (1 e), which gave better results at a slightly elevated temperature of 35 °C, took place selectively at the methyl group and not in the activated benzylic position. Although the yields, obtained from reactions of N,N‐dimethylethylamine (1 f) or N,N‐dimethylisopropylamine (1 g) are not satifactory yet, the results clearly support the impression that cationic titanium catalysts might be useful for potential industrial applications, for example, hydroaminoalkylation reactions using gaseous trimethylamine as a substrate. In the case of N,N‐dimethylisopropylamine (1 g), two products, a mono‐ (9 b) and a dialkylated product (10 b) were isolated in a combined yield of 42 % from a reaction with 1‐octene (2 c). This yield could already be improved to 57 % (9 b, 47 % and 10 b, 10 %) by using a catalyst loading of 20 mol % at a slightly evevated temperature of 35 °C. Finally, it should be noted that all attempts to react N,N‐dimethylaniline with 4‐phenylbutene (2 a) or 1‐octene (2 c) failed so far.
Scheme 4.
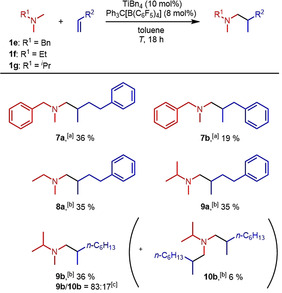
Additional successful hydroaminoalkylation reactions of alkenes with tertiary amines. Reaction conditions: amine (1.00 mmol), alkene (1.50 mmol), TiBn4 (10 mol %), Ph3C[B(C6F5)4] (8 mol %), toluene (5 mL), T, 18 h. [a] 35 °C, 100 mL Schlenk tube. [b] 28 °C, 25 mL vial. [c] Selectivity was determined by GC analysis prior to chromatography.
In summary, we have presented the first examples of titanium‐catalyzed hydroaminoalkylation reactions of alkenes that use tertiary amines as substrates. The cationic titanium catalyst is generated in situ from readily available TiBn4 [5f] and Ph3C[B(C6F5)4]. Successful hydroaminoalkylation could be achieved with heterocyclic and non‐heterocyclic aliphatic amines as well as mono‐ and 1,1‐ or 1,2‐disubstituted alkenes at temperatures close to room temperature. Particularly interesting is the fact that occasionally, α‐CH2‐alkylation of the amine is preferred over a competing α‐CH3‐alkylation. Further opimization studies to take advantage of this promising result and to expand the scope of the reaction towards aromatic amines are presently underway in our laboratories.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Research Training Group “Chemical Bond Activation” (GRK 2226) funded by the Deutsche Forschungsgemeinschaft for financial support of our research. Open access funding enabled and organized by Projekt DEAL.
D. Geik, M. Rosien, J. Bielefeld, M. Schmidtmann, S. Doye, Angew. Chem. Int. Ed. 2021, 60, 9936.
References
- 1.
- 1a. McGrath N. A., Brichacek M., Njardarson J. T., J. Chem. Educ. 2010, 87, 1348–1349; [Google Scholar]
- 1b.Top Pharmaceuticals Poster | Njarðarson (arizona.edu): https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster.
- 2.Selected reviews:
- 2a. Roesky P. W., Angew. Chem. Int. Ed. 2009, 48, 4892–4894; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4988–4991; [Google Scholar]
- 2b. Chong E., Garcia P., Schafer L. L., Synthesis 2014, 46, 2884–2896; [Google Scholar]
- 2c. Edwards P. M., Schafer L. L., Chem. Commun. 2018, 54, 12543–12560; [DOI] [PubMed] [Google Scholar]
- 2d. Hannedouche J., Schulz E., Organometallics 2018, 37, 4313–4326. [Google Scholar]
- 3.Selected reviews:
- 3a. Dong Z., Ren Z., Thompson S. J., Xu Y., Dong G., Chem. Rev. 2017, 117, 9333–9403; [DOI] [PubMed] [Google Scholar]
- 3b. Nakajima K., Miyake Y., Nishibayashi Y., Acc. Chem. Res. 2016, 49, 1946–1956. [DOI] [PubMed] [Google Scholar]
- 4.Selected recent examples:
- 4a. Leng L., Fu Y., Liu P., Ready J. M., J. Am. Chem. Soc. 2020, 142, 11972–11977; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Trowbridge A., Reich D., Gaunt M. J., Nature 2018, 561, 522–527; [DOI] [PubMed] [Google Scholar]
- 4c. Thullen S. M., Rovis T. A., J. Am. Chem. Soc. 2017, 139, 15504–15508. [DOI] [PubMed] [Google Scholar]
- 5.Selected examples of group 4 metal catalysts:
- 5a. Kubiak R., Prochnow I., Doye S., Angew. Chem. Int. Ed. 2009, 48, 1153–1156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1173–1176; [Google Scholar]
- 5b. Prochnow I., Kubiak R., Frey O. N., Beckhaus R., Doye S., ChemCatChem 2009, 1, 162–172; [Google Scholar]
- 5c. Kubiak R., Prochnow I., Doye S., Angew. Chem. Int. Ed. 2010, 49, 2626–2629; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2683–2686; [Google Scholar]
- 5d. Dörfler J., Preuß T., Schischko A., Schmidtmann M., Doye S., Angew. Chem. Int. Ed. 2014, 53, 7918–7922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8052–8056; [Google Scholar]
- 5e. Koperniku A., Foth P. J., Sammis G. M., Schafer L. L., J. Am. Chem. Soc. 2019, 141, 18944–18948; [DOI] [PubMed] [Google Scholar]
- 5f. Bielefeld J., Doye S., Angew. Chem. Int. Ed. 2020, 59, 6138–6143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6194–6199. [Google Scholar]
- 6.Selected examples of group 5 metal catalysts:
- 6a. Clerici M. G., Maspero F., Synthesis 1980, 305–306; [Google Scholar]
- 6b. Herzon S. B., Hartwig J. F., J. Am. Chem. Soc. 2007, 129, 6690–6691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Eisenberger P., Ayinla R. O., Lauzon J. M. P., Schafer L. L., Angew. Chem. Int. Ed. 2009, 48, 8361–8365; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8511–8515; [Google Scholar]
- 6d. Zi G., Zhang F., Song H., Chem. Commun. 2010, 46, 6296–6298; [DOI] [PubMed] [Google Scholar]
- 6e. Reznichenko A. L., Hultzsch K. C., J. Am. Chem. Soc. 2012, 134, 3300–3311; [DOI] [PubMed] [Google Scholar]
- 6f. Chong E., Brandt J. W., Schafer L. L., J. Am. Chem. Soc. 2014, 136, 10898–10901; [DOI] [PubMed] [Google Scholar]
- 6g. Daneshmand P., Roşca S.-C., Dalhoff R., Yin K., DiPucchio R. C., Ivanovich R. A., Polat D. E., Beauchemin A. M., Schafer L. L., J. Am. Chem. Soc. 2020, 142, 15740–15750. [DOI] [PubMed] [Google Scholar]
- 7.Scandium catalysts:
- 7a. Nako A. E., Oyamada J., Nishiura M., Hou Z., Chem. Sci. 2016, 7, 6429–6434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Liu F., Luo G., Hou Z., Luo Y., Organometallics 2017, 36, 1557–1565; [Google Scholar]
- 7c. Gao H., Su J., Xu P., Xu X., Org. Chem. Front. 2018, 5, 59–63; [Google Scholar]
- 7d. Luo G., Liu F., Luo Y., Zhou G., Kang X., Hou Z., Luo L., Organometallics 2019, 38, 1887–1896; [Google Scholar]
- 7e. Su J., Zhou Y., Xu X., Org. Biomol. Chem. 2019, 17, 2013–2019. [DOI] [PubMed] [Google Scholar]
- 8.J. Gambogi, Mineral commodity summaries 2020, Scandium, U.S. Geological Survey 2020, pp. 144–145.
- 9. Kaper T., Fischer M., Warsitz M., Zimmering R., Beckhaus R., Doye S., Chem. Eur. J. 2020, 26, 14300–14304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaper T., Fischer M., Thye H., Geik D., Schmidtmann M., Beckhaus R., Doye S., Chem. Eur. J. 2021, 27, 10.1002/chem.202100238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hunt A. J., Farmer T. J. in Sustainable Catalysis: With Non-endangered Metals, Part 1 (Ed.: North M.), Royal Society of Chemistry, Cambridge, 2015, pp. 1–14. [Google Scholar]
- 12.
- 12a. Prochnow I., Zark P., Müller T., Doye S., Angew. Chem. Int. Ed. 2011, 50, 6401–6405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6525–6529; [Google Scholar]
- 12b. Manßen M., Lauterbach N., Dörfler J., Schmidtmann M., Saak W., Doye S., Beckhaus R., Angew. Chem. Int. Ed. 2015, 54, 4383–4387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4458–4462. [Google Scholar]
- 13. Lin M., Baird M. C., J. Organomet. Chem. 2001, 619, 62–73. [Google Scholar]
- 14.In the absence of Ph3C[B(C6F5)4], no reaction occurs.
- 15.For details, see the Supporting Information.
- 16. Deposition Number 2049498 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary