

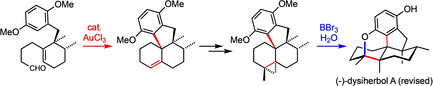
Abstract
A 12‐step total synthesis of the natural product dysiherbol A, a strongly anti‐inflammatory and anti‐tumor avarane meroterpene isolated from the marine sponge Dysidea sp., was elaborated. As key steps, the synthesis features an enantioselective Cu‐catalyzed 1,4‐addition/enolate‐trapping opening move, an Au‐catalyzed double cyclization to build up the tetracyclic core‐carbon skeleton, and a late installation of the C5‐bridgehead methyl group via proton‐induced cyclopropane opening associated with spontaneous cyclic ether formation. The obtained pentacyclic compound (corresponding to an anhydride of the originally suggested structure for dysiherbol A) showed identical spectroscopic data as the natural product, but an opposite molecular rotation. CD‐spectroscopic measurements finally confirmed that both the constitution and the absolute configuration of the originally proposed structure of (+)‐dysiherbol A need to be revised.
Keywords: cyclization, enantioselectivity, gold catalysis, natural products, rearrangement
In a key step of the first enantioselective total synthesis of the anti‐inflammatory marine meroterpene dysiherbol A, the tetra‐carbocyclic core skeleton is set‐up in a unique gold‐catalyzed double cyclization, and the ether bridge of the pentacyclic target structure is spontaneously formed under cyclopropane opening upon deprotection of a late intermediate.
In 2016 Jiao et al. reported the isolation and structural elucidation of dysiherbols A–C (1–3) and dysideanone E (4) as metabolites of the sponge Dysidea sp. collected in the South China Sea (Figure 1). [1] Initial biological testing revealed dysiherbol A (as the most active of these compounds) to exhibit cytotoxic activity towards the cancer cell line NCI H‐929 at sub‐micromolar concentrations. Furthermore, 1 showed strong inhibition of NF‐κB, a protein involved in the regulation of inflammatory, immunological and carcinogenic processes.[ 1 , 2 ] From a structural point of view, dysiherbol A (1) and its congeners 2 and 3 belong to the hydroquinone sesquiterpenes, a large family of meroterpenes of mixed biosynthetic origin. [3] More specifically, the dysiherbols are members of the avarane sub‐family, according to the substitution pattern of the decalin AB ring system. Since the discovery of avarol (5) in 1974 [4] as a multi‐bioactive compound, [5] a variety of avarane meroterpenoids have been described, among them aureol B (6) [6] and the cycloaurenones (such as 7 and 8), [7] the latter featuring the same carbon skeleton as the dysiherbols, however, with a cis‐configuration of the decalin system.
Figure 1.
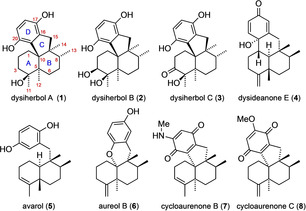
Proposed structure of dysiherbols A–C, dysideanone E and selected related meroterpenoids with an avarane skeleton.
Due to their interesting structural and biological properties, meroterpenoids of the avarane family have attracted much attention of synthetic chemists.[ 8 , 9 , 10 ] In 2017, Echavarren and co‐workers succeeded in synthesizing simplified compounds displaying the tetracyclic carbon ring system of dysiherbols and cycloaurenones. [11] However, the total synthesis of these natural products remained an unsolved challenge.
Considering the promising biological activities of dysiherbol A and the challenge of stereoselectively constructing the tetracyclic 6/6/5/6‐fused carbon skeleton with five adjacent (mostly quaternary) stereocenters, we decided to tackle its synthesis. We report here the first enantioselective total synthesis of dysiherbol A, which also led to a revision of both the constitution and the absolute configuration of the natural product.
As outlined in Scheme 1, our strategic plan was to synthesize dysiherbol A (1) from the simplified tetracyclic precursor 9 by late‐stage (diastereoselective) introduction of the methyl groups in ring A. We envisioned that ketone 9 could possibly be obtained from aldehyde 10 through Lewis acid‐mediated twofold (cationic) cyclization and subsequent oxidation. In this key step, the tetracyclic ring skeleton would be diastereoselectively built up under concomitant formation of two strategic C−C bonds.
Scheme 1.
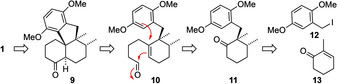
Retrosynthetic analysis of dysiherbol A.
We further reasoned that the synthesis of the required cyclization precursor 10 could be achieved from the cyclohexanone derivative 11 which in turn could be prepared through enantioselective Cu‐catalyzed conjugate addition of a methyl anion equivalent to 2‐methyl‐2‐cyclohexenone (13) [12] followed by diastereoselective alkylation of the resulting enolate with the benzylic iodide 12, in analogy to a report by Cramer and co‐workers. [13]
According to this plan, we started our investigation by reacting 13 [14] with AlMe3 in the presence of a catalyst generated in situ from copper(I) thiophene‐2‐carboxylate (CuTC) and the (R)‐BINOL‐derived chiral phosphoramidite ligand L*. [15] After activating the resulting aluminum enolate as an ate‐complex by addition of methyllithium, the subsequent C‐alkylation with iodide 12 [16] proceeded smoothly in the presence of tripyrrolidinophosphoric acid triamide (TPPA) as a cosolvent [17] to afford ketone 11 (96 % ee, HPLC) in 59 % yield as a single diastereomer after chromatographic purification (Scheme 2).
Scheme 2.
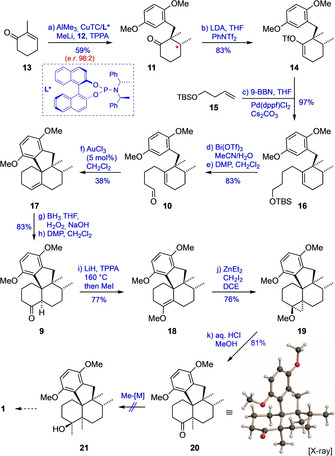
Synthesis of key intermediate 9 and unsuccessful attempts at its conversion to 1. Reagents and conditions: a) AlMe3, CuTC/L1* (2.4 mol %), Et2O, −30 °C, 4.5 h; then MeLi, 12, TPPA, −30→25 °C, 17 h; b) LDA, THF, PhNTf2, −78→25 °C, 3 h; c) 15, 9‐BBN, THF, 25 °C, 2 h; then 14, Pd(dppf)Cl2 (3 mol %), Cs2CO3, H2O, DMF, 25 °C, 1 h; d) Bi(OTf)3 (4 mol %), CH3CN/H2O, 25 °C, 1.5 h, 97 %; e) DMP, CH2Cl2, 0→25 °C, 2 h, 86 %; f) AuCl3 (5 mol %), CH2Cl2, 0 °C, 11 min; g) BH3 ⋅THF, THF, 0→30 °C, 10 h; then NaOH, H2O2, THF/H2O, 0→25 °C, 14 h; h) DMP, CH2Cl2, 0→30 °C, 2 h; i) LiH, TPPA, 160 °C, 1.5 h; then MeI, 25 °C, 20 h; j) ZnEt2, CH2I2, DCE, 25 °C, 35 min; k) aq. HCl, MeOH, reflux, 35 min. CuTC=copper(I) thiophene‐2‐carboxylate, TPPA=tripyrrolidinophosphoric acid triamide, LDA=lithium diisopropylamide, TBS=tert‐butyldimethylsilyl, 9‐BBN=9‐borabicyclo[3.3.1]nonane, dppf=1,1′‐bis(diphenylphosphino)ferrocene, DMP=Dess–Martin periodinane, DCE=dichloroethane.
Having thus achieved the chirogenic opening step with high enantioselectivity on a multi‐gram scale, the next task was the conversion of ketone 11 into aldehyde 10 under introduction of a C4 chain. For this purpose, ketone 11 was first converted into the enol triflate 14 by treatment with LDA and N‐phenyl triflimide. Suzuki–Miyaura cross‐coupling of 14 with the borane prepared in situ from TBS‐protected homoallylic alcohol 15 and 9‐BBN then proceeded cleanly under the chosen conditions (cat. Pd(dppf)Cl2, Cs2CO3, H2O, DMF) [18] to deliver 16 in excellent yield. The cleavage of the TBS group was achieved with catalytic amounts of Bi(OTf)3 in the presence of water, [19] and the resulting alcohol was directly subjected to oxidation with the Dess–Martin periodinane (DMP) to give 10 in high yield (68 % overall from 11).
With substantial amounts of the aldehyde 10 in our hands, we went on to investigate the planned twofold cyclization. Initial experiments revealed that treatment of 10 with common Lewis acids (such as BF3 ⋅OEt2, AlCl3 or SnCl4) only resulted in the formation of complex product mixtures usually containing the ketone 22 as a major component. However, after extensive screening of various Lewis acids, we succeeded in identifying AuCl3 as the only catalyst capable of achieving the desired transformation (under elimination of water) to give the olefin 17 as a sole major product according to GC–MS analysis. On a preparative scale, this unique reaction proved to be insensitive towards air and moisture and proceeded within minutes in the presence of only 5 mol % of AuCl3. After chromatographic purification, 17 was obtained in 38 % yield, and its structure was secured by X‐ray crystallography (Scheme 3). To explain the unique performance of AuCl3 in this transformation, we suppose that, in contrast to all other Lewis acids tested, AuCl3 is able to convert the primary cyclization intermediate 23 rapidly into a more stable allylic cation (24) under formation of the AuCl3(OH)− anion. The cation 24 is then attacked by the electron‐rich aromatic ring to yield the observed product 17 in a SEAr cyclization under regeneration of the catalyst (AuCl3). To support this mechanistic proposal, we separately treated a sample of compound 25 [20] with 4 mol % of AuCl3, which is known to catalyze substitution reactions of allylic alcohols.[ 21 , 22 ] And indeed, in this case the tetracyclic olefin 17 was again formed, even in an improved yield of 50 %.
Scheme 3.
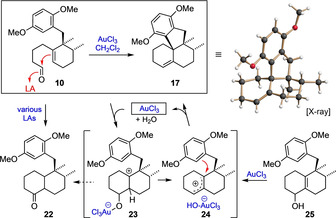
A mechanistic proposal for the AuCl3‐catalyzed twofold cyclization of aldehyde 10 to the tetracyclic olefin 17.
Having developed a reliable protocol for constructing the tetracyclic core structure of the dysiherbols by double cyclization of aldehyde 10, the remaining challenge was the installation of the two missing methyl groups as well as the hydroxy function at ring A. To prepare for this, we first converted 17 into the originally devised ketone 9 through hydroboration/oxidation and subsequent DMP oxidation (Scheme 2). As the introduction of the bridgehead methyl group (C12) could not be achieved by direct α‐methylation of the ketone (due to the steric hindrance), we had to take a deviation via opening of an enolether‐derived cyclopropane intermediate. [23] Remarkably, the regioselective, thermodynamically controlled ketone deprotonation was achieved only by treatment of 9 with LiH in TPPA at 160 °C, and the resulting lithium enolate was O‐alkylated with MeI to give the enolether 18. The subsequent Simmons–Smith cyclopropanation [24] proceeded diastereoselectively to give 19, from which the desired α‐methyl ketone 20 was obtained under acidic conditions (47 % overall yield from 9). To our disappointment, all attempts to convert 20 into the tertiary alcohol 21, that is, “dysiherbol A dimethyl ether”, by nucleophilic introduction of a methyl anion equivalent (MeMgBr, MeLi, MeLi/CeCl3, [25] AlMe3 or ZrMe4 [26] ) failed. This result, however, was not completely unexpected, as the crystal structure of 20 not only confirmed the steric shielding of both faces of the carbonyl group but also indicated an electronic deactivation, as reflected by a distance of only 2.51 Å between the carbonyl C‐atom and the adjacent methoxy oxygen (n→π* interaction). [27]
In contrast to 20, the non‐methylated ketone 9 reacted smoothly with MeLi/CeCl3 to give alcohol 26 as a single diastereomer in virtually quantitative yield without the need of chromatographic purification (Scheme 4). Notably, the crystal structure of 26 displays an intramolecular hydrogen bond between the OH and the methoxy group, once again pinpointing the spatial proximity and the interaction of the functionalities at C4 and C20. The close structural relationship of 26 with the target molecule 1 prompted us to investigate its deprotection. The triol 27, that is, putative 12‐nor‐dysiherbol A, was obtained in high yield by heating 26 with an excess of LiSEt in TPPA to 170 °C. [28] In contrast, reaction of 26 with BBr3 in CH2Cl2 followed by hydrolytic workup resulted in the formation of 28 under spontaneous dehydrative cyclization. Besides characteristic changes in the NMR spectrum, the structure of 28 was supported by its mass spectrum which indicated the loss of one water molecule. It is noteworthy that comparison of the NMR data for compounds 27 and 28 with those reported for dysiherbol A showed a much closer relationship of the natural product to 28 compared to 27 (Table 1).
Scheme 4.
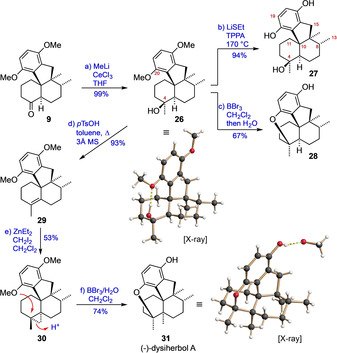
Completion of the synthesis of (−)‐dysiherbol A. Reagents and conditions: a) CeCl3, MeLi, THF, −78 °C→25 °C, 18 h; b) LiSEt (10 equiv), TPPA, 170 °C, 4 h; c) BBr3, CH2Cl2, 25 °C, 25 h, then H2O, 25 °C, 1 h; d) pTsOH⋅H2O, 3 Å MS, toluene, reflux, 4 h; e) ZnEt2, CH2I2, CH2Cl2, 25 °C, 2 h, 2 cycles; f) BBr3, H2O, CH2Cl2, 27 °C, 40 min. pTsOH=p‐toluenesulfonic acid, 3 Å MS=molecular sieves (pore size: 3 Å).
Table 1.
Comparison of selected 1H and 13C NMR signals[a] of compounds 27 and 28 with those of dysiherbol A (taken from ref. [1])
|
δ [ppm] |
27 [b] |
28 [b] |
dysiherbol A[c] |
|---|---|---|---|
|
C4 |
72.5 |
79.0 |
82.4 |
|
C10 |
58.0 |
47.7 |
49.1 |
|
C11 |
31.4 |
25.9 |
21.9 |
|
C19 |
116.3 |
111.3 |
111.1 |
|
H‐8 |
1.43 |
1.13 |
1.24 |
|
H‐13 |
0.76 |
0.82 |
0.83 |
|
H‐15a H‐15b |
2.66 2.42 |
2.64 2.59 |
2.57 2.54 |
[a] All spectra in CDCl3 (77.00 ppm). [b] 500 MHz (1H)/126 MHz (13C). [c] 600 MHz (1H)/150 MHz (13C).
We therefore reasoned that dysiherbol A might actually have a similar pentacyclic structure as 28, which also opened a new option for the end game of the synthesis, as the protolytic opening of a cyclopropane (30) could possibly be linked to the installation of the ether bridge (Scheme 4). Therefore, we proceeded with the conversion of 26 to 30, which was achieved in two steps by acid‐mediated elimination of water and subsequent cyclopropanation (ZnEt2, CH2I2) of the olefin 29. Much to our satisfaction, we then succeeded in realizing the final key step. Under optimized conditions, the reaction of cyclopropane 30 with an excess of BBr3 in the presence of water afforded compound 31 in 74 % yield, the structure of which (crystallized as an MeOH adduct) was secured by X‐ray crystallography. The spectroscopic data of 31 agreed perfectly with those reported for natural dysiherbol A, [1] thus confirming the necessary revision of the originally proposed structure.
To gain insight into the proton‐induced cyclopropane opening step, we also investigated the reaction of 30 with methanesulfonic acid (MsOH) in toluene (Scheme 5). Interestingly, the aureane derivative 33 was formed in high yield under comparably mild conditions (2 equiv MsOH, 25 °C) while the pentacyclic avarane 32 (i.e. dysiherbol A methyl ether) was formed as the only main product in the presence of a large excess of MsOH at 55 °C. This result indicates the regioselectivity of the primary cyclopropane opening step being unimportant due to a Wagner–Meerwein rearrangement equilibrium between the two cationic intermediates 35 and 36. While at lower temperature the proton elimination of 36 (to 33) is favored, the cyclization of 35 to 34 and the subsequent cleavage of the O−Me bond occur only under harsher conditions, under which, incidentally, 33 is converted to 32 in 40 % yield, as shown in a separate experiment. Noteworthy, the carbon skeleton of the tetracyclic aureane 33, has never been found in nature (yet).
Scheme 5.
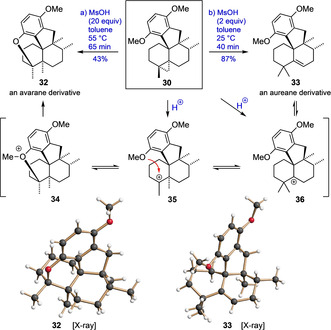
Acid‐mediated conversion of cyclopropane 30 to either the pentacyclic avarane 32 or the aureane 33. MsOH=methanesulfonic acid.
Another surprise was that the molecular rotation of synthetic dysiherbol A (31) ([α]D=−23°; c=0.5 in MeOH) did not match the one reported for the natural product ([α]D=+23°; c=0.1 in MeOH). [1] While the absolute configuration of our sample was secured by X‐ray crystal structure analysis, the original configurational assignment was based on the comparison of the experimental CD spectrum with those calculated for 1. We therefore took a CD spectrum of 31 which clearly confirmed it being the enantiomer of natural dysiherbol A (Figure 2).
Figure 2.
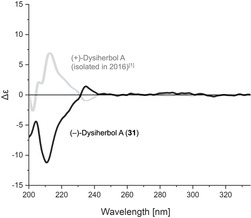
Experimental CD spectra of natural (+)‐dysiherbol A (gray) [1] and synthetic (−)‐dysiherbol A (black).
In conclusion, we have elaborated a reliable and efficient enantioselective synthesis of the unnatural enantiomer of dysiherbol A (31). Structural assignments were confirmed by X‐ray crystal structure analyses. [29] The elaborated sequence (12 steps, 5 % overall yield from 13) features some remarkable steps, such as the Au‐catalyzed two‐fold cyclization of 10 to build up the carbocyclic core, and the single‐step conversion of cyclopropane 30 to the target. [30] As the enantiomer of the chiral ligand used in the chirogenic opening step is readily available, we are now going to use the developed protocol to also prepare substantial amounts of the natural enantiomer of dysiherbol A in order to further explore its biological properties.
The work presented here furthermore demonstrates the importance of total synthesis in the context of structure elucidation. Specifically, we have proven that the structures of the dysiherbols have to be revised, both with respect to their constitution (as cyclic ethers) and their absolute configuration. Noteworthy, the revised structures (Figure 3) now display the same absolute configuration at the stereocenters C8 and C9 as virtually all other avarane meroterpenes from Dysidea sp. including the dysideanones. [3]
Figure 3.
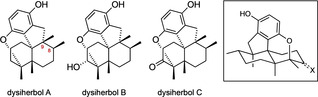
Revised structures of dysiherbols A–C and a general 3D representation idealizing the chair conformation of the trans‐decalin ring system.
Last but not least we would like to mention that while we were in the process of finalizing this manuscript, a publication by Lu and co‐workers appeared describing a total synthesis of (racemic) (±)‐dysiherbol A following a different strategy. [31]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (doctoral fellowship to J.B.), the Jürgen Manchot Stiftung (doctoral fellowship to I.G.) and the University of Cologne. Open access funding enabled and organized by Projekt DEAL.
J. Baars, I. Grimm, D. Blunk, J.-M. Neudörfl, H.-G. Schmalz, Angew. Chem. Int. Ed. 2021, 60, 14915.
Dedicated to Professor K. Barry Sharpless on the occasion of his 80th birthday
References
- 1. Jiao W. H., Shi G. H., Xu T. T., Chen G. D., Gu B. B., Wang Z., Peng S., Wang S. P., Li J., Han B. N., Zhang W., Lin H. W., J. Nat. Prod. 2016, 79, 406–411. [DOI] [PubMed] [Google Scholar]
- 2. Faustman D., Davis M., Nat. Rev. Drug Discovery 2010, 9, 482–493. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. García P. A., Hernández Á. P., San Feliciano A., Castro M. Á., Mar. Drugs 2018, 16, 292; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Menna M., Imperatore C., D′Aniello F., Aiello A., Mar. Drugs 2013, 11, 1602–1643; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Marcos I. S., Conde A., Moro R. F., Basabe P., Diez D., Urones J. G., Mini-Rev. Org. Chem. 2010, 7, 230–254. [Google Scholar]
- 4. Minale L., Riccio R., Sodano G., Tetrahedron Lett. 1974, 15, 3401–3404. [Google Scholar]
- 5.
- 5a. Tommonaro G., García-Font N., Vitale R. M., Pejin B., Iodice C., Cañadas S., Marco-Contelles J., Oset-Gasque M. J., Eur. J. Med. Chem. 2016, 122, 326–338; [DOI] [PubMed] [Google Scholar]
- 5b. Namba T., Kodama R., Mar. Drugs 2015, 13, 2376–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Djura P., Stierle D. B., Sullivan B., Faulkner D. J., Arnold E. V., Clardy J., J. Org. Chem. 1980, 45, 1435–1441. [Google Scholar]
- 7. Kim C.-K., Woo J.-K., Kim S.-H., Cho E., Lee Y.-J., Lee H. S., Sim C. J., Oh D.-C., Oh K.-B., Shin J., J. Nat. Prod. 2015, 78, 2814–2821. [DOI] [PubMed] [Google Scholar]
- 8.For previous total syntheses of avarane meroterpenoids, see:
- 8a. Sarma A. S., Chattopadhyay P., J. Org. Chem. 1982, 47, 1727–1731; [Google Scholar]
- 8b. Bruner S. D., Radeke H. S., Tallarico J. A., Snapper M. L., J. Org. Chem. 1995, 60, 1114–1115; [Google Scholar]
- 8c. Locke E. P., Hecht S. M., Chem. Commun. 1996, 2717–2718; [Google Scholar]
- 8d. An J., Wiemer D. F., J. Org. Chem. 1996, 61, 8775–8779; [DOI] [PubMed] [Google Scholar]
- 8e. Ling T., Xiang A. X., Theodorakis E. A., Angew. Chem. Int. Ed. 1999, 38, 3089–3091; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 3277–3279; [Google Scholar]
- 8f. Nakatani M., Nakamura M., Suzuki A., Inoue M., Katoh T., Org. Lett. 2002, 4, 4483–4486; [DOI] [PubMed] [Google Scholar]
- 8g. Nakamura M., Suzuki A., Nakatani M., Fuchikami T., Inoue M., Katoh T., Tetrahedron Lett. 2002, 43, 6929–6932; [Google Scholar]
- 8h. Sakurai J., Oguchi T., Watanabe K., Abe H., Kanno S.-i., Ishikawa M., Katoh T., Chem. Eur. J. 2008, 14, 829–837; [DOI] [PubMed] [Google Scholar]
- 8i. Watanabe K., Sakurai J., Abe H., Katoh T., Chem. Commun. 2010, 46, 4055–4057; [DOI] [PubMed] [Google Scholar]
- 8j. Sakurai J., Kikuchi T., Takahashi O., Watanabe K., Katoh T., Eur. J. Org. Chem. 2011, 2948–2957; [Google Scholar]
- 8k. Katoh T., Heterocycles 2013, 87, 2199–2224; [Google Scholar]
- 8l. Sumii Y., Kotoku N., Fukuda A., Kawachi T., Sumii Y., Arai M., Kobayashi M., Bioorg. Med. Chem. 2015, 23, 966–975; [DOI] [PubMed] [Google Scholar]
- 8m. Katoh T., Atsumi S., Saito R., Narita K., Katoh T., Eur. J. Org. Chem. 2017, 3837–3849; [Google Scholar]
- 8n. Takeda Y., Nakai K., Narita K., Katoh T., Org. Biomol. Chem. 2018, 16, 3639–3647; for a general review on meroterpene total synthesis, see: [DOI] [PubMed] [Google Scholar]
- 8o. Gordaliza M., Mar. Drugs 2012, 10, 358–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For recent syntheses of polycyclic aureane meroterpenes, see:
- 9a. Rosales A., Muñoz-Bascón J., Roldan-Molina E., Rivas-Bascón N., Padial N. M., Rodríguez-Maecker R., Rodríguez-García I., Oltra J. E., J. Org. Chem. 2015, 80, 1866–1870; [DOI] [PubMed] [Google Scholar]
- 9b. Speck K., Wildermuth R., Magauer T., Angew. Chem. Int. Ed. 2016, 55, 14131–14135; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14337–14341; [Google Scholar]
- 9c. Wildermuth R., Speck K., Haut F. L., Mayer P., Karge B., Bronstrup M., Magauer T., Nat. Commun. 2017, 8, 2083; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Speck K., Magauer T., Chem. Eur. J. 2017, 23, 1157–1165; [DOI] [PubMed] [Google Scholar]
- 9e. Haut F.-L., Speck K., Wildermuth R., Möller K., Mayer P., Magauer T., Tetrahedron 2018, 74, 3348–3357. [Google Scholar]
- 10.Synthesis of carbotetracycles related to dysideanone: Haque M. A., Jana C. K., Chem. Eur. J. 2017, 23, 13300–13304. [DOI] [PubMed] [Google Scholar]
- 11.Synthesis of carbotetracycles related to dysiherbol and cycloaurenone: Yin X., Mato M., Echavarren A. M., Angew. Chem. Int. Ed. 2017, 56, 14591–14595; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14783–14787. [Google Scholar]
- 12.
- 12a. Vuagnoux-d′Augustin M., Alexakis A., Chem. Eur. J. 2007, 13, 9647–9662; [DOI] [PubMed] [Google Scholar]
- 12b. Alexakis A., Albrow V., Biswas K., Augustin M., Prieto O., Woodward S., Chem. Commun. 2005, 2843. [DOI] [PubMed] [Google Scholar]
- 13. Ngoc D. T., Albicker M., Schneider L., Cramer N., Org. Biomol. Chem. 2010, 8, 1781–1784. [DOI] [PubMed] [Google Scholar]
- 14.2-Methyl-2-cyclohexenone (13) was prepared from 2-methylcyclohexanone by α-bromination and elimination as described by: Hua D. H., Chen Y., Sin H.-S., Maroto M. J., Robinson P. D., Newell S. W., Perchellet E. M., Ladesich J. B., Freeman J. A., Perchellet J.-P., Chiang P. K., J. Org. Chem. 1997, 62, 6888–6896. [Google Scholar]
- 15. Feringa B. L., Acc. Chem. Res. 2000, 33, 346–353. [DOI] [PubMed] [Google Scholar]
- 16.Compound 12 was prepared according to:
- 16a. Sudhir U., James B., Joly S., Nair M. S., Res. Chem. Intermed. 2003, 29, 523–532; [Google Scholar]
- 16b. Kaplan H. Z., Rendina V. L., Kingsbury J. S., J. Org. Chem. 2013, 78, 4620–4626. [DOI] [PubMed] [Google Scholar]
- 17.For the use of TPPA as a substitute for carcinogenic HMPA, see:
- 17a. Ozari Y., Jagur-Grodzinski J., J. Chem. Soc. Chem. Commun. 1974, 295–296; [Google Scholar]
- 17b. McDonald C. E., Ramsey J. D., Sampsell D. G., Butler J. A., Cecchini M. R., Org. Lett. 2010, 12, 5178–5181; [DOI] [PubMed] [Google Scholar]
- 17c. Berndt M., Hölemann A., Niermann A., Bentz C., Zimmer R., Reissig H.-U., Eur. J. Org. Chem. 2012, 1299–1302. [Google Scholar]
- 18. Kim D. E., Zhu Y., Newhouse T. R., Org. Biomol. Chem. 2019, 17, 1796–1799. [DOI] [PubMed] [Google Scholar]
- 19. Barnych B., Vatèle J.-M., Synlett 2011, 22, 2048–2052. [Google Scholar]
- 20.Compound 25 was obtained as a mixture of diastereomers by reduction of the corresponding enone with NaBH4/CeCl3 (see the Supporting Information).
- 21.Selected reviews on Au catalysis:
- 21a. Hashmi A. S. K., Chem. Rev. 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]
- 21b. Fürstner A., Davies P. W., Angew. Chem. Int. Ed. 2007, 46, 3410–3449; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3478–3519; [Google Scholar]
- 21c. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Zi W., Toste F. D., Chem. Soc. Rev. 2016, 45, 4567–4589; [DOI] [PubMed] [Google Scholar]
- 21e. Pflästerer D., Hashmi A. S. K., Chem. Soc. Rev. 2016, 45, 1331–1367. [DOI] [PubMed] [Google Scholar]
- 22.For examples of Au-catalyzed allylic substitution reactions, see:
- 22a. Guo S., Song F., Liu Y., Synlett 2007, 964–968; [Google Scholar]
- 22b. Rao W., Chan P. W. H., Org. Biomol. Chem. 2008, 6, 2426–2433; [DOI] [PubMed] [Google Scholar]
- 22c. Kothandaraman P., Rao W., Zhang X., Chan P. W. H., Tetrahedron 2009, 65, 1833–1838; [Google Scholar]
- 22d. Biannic B., Ghebreghiorgis T., Aponick A., Beilstein J. Org. Chem. 2011, 7, 802–807; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22e. Mukherjee P., Widenhoefer R. A., Org. Lett. 2011, 13, 1334–1337; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22f. Uria U., Vila C., Lin M. Y., Rueping M., Chem. Eur. J. 2014, 20, 13913–13917; for a review, see: [DOI] [PubMed] [Google Scholar]
- 22g. Quintavalla A., Bandini M., ChemCatChem 2016, 8, 1437–1453. [Google Scholar]
- 23.
- 23a. Wenkert E., Mueller R. A., E. J. Reardon, Jr. , Sathe S. S., Scharf D. J., Tosi G., J. Am. Chem. Soc. 1970, 92, 7428–7435; [Google Scholar]
- 23b. Corey E. J., Lee J., Liu D. R., Tetrahedron Lett. 1994, 35, 9149–9152. [Google Scholar]
- 24. Furukawa J., Kawabata N., Nishimura J., Tetrahedron 1968, 24, 53–58. [Google Scholar]
- 25. Imamoto T., Sugiura Y., Takiyama N., Tetrahedron Lett. 1984, 25, 4233–4423. [Google Scholar]
- 26. Reetz M. T., Steinbach R., Westermann J., Urz R., Wenderoth B., Peter R., Angew. Chem. Int. Ed. Engl. 1982, 21, 135; [Google Scholar]; Angew. Chem. 1982, 94, 133. [Google Scholar]
- 27.
- 27a. Newberry R. W., Raines R. T., Acc. Chem. Res. 2017, 50, 1838–1846; a distance of 2.51 Å is clearly below the sum of the van der Waals radii, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Batsanov S. S., Inorg. Mater. 2001, 37, 871–885. [Google Scholar]
- 28. Cvengros J., Neufeind S., Becker A., Schmalz H., Synlett 2008, 2008, 1993–1998. [Google Scholar]
- 29.Deposition numbers 2077903 (for 17), 2077904 (for 29), 2077905 (for 11), 2077906 (32), 2077907 (for 18), 2077908 (for 20), 2077909 (for 33), 2077910 (for 26), 2077911 (for 19), 2077912 (for epi-11), 2077913 (for 31), 2077914 (for 9), and 2082956 (for 27) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 30.For an unsuccessful attempt to apply an Au-catalyzed domino process in the synthesis of polycyclic auranes, see: Wildermuth R., Speck K., Magauer T., Synthesis 2016, 48, 1814–1824. [Google Scholar]
- 31. Chong C., Zhang Q., Ke J., Zhang H., Yang X., Wang B., Ding W., Lu Z., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202100541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 10.1002/ange.202100541. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary