

Abstract
Tubulin plays essential roles in vital cellular activities and is the target of a wide range of proteins and ligands. Here, using a combined computational and crystallographic fragment screening approach, we addressed the question of how many binding sites exist in tubulin. We identified 27 distinct sites, of which 11 have not been described previously, and analyzed their relationship to known tubulin–protein and tubulin–ligand interactions. We further observed an intricate pocket communication network and identified 56 chemically diverse fragments that bound to 10 distinct tubulin sites. Our results offer a unique structural basis for the development of novel small molecules for use as tubulin modulators in basic research applications or as drugs. Furthermore, our method lays down a framework that may help to discover new pockets in other pharmaceutically important targets and characterize them in terms of chemical tractability and allosteric modulation.
Keywords: crystallographic fragment screening, microtubules, molecular dynamics simulation, protein–ligand interactions, tubulin
How many binding sites exist in the vital anticancer protein target tubulin? A combined computational and crystallographic fragment screening approach now answers this question and reveals 11 novel sites as well as 56 novel small‐molecule ligands in tubulin.
Introduction
Microtubules are dynamic cytoskeletal filaments, which are assembled from and disassembled into their αβ‐tubulin heterodimeric building blocks. An outstanding property of tubulin is its capacity to bind a plethora of regulators, whose main activities are to modulate microtubule dynamics and organization, and consequently microtubule function. In cells, it is targeted by diverse proteins that enable microtubules to control fundamental physiological processes in all eukaryotes ranging from cell division, cell motility, cell polarity to intracellular trafficking (reviewed in ref. [1]). In addition, a large number of chemically diverse, small molecule ligands bind to six so far identified, distinct binding sites in tubulin (reviewed in ref. [2]). Notably, compounds that interfere with the function of tubulin have been very successfully used to treat human pathologies including cancer, infectious diseases and neurological disorders, but also in basic research studies aimed at understanding cell physiology (reviewed in ref. [3]).
The observation that tubulin can bind so many different proteins and ligands raises the intriguing question of how many different binding sites do exist in tubulin. Here, we addressed this question using a combined computational and crystallographic fragment screening approach. Our study provides a comprehensive analysis of binding sites in tubulin, and offers a unique structural and mechanistic framework for novel antitubulin ligand design and engineering approaches.
Results
Computational Analysis
We initially performed a 1.1 μs‐long molecular dynamics (MD) simulation in explicit solvent with a high‐resolution crystal structure of the bovine brain αβ‐tubulin heterodimer, which is predominantly composed of the α1‐ and β2‐tubulin isotypes. [4] The root mean square deviation (RMSD) of the Cα carbon atoms of the tubulin structure compared to the starting structure stabilized after 400 ns of the simulation and oscillated around 2.7 Å for the remaining time of the simulation (Figure S1). However, without taking the long loops H1‐S2, S7‐H9 (M‐loop), and S9‐S10 of both the α‐ and β‐tubulin monomers into account (see ref. [5] for designation of secondary structure elements and residue numbering), the stability was achieved after only 100 ns of simulation with an average RMSD of 1.8 Å (Figure S1).
Next, we computationally identified pockets in tubulin, analyzed their relative dynamics and persistency, and assessed their communication networks by tracking the exchange of atoms between adjacent pockets during the entire course of the simulation. For the description of the predicted pockets, we arbitrarily gave them an identifier (pID) by numbering them consecutively with roman numbers in both the α‐ and β‐tubulin monomers. An overview of the location of the pockets in α‐ and β‐tubulin is given in Figure 1 a,c and selected features of all pockets are collected in Table 1 and Table S1. An illustration of the dynamic crosstalk between pockets is illustrated in Figure 1 b,d and the involved residues are highlighted in Table S1. In the following, we first describe the pockets observed in β‐tubulin and then those in α‐tubulin.
Figure 1.
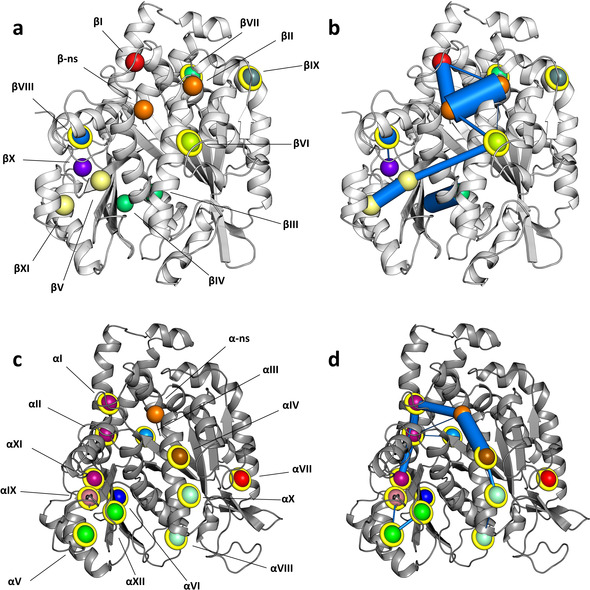
Tubulin pockets and their communication networks predicted by MD simulation. a, c) Predicted pockets in β‐tubulin ((a), light gray ribbon representation) and α‐tubulin ((c), dark gray ribbon representation). b, d) Predicted pocket communication networks in β‐tubulin (b) and α‐tubulin (d). Marine blue lines depict connected network nodes; their widths are displayed proportional to the respective communication frequency between two nodes. Spheres represent center of masses of the pockets (corresponding to network nodes) and are shown in different colors. Identical colors indicate pockets that are often merged during the simulation. Spheres coated with yellow rings highlight novel sites. See also Table S1.
Table 1.
Pockets and sites identified by MD and crystallographic fragment screening, respectively.
|
Pocket[a] |
Site[b] |
Shared SS[c] |
Notes[d] |
|---|---|---|---|
|
pID βI |
sID βαIII (22) |
βT5, βH5, βH11 * |
Extension of the vinblastine site; targeted by the C15‐C33 moiety of plocabulin |
|
pID βII |
– |
– |
γ‐phosphate site of the guanosine nucleotide |
|
pID βIII |
sID βIV (16) |
βS4, βS5, βH5‐S6, βS6, βH7, βT7, βH8, βS7, βS8, βS10 |
Colchicine site |
|
pID βIV |
sID βIV (16) |
βT7, βH8, βS8, βS9 |
Colchicine site; equivalent to pID αVI |
|
pID βV |
sID βII (2) |
βH1, βS7, βM, βS8, βS9‐S10, βS10 |
Taxane site |
|
pID βVI |
– |
– |
Novel; mediating communication between β‐ns and the taxane site; equivalent to pID αIV |
|
pID βVII |
– |
– |
Novel |
|
pID βVIII |
sID βI (2) |
βH6, βH9‐S8 |
Targeted by TPX2; equivalent to pID αIX |
|
pID βIX |
– |
– |
Novel |
|
pID βX |
– |
– |
Part of the laulimalide/peloruside site |
|
pID βXI |
sID βII (2) |
βM, βS9‐S10, βS10 * |
Taxane site |
|
– |
sID βIII (1) |
– |
Novel |
|
– |
sID βV (8) |
– |
Targeted by dynein and CPAP |
|
– |
sID βαI (3) |
– |
Targeted by tau, TPX2, kinesin‐13, Ustilago maydis kinesin‐5 and iE5 alphaRep |
|
– |
sID βαII (16) |
– |
Involved in longitudinal inter‐tubulin dimer contacts in microtubules |
|
– |
sID βαIII (22) |
– |
Extended vinblastine site; involved in longitudinal inter‐tubulin dimer contacts in microtubules; targeted by DARPin 1/2 and RB3 |
|
pID αI |
– |
– |
Involved in longitudinal intra tubulin contact; targeted by TTL; equivalent to pID βI |
|
pID αII |
– |
– |
Merges with pID αI |
|
pID αIII |
– |
– |
Novel |
|
pID αIV |
sID αII (3) |
αH1, αH2, αH2‐S3, αH7 |
Novel; communicates with α‐ns |
|
pID αV |
– |
– |
Targeted by Alp14; communicates with pID αXII |
|
pID αVI |
sID βαIII (22) |
αH10‐αS9, αH8, αS9 * |
Extension of the vinblastine site; targeted by RB3; equivalent to βIV |
|
pID αVII |
– |
– |
Involved in lateral inter‐tubulin dimer contacts in microtubules; targeted by iiiA5 alphaRep |
|
pID αVIII |
– |
– |
Novel |
|
pID αIX |
sID αI (1) |
αH6, αM, αH9 |
Novel; equivalent to pID βVIII |
|
pID αX |
– |
– |
Novel |
|
pID αXI |
– |
– |
Novel |
|
pID αXII |
– |
– |
Novel; communicates with αV; equivalent to βV |
[a] Identifier of pockets predicted computationally. [b] Identifier of sites identified by the crystallographic fragment screen. The number of fragments targeting a particular site is given in parenthesis. [c] Shared tubulin secondary structural elements between corresponding pockets and sites. Asterisks indicate partial overlap. [d] For additional notes, see Supplementary Table S1, Table S3, Table S4, and Table S5. ns, nucleotide site; Novel, site that has not been described to be targeted by any structurally characterized ligands or protein partners.
The first pocket on β‐tubulin that attracted our attention is pID βII, which displays a persistency value (p) of 99 %. Even though the presence of this pocket may also be influenced by the adjacent β‐phosphate group of the bound GDP, its boundaries leave enough space to accommodate the γ‐phosphate group when the β‐nucleotide site is occupied by GTP or an inorganic phosphate molecule in the case of GDP‐Pi. Two pockets, pID βV and βXI (p=98 % and 41 %, respectively) belong to the structurally well‐characterized taxane site of β‐tubulin.[ 4 , 6 ] They merged along 30 % of the simulation to give raise to a single, wider pocket. Other pockets that belong to known β‐tubulin sites comprise the colchicine site [7] (pID βIII and βIV; both with p=100 %), an allosteric pocket of the maytansine site [8] (pID βI; p=58 %), which accommodates the C15‐C33 moiety of the phase II anticancer drug plocabulin (PM060184[ 8 , 9 ]), and the laulimalide/peloruside site [10] (pID βX, p=46 %).
A prominent pocket in β‐tubulin is pID βVI (p=52 %), which is formed by residues of helices βH1, βH2, and βH7. Intriguingly, it acts as a bridge between the taxane‐site pocket pID βV and the β‐nucleotide site. This predicted crosstalk between these two sites is in line with biochemical results demonstrating that the presence of a ligand in the taxane site affects the interaction of GDP or GTP with the β‐nucleotide site. [11] In addition, pocket pID βVIII (p=57 %), which is formed by residues of helix βH6 and the loop βH9‐βS8 weakly communicates with pID βX of the laulimalide/peloruside site. Finally, pID βVII and βIX (p=32 % and 31 %, respectively), which are formed by residues of helices βH3, βH5, βH12, and helix βH3 and loop βS3‐βH3 respectively, do not show any contribution to the pocket communication network.
The overall pocket distribution in α‐tubulin is somewhat similar to that detected in β‐tubulin. A distinctive pocket is pID αVI (p=52 %) that resembles the entrance of the colchicine site of β‐tubulin (equivalent to pocket pID βIV). Interestingly, despite the fact that the αS9‐αS10 loop is longer than the corresponding one in β‐tubulin, which is part of the taxane site, two pockets, pID αV (p=51 %) and pID αXII (p=32 %), were identified in this region. Close to them and separated by the αM‐loop, we found pocket pID αIX (p=30 %), which is formed by residues of helices αH6, αH9 and loop αH9‐αS8.
Pocket pID αIV (p=69 %), which is formed by residues of helices αH1, αH2, αH7 and loop αH2‐αS3, is in a communication pathway that includes also the α‐nucleotide site but not pockets pID αV and αXII, which are located close to the αM‐loop. In contrast, the equivalent pID αIV pocket in β‐tubulin (pID βVI) connects the βM‐loop containing taxane site of β‐tubulin with the β‐nucleotide site (see above). The pID αIV‐mediated communication network that is located close to the intra‐dimer interface includes in addition pockets pID αI (equivalent to pID βI and in reach of the βH10‐βS9 loop of the β‐tubulin monomer), αII and αXI (p=86 %, 77 % and 51 %, respectively). These additional pockets are often merged together to form a single, larger cavity formed by residues of helices αH5, αH6, and αH11, and loops αH9‐αS8 and αS5‐αH5. The network is interrupted by pockets pID αX (p=30 %) and αVIII (p=51 %), which are formed by residues of helix αH1 and loop αH1‐αH1′, and helix αH1 and loops αH1‐αH1′ and αS9‐αS10, respectively. Finally, pocket pID αIII (p=62 %), which is shaped by residues of loop αH8‐αS7 and helices αH5 and αH11, and pID αVII (p=85 %), which is surrounded by helix αH3 and the loops αH1‐αS2 and αH2‐αS3 appeared to be both isolated and are not involved in any communication network.
Crystallographic Fragment Screening
With the two‐fold objective to experimentally validate our computational predictions and to identify potential ligands able to bind into novel pockets in tubulin, we conducted an X‐ray crystallography‐based fragment screen. A fragment is a small, ≈200 Da chemical entity that in combination with a crystal structure of the fragment complexed to its target has been recognized as a powerful tool for structure‐based drug design. [12] To this end, we used a well‐established crystal system composed of two bovine brain αβ‐tubulin heterodimers (the monomers are denoted αTub1, βTub1, αTub2, and βTub2), the stathmin‐like domain of rat RB3 and chicken tubulin tyrosine ligase (TTL); the complex is denoted T2R‐TTL.[ 4 , 13 ] We soaked individual crystals with 708 different fragments, collected 672 X‐ray diffraction data sets, and solved 503 structures with a resolution better than 4.0 Å. In these structures, unambiguous electron densities for 59 unique fragments were identified (resolution range of 1.9–3.1 Å; Table S2 and Figure S2). From these, 15 bound simultaneously to two or more different tubulin sites and 10 bound as pairs two times to the same site. Three fragments were not considered for further analysis, as they were bound to sites involving RB3 residues or crystal contacts.
For their description, we arbitrarily gave the fragment sites an identifier (sID) by numbering them consecutively according to their location in the T2R‐TTL complex, i.e., β‐tubulin, β‐tubulin‐α‐tubulin inter‐dimer interface (i.e., βTub1‐αTub2), and α‐tubulin. We also use the term “site” in this section to distinguish it from a computationally predicted “pocket” (see above). An overview of the location of the fragment sites in the two αβ‐tubulin heterodimers of T2R‐TTL is given in Figure 2 and selected features of them are collected in Table 1 and Table S3. The chemical structures of the 59 identified fragments and some of their structural particularities that are important for binding are shown in Figure S3 and Figure 3, respectively. In the following, we first describe the sites in β‐tubulin, move to the ones located at the β‐tubulin‐α‐tubulin inter‐dimer interface and finally to the ones in α‐tubulin. For simplicity, we only explicitly describe interactions of common fragment motifs with tubulin residues.
Figure 2.
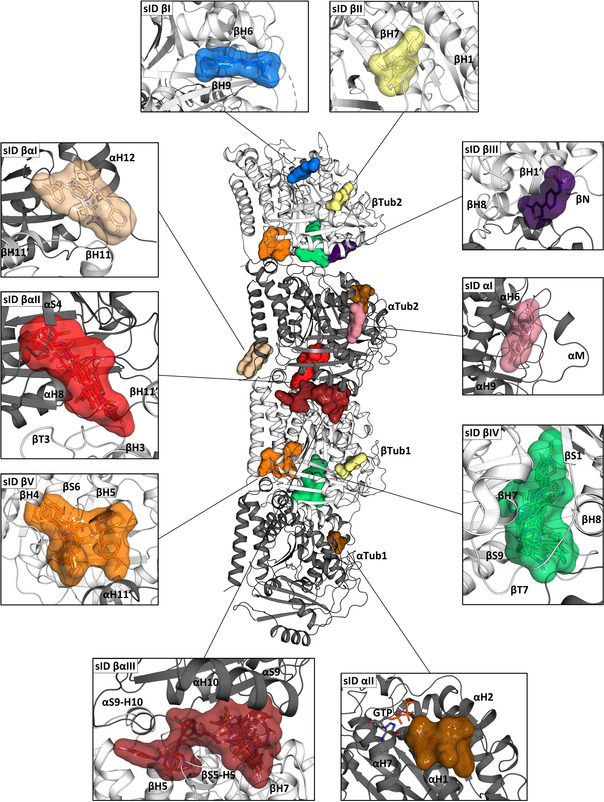
Fragment‐binding sites in tubulin determined by X‐ray crystallography. In the center of the figure, the structure of the two tubulin heterodimers αTub1‐βTub1 and αTub2‐βTub2 are depicted as they are observed in the T2R‐TTL complex. For simplicity, the RB3 and TTL molecules have been omitted. The α‐ and β‐tubulin monomers are shown in dark and light gray ribbon representation, respectively. The surrounding panels show close up views of the revealed fragment sites; the views in the individual panels differ in orientation from the central overview. Only one site is shown in cases where equivalent ones were found in both tubulin dimers. Secondary structural elements defining the sites are labelled. See also Table S3.
Figure 3.
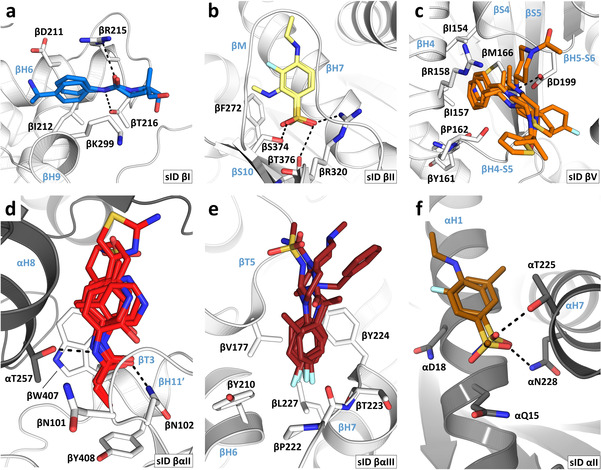
Interaction modes and common binding motifs of fragments. a) Fragments 01 and 53 in sID βI. b) Fragments 02 and 03 in sID βII. c) Fragments 20, 21, 22, 23 and 24 in sID βV. d) Fragments 04, 11 31, 32, 35, 40 and 43 in sID βαII. e) Fragments 22, 44, 45, 49, 51 and 54 in sID βαIII. f) Fragments 02 and 25 in sID αII. For all panels, the α‐ and β‐tubulin monomers are depicted in dark and light gray ribbon representation, respectively. Side chains interacting with common fragment binding motifs are shown in stick representation. Secondary structural elements are labeled in blue. Fragments are shown in stick representation using the same color code for their carbon atoms as in Figure 2. Oxygen, nitrogen, sulfur, fluorine and bromine atoms are colored red, blue, yellow, cyan, and brown, respectively. The chemical structures of all 59 fragments identified in this study are shown in Figure S3.
A well‐populated site on β‐tubulin is sID βIV, which corresponds to the colchicine site. It is targeted by 16 chemically diverse fragments that populate the three zones of the site [14] and which fill a total volume, Vf, of 780 Å3. They interacted with the protein through either hydrophobic or mixed hydrophobic and polar contacts mediated by residues of helices βH7 and βH8, strands βS1, βS4, βS5, βS6, βS8, βS9, and βS10, and loops βT7 and αT5. In comparison to the apo tubulin structure, [13] all fragments were able to displace loop βT7, an induced structural change that is characteristic of colchicine‐site ligands. [7] Five of them bound twice pairwise to the same site. To our surprise, we found only two fragments in the large taxane site of β‐tubulin (sID βII; V f=207 Å3). This site is formed by residues of helices βH1 and βH7, strands βS7, βS8, and βS10 and loops βM and βS9‐βS10. The two fragments share a methylsulfonyl‐benzene moiety as a common binding motif. Its sulfonyl group interacted with residues βR320, βS374, and βT376, and its benzene group interacted with βF272 and is wrapped around by the βM‐loop. An additional site on β‐tubulin is sID βI (V f=173 Å3), which is formed by helices βH6 and βH9, and loop βH9‐βS8 and is targeted by two fragments. The two fragments share an anilide core as a common binding motif. This core established two hydrogen bonds to βR215 and βT216 whereas the aromatic moiety filled a small hydrophobic cavity that is located between helices βH6 and βH9 and which is formed by βD211, βI212, and βK299. The two varying amide extensions of the fragments were found to be fully solvent exposed. Notably, sID βI is located adjacent to the laulimalide/peloruside site.
An intriguing site on β‐tubulin is sID βV, which is targeted by eight fragments (V f=669 Å3). The site is formed by residues of helices βH4, βH5, βH8, and αH11′, strands βS4 and βS5, and loops βH4‐βS5, βH5‐βS6, and βH8‐βS7. Interestingly, it is consistently occupied with a 2‐(N‐morpholino)ethanesulfonic acid (MES) molecule in various T2R‐TTL crystal structures (e.g., PDB ID 5LXT). In the absence of a ligand, access to this site is occluded due to the formation of a salt bridge between βD199 and βR158, which is broken up upon ligand binding. Five out of the eight fragments that bound to this site established an interaction with βD199 through a nitrogen atom, which is otherwise occupied by the side chain of βR158 or a MES molecule. Besides this nitrogen atom, the fragments are extended by diverse aliphatic or aromatic moieties that interacted differently with the protein. Fragments with aromatic moieties that are connected by an aliphatic linker to the nitrogen atom are able to penetrate into a deep hydrophobic cavity formed by residues βI154, βI157, βY161, βP162, and βM166. Fragments lacking this aromatic extension do not bind this cavity, but interact with residues surrounding its entrance. The last site on β‐tubulin is sID βIII (V f=179 Å3), which is targeted by one fragment and formed by residues of helix βH1′, the βN‐terminus and loop βT7. This fragment also targeted site sID βαII.
A large site located at the β‐tubulin‐α‐tubulin inter‐dimer interface of T2R‐TTL is sID βαIII that corresponds to an extended vinca site. [15] It is targeted by 22 fragments (V f=1139 Å3) and is formed by residues of helices βH5, βH6, βH7, βH11, αH8, and αH10 and loops βT5, βH6‐βH7, αT7, and αH10‐αS9. Six of them contain a para‐substituted fluorobenzene moiety as a common binding motif, which is buried in a small hydrophobic cavity formed by the residues βV177, βY210, βP222, βT223, βY224, and βL227. The remaining moieties of these six fragments, as well as the other fragments, differently exploit the large volume of sID βαIII through hydrophobic or mixed hydrophobic and polar contacts. Notably, ten out of the 22 fragments also bound to an additional site in tubulin and three fragments bound pairwise twice to the same site.
Another interesting site observed at the β‐tubulin‐α‐tubulin inter‐dimer interface is sID βαII, which is located between the maytansine and pironetin [16] sites. It is targeted by 16 fragments (V f=632 Å3) and is formed by residues of helices βH3′, βH11′, and αH8, strand αS4, and loops βT3, βT5, αH3‐S4, and αH4‐αS5. Five out of the 16 fragments share an acetanilide group and two fragments a propionanilide group as common binding motif, which bound in a small cavity by forming two hydrogen‐bonds to βN102 and αT257, and by establishing a π‐stacking interaction with βW407. Furthermore, due to this common π‐stacking interaction the varying moieties of these seven fragments are all oriented in the same direction towards the α‐tubulin monomer. The remaining eight fragments share little chemical similarities and interacted with tubulin through hydrophobic or mixed hydrophobic and polar contacts. A third site that we observed at the β‐tubulin‐α‐tubulin inter‐dimer interface is the shallow surface site sID βαI (V f=511 Å3), which is formed by residues of helix αH12 and loops βH11‐βH11′ and αH8‐αS7. It is targeted by three fragments that mainly bound the protein through hydrogen bonding interactions. Notably, they replaced structural water molecules present in the apo T2R‐TTL structure upon binding. One fragment bound pairwise twice to the same site.
Compared to β‐tubulin that interacted with 56 fragments, only four different fragments were found in α‐tubulin, all of which also bound to a second site in the protein. Site sID αII (V f=364 Å3) is located close to the α‐nucleotide site. It is formed by residues of αH1, αH2, and αH7 and the loop αH2‐αS3. Three fragments targeted sID αII, whereby two of them share a sulfonyl group as a common motif that interacted with residues αT225 and αN228. In addition, a single fragment bound to a site formed by residues of helices αH6 and αH9 and the αM‐loop (sID αI, V f=185 Å3). This fragment also targeted site sID βIV (colchicine site).
It is well known that human cells express different αβ‐tubulin isotypes encoded by several α‐ and β‐tubulin genes (reviewed in ref. [17]). We thus wondered whether the residues that form our identified fragment sites differ between tubulin isotypes. Interestingly and as documented in Table S3, we found at least one isotype‐specific residue substitution in all fragment‐binding sites.
Comparison between Pockets and Sites
How do the results of our computational‐ and crystallography‐based approaches compare? As documented in Table 1, Table S1 and Table S3, we obtained a good agreement for most of the pockets and sites. In particular: pID βI → β‐tubulin half‐site of sID βαIII, pID βIII and βIV → sID βIV, pID βV and βXI → sID βII, pID βVIII → sID βI, pID αIV → sID αII, pID αVI → β‐tubulin half‐site of the sID βαIII, and pID αIX → sID αI. However, others were revealed only by either of the two methods, which can be explained, for example, by the following reasons: (i) pockets whose access is hindered by the RB3 and TTL bound to the tubulin dimers in the T2R‐TTL complex (pID βVII, αI and αII) are occluded for fragment binding; (ii) pockets that are too small (pID βIX) cannot accommodate the average ≈200 Da size of the fragments tested; (iii) composite sites that are located at the β‐tubulin‐α‐tubulin inter‐dimer interface in T2R‐TTL and which involve binding of elements from both tubulin monomers (sIDs βαI, βαII, and parts of βαIII) are not considered in our computational strategy; (iv) shallow surface sites (sID βIII) are not detected by our computational algorithm due to the selected probe radii; (v) sites that are induced upon fragment binding (sID βV) and do not persist in the absence of a ligand are not readily detectable in MD simulations.
We observed differences in how the two methods detected known drug‐binding sites in tubulin (Table 1, Table S1 and Table S3). For example, our crystallographic fragment screen did not detect the maytansine site pharmacophore [8] and the laulimalide/peloruside site. This could be due to a possible mismatch in terms of chemical properties, sizes, and shapes between the fragments and these two particular sites. Furthermore, the pironetin site was not detected by both methods, which can be explained by the fact that pironetin binds covalently to α‐tubulin by an induced fit mechanism.[ 16 , 18 ] Nine out of the 12 computationally predicted pockets in α‐tubulin were not targeted by any of the fragments, which can be explained, for example, that the chemical space of the fragment library used was limited, that these pockets are not druggable, and/or that the crystallization conditions used prevented fragment binding.
Analysis of Tubulin–Tubulin and Tubulin–Protein Interactions
We wondered whether our combined computational and crystallographic approach also detected structurally characterized interactions between tubulin subunits and between tubulin and protein partners. For this, we use the term “contact point” to distinguish it from a computationally predicted “pocket” or a crystallographically determined “site” (see above). To this end, we first analyzed homotypic interactions between tubulin dimers as they occur in the microtubule lattice. [19] We then inspected heterotypic interactions of tubulin and microtubules with protein partners, whose complex structures were determined to high resolution either by X‐ray crystallography or by cryo‐electron microscopy (http://www.rcsb.org). The results of our analysis are illustrated in Figure 4, Figure 5 and Figure 6, and summarized in Table 1, Table S4 and Table S5.
Figure 4.
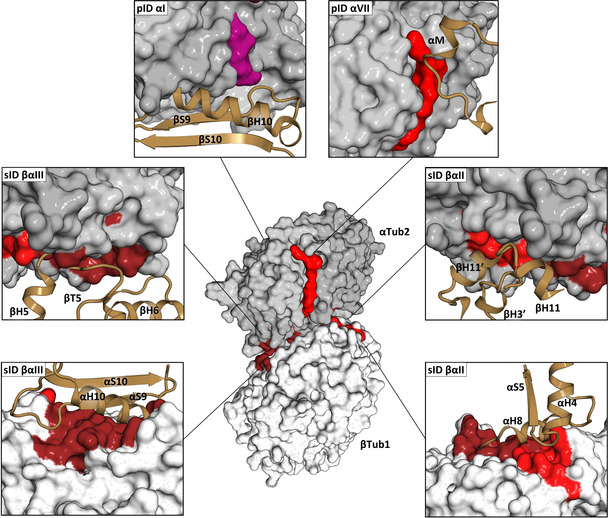
Analysis of tubulin–tubulin contact points. In the center of the Figure, β‐tubulin (βTub1, light gray) and α‐tubulin (αTub2, dark gray) monomers forming a longitudinal inter‐dimer contact along a protofilament in a microtubule (PDB ID 3JAR) are shown in surface representation. The computationally predicted pockets and experimentally determined fragment sites, which are involved in tubulin‐tubulin contacts either along or across protofilaments in microtubules are highlighted in the same color as in Figure 1 and Figure 2. The surrounding panels show close up views of all contact points. The interacting secondary structural elements of neighboring tubulin monomers in the microtubule lattice are shown in brown ribbon representation. See also Table S4.
Figure 5.
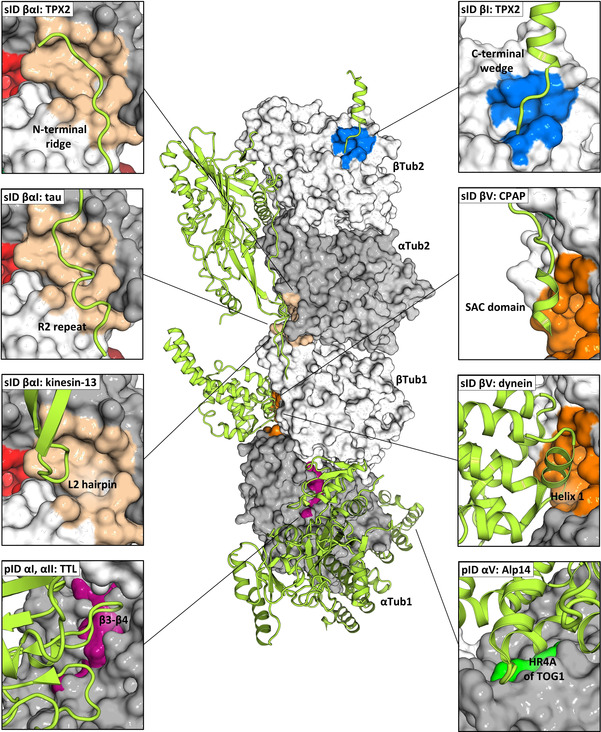
Analysis of tubulin–protein contact points (part 1). In the center of the panel, the structure of the two tubulin heterodimers αTub1‐βTub1 and αTub2‐βTub2 of the T2R‐TTL complex are depicted in surface representation; the α‐ and β‐tubulin monomers are colored in dark and light gray, respectively. The computationally predicted pockets and experimentally determined fragment sites, which are targeted by protein partners are represented and colored as in Figure 1 and Figure 2. Protein partners are shown in light green ribbon representation. The surrounding panels show close up views of all interaction sites; the views in the individual panels differ in orientation from the central overview. The following PDB IDs were used for the analysis: 5ITZ (CPAP), 6B0I (kinesin‐13), 6MZG (Alp14), 5LXT (TTL), 6BJC (TPX2), 6CVN (tau), and 6RZA (dynein). See also Table S5.
Figure 6.
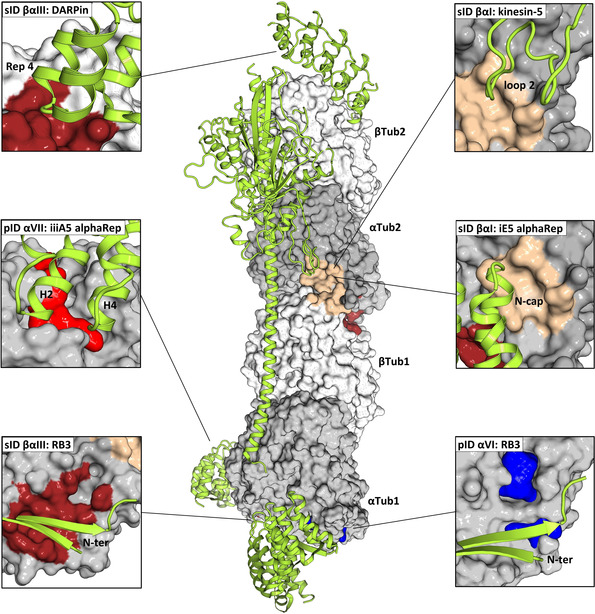
Analysis of tubulin–protein contact points (part 2). In the center of the panel, the structure of the two tubulin heterodimers αTub1‐βTub1 and αTub2‐βTub2 of the T2R‐TTL complex are depicted in surface representation; the α‐ and β‐tubulin monomers are colored in dark and light gray, respectively. The computationally predicted pockets and experimentally determined fragment sites, which are targeted by protein partners are represented and colored as in Figure 1 and Figure 2. Protein partners are shown in light green ribbon representation. The surrounding panels show close up views of all interaction sites; the views in the individual panels differ in orientation from the central overview. The following PDB IDs were used for the analysis: 5EYP (DARPin), 5MM7 (kinesin‐5), 5LXT (RB3), 6GX7 (iiiA5 alphaREP), and 6GWC (iE5 alphaRep). See also Table S5.
A computationally predicted pocket involved in a major lateral inter‐tubulin dimer interaction is pID αVII, which represents the contact point accommodating the αM‐loop of an adjacent α‐tubulin monomer across protofilaments in the microtubule lattice. Pocket pID αI in turn is involved in a longitudinal intra‐tubulin dimer contact by accommodating the βH10‐βS9 loop of a neighboring β‐tubulin monomer along a protofilament. Two fragment sites were identified as contact points mediating major longitudinal inter‐tubulin dimer interactions. (i) The α‐ and β‐tubulin half sites of sID βαII accommodate helix βH3′ and loop βT3, and helix αH8 of neighboring β‐ and α‐tubulin monomers, respectively. (ii) The α‐ and β‐tubulin half sites of sID βαIII accommodate loop βT5, and helix αH10, strand αS9, and loop αH10‐αS9 of neighboring β‐ and α‐tubulin monomers, respectively.
Concerning contact points of tubulin and microtubules with protein partners, we noted that the computationally predicted pockets pID αI and αII are located close to the region that is bound by the tubulin modifying enzyme TTL. [13] In addition, pockets pID αV and αVI are targeted by the TOG domain of the microtubule polymerase Stu2/Alp14 [20] and the N‐terminal β‐hairpin of the stathmin‐like domain of the tubulin sequestering protein RB3, [7b] respectively. Two fragment sites, sID βI and βαI interact with the wedge and ridge domain of the spindle assembly factor TPX2, [21] respectively. Site sID βV is targeted by the microtubule‐binding domain of the dynein motor heavy chain [22] and the PN2‐3 domain of the centrosomal protein CPAP. [23] We further found that sID βαI interacts with the motor domains of Ustilago maydis kinesin‐5 [24] and kinesin‐13, [25] two family members of kinesin microtubule depolymerases, as well as with the R1 and R2 repeats of the microtubule‐stabilizing protein tau. [26] Finally, the synthetic protein binders iE5 alphaRep [27] and DARPins 1 and 2 [28] bind to the α‐tubulin half site of sID βαI and the β‐tubulin half site of sID βαIII, respectively, and pocket pID αVII is targeted by the artificial protein binder iiiA5 alphaRep. [29] The α‐tubulin half site of sID βαIII is in addition also targeted by the stathmin‐like domain of RB3. [7b]
We found a few cases where protein partners did not bind to one of the computationally predicted tubulin pockets or crystallographic fragment sites. These are the motor domains of motile kinesins, [30] the CH domains of end binding proteins (EBs),[ 19 , 31 ] the CKK domain of the microtubule minus‐end‐targeting proteins CAMSAPs, [32] the spectrin domain of the protein regulator of cytokinesis 1 (PRC1), [33] the second and third helical motifs of the chlamydial type three secretion effector protein CopN, [29] the vasohibin‐SVBP complex, [34] and two synthetic protein binders.[ 27 , 29 ] With the exception of latter, all these interactions are mediated through large shallow, composite binding sites formed either at the intra‐tubulin dimer interface or at inter‐tubulin dimer interfaces formed between two or four tubulin dimers in the microtubule lattice. They are thus difficult to be detected by either of our two methods used.
Discussion
Our combined computational and crystallographic fragment screening approach identified a total of 27 distinct binding sites in tubulin. Notably, all major known tubulin‐drug binding sites were readily detected. Furthermore, several key contact points between tubulin dimers in the microtubule lattice as well as between tubulin dimers and secondary structural elements of regulatory protein partners were revealed. Importantly, our analysis disclosed 18 sites that are not targeted by any of the antitubulin drugs that have been structurally characterized to date. 11 out of those (four in α‐tubulin and seven in β‐tubulin) are also not targeted by any structurally characterized protein partner and thus represent completely new sites. Our method further revealed an intricate, dynamic communication network between different pockets located also remote from each other in both tubulin monomers. Finally, we identified 56 chemically diverse fragments that target a total of 10 different sites in tubulin.
It is notable that the vast majority of structurally characterized ligands and protein partners were found to target β‐tubulin. The preference for β‐tubulin over α‐tubulin could be explained by the fact that the GTP hydrolysis cycle takes place on β‐tubulin, making this monomer a favorable target for interfering with microtubule dynamics. A question has thus been whether α‐tubulin can also be considered as a target for the development of small molecule modulators of microtubule dynamics. Our analysis indeed identified several sites in and fragments able to bind to α‐tubulin. It revealed further several fragment sites whose residue composition differ amongst tubulin isotypes, which offers a basis for isotype‐selective ligand design. This finding is particularly interesting in the context of chemotherapy since a widely recognized resistance mechanism against anticancer tubulin‐targeting agents is the upregulation of specific tubulin isotypes by cancer cells. [35] Finally, to the best of our knowledge the so far structurally characterized ligands and proteins that target tubulin do not display any overlapping binding sites, which is rather surprising. Our crystallographic fragment screen now revealed four sites that are targeted by both fragments and secondary structural elements of major cellular microtubule regulators including tau, dynein, kinesin‐13, kinesin‐5, TPX2, and CPAP/SAS‐4.
In conclusion, our analysis provides a comprehensive description of the shape, chemical property and dynamics of small molecule‐binding sites in tubulin. Until now, drug discovery efforts were directed towards interfering with microtubule dynamics. Our results not only offer a platform for the innovative design of more selective antitubulin ligands with novel mechanisms of action, they also provide a structural basis for the design of inhibitors of tubulin‐protein interactions. In more general terms, our combined computational and experimental approach offers a framework that may help identifying new ligand‐binding sites in any other pharmaceutically relevant target and characterize them in terms of chemical tractability and allosteric modulation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
We are indebted to A. Douangamath and R. Zhang for excellent support with the crystallographic fragment screening, and acknowledge the Diamond Light Source for access to the fragment screening facility XChem and for beamtime on beamline I04‐1 under proposal LB17334 (to M.O.S.). This work has been supported by iNEXT, grant number PID2692 (to M.O.S.), funded by the Horizon 2020 program of the European Union, and by grants from the Regione Lombardia (ID 239047 NEON; to A.C.) and from the Swiss National Science Foundation (31003A_166608 and 310030_192566; to M.O.S.). Coordinates of the X‐ray crystal structures have been deposited in the RCSB PDB (http://www.rcsb.org); all accession numbers are given in Table S3.
T. Mühlethaler, D. Gioia, A. E. Prota, M. E. Sharpe, A. Cavalli, M. O. Steinmetz, Angew. Chem. Int. Ed. 2021, 60, 13331.
Contributor Information
Andrea Cavalli, Email: andrea.cavalli@iit.it.
Michel O. Steinmetz, Email: michel.steinmetz@psi.ch.
References
- 1. Downing K. H., Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [DOI] [PubMed] [Google Scholar]
- 2. Steinmetz M. O., Prota A. E., Trends Cell Biol. 2018, 28, 776–792. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Baas P. W., Ahmad F. J., Brain 2013, 136, 2937–2951; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Dumontet C., Jordan M. A., Nat. Rev. Drug Discovery 2010, 9, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prota A. E., Bargsten K., Zurwerra D., Field J. J., Diaz J. F., Altmann K. H., Steinmetz M. O., Science 2013, 339, 587–590. [DOI] [PubMed] [Google Scholar]
- 5. Löwe J., Li H., Downing K. H., Nogales E., J. Mol. Biol. 2001, 313, 1045–1057. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Alushin G. M., Lander G. C., Kellogg E. H., Zhang R., Baker D., Nogales E., Cell 2014, 157, 1117–1129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Nogales E., Wolf S. G., Downing K. H., Nature 1998, 391, 199–203. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Prota A. E., Danel F., Bachmann F., Bargsten K., Buey R. M., Pohlmann J., Reinelt S., Lane H., Steinmetz M. O., J. Mol. Biol. 2014, 426, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 7b. Ravelli R. B., Gigant B., Curmi P. A., Jourdain I., Lachkar S., Sobel A., Knossow M., Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- 8. Prota A. E., Bargsten K., Diaz J. F., Marsh M., Cuevas C., Liniger M., Neuhaus C., Andreu J. M., Altmann K. H., Steinmetz M. O., Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galmarini C. M., Martin M., Bouchet B. P., Guillen-Navarro M. J., Martinez-Diez M., Martinez-Leal J. F., Akhmanova A., Aviles P., BMC Cancer 2018, 18, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prota A. E., Bargsten K., Northcote P. T., Marsh M., Altmann K. H., Miller J. H., Diaz J. F., Steinmetz M. O., Angew. Chem. Int. Ed. 2014, 53, 1621–1625; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1647–1651. [Google Scholar]
- 11. Field J. J., Pera B., Gallego J. E., Calvo E., Rodriguez-Salarichs J., Saez-Calvo G., Zuwerra D., Jordi M., Andreu J. M., Prota A. E., Menchon G., Miller J. H., Altmann K. H., Diaz J. F., J. Nat. Prod. 2018, 81, 494–505. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Patel D., Bauman J. D., Arnold E., Prog. Biophys. Mol. Biol. 2014, 116, 92–100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Hartshorn M. J., Murray C. W., Cleasby A., Frederickson M., Tickle I. J., Jhoti H., J. Med. Chem. 2005, 48, 403–413; [DOI] [PubMed] [Google Scholar]
- 12c. O'Reilly M., Cleasby A., Davies T. G., Hall R. J., Ludlow R. F., Murray C. W., Tisi D., Jhoti H., Drug Discovery Today 2019, 24, 1081–1086. [DOI] [PubMed] [Google Scholar]
- 13. Prota A. E., Magiera M. M., Kuijpers M., Bargsten K., Frey D., Wieser M., Jaussi R., Hoogenraad C. C., Kammerer R. A., Janke C., Steinmetz M. O., J. Cell Biol. 2013, 200, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Massarotti A., Coluccia A., Silvestri R., Sorba G., Brancale A., ChemMedChem 2012, 7, 33–42. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Gigant B., Wang C., Ravelli R. B., Roussi F., Steinmetz M. O., Curmi P. A., Sobel A., Knossow M., Nature 2005, 435, 519–522; [DOI] [PubMed] [Google Scholar]
- 15b. Doodhi H., Prota A. E., Rodriguez-Garcia R., Xiao H., Custar D. W., Bargsten K., Katrukha E. A., Hilbert M., Hua S., Jiang K., Grigoriev I., Yang C. P., Cox D., Horwitz S. B., Kapitein L. C., Akhmanova A., Steinmetz M. O., Curr. Biol. 2016, 26, 1713–1721. [DOI] [PubMed] [Google Scholar]
- 16. Yang J., Wang Y., Wang T., Jiang J., Botting C. H., Liu H., Chen Q., Naismith J. H., Zhu X., Chen L., Nat. Commun. 2016, 7, 12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ludueña R. F., Int. Rev. Cytol. 1997, 178, 207–275. [DOI] [PubMed] [Google Scholar]
- 18. Prota A. E., Setter J., Waight A. B., Bargsten K., Murga J., Diaz J. F., Steinmetz M. O., J. Mol. Biol. 2016, 428, 2981–2988. [DOI] [PubMed] [Google Scholar]
- 19. Zhang R., Alushin G. M., Brown A., Nogales E., Cell 2015, 162, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nithianantham S., Cook B. D., Beans M., Guo F., Chang F., Al-Bassam J., eLife 2018, 7, e38922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang R., Roostalu J., Surrey T., Nogales E., eLife 2017, 6, e30959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Lacey S. E., He S., Scheres S. H., Carter A. P., eLife 2019, 8, e47145; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Redwine W. B., Hernandez-Lopez R., Zou S., Huang J., Reck-Peterson S. L., Leschziner A. E., Science 2012, 337, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma A., Aher A., Dynes N. J., Frey D., Katrukha E. A., Jaussi R., Grigoriev I., Croisier M., Kammerer R. A., Akhmanova A., Gonczy P., Steinmetz M. O., Dev. Cell 2016, 37, 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. von Loeffelholz O., Moores C. A., J. Struct. Biol. 2019, 207, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Benoit M., Asenjo A. B., Sosa H., Nat. Commun. 2018, 9, 1662; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Trofimova D., Paydar M., Zara A., Talje L., Kwok B. H., Allingham J. S., Nat. Commun. 2018, 9, 2628; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Wang W., Cantos-Fernandes S., Lv Y., Kuerban H., Ahmad S., Wang C., Gigant B., Nat. Commun. 2017, 8, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kellogg E. H., Hejab N. M. A., Poepsel S., Downing K. H., DiMaio F., Nogales E., Science 2018, 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Campanacci V., Urvoas A., Consolati T., Cantos-Fernandes S., Aumont-Nicaise M., Valerio-Lepiniec M., Surrey T., Minard P., Gigant B., Structure 2019, 27, 497–506, e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Pecqueur L., Duellberg C., Dreier B., Jiang Q., Wang C., Pluckthun A., Surrey T., Gigant B., Knossow M., Proc. Natl. Acad. Sci. USA 2012, 109, 12011–12016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Ahmad S., Pecqueur L., Dreier B., Hamdane D., Aumont-Nicaise M., Pluckthun A., Knossow M., Gigant B., Sci. Rep. 2016, 6, 28922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campanacci V., Urvoas A., Cantos-Fernandes S., Aumont-Nicaise M., Arteni A. A., Velours C., Valerio-Lepiniec M., Dreier B., Pluckthun A., Pilon A., Pous C., Minard P., Gigant B., Proc. Natl. Acad. Sci. USA 2019, 116, 9859–9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Gigant B., Wang W., Dreier B., Jiang Q., Pecqueur L., Pluckthun A., Wang C., Knossow M., Nat. Struct. Mol. Biol. 2013, 20, 1001–1007; [DOI] [PubMed] [Google Scholar]
- 30b. Atherton J., Farabella I., Yu I. M., Rosenfeld S. S., Houdusse A., Topf M., Moores C. A., eLife 2014, 3, e03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maurer S. P., Fourniol F. J., Bohner G., Moores C. A., Surrey T., Cell 2012, 149, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Atherton J., Jiang K., Stangier M. M., Luo Y., Hua S., Houben K., van Hooff J. J. E., Joseph A. P., Scarabelli G., Grant B. J., Roberts A. J., Topf M., Steinmetz M. O., Baldus M., Moores C. A., Akhmanova A., Nat. Struct. Mol. Biol. 2017, 24, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellogg E. H., Howes S., Ti S. C., Ramirez-Aportela E., Kapoor T. M., Chacon P., Nogales E., Proc. Natl. Acad. Sci. USA 2016, 113, 9430–9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li F., Li Y., Ye X., Gao H., Shi Z., Luo X., Rice L. M., Yu H., eLife 2020, 9, e58157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kavallaris M., Nat. Rev. Cancer 2010, 10, 194–204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary