

Abstract
Cp2Ti(TFA) is a broadly applicable catalyst for the [2+2] cycloaddition of bisenones by inner‐sphere electron transfer. The attractiveness of this mechanism is shown by comparison with outer‐sphere ET methods. DFT calculations show that the reaction proceeds through a unique unfavorable 5‐exo (the rate‐determining step) and a favorable 4‐exo cyclization.
Keywords: cyclization, electron transfer, homogeneous catalysis, radicals, titanium
For [2+2] cycloaddition reactions of bisenones, higher yields and diastereoselectivities as well as greater substrate scope were observed under catalysis by Cp2Ti(TFA) than with previously established methods (see scheme). The mechanism is characterized by a highly unusual combination of an unfavorable and rate‐determining 5‐exo and favorable 4‐exo cyclization, as established by DFT calculations.

Catalysis in single electron steps [1] or metalloradical catalysis (MRC) [2] is a strategy for merging the advantages of radical chemistry, such as the ease of radical generation, high functional group tolerance, and mildness of reaction conditions with those of transition metal catalysis, such as control of selectivity and efficiency. The titanocene(III)/titanocene(IV) couple has been successfully applied in catalysis in single electron steps because of its efficient shuttling between the two neighboring oxidation states, other ligands for titanium (salen) and other metal complexes (Sm) have been successfully used in a similar manner. [3] The use of epoxides as radical precursors highlights one important advantage of catalysis in single electron steps. The requirement of substrate binding by the catalyst prior to ET allows highly regioselective epoxide opening by an inner‐sphere electron transfer (ET). [4] In analogy, in MRC with cobalt–carbene radicals, reductive eliminations have been carried out enantioselectively. [5]
Photoredox catalysis (PRC) that has been used in a myriad of applications is an exceptionally successful approach to enable radical reactions by electron transfer. [6] The conceptual differences to catalysis in single electron steps and PRC is that ET from the photoexcited complexes typically occurs without substrate coordination to the catalyst through outer‐sphere electron transfer and, hence, without catalyst control of radical generation.
For highlighting the differences of inner‐ and outer‐sphere electron transfer reagents in catalytic radical chemistry, we analyzed the [2+2] cycloaddition of bisenones as a case example (Scheme 1).
Scheme 1.
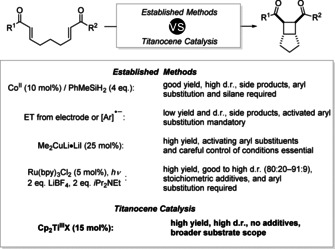
Comparison of catalysis in single electron steps with reported conditions for the [2+2] cycloaddition of bisenones. bpy=2,2′‐bipyridine.
The Krische group was the first to report the reaction. [7] They reported cobalt catalysts in the presence of silanes for a cycloaddition through hydro‐ and cyclometallation, and electrochemical methods, arene radical anions, and Me2CuLi for outer‐sphere ETs to the bisenones. In a landmark paper, Yoon and his group showed how the reaction can be carried out under PRC with Ru(bpy)3Cl2 as photoredox catalyst. [8] The addition of LiBF4 (2 equiv) to activate the substrates for outer‐sphere ET was essential, as well as iPr2NEt (2 equiv) for the reductive quenching of photoexcited Ru.
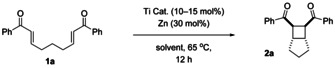
Herein, we show that the catalytic intramolecular [2+2] cycloaddition of bisenones can also be carried out through inner‐sphere ET from Cp2Ti(III)X catalysts after complexation of the substrates. As will be shown, this allows a broader substrate scope and often higher stereoselectivity than in the outer‐sphere ET methods. Our results of the initial catalyst and condition screening for substrate 1 a are summarized in Table 1.
Table 1.
Identification of suitable reaction conditions for the titanocene‐catalyzed [2+2] cycloaddition of 1 a.[a]
|
Entry |
Solvent |
Catalyst (mol %) |
Yield [%][b] |
|---|---|---|---|
|
1 |
THF |
Cp2TiCl2 (10) |
n.r. |
|
2 |
THF |
Cp2Ti(OMs)2 (10) |
45[c] |
|
3 |
THF |
Cp2Ti(TFA)2 (10) |
78 |
|
4 |
THF |
Cp*2TiCl2 (10) |
n.r. |
|
5 |
THF |
Cp2Ti(TFA)2 (15) |
85 |
|
6 |
ethyl acetate |
Cp2Ti(TFA)2 (15) |
85 |
|
7 |
dioxane |
Cp2Ti(TFA)2 (15) |
85[d] |
[a] Reaction conditions: Ti catalyst (10–15 mol %), Zn (30 mol %), 0.05 M 1 a in solvent, 65 °C. [b] Yield of the isolated product, only diastereomer shown formed (d.r. >99:<1). [c] Yield determined by 1H NMR spectroscopy against an internal standard (trimethoxybenzene). [d] Yield after 8 h.
The reaction can be carried out under titanocene catalysis (10 mol % Cp2TiX at 65 °C in THF with Cp2TiX prepared through zinc reduction of Cp2TiX2). [9] It is essential that more electron deficient ligands X than Cl are used. With Cp2TiCl, none of the desired 2 a was obtained, Cp2TiOMs (‐OMs= ‐O3SCH3) gave 2 a in 45 % yield and our best catalyst Cp2Ti(TFA) (‐TFA=‐O2CCF3) resulted in an isolated yield of 78 % of 2 a in THF. A slightly better yield (85 %) was obtained with 15 mol % of Cp2Ti(TFA). [10] Curiously, the weakest reductant Cp2Ti(TFA) [E p for the anodic oxidation of Cp2TiX from CV experiments (THF vs. Fc/Fc+): X=Cl: −0.83 V; X=OMs: −0.62 V, X=TFA: −0.58 V] gives the most active catalyst! This is rather surprising as the results from the established methods suggest that an easier initial ET to the bisenone leads to higher yields.
In all cases, 2 a was obtained exclusively as the diastereoisomer shown. 1,4‐THF, 1,4‐dioxane and EtOAc as solvent gave essentially identical results. For reasons of comparability, all reactions were run for 12 h. However, the same yield was obtained after 8 h (entry 7) and no further attempts were undertaken to investigate even shorter reaction times.
We examined the scope of the reaction by varying the substituents of the bisenone system next (Table 2). With R1=R2=aryl, the desired products are obtained in high yields and essentially as single diastereoisomers [d.r. >99:<1 except 2 b (96:4)]. An exception is 1 f with two NO2 substituents that are reduced by Cp2Ti(TFA) resulting in catalyst deactivation. Yields are higher than in the cobalt‐catalyzed reactions developed by Krische and slightly lower than in Yoon's excellent PRC. Our diastereoselectivities are as high as in Krische's cobalt‐catalyzed reactions and higher than under Yoon's conditions (>91:<9 and 91:9 for 2 i). Substrates with only one aryl group (1 g–i) worked well.
Table 2.
Substrate scope of the titanocene‐catalyzed [2+2] cycloaddition of bisenones.
|
|
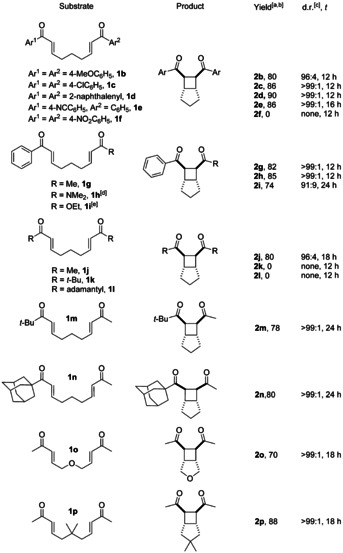
[a] Reaction conditions: 0.05 M 1 in 1,4‐dioxane, 80 °C. [b] Yield of the isolated product. [c] The diastereomeric ratio was determined by 1H NMR spectroscopy. [d] The reaction was carried out with 0.05 M 1 h in THF at 65 °C. [e] The reaction was carried out with 20 mol % Cp2Ti(TFA)2.
Substrates with R1 and R2 being alkyl (1 j and 1 m–p) give the desired products in yields between 70 % and 88 % as single isomers except 1 j where the d.r. is only slightly lower (d.r. 96:4). The two substrates that are not turned over (1 k and 1 l) because both bulky substituents R seem to prevent binding of the active titanocene(III) catalyst. One coordinating carbonyl group (1 m, 1 n) is mandatory for successful catalysis. This strongly suggests that an inner‐sphere SET occurs with Cp2Ti(TFA) that leads to the broadest substrate scope in the [2+2] cycloaddition, as none of the previously described systems provide products with two alkyl substituents (2 j and 2 m–p).
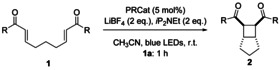
After the groundbreaking work with Ru(bpy)3Cl2 by Yoon and co‐workers, many other PRC catalysts have been devised. To further study the difference in reactivity between titanocene(III)‐mediated inner‐sphere electron transfer and PRC‐mediated outer‐sphere electron transfer in the [2+2] cycloaddition, we decided to investigate the performance of two more reducing iridium complexes [6f] (with respect to Ru(bpy)3Cl2 after reductive quenching) and the less reducing organic dye 4CzIPN [6g] (with respect to Ru(bpy)3Cl2 after reductive quenching) to understand the influence of the PRC′s redox potential on the [2+2] cycloaddition under Yoon's conditions (Table 3) to ensure a fair comparison.
Table 3.
Investigation of other photoredox catalysts under previously reported conditions.
|
Entry |
R |
PRCat |
Yield of 2 [%][a,b] |
|---|---|---|---|
|
1 |
C6H5, 1 a |
[Ir{dF(CF3)ppy}2(dtbpy)]PF6 |
88 |
|
2 |
Me, 1 j |
[Ir{dF(CF3)ppy}2(dtbpy)]PF6 |
0 |
|
3 |
C6H5, 1 a |
fac‐Ir(ppy)3 |
90 |
|
4 |
Me, 1 j |
fac‐Ir(ppy)3 |
0 |
|
5 |
C6H5, 1 a |
4CzIPN |
85 |
|
6 |
Me, 1 j |
4CzIPN |
0 |
[a] Reaction conditions: (PRCat, 5 mol %), LiBF4 (2 equiv), iP2NEt (2 equiv), 0.1 M 1 in CH3CN, blue LEDs, room temperature, 1 h. [b] The yield was determined by 1H NMR spectroscopy against the internal standard 1,3,5‐trimethoxybenzene. ppy=2‐phenylpyridine.
With 1 a high yields of 2 a were obtained with all catalysts. However, none of the three photoredox catalysts (PRCats) led to the formation of 1 j. This indicates that the success of the reaction is not only dependent on the redox potential of the catalyst. The coordination between the catalyst and the substrate is mandatory for the efficient transformation.
Thus, of all catalysts reported or investigated here, Cp2Ti(TFA) is the only complex to efficiently transform the alkyl substituted bisenones (1 j and 1 m–p) to the desired products. Complexation of the substrate to enforce an inner‐sphere SET seems mandatory. Curiously, it is the least reducing of all catalysts investigated by Krische, Yoon, and us.
To understand the substrate scope of titanocene catalysis and the counterintuitive dependence of catalyst performance on its redox potential, we studied the proposed mechanism reaction of the [2+2] cycloaddition of 1 j by computational means. The anticipated catalytic cycle (for all substrates) is shown in Scheme 2.
Scheme 2.
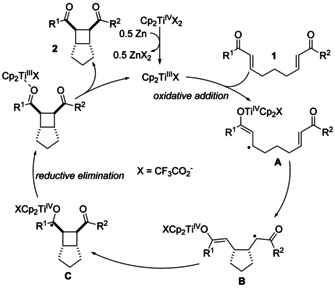
Proposed mechanism of the titanocene‐catalyzed [2+2] cycloaddition.
The reaction starts with an inner‐sphere ET to the enone complexed to TiIII that delivers the stabilized radical A. This step constitutes the oxidative addition. Enones have been used as radical precursors in Cp2TiCl‐mediated and ‐catalyzed umpolung reactions. [11] Radical generation is followed by a 5‐exo cyclization of the stabilized radical anion A to yield B. Radical C is formed by a 4‐exo cyclization of the enoyl radical in B to the titanocene enolate. From C, the product is liberated and the catalyst regenerated by back electron transfer from the ketyl radical to TiIV and dissociation of 2*Cp2TiIIIX. Both steps formally constitute a reductive elimination. As an alternative for closing the cycle, one can also imagine a direct intermolecular reduction of 1 by C in a process reminiscent of a chain reaction. While this option cannot be strictly ruled out, we believe the high dilution necessary and the exergonic nature of the reductive elimination (see discussion of Scheme 3 below) support our intramolecular reductive elimination.
Scheme 3.
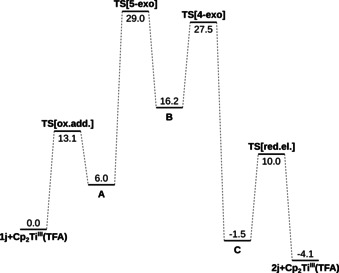
Energy profile of the conversion of 1 j into 2 j in the presence of Cp2Ti(TFA) in 1,4‐dioxane at 353.15 K. All Gibbs free energies are shown in kcal mol−1. The complex labels A–C refer to those given in Scheme 2. All structures were investigated at the PW6B95‐D4/def2‐QZVP+COMSO‐RS[1,4‐dioxane]//r2SCAN‐3c(COSMO[1,4‐dioxane]) level of theory.
In order to validate this mechanistic proposal, we studied the reaction of 1 j to 2 j computationally for Cp2Ti(TFA) in 1,4‐dioxane. The conformational spaces of all minimum structures and transition states were therefore examined and all final structures were investigated on the PW6B95‐D4/def2‐QZVP+COMSO‐RS[1,4‐dioxane]//r2SCAN‐3c(COSMO[1,4‐dioxane]) level of theory. [12]
At 353.15 K, the ΔG 353.15 of the overall reaction is −4.1 kcal mol−1. This relatively low value is due to the strain of the four‐membered ring. The oxidative addition is thermodynamically unfavorable (ΔG 353.15=+6.0 kcal mol−1) with an activation energy ΔG*353.15 of +13.1 kcal mol−1. The 5‐exo cyclization leading to B is most unfavorable step of the cycle (ΔG 353.15=+10.2 kcal mol−1 and ΔG*353.15=+23.0 kcal mol−1) because from the resonance stabilized ketyl radical and enone in A, the relatively unstabilized enoyl radical and a titanium enolate are formed in B.
The ensuing 4‐exo cyclization is energetically remarkably advantageous (ΔG 353.15=−17.7 kcal mol−1 and ΔG*353.15= +11.3 kcal mol−1). This reflects the high affinity of the electron rich enolate towards addition of the electron deficient enoyl radical. The barrier for the 4‐exo cyclization [14] is relatively low because in B, the enolate and enoyl radicals are in relatively close proximity in the cis‐disubstituted cyclopentane. From ketyl radical C the back electron transfer to TiIV liberates the products and regenerates the active catalyst Cp2Ti(TFA). This step is slightly exergonic (ΔG 353.15=−2.6 kcal mol−1 and ΔG*353.15=+11.5 kcal mol−1).
We also studied the [2+2] cycloaddition with Cp2Ti+ as catalyst. Such cationic complexes can form from Cp2TiX, especially with weaker ligands than Cl or in suitable solvents. [15] However, formation of A and especially the 5‐exo cyclization are noticeably less favorable than with Cp2Ti(TFA). However, the chelated radical cationic analogue B+ of B is relatively close to B in energy and its 4‐exo cyclization via the ideally preorganized TS[4‐exo]+ is almost barrierless (see SI for details and Figure 1 for the structures). Therefore, it is not possible to exclude that the 4‐exo cyclization occurs at least in part via B+ and TS[4‐exo]+.
Figure 1.
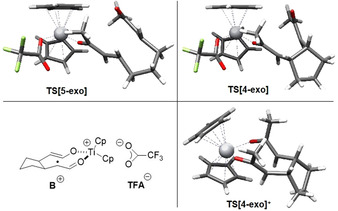
Structures of TS[5‐exo], TS[4‐exo], B+ and the chelated TS[4‐exo]+.
The identification of the 5‐exo cyclization as the rate‐determining step of the catalytic cycle is quite unusual, as 5‐exo cyclizations of nonstabilized radicals are typically fast reactions in titanocene catalysis. [16] However, an observation important for the interpretation of our results has been reported earlier by Streuff.[ 11b , 11c , 11d ] He noted that in titanocene‐catalyzed reductive cross‐coupling reactions of enones only acrylonitriles lead to fast conjugate radical additions due to a favorable HOMO–LUMO gap. With other acceptors, such as acrylates or acrylamides, the conjugate additions were too slow and dimerization or over‐reduction of the enone substrates occurred.
In accordance, we suggest that in the [2+2] cycloadditions, only the weakly reducing Cp2Ti(TFA) is unable to reductively intercept radical A. This provides sufficient lifetime to A to allow the pivotal 5‐exo cyclization to B. The computational analysis of the reaction of 1 j with Cp2TiCl supports this notion. The potential energy surface is similar to that for the Cp2Ti(TFA) with the 5‐exo cyclization being rate‐determining (see the Supporting Information, Scheme S1 for details). Thus, the failure of Cp2TiCl as catalyst must be due to “off‐cycle events”. We suggest that these events are the reductive trapping of A and the lower thermal stability of Cp2TiCl.[ 10a , 10b , 10e ] Cp*2TiCl (Table 1, entry 4) is thermally stable but gives no product. This suggests that a catalyst with high reducing power leads to the trapping of A.
The 5‐exo cyclization as the rate‐determining step also explains why the [2+2] cycloaddition requires harsher conditions than the titanium‐ and samarium‐catalyzed inter‐ and intramolecular radical redox relay reactions of cyclopropyl ketones (Scheme 4).[ 3f , 3h , 3j ]
Scheme 4.
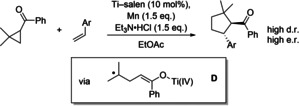
TiIII–salen‐catalyzed radical relay [3+2] cycloaddition of cyclopropyl ketones.
After ketyl radical formation and cyclopropane opening radical D is formed that is not stabilized by conjugation with the enolate. Therefore, addition to the olefin, especially to styrenes, is much more favorable than that in the case of A (Scheme 2).
In summary, Cp2Ti(TFA) is a broadly applicable catalyst in the [2+2] cycloaddition of bisenones. It leads to a broader substrate scope than the methods developed previously. We attribute this feature in the mandatory binding of the substrate to the TiIII center before ET. Compared to PRC this renders the external activation by a Lewis acid for an outer‐sphere ET superfluous. Moreover, Cp2Ti(TFA) catalysis proceeds under highly sustainable conditions because only Zn dust for the reduction of Cp2Ti(TFA)2 and the substrate is needed. No further additives are required.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the DFG (Ga 619/12‐2 to A.G.) and in the framework of the “Gottfried Wilhelm Leibniz Prize” to S.G. J.B.S. is most grateful for financial support by the “Fonds der Chemischen Industrie (FCI)”. Open access funding enabled and organized by Projekt DEAL.
Z. Zhang, J. B. Stückrath, S. Grimme, A. Gansäuer, Angew. Chem. Int. Ed. 2021, 60, 14339.
References
- 1.
- 1a. Gansäuer A., Fleckhaus A., Alejandre Lafont M., Okkel A., Kotsis K., Anoop A., Neese F., J. Am. Chem. Soc. 2009, 131, 16989–16999; [DOI] [PubMed] [Google Scholar]
- 1b. Gansäuer A., Hildebrandt S., Vogelsang E., R. A. Flowers II , Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. van der Vlugt J. I., Chem. Eur. J. 2019, 25, 2651–2662; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Huang H.-M., Garduño-Castro M. H., Morill C., Procter D. J., Chem. Soc. Rev. 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Gansäuer A., Rinker B., Pierobon M., Grimme S., Gerenkamp M., Mück-Lichtenfeld C., Angew. Chem. Int. Ed. 2003, 42, 3687–3690; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3815–3818; [Google Scholar]
- 3b. Gansäuer A., Rinker B., Ndene-Schiffer N., Pierobon M., Grimme S., Gerenkamp M., Mück-Lichtenfeld C., Eur. J. Org. Chem. 2004, 2337–2351; [Google Scholar]
- 3c. Gansäuer A., Klatte M., Brändle G. M., Friedrich J., Angew. Chem. Int. Ed. 2012, 51, 8891–8894; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9021–9024; [Google Scholar]
- 3d. Henriques D. S. G., Zimmer K., Klare S., Meyer A., Rojo-Wiechel E., Bauer M., Sure R., Grimme S., Schiemann O., R. A. Flowers II , Gansäuer A., Angew. Chem. Int. Ed. 2016, 55, 7671–7675; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7801–7805; [Google Scholar]
- 3e. Hao W., Wu X., Sun J. Z., Siu J. C., MacMillan S. N., Lin S., J. Am. Chem. Soc. 2017, 139, 12141–12144; [DOI] [PubMed] [Google Scholar]
- 3f. Hao W., Harenberg J. H., MacMillan S. N., Lin S., J. Am. Chem. Soc. 2018, 140, 3514–3517; [DOI] [PubMed] [Google Scholar]
- 3g. Yao C., Dahmen T., Gansäuer A., Norton J., Science 2019, 364, 764–767; [DOI] [PubMed] [Google Scholar]
- 3h. Huang H.-M., McDouall J. J. W., Procter D. J., Nat. Catal. 2019, 2, 211–218; [Google Scholar]
- 3i. Funk P., Richrath R. B., Bohle F., Grimme S., Gansäuer A., Angew. Chem. Int. Ed. 2021, 60, 5482–5488; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 5542–5548; [Google Scholar]
- 3j. Agasti S., Beattie N. A., McDouall J. J. W., Procter D. J., J. Am. Chem. Soc. 2021, 143, 3655–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Funken N., Zhang Y.-Q., Gansäuer A., Chem. Eur. J. 2017, 23, 19–32; [DOI] [PubMed] [Google Scholar]
- 4b. Gansäuer A., Fan C.-A., Keller F., Keil J., J. Am. Chem. Soc. 2007, 129, 3484–3485; [DOI] [PubMed] [Google Scholar]
- 4c. Gansäuer A., Shi L., Otte M., J. Am. Chem. Soc. 2010, 132, 11858–11859; [DOI] [PubMed] [Google Scholar]
- 4d. Gansäuer A., Karbaum P., Schmauch D., Einig M., Shi L., Anoop A., Neese F., Chem. Asian J. 2014, 9, 2289–2294; [DOI] [PubMed] [Google Scholar]
- 4e. Funken N., Mühlhaus F., Gansäuer A., Angew. Chem. Int. Ed. 2016, 55, 12030–12034; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12209–12213; [Google Scholar]
- 4f. Mühlhaus F., Weißbarth H., Dahmen T., Schnakenburg G., Gansäuer A., Angew. Chem. Int. Ed. 2019, 58, 14208–14212; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14346–14350. [Google Scholar]
- 5.
- 5a. Karns A. S., Goswami M., de Bruin B., Chem. Eur. J. 2018, 24, 5253–5258; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Li C., Lang K., Lu H., Hu Y., Cui X., Wojtas L., Zhang X. P., Angew. Chem. Int. Ed. 2018, 57, 16837–16841; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17079–17083; [Google Scholar]
- 5c. Lang K., Torker S., Wojtas L., Zhang X. P., J. Am. Chem. Soc. 2019, 141, 12388–12396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. jin L.-M., Xu P., Xie J., Zhang X. P., J. Am. Chem. Soc. 2020, 142, 20828–20836; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Lang K., Li C., Kim I., Zhang X. P., J. Am. Chem. Soc. 2020, 142, 20902–20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected reviews, see:
- 6a. Poplata S., Tröster A., Zou Y.-Q., Bach T., Chem. Rev. 2016, 116, 9748–9815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Rev. Chem. 2017, 1, 0052; [Google Scholar]
- 6c. McAtee R. C., McClain E. J., Stephenson C. R. J., Trends Chem. 2019, 1, 111–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Lipp A., Badir S. O., Molander G. A., Angew. Chem. Int. Ed. 2021, 60, 1714–1726; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1738–1750; [Google Scholar]
- 6e. Yu X.-Y., Chen J.-R., Xiao W.-J., Chem. Rev. 2021, 121, 506–561; [DOI] [PubMed] [Google Scholar]
- 6f. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; Organic dyes: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6g. Luo J., Zhang J., ACS Catal. 2016, 6, 873–877. [Google Scholar]
- 7.
- 7a. Baik T.-G., Luis A. L., Wang L.-C., Krische M. J., J. Am. Chem. Soc. 2001, 123, 6716–6717; [DOI] [PubMed] [Google Scholar]
- 7b. Roh Y., Jang H.-Y., Lynch V., Bauld N. L., Krische M. J., Org. Lett. 2002, 4, 611–613; [DOI] [PubMed] [Google Scholar]
- 7c. Wang L.-C., Jang H.-Y., Roh Y., Lynch V., Schultz A. J., Wang X., Krische M. J., J. Am. Chem. Soc. 2002, 124, 9448–9453; [DOI] [PubMed] [Google Scholar]
- 7d. Yang J., Cauble D. F., Berro A. J., Bauld N. L., Krische M. J., J. Org. Chem. 2004, 69, 7979–7984; [DOI] [PubMed] [Google Scholar]
- 7e. Yang J., Felton G. A. N., Bauld N. L., Krische M. J., J. Am. Chem. Soc. 2004, 126, 1634–1635. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Ischay M. A., Anzovino M. E., Du J., Yoon T. P., J. Am. Chem. Soc. 2008, 130, 12886–12887; [DOI] [PubMed] [Google Scholar]
- 8b. Du J., Yoon T. P., J. Am. Chem. Soc. 2009, 131, 14604–14605; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Ischay M. A., Lu Z., Yoon T. P., J. Am. Chem. Soc. 2010, 132, 8572–8574; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Ischay M. A., Yoon T. P., Eur. J. Org. Chem. 2012, 3359–3372; [Google Scholar]
- 8e. Tyson E. L., Farney E. P., Yoon T. P., Org. Lett. 2012, 14, 1110–1113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8f. Yoon T. P., ACS Catal. 2013, 3, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. RajanBabu T. V., Nugent W. A., J. Am. Chem. Soc. 1994, 116, 986–997; [Google Scholar]
- 9b. Gansäuer A., Bluhm H., Pierobon M., J. Am. Chem. Soc. 1998, 120, 12849–12859. [Google Scholar]
- 10.
- 10a. Gansäuer A., Behlendorf M., von Laufenberg D., Fleckhaus A., Kube C., Sadasivam D. V., R. A. Flowers II , Angew. Chem. Int. Ed. 2012, 51, 4739–4742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4819–4823; [Google Scholar]
- 10b. Gansäuer A., Kube C., Daasbjerg K., Sure R., Grimme S., Fianu G. D., Sadasivam D. V., R. A. Flowers II , J. Am. Chem. Soc. 2014, 136, 1663–1671; [DOI] [PubMed] [Google Scholar]
- 10c. Gansäuer A., von Laufenberg D., Kube C., Dahmen T., Michelmann A., Behlendorf M., Sure R., Seddiqzai M., Grimme S., Sadasivam D. V., Fianu G. D., R. A. Flowers II , Chem. Eur. J. 2015, 21, 280–289; [DOI] [PubMed] [Google Scholar]
- 10d. Gansäuer A., Hildebrandt S., Michelmann A., Dahmen T., von Laufenberg D., Kube C., Fianu G. D., R. A. Flowers II , Angew. Chem. Int. Ed. 2015, 54, 7003–7006; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7109–7112; [Google Scholar]
- 10e. Richrath R. B., Olyschläger T., Hildebrandt S., Enny D. G., Fianu G. D., R. A. Flowers II , Gansäuer A., Chem. Eur. J. 2018, 24, 6371–6379. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Estévez R. E., Oller-López J. E., Robles R., Melgarejo C. R., Gansäuer A., Cuerva J. M., Oltra J. E., Org. Lett. 2006, 8, 5433–5436; [DOI] [PubMed] [Google Scholar]
- 11b. Streuff J., Chem. Eur. J. 2011, 17, 5507–5510; [DOI] [PubMed] [Google Scholar]
- 11c. Bichovski T., Haas T. M., Kratzert D., Streuff J., Chem. Eur. J. 2015, 21, 2339–2342; [DOI] [PubMed] [Google Scholar]
- 11d.for a review, see: Streuff J., Synthesis 2013, 45, 281–307. [Google Scholar]
- 12.The conformations of all structures in the reaction of 1 j to 2 j for Cp2Ti(TFA) in 1,4-dioxane were investigated with the CREST 2.11[13a–c] program using the GFN2-xTB(ALPB[1,4-dioxane])[13d] semiempirical level of theory implemented in the xtb code.[13e,f] Further refinement was done with the CENSO 1.0.0[13g,h] program and the recently developed r2SCAN-3c composite method[13i] based on the r2SCAN meta-GGA density functional[13j] with the def2-mTZVPP basis set. All further geometry optimizations and single-point calculations were performed with the TURBOMOLE 7.4.1 and 7.5.1 program packages.[13k,l] All minimum structures were optimized at the r2SCAN-3c(COSMO[1,4-dioxane])[13i,m] level, and thermostatistical contributions (G mRRHO)[13n,o] at 353.15 K were obtained from subsequent frequency calculations. Single-point energy calculations were performed with PW6B95-D4/def2-QZVP[13p–r], and solvation free energies (δG solv) were calculated with COSMO-RS[13s,t] at 353.15 K using the COSMOtherm program package[15u] with the 2019 parametrization. Reaction-path searches for all steps of the catalytic cycle were performed with GSM[13v–x] at the GFN2-xTB(ALPB[1,4-dioxane]) level of theory. The remaining procedure was conducted for transition states in the same way as mentioned above. Further technical details and molecular structures are given in the Supporting Information.
- 13.
- 13a. Pracht P., Bohle F., Grimme S., Phys. Chem. Chem. Phys. 2020, 22, 7169–7192; [DOI] [PubMed] [Google Scholar]
- 13b. Grimme S., J. Chem. Theory Comput. 2019, 15, 2847–2862; [DOI] [PubMed] [Google Scholar]
- 13c. CREST version 2.11, Universität Bonn, Mulliken Center for Theoretical Chemistry, Bonn, Germany 2021, https://github.com/grimme-lab/crest/releases;
- 13d. Bannwarth C., Ehlert S., Grimme S., J. Chem. Theory Comput. 2019, 15, 1652–1671; [DOI] [PubMed] [Google Scholar]
- 13e. Bannwarth C., Caldeweyher E., Ehlert S., Hansen A., Pracht P., Seibert J., Spicher S., Grimme S., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2021, 11, e1493; [Google Scholar]
- 13f. xtb version 6.3.3, Universität Bonn, Mulliken Center for Theoretical Chemistry, Bonn, Germany 2020, https://github.com/grimme-lab/xtb;
- 13g. CENSO version 1.0.0, Universität Bonn, Mulliken Center for Theoretical Chemistry, Bonn, Germany 2021, https://github.com/grimme-lab/CENSO/releases;
- 13h. Grimme S., Bannwarth C., Dohm S., Hansen A., Pisarek J., Pracht P., Seibert J., Neese F., Angew. Chem. Int. Ed. 2017, 56, 14763–14769; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14958–14964; [Google Scholar]
- 13i. Grimme S., Hansen A., Ehlert S., Mewes J.-M., J. Chem. Phys. 2021, 154, 064103; [DOI] [PubMed] [Google Scholar]
- 13j. Furness J. W., Kaplan A. D., Ning J., Perdew J. P., Sun J., J. Phys. Chem. Lett. 2020, 11, 8208–8215; [DOI] [PubMed] [Google Scholar]
- 13k. Furche F., Ahlrichs R., Hättig C., Klopper W., Sierka M., Weigend F., Wiley Interdiscip. Rev.: Comput. Mol. Sci 2014, 4, 91–100; [Google Scholar]
- 13l. TURBOMOLE 7.4.1/7.5.1, Universität Karlsruhe and Forschungszentrum Karlsruhe GmbH, Karlsruhe, Germany 2019/2020, https://www.turbomole.org;
- 13m. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805; [Google Scholar]
- 13n. Grimme S., Chem. Eur. J. 2012, 18, 9955–9964; [DOI] [PubMed] [Google Scholar]
- 13o. Spicher S., Grimme S., J. Phys. Chem. Lett. 2020, 11, 6606–6611; [DOI] [PubMed] [Google Scholar]
- 13p. Zhao Y., Truhlar D. G., J. Phys. Chem. A 2005, 109, 5656–5667; [DOI] [PubMed] [Google Scholar]
- 13q. Caldeweyher E., Ehlert S., Hansen A., Neugebauer H., Spicher S., Bannwarth C., Grimme S., J. Chem. Phys. 2019, 150, 154122; [DOI] [PubMed] [Google Scholar]
- 13r. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297; [DOI] [PubMed] [Google Scholar]
- 13s. Klamt A., J. Phys. Chem. 1995, 99, 2224–2235; [Google Scholar]
- 13t. Klamt A., Jonas V., Bürger T., Lohrenz J. C. W., J. Phys. Chem. A 1998, 102, 5074–5085; [Google Scholar]
- 13u. Eckert F., Klamt A., AIChE J. 2002, 48, 369–385; [Google Scholar]
- 13v. Zimmerman P. M., J. Chem. Phys. 2013, 138, 184102; [DOI] [PubMed] [Google Scholar]
- 13w. Zimmerman P., J. Chem. Theory Comput. 2013, 9, 3043–3050; [DOI] [PubMed] [Google Scholar]
- 13x. Dohm S., Bursch M., Hansen A., Grimme S., J. Chem. Theory Comput. 2020, 16, 2002–2012. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Gansäuer A., Lauterbach T., Geich-Gimbel D., Chem. Eur. J. 2004, 10, 4983–4990; [DOI] [PubMed] [Google Scholar]
- 14b. Friedrich J., Walczak K., Dolg M., Piestert F., Lauterbach T., Worgull D., Gansäuer A., J. Am. Chem. Soc. 2008, 130, 1788–1796; [DOI] [PubMed] [Google Scholar]
- 14c. Gansäuer A., Worgull D., Knebel K., Huth I., Schnakenburg G., Angew. Chem. Int. Ed. 2009, 48, 8882–8885; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9044–9047; [Google Scholar]
- 14d. Gansäuer A., Knebel K., Kube C., van Gastel M., Cangönül A., Daasbjerg K., Hangele T., Hülsen M., Dolg M., Friedrich J., Chem. Eur. J. 2012, 18, 2591–2599. [DOI] [PubMed] [Google Scholar]
- 15. Gansäuer A., Behlendorf M., Cangönül A., Kube C., Cuerva J. M., Friedrich J., van Gastel M., Angew. Chem. Int. Ed. 2012, 51, 3266–3270; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3320–3324. [Google Scholar]
- 16.
- 16a. Gansäuer A., Pierobon M., Synlett 2000, 1357–1359; [Google Scholar]
- 16b. Gansäuer A., Pierobon M., Bluhm H., Synthesis 2001, 2500–2520; [Google Scholar]
- 16c. Gansäuer A., Otte M., Shi L., J. Am. Chem. Soc. 2011, 133, 417–418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary