

Abstract
The ever‐growing interest in sustainable energy sources leads to a search for an efficient, stable, and inexpensive homogeneous water oxidation catalyst (WOC). Herein, the PO4 3− templated synthesis of three abundant‐metal‐based germanotungstate (GT) clusters Na15[Ge4PCo4(H2O)2W24O94] ⋅ 38H2O (Co4), Na2.5K17.5[Ge3PCo9(OH)5(H2O)4W30O115] ⋅ 45H2O (Co9), Na6K16[Ge4P4Co20(OH)14(H2O)18W36O150] ⋅ 61H2O (Co20) with non‐, quasi‐, or full cubane motifs structurally strongly reminiscent of the naturally occurring {Mn4Ca} oxygen evolving complex (OEC) in photosystem II was achieved. Under the conditions tested, all three GT‐scaffolds were active molecular WOCs, with Co9 and Co20 outperforming the well‐known Na10[Co4(H2O)2(PW9O34)2] {Co4P2W18} by a factor of 2 as shown by a direct comparison of their turnover numbers (TONs). With TONs up to 159.9 and a turnover frequency of 0.608 s−1 Co9 currently represents the fastest Co‐GT‐based WOC, and photoluminescence emission spectroscopy provided insights into its photocatalytic WOC mechanism. Cyclic voltammetry, dynamic light scattering, UV/Vis and IR spectroscopy showed recyclability and integrity of the catalysts under the applied conditions. The experimental results were supported by computational studies, which highlighted that the facilitated oxidation of Co9 was due to the higher energy of its highest occupied molecular orbital electrons as compared to Co4.
Keywords: cobalt, homogeneous catalysis, nanoclusters, photochemistry, sustainable energy
Water oxidation: The stabilization of non‐, quasi‐ and full‐cubane topologies in three CoII germanotungstates (Co‐GT) [Ge4PCo4(H2O)2W24O94]15− (Co4), [Ge3PCo9(OH)5(H2O)4W30O115]20− (Co9), [Ge4P4Co20(OH)14(H2O)18W36O150]22− (Co20) via PO4 3− and GT lacunary ligands is reported. The Co‐GTs are active molecular water oxidation catalysts with Co9 and Co20 outperforming the non‐cubane [Co4(H2O)2(PW9O34)2]10− by a factor of 2, showing the structural and functional similarity of the GT‐encapsulated cubanes to the {Mn4Ca} oxygen evolving complex.
Introduction
The development of an artificial, efficient, stable, and inexpensive homogeneous water oxidation catalyst (WOC), which can mimic the natural photosynthesis process to meet mankind's growing energy demands, is of utmost interest. [1] Over the past decade, 3d‐ and 4d‐doped polyoxotungstates (POTs) have been reported as promising all‐inorganic water oxidation catalysts (POT‐WOCs), which, contrary to their organic counterparts, can withstand the harsh oxidizing conditions of the water oxidation half‐reaction. [2] Starting in 2008, Bonchio and co‐workers [3] and Hill and co‐workers [4] independently reported the ruthenium‐containing POT [{Ru4O4(OH)2(H2O)4}(γ‐SiW10O36)2]10− (Ru4) as the first efficient POT for visible‐light‐driven homogeneous water oxidation catalysis [turnover frequency (TOF)=0.25 s−1]. In Ru4, the WOC active metal core consists of a tetrahedron of four ruthenium centers sandwiched between two dilacunary [γ‐SiW10O36]8−[5] POT units, thereby resembling the core {Mn4Ca} in the oxygen‐evolving complex (OEC) in photosystem II (Figure S6). [6] In 2010, the activity of the cobalt‐containing Weakley dimer [Co4(H2O)2(PW9O34)2]10− as the first non‐precious metal‐based homogeneous POT‐WOC was reported by Hill and co‐workers. [7] Under visible‐light irradiation and in the presence of [Ru(bpy)3]2+ as a photosensitizer and S2O8 2− as an oxidant, WOC activity with turnover numbers (TONs) up to 220 and TOFs up to 5 s−1 was observed. Since then, attention was given to Co‐containing polyoxometalates (POMs) in terms of their stability and activity as WOCs under various reaction conditions with special interest in cobalt‐cubane (Table S2) scaffolds as bio‐inspired cost‐effective WOCs with enhanced photocatalytic activity.[ 8 , 9 ] Significant progress was achieved by Wei et al., who reported the octa‐cobalt‐substituted silicotungstate [Co8(OH)6(H2O)2(CO3)3(A‐α‐SiW9O34)2]16− as the currently fastest Co‐substituted bio‐inspired POT‐WOC (TOF=10 s−1, Table S3). [10] At the same time, Wang and co‐workers investigated the photocatalytic WOC performance of a series of isostructural cubane‐incorporating hexadecanuclear Co‐POTs [{Co4(OH)3PO4}4(XW9O34)4]n− (X=PV, AsV; n=28; X=SiIV, GeIV; n=32) showing that the type of primary heteroatom in the lacunary POM unit efficiently modulates their overall redox properties and WOC activity with GeIV (TOF=0.105 s−1) and AsV (TOF=0.053 s−1) containing representatives exhibiting the highest WOC activity. [11] Taken together the structural and compositional features of the most promising POT‐WOCs recently reported in literature and considering the low number of two cobalt cubane germanotungstates with proven WOC activity reported so far (Table S2), we chose the structurally simple yet versatile PO4 3− group for the synthesis of all‐inorganic cobalt germanotungstate (GT) clusters as OEC models. Herein, we report the PO4 3−‐templated stabilization of a series of all‐inorganic Co‐GT, Na15[Ge4PCo4(H2O)2W24O94] ⋅ 38H2O (Co4), Na2.5K17.5[Ge3PCo9(OH)5(H2O)4W30O115] ⋅ 45H2O (Co9) and Na6K16[Ge4P4Co20(OH)14(H2O)18W36O150] ⋅ 61H2O (Co20) with non‐, quasi‐, or full {CoII 4O4} cubane motifs and their activity in visible‐light‐driven water oxidation. With 20 incorporated CoII metal centers Co20 comprises the highest reported number of incorporated metal centers in a cubane‐encapsulating POT (Table S2). Under visible‐light irradiation and in the presence of [Ru(bpy)3]2+ as a photosensitizer and S2O8 2− as an oxidant, WOC activity with TONs up to 159.9 and TOFs up to 0.608 s−1 for Co9 in borate buffer at pH 8.0 was detected, which to the best of our knowledge represents the currently fastest Co‐GT‐based homogeneous POT‐WOC (Table S3).
Results and Discussion
Synthesis and structure
Co4, Co9 and Co20 are synthesized in a non‐buffered aqueous solution by adjusting the pH of a reaction mixture containing the corresponding tungsten source (GeO2 and Na2WO4 for Co4, K8[γ‐GeW10O36] ⋅ 6H2O [12] for Co9 and K8Na2[A‐α‐GeW9O34] ⋅ 25H2O [13] for Co20) and CoCl2 to pH 7.6 via addition of Na3PO4. Heat activation of the PO4 3− group enabling its coordination to the Co‐oxo cores and subsequent filtration of the cooled reaction mixture results in single crystals of Co4, Co9 and Co20 at 20 °C (CCDC 1876468‐1876470, Figure 1A–F). Single‐crystal X‐ray diffraction (SXRD) studies revealed that Co4 and Co20 crystallize in the monoclinic space groups P21/c and C2/c, whereas Co9 crystallizes in the triclinic space group P (Tables S5–S10). In all three compounds at least one PO4 3− group stabilizes the POT scaffold. For Co4 a PO4 3− group connects two trigonal edge‐shared {W3O13} fragments to the four octahedrally coordinated CoII centers, which are encapsulated by two B‐α‐{GeW9} units and two germanium octahedrons located externally in the structure (Figure 1D, inset). Note, Co4 is the first reported structure with germanium octahedrons in a pure inorganic GT. In Co9, the PO4 3− group connects two α‐{Co2GeW10} Keggin moieties with one exchangeable CoII‐coordinated aqua ligand per α‐{Co2GeW10} unit and one virtual “γ‐{Co3GeW9}” building block (Figure S7) to a single {WO6} octahedron. Two octahedrally coordinated CoIIO5(H2O) centers encapsulated by the PO4 3− and the single {WO6} unit complete the trimeric polyanion by forming a {CoII 3O4(H2O)2} quasi‐cubane (Figure 1E, inset) featuring two exchangeable aqua ligands, thereby resulting in a total number of four CoII positions exhibiting exchangeable aqua ligands, suitable for water molecules to supposedly coordinate and subsequently get oxidized. The architecture of Co20 presents a tetrameric aggregate of four α‐{Co3GeW9} units linking to a central {CoII 4O4} cubane, which is geometrically closely related to the {Mn3CaO4} cubane of the OEC in PSII [6b] (Figure S6) and stabilized by four PO4 3− groups (Figure 1F, inset). Four covalently bound CoII octahedra located in the POT‐encapsulated Co‐oxo core and externally as antenna‐like metal centers [14] complete the Co20 framework. The compounds’ elemental composition and homogeneity was determined by elemental analysis, IR spectroscopy (Figure S2), thermogravimetric analysis (TGA; Figures S3–S5, Table S4), and powder XRD (PXRD; Figures S8–S10). To probe the solution stability of Co4, Co9 and Co20, time‐dependent UV/Vis spectra were recorded at various pH conditions. It should be mentioned that UV/Vis experiments on Co4, Co9 and Co20 in the absorption range for octahedrally coordinated CoII centers could not be performed due to the low solubility of the Co‐GTs and the strong domination of the pπ(Ot)→dπ*(W) ligand‐to‐metal charge‐transfer (LMCT) transitions, which is a problem commonly encountered in POM chemistry. [15] Therefore, the precatalytic POT stability was assessed by investigating the LMCT transitions in the tungsten absorption range. The UV/Vis spectra of Co4, Co9 and Co20 display an absorption maximum at approximately 205 nm with a shoulder at approximately 250 nm corresponding to the Keggin‐type framework in aqueous solution (Figures S17–S19). [16]
Figure 1.
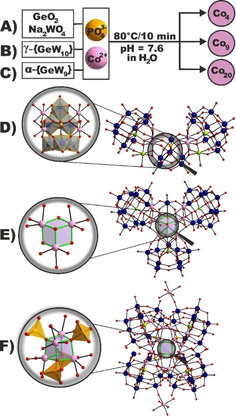
Schematic representation of the building block dependent synthesis of (A) Co4, (B) Co9, and (C) Co20, which are templated by PO4 3− after heat activation. Ball‐and‐stick representation of (D) Co4, (E) Co9 enclosing the quasi‐ {CoII 3O4} cubane, and (F) Co20 incorporating the PO4 3−‐stabilized {CoII 4O4} cubane. Color code: {WO6}, dark blue; CoII, pink; GeIV, lime; PV, light orange; O, red, {W3O13} triads of Co4, grey octahedra.
Acidification of an unbuffered solution of Co20 (pH=0.7) led to stepwise degradation of the POT framework shown by the disappearance of the shoulder at approximately 250 nm (Figure S20), whereas all UV/Vis spectra remain unchanged in 80 mm sodium borate buffer (pH=7.5–9) solutions for at least 2 h mimicking the photocatalytic conditions (Figures S17–S19), which suggests pre‐catalytic stability of Co4, Co9 and Co20 until O2 saturation is reached in the WOC experiments (Figure 2A).
Figure 2.
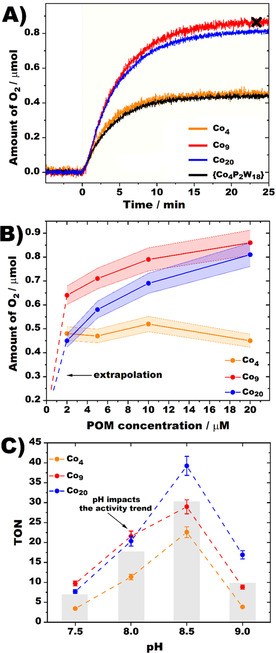
(A) As‐recorded O2 evolution profiles for Co4, Co9 and Co20 compounds along with the reference {Co4P2W18}, measured from 20 μm POM solutions buffered in 80 mm borate buffer at pH 8 and containing Na2S2O8 (5 mm) and [Ru(bpy)3]2+ (1 mm) as an oxidant and a photosensitizer, respectively. (B) Recorded oxygen evolution amounts as a function of POM catalyst concentrations measured for 2, 5, 10 and 20 μm values. A summary of as‐recorded profiles is shown in Figure S23. (C) Amount of generated O2 plotted as TONs as a function of pH (7.5–9). The bars indicate an average O2 amount generated by all three catalysts at a certain pH to demonstrate a cumulative effect of pH on activity. For all conditions, a monochromatic visible LED (λ max=445±13 nm) was chosen as light source to trigger photocatalytic reaction.
Cyclic voltametric measurements were performed on Co4, Co9 and Co20 (vs. Ag/AgCl, in 80 mm sodium borate buffer pH=8) thereby revealing Co2+/Co3+ oxidation waves in the range E≈0.73–0.89 V [17] (Figure S21) occurring at lower potentials than those for other Co‐POM‐WOC systems at pH≥8 (>0.9 V),[ 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 ] whereas the Co(NO3)2 reference solution gives the most intense peaks for Co2+/Co3+ oxidation (at 0.84 V) as well as for water oxidation (at 1.37 V) (Figure S22) showing that the cobalt centers incorporated in the germanotungstates do not behave like free cobalt ions (Table S12) and additionally indicating their pre‐catalytic stability.
Visible light‐driven water oxidation
To probe the photocatalytic WOC activity of Co4, Co9 and Co20, 80 mm aqueous borate buffer solutions (pH=7.5–9) of POTs (concentrations ranging from 2 to 20 μm) containing 1 mm Ru[bpy]3Cl2 (bpy=2,2’‐bipyridine) as the photosensitizer (PS) and 5 mm Na2S2O8 as a sacrificial agent (SA) were used. Note that various parameters such as the shape of the reaction vessel, light intensity, stirring rate as well as the ratio of gaseous head space to total volume render a direct comparison of the WOC performance of POTs tested in different catalytic systems difficult. [19c] Hence, the literature‐known Weakley‐type [Co4(H2O)2(PW9O34)2]10− {Co4P2W18} was used as a benchmark POT‐WOC.[ 7 , 11 , 19 ] Figure 2A shows as‐recorded O2 evolution profiles of 20 μm Co4, Co9, Co20 and {Co4P2W18} solutions at pH 8 and indicates WOC activity for all compounds, with Co9 and Co20 reaching TONs of 21.6 and 20.4, thus outperforming {Co4P2W18} (TON=10.9 under elsewise identical conditions) by a factor of 2 (Table 1). Initial TOF values of 0.047 (Co4), 0.069 (Co9), and 0.068 s−1 (Co20), respectively, are determined from the derivative plots, which are comparable to those of other reported Co‐POM‐WOCs like [{Co4(OH)3(PO4)}4(SiW9O34)4]32− (TOF=0.053 s−1) and [{Co4(OH)3(PO4)}4(GeW9O34)4]32− (TOF=0.105 s−1), both exhibiting structurally similar attributes like an incorporated {CoII 4O4} cubane motif (Table S3). [11] Figure 2B demonstrates the amount of generated O2 as a function of POM concentration and reveals a direct activity correlation with increasing concentration for Co9 and Co20 (red and blue trends), in contrast to Co4 (orange trend), which indicates that factors other than the catalyst concentration, such as the amount of sacrificial agent or photosensitizer, control its WOC performance (Figures S23, S24). At pH=8 Co9 is the most active WOC species with a rather small advantage over Co20, which gets more pronounced at lower catalyst concentrations (Table 1) with Co9 reaching TONs up to 159 and a TOF=0.608 s−1, currently representing the highest TOF for a Co‐GT‐based homogeneous POM‐WOC. The higher WOC activity of Co9 compared to both Co4 and Co20 may have its origin in the enhanced accessibility of the CoII centers, which leads to supposedly facilitated coordination and oxidation of water molecules on the catalytic centers (Figures 1E, S7, Table S11). [10] pH‐dependent WOC studies (pH=7.5–9) revealed higher O2 evolution amounts and TOF values with increasing pH values from 7.5 to 8.5 (Figure 2C), which is related to thermodynamic aspects of water oxidation catalysis according to the Nernst equation, [20] and partial POM deprotonation that results in its enhanced interaction with the photosensitizer. [21] A strong activity drop at pH=9 (indicated with grey bars in Figure 2C) is related to [Ru(bpy)3]2+ degradation. [2a] Overall, the highest WOC activity was determined at pH=8.5, indicating a trade‐off between activity and PS‐stability at these conditions. At pH=8.5 (in contrast to the data obtained at pH=8) Co20 (Table 1) greatly outperforms Co9. According to bond valence sum (BVS) studies, Co20 is the most protonated species in the solid state of the three (H25Co20, H13Co9, H4Co4, Table S11), which in turn means that the effect of its deprotonation at higher pH values is more pronounced leading to a stronger WOC increase. This activity trend implies that the ultimate performance of Co4, Co9 and Co20 is affected by the number of CoII centers, their accessibility, and the extent of POM‐PS pairing, and can be governed by each of these factors depending on the pH.
Table 1.
Summary table showing average TON and initial TOF values for Co4, Co9, Co20 and the Weakley‐type POM {Co4P2W18} as well as the O2 yield generated by the respective POM with varying concentration (2–20 μm) in borate buffer [80 mm], pH=8.0.
|
Co‐POM (μm) |
O2 [μmol] (TON) |
TOF [s−1] |
O2 yield [%] |
|---|---|---|---|
|
Co4 (2) |
0.480 (120.50) |
0.422 |
9.60 |
|
Co9 (2) |
0.640 (159.90) |
0.608 |
12.80 |
|
Co20 (2) |
0.450 (111.40) |
0.405 |
4.50 |
|
Co4 (5) |
0.470 (46.90) |
0.148 |
9.40 |
|
Co9 (5) |
0.710 (71.30) |
0.263 |
14.20 |
|
Co20 (5) |
0.575 (57.50) |
0.189 |
28.40 |
|
Co4 (10) |
0.520 (25.90) |
0.091 |
10.40 |
|
Co9 (10) |
0.790 (39.30) |
0.125 |
15.80 |
|
Co20 (10) |
0.690 (34.40) |
0.105 |
13.80 |
|
Co4 (20) |
0.450 (11.30) |
0.047 |
9.00 |
|
Co9 (20) |
0.860 (21.60) |
0.069 |
17.20 |
|
Co20 (20) |
0.810 (20.40) |
0.068 |
16.20 |
|
{Co4P2W18} (20) |
0.430 (10.90) |
0.015 |
8.60 |
Post‐catalytic stability studies
The stability of molecular WOCs is a topic of current interest due to the possibility of the WOCs to decompose into catalytically active oxide nanoparticles. [22] Note that 183W or 31P NMR measurements on Co4, Co9 and Co20 could not be performed, due to the low solubility and strong paramagnetic nature of the incorporated CoII metal centers. [23] Hence, various reloading experiments and post‐catalytic characterizations were conducted to demonstrate that O2 evolution is indeed triggered by the investigated Co‐GT and to verify their integrity under turnover conditions. [24]
First, blank WOC experiments in pure water and those performed in the absence of PS and SA showed no activity (Figure S25A), indicating the validity of the experimental setup. Only a small activity was recorded in the absence of any Co‐GT (≈20 % of the activity of Co9 at pH=8) (Figure S25A), which suggests a negligible contribution of direct water oxidation by the PS* as a side reaction and is in line with previous reports. [25] Moreover, a reference WOC experiment using the unsubstituted [PW12O40]3− Keggin [26] POT instead of Co4, Co9 or Co20 yielded a similarly low activity (Figure S25B) thereby excluding in‐situ formed tungsten‐based species to be responsible for the observed WOC activity. Second, after O2 level reached saturation (point x in Figure 2A), the reaction solution was re‐loaded with PS and SA. Figure S26 containing the summary WOC data demonstrates that this second illumination cycle triggers additional O2 evolution, which suggests that the observed WOC saturation is not a result of POM deactivation or degradation but can rather be related to the depletion of the other WOC‐solution components such as the [Ru(bpy)3]2+ or the S2O8 2−, and additionally indicates recyclability of the POM‐WOCs. Third, following an established procedure, [24c] a toluene solution of tetra‐n‐heptylammonium nitrate (THpANO3) was used to quantitatively extract Co4, Co9 and Co20 from the respective post‐catalytic solutions (see Figure S27 along with discussion). As this selective procedure does not extract CoOx or Co2+ aq, we analyzed the remaining aqueous phases with X‐ray fluorescence spectroscopy (XRF) to elucidate the potential leaching of Co into the reaction mixture under photocatalytic conditions. The XRF spectra of the solutions look similar and did not show any Co traces (Figure S27, Table S13), which indicates that the Co‐GT underwent neither decomposition (e. g., into Co2+) nor degradation (e. g., into CoOx), processes that have been previously identified to be responsible for WOC performance of other POMs and under different experimental conditions. [22] Fourth, dynamic light scattering (DLS) was performed on 20 μm solutions of Co4, Co9 or Co20, [Ru(bpy)3]2+ (1 mm) and S2O8 2‐ (5 mm) in 80 mm borate buffer (pH=8) after 30 min irradiation. The DLS measurements showed no nanoparticles after photocatalytic water oxidation (Figure S28). In addition, the same experiments were conducted using 20 μm Co(NO3)2 ⋅ 6H2O instead of the corresponding Co‐GT and here nanoparticles with a diameter of approximately 26.4 nm were detected (Figure S28). This is in line with the XRF experiments (Figure S27, Table S13) and confirms that, in contrast to the Co(NO3)2 ⋅ 6H2O system, no metal hydroxide/oxide nanoparticles (especially cobalt hydroxide/oxide nanoparticles) are generated via hydrolytic decomposition of Co4, Co9 and Co20, nor through detachment of the four covalently bound CoII antenna ligands present in Co20 after the photocatalytic experiments. Finally, attenuated total reflectance (ATR)‐IR spectra of Co4, Co9 and Co20 were recorded after the photocatalytic experiments and subsequent precipitation with cesium chloride. These clearly show the characteristic W−O−W bridging and terminal W=O vibrations in the tungsten fingerprint area from 300–1000 cm−1 (Figures S29–S31), which indicates the solution of the polyanions is stable under turnover conditions and represents an established method frequently used for the post‐catalytic study of POMs.[ 23 , 27 ]
Mechanistic studies
Photoluminescence (PL) emission spectroscopy was employed to investigate the photocatalytic WOC mechanism and to understand the electron transfer kinetics between the reaction solution components. Figure S32 reveals that the PL emission of [Ru(bpy)3]2+ is quenched by both Na2S2O8 and Co9 in a linear Stern‐Volmer behavior depending on their concentrations (Figure S33). The calculated rate constant for the oxidative quenching by Na2S2O8 is 45 times lower than that of the reductive quenching by Co9. Considering (a) the much higher Na2S2O8 concentration (5 mm) as compared to Co9 (2–20 μm) present in the WOC reaction solution, and (b) the use of the borate buffer that weakens the formation of an ion pair between polyanionic Co9 and cationic [Ru(bpy)3]2+, [17] oxidative quenching dominates under the photocatalytic conditions. This conclusion is strongly confirmed by time‐resolved PL decay profiles. Figure S34 indicates that in the presence of 10 mm Na2S2O8 and 20 μm of Co9, the original PS* lifetime of approximately 395 ns decreases to approximately 264 and 339 ns, respectively, illustrating that both quenchers speed up the decay kinetics of *Ru(bpy)3 2+ and that S2O8 2− takes up electrons more efficiently. Thus, it can be suggested that during the photocatalytic process, Co9 is oxidized by the oxidized form of the PS (Scheme S1), which is in accordance with previously reported POM‐WOCs.[ 17 , 28 ]
Computational studies
The electronic structures of Co4 and Co9 were analyzed by means of density functional theory (DFT) simulations. [29] All cobalt atoms in Co4 and Co9 were considered in the +2 oxidation state with a formal d7 high‐spin configuration. Both polyanions exhibit a complex electronic structure with 12 (Co4) and 27 (Co9) unpaired electrons, respectively. Therefore, the corresponding models were simplified by replacing all except one of the CoII ions with ZnII resulting in a single catalytic site with a formal spin state characterized by three unpaired electrons. It has been recently shown that such an approximation does not affect the eigenvalues of the frontier molecular orbitals considerably, thereby representing a good trade‐off between the computational costs involved for the study of such systems and a reasonable level of accuracy. [30] The Co active site in the high spin state forms a distorted octahedron with the neighboring O atoms. This local environment is very similar in Co4 and Co9 and the calculated results are in good agreement with the values measured by XRD. In Co4, the calculated values of the Co−O bond lengths present in the octahedron are around 2.10±0.08 Å (range 2.00–2.19 Å), in good agreement with the value of 2.10±0.05 Å (range 2.00–2.20 Å) observed in the crystal structures. In Co9, these bonds have a comparable length of approximately 2.12±0.05 Å (range 2.06–2.20 Å), compared with the experimental value of 2.11±0.06 Å (range 1.96–2.27 Å). Considering the electronic configuration of Co4 and Co9, Figure 3 shows the calculated density of states for the alpha and beta electrons obtained from the calculated molecular orbitals eigenvalues after applying a Gaussian smearing. The highest occupied molecular orbitals (HOMO) of Co9 lie higher in energy than those of the Co4 system, while the lowest unoccupied molecular orbitals (LUMO) have comparable energies. In particular, the calculated eigenvalues for the HOMO electrons in Co4 have values of −6.64 and −6.21 eV for the α and β electrons, respectively, while for Co9 both alpha and beta HOMO electrons have an energy of around −5.71 eV (Figures 3, 4). Regarding the LUMO orbitals, they are almost degenerate for both systems: for Co4 the α and β energies have values of −2.48 and −2.50 eV, respectively, while for Co9 both α and β LUMO electrons have an energy of −2.46 eV. Consequently, the HOMO‐LUMO gap for the Co4 system is 4.16 eV for the α electrons and 3.71 eV for the β ones. For Co9, the HOMO‐LUMO gap is around 3.25 eV for both spin orientations. The observed HOMO‐LUMO gap trends are additionally supported by experimental estimations done by performing diffuse reflectance spectroscopy (Figures S11–S14) and cyclic voltammetry measurements (Figures S15, S16) on Co4 and Co9.
Figure 3.
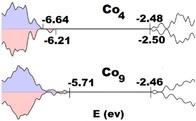
Density of states for the α (blue) and β (red) electrons in the Co4 and Co9. Shaded areas represent occupied states.
Figure 4.
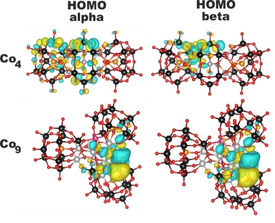
Representation of the α and β HOMO for Co4 and Co9. Color code: WVI, black; CoII, pink; GeIV, orange; PV, light purple; O, red; ZnII, silver.
Our mechanistic photoluminescence findings suggested oxidative quenching to be the dominant WOC pathway, which implies that Co9 undergoes oxidation by [Ru(bpy)3]3+ on the third stage of the catalytic cycle (Scheme S1). According to DFT calculations, the higher energy of the HOMO electrons in Co9 indicates that it is thermodynamically more prone to oxidation as compared to Co4. Considering the higher TOF values obtained for the Co9, these complementary data suggest that POT oxidation may be one of the rate‐limiting processes and that a careful adjustment of the POM/PS redox properties can be used as a tool to optimize the ultimate WOC performance.
Conclusion
The phosphate templated stabilization of non‐ (Co4), {CoII 3O4} (Co9) quasi‐ and {CoII 4O4} (Co20) full cubane structural motifs in germanotungstates as cost‐effective, active, and stable molecular water oxidation catalysts (WOCs) mimicking the naturally occurring oxygen‐evolving complex (OEC) is highlighted. The functional properties of the PO4 3−‐stabilized cubane motifs are shown by the doubled WOC activity of Co9 and Co20 compared to the Weakley‐type (non‐cubane) benchmark {Co4P2W18}. Comprehensive mechanistic and electronic structure studies by photoluminescence and density functional theory suggest an oxidative quenching WOC mechanism and relate the ultimate performance of Co9 to its redox levels. The confirmed stability and recyclability of Co9 and Co20 encourage their immobilization in matrices leading to heterogeneous photocatalytic materials as a subject of future applications.
Experimental Section
Preparation of Na15[Ge4PCo4(H2O)2W24O94] ⋅ 38H2O (Co4)
Na2WO4 ⋅ 2H2O (1.82 g, 5.52 mmol) and GeO2 (0.054 g, 0.52 mmol) were dissolved in 40 mL water and subsequently CoCl2 ⋅ 6H2O (0.073 g, 0.31 mmol) was added. The pH was quickly adjusted from approximately 8.5 to 7.4 by the addition of 1 m HCl (≈5 mL) and then further to approximately 4.8 by 37 % HCl. The pH of the clear red solution was then raised to approximately 7.9 by adding solid Na3PO4 (≈0.28 g, 1.708 mmol) and adjusted to approximately 7.6 with 1 m HCl. The reaction mixture was then heated for approximately 10 min at 80 °C and filtered. The dark red solution was stored in an open beaker at room temperature. After about 2–3 weeks, red rhombohedral‐shaped crystals formed. After recrystallizing in water for several times some X‐ray‐suitable crystals were obtained. Yield: 0.015 g (1 % based on W). Anal. Calcd. [%] for Co4: Na, 4.62; Co, 3.89; P, 0.41; W, 59.09; Ge, 3.81; Found: Na, 4.70; Co, 3.80; P, 0.15; W, 53.1; Ge, 2.15. Selected Fourier‐transform (FT)IR bands: =3340 (br), 1617 (m), 1088 (w), 1038 (w), 934 (m), 865 (s), 820 (m), 759 (s), 650 (s), 583 (s), 509 (s), 437 cm−1 (s).
Preparation of Na2.5K17.5[Ge3PCo9(OH)5(H2O)4W30O115] ⋅ 45H2O (Co9)
K8[γ‐GeW10O36] ⋅ 6H2O [12] (0.5 g, 0,172 mmol) was dissolved in 15 mL H2O and subsequently CoCl2 ⋅ 6H2O (0.119 g, 0.5 mmol) was added. The pH of the clear dark red solution was then adjusted to 7.7 by adding solid Na3PO4 (≈0.07 g, 0.427 mmol). A color change to magenta occurred and slight precipitate formed. The reaction mixture was then heated for approximately 10 min at 80 °C, filtered and stored partially closed in a temperature‐controlled crystallization room (19±1 °C). After about one month, dark red‐violet block‐shaped for X‐ray‐suitable crystals formed in addition to very lightly colored crystalline precipitate. Yield: 0.14 g (25 % based on W). Anal. Calcd. [%] for Co9: Na, 0.71; K, 6.85; Co, 5.46; P, 0.32; W, 56.8; Ge, 2.24; Found: Na, 0.94; K, 6.42; Co, 4.15; P, 0.53; W, 49.5; Ge, 2.00. Selected FTIR bands: =3364 (s), 3217 (s), 1620 (m), 1096 (w), 1033 (w), 1004 (w), 935 (m), 860 (m), 811 (m), 748 (m), 670 (m), 509 (m), 434 cm−1 (m).
Preparation of Na6K16[Ge4P4Co20(OH)14(H2O)18W36O150] ⋅ 61H2O (Co20)
K8Na2[A‐α‐GeW9O34] ⋅ 25H2O [13] (0.5 g, 0.16 mmol) was dissolved in 40 mL H2O and subsequently CoCl2 ⋅ 6H2O (0.119 g, 0.47 mmol) was added. After stirring for approximately 20 min at room temperature, the pH of the clear red solution was then raised to 7.6 by adding solid Na3PO4 (≈0.07 g, 0.427 mmol). The reaction mixture was then heated for approximately 10 min at 80–85 °C, filtered and stored in an open beaker in a temperature‐controlled crystallization room (19±1 °C). After 2–3 days, red needle‐shaped crystals formed. After recrystallizing in water and slow evaporation at +4 °C X‐ray‐suitable crystals were obtained. Yield: 0.08 g (15 % based on W). Anal. Calcd. [%] for Co20: Na, 0.51; K, 3.79; Co, 10.11; P, 0.92; W, 49.37; Ge, 2.17; Found: Na, 1.02; K, 4.64; Co, 7.09; P, 0.88; W, 45.1; Ge, 2.06. Selected FTIR bands: =3340 (br), 1614 (w), 1088 (w), 927 (m), 863 (m), 783 (s), 650 (s), 584 (s), 518 (s), 452 (s), 438 cm−1 (s).
Preparation and characterization of Na10[Co4(H2O)2(α‐PW9O34)2] ⋅ 27H2O ({Co4P2W18})
{Co4P2W18} was prepared according to the literature procedure reported by Hill and co‐workers. [7] The identity of {Co4P2W18} was proven by single‐crystal XRD (Table S1) and electrospray ionization mass spectrometry (ESI‐MS) (Figure S1).
Visible‐light‐driven water oxidation
A homogeneous solution of 80 mm aqueous borate buffer (pH=7.5, 8, 8.5 or 9) containing 1.0 mm of [Ru(bpy)3]Cl2, 5.0 mm of Na2S2O8 and POM catalyst with concentrations varying from 2–20 μm was prepared in a two‐necked closed glass reactor equipped with an outer water‐cooling jacket. This solution was then deaerated using a flow of argon and later irradiated with a monochromatic visible LED (λ max=445±13 nm) to trigger a photocatalytic reaction. The oxygen evolution was followed in situ using an optical oxygen meter (FireStingO2, Pyroscience, Germany) and a needle‐like oxygen‐sensitive optical sensor (OXF900PT‐OI) with a working principle based on the quenching of the REDFLASH indicator (immobilized on the sensor tip) luminescence caused by a collision between oxygen molecules and the indicator. In a single experiment, the oxygen sensor was inserted through a Viton septum placed in a screw cap on one of the necks of the reactor. The O2 concentration was measured directly in %O2 and was later converted to μmol and TONs based on the control experiments and the ideal gas equation. Initial TOFs were calculated as the maximum derivative (obtained from the “μmol of O2 vs. time” plots) divided by the number of moles of the catalyst.
Computational details
All DFT calculations were performed employing the computer code NWChen. [31] The exchange‐correlation functional was approximated by employing the Becke‐3‐parameter‐Lee–Yang–Parr functional (B3LYP). [32] The core electrons of the Co, P, Ge, W, and Zn atoms were described by the LANL2DZ effective core potential [33] and the corresponding basis set was used for the valence electrons. Electrons of the H atoms were described by the 6–31G basis set and the 3–21G basis set was used for the O atoms. [34] Employing the larger 6–31G basis set for the O atoms affects the HOMO‐LUMO gap of Co4 by around the 0.2 %, while the eigenvalues corresponding to the HOMO molecular orbitals change by slightly less than the 5 %. We therefore performed all calculations employing the smaller basis set. In both Co4 and Co9, all Co atoms except one were substituted by Zn atoms. This procedure was performed to simplify the complex magnetic structure endowed by the presence of multiple Co2+ cations. Haider et al. have shown that for similar Co‐containing polyanions, such substitution affects the eigenvalues of the frontier molecular orbitals only marginally. [30] The remaining Co atom in the polyanion was considered to be in the high‐spin quadruplet configuration. All structures were optimized in water, described with the conductor‐like screening (COSMO) continuum solvation model. [35] The permittivity of water was set to 78.36 and the radii of the atomic‐centered spheres used to construct the molecule‐shaped cavity were set to the corresponding atomic Van der Waals radii. The structures of all compounds were relaxed within the solvent model to a minimum of the potential energy surface employing a quasi‐Newton optimization method.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was funded by the Austrian Science Fund (FWF): P33089 (to A.R.) and P32801 (to A.C.) and the University of Vienna. E.T. and A.R. acknowledge the University of Vienna for awarding a Uni:docs fellowship to E.T. We are grateful to Ao.Univ.‐Prof. Christian L. Lengauer for support with TGA measurements at the Institut für Mineralogie und Kristallographie, University of Vienna and Dr. Marek Bujdos for ICP‐MS and Flame AAS measurements, Institute of Geology, Comenius University in Bratislava. We would like to acknowledge Matthias Preidl and Pablo Ayala for conducting preliminary WOC studies and XRF measurements. Lastly, we thank Dipl.‐Ing. Alexander Prado‐Roller for valuable discussions concerning X‐ray data refinements and help with PXRD measurements as well as Anna Fabisikova, MSc and Nadiia Gumerova Ph.D. for ESI‐MS data interpretation and Niusha Lasemi Ph.D. for help with DLS measurements.
E. Al-Sayed, S. P. Nandan, E. Tanuhadi, G. Giester, M. Arrigoni, G. K. H. Madsen, A. Cherevan, D. Eder, A. Rompel, ChemSusChem 2021, 14, 2529.
Contributor Information
Emir Al‐Sayed, http://www.bpc.univie.ac.at.
Sreejith P. Nandan, https://www.imc.tuwien.ac.at/division_molecular_materials_chemistry/.
Prof. Dr. Dominik Eder, Email: dominik.eder@tuwien.ac.at.
Prof. Dr. Annette Rompel, Email: annette.rompel@univie.ac.at.
References
- 1.
- 1a. Balzani V., Credi A., Venturi M., ChemSusChem 2008, 1, 26–58; [DOI] [PubMed] [Google Scholar]
- 1b. Ye S., Ding C., Liu M., Wang A., Huang Q., Li C., Adv. Mater. 2019, 31, 1902069. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Kuznetsov A. E., Geletii Y. V., Hill C. L., Morokuma K., Musaev D. G., J. Am. Chem. Soc. 2009, 131, 6844–6854; [DOI] [PubMed] [Google Scholar]
- 2b. Quiñonero D., Kaledin A. L., Kuznetsov A. E., Geletii Y. V., Besson C., Hill C. L., Musaev D. G., J. Phys. Chem. A 2010, 114, 535–542; [DOI] [PubMed] [Google Scholar]
- 2c. Sartorel A., Miró P., Salvadori E., Romain S., Carraro M., Scorrano G., Valentin M. D., Llobet A., Bo C., Bonchio M., J. Am. Chem. Soc. 2009, 131, 16051–16053; [DOI] [PubMed] [Google Scholar]
- 2d. Chen R., Yan Z. H., Kong X. J., ChemPhotoChem 2020, 4, 157–167; [Google Scholar]
- 2e. Han Q., Ding Y., Dalton Trans. 2018, 47, 8180–8188; [DOI] [PubMed] [Google Scholar]
- 2f. Li N., Liu J., Dong B.-X., Lan Y.-Q., Angew. Chem. Int. Ed. 2020, 59, 20779–20793; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 20963–20977. [Google Scholar]
- 3. Sartorel A., Carraro M., Scorrano G., Zorzi R. D., Geremia S., McDaniel N. D., Bernhard S., Bonchio M., J. Am. Chem. Soc. 2008, 130, 5006–5007. [DOI] [PubMed] [Google Scholar]
- 4. Geletii Y. V., Botar B., Kögerler P., Hillesheim D. A., Musaev D. G., Hill C. L., Angew. Chem. Int. Ed. 2008, 47, 3896–3899. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3960–3963. [Google Scholar]
- 5. Canny J., Tezé A., Thouvenot R., Hervé G., Inorg. Chem. 1986, 25, 2114–2119. [Google Scholar]
- 6.
- 6a. Cinco R. M., Robblee J. H., Rompel A., Fernandez C., Yachandra V. K., Sauer K., Klein M. P., J. Phys. Chem. B 1998, 102, 8248–8256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Yano J., Yachandra V. K., Chem. Rev. 2014, 114, 4175–4205; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Mukherjee S., Stull J. A., Yano J., Stamatatos T. C., Pringouri K., Stich T. A., Abboud K. A., Britt R. D., Yachandra V. K., Christou G., Proc. Natl. Acad. Sci. USA 2012, 109, 2257–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin Q., Tan J. M., Besson C., Geletii Y. V., Musaev D. G., Kuznetsov A. E., Luo Z., Hardcastle K. I., Hill C. L., Science 2010, 328, 342–345. [DOI] [PubMed] [Google Scholar]
- 8. Liu Z.-J., Wang X.-L., Qin C., Zhang Z.-M., Li Y.-G., Chen W.-L., Wang E.-B., Coord. Chem. Rev. 2016, 313, 94–110. [Google Scholar]
- 9. Fukuzumi S., Lee Y.-M., Nam W., Dalton Trans. 2019, 48, 779–798. [DOI] [PubMed] [Google Scholar]
- 10. Wei J., Feng Y., Zhou P., Liu Y., Xu J., Xiang R., Ding Y., Zhao C., Fan L., Hu C., ChemSusChem 2015, 8, 2630–2634. [DOI] [PubMed] [Google Scholar]
- 11. Han X.-B., Zhang Z.-M., Zhang T., Li Y.-G., Lin W., You W., Su Z.-M., Wang E.-B., J. Am. Chem. Soc. 2014, 136, 5359–5366. [DOI] [PubMed] [Google Scholar]
- 12. Nsouli N. H., Bassil B. S., Dickman M. H., Kortz U., Keita B., Nadjo L., Inorg. Chem. 2006, 45, 3858–3860. [DOI] [PubMed] [Google Scholar]
- 13. Bi L.-H., Kortz U., Nellutla S., Stowe A. C., van Tol J., Dalal N. S., Keita B., Nadjo L., Inorg. Chem. 2005, 44, 896–903. [DOI] [PubMed] [Google Scholar]
- 14. Bassil B. S., Nellutla S., Kortz U., Stowe A. C., van Tol J., Dalal N. S., Keita B., Nadjo L., Inorg. Chem. 2005, 44, 2659–2665. [DOI] [PubMed] [Google Scholar]
- 15. Pope M. T., Heteropoly and Isopoly Oxometalates, Vol. 2, Springer Verlag, Berlin, 1983, pp. 10–26. [Google Scholar]
- 16. Bi L., Li B., Wu L., Bao Y., Inorg. Chim. Acta 2009, 362, 3309–3313. [Google Scholar]
- 17. Natali M., Bazzan I., Goberna-Ferrón S., Al-Oweini R., Ibrahim M., Bassil B. S., Dau H., Scandola F., Galán-Mascarós J. R., Kortz U., Green Chem. 2017, 19, 2416–2426. [Google Scholar]
- 18.
- 18a. Chen W.-C., Wang X.-L., Qin C., Shao K.-Z., Su Z.-M., Wang E.-B., Chem. Commun. 2016, 52, 9514–9517; [DOI] [PubMed] [Google Scholar]
- 18b. Tanaka S., Annaka M., Sakai K., Chem. Commun. 2012, 48, 1653–1655. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Huang Z., Luo Z., Geletii Y. V., Vickers J. W., Yin Q., Wu D., Hou Y., Ding Y., Song J., Musaev D. G., J. Am. Chem. Soc. 2011, 133, 2068–2071; [DOI] [PubMed] [Google Scholar]
- 19b. Berardi S., La Ganga G., Natali M., Bazzan I., Puntoriero F., Sartorel A., Scandola F., Campagna S., Bonchio M., J. Am. Chem. Soc. 2012, 134, 11104–11107; [DOI] [PubMed] [Google Scholar]
- 19c. Cao S., Piao L., Angew. Chem. Int. Ed. 2020, 59, 18312–18320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18468–18476. [Google Scholar]
- 20. Lv H., Geletii Y. V., Zhao C., Vickers J. W., Zhu G., Luo Z., Song J., Lian T., Musaev D. G., Hill C. L., Chem. Soc. Rev. 2012, 41, 7572–7589. [DOI] [PubMed] [Google Scholar]
- 21. Soriano-López J., Song F., Patzke G. R., Galán-Mascarós J. R., Front. Chem. 2018, 6, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Folkman S. J., Finke R. G., ACS Catal. 2017, 7, 7–16; [Google Scholar]
- 22b. Folkman S. J., Soriano-Lopez J., Galán-Mascarós J. R., Finke R. G., J. Am. Chem. Soc. 2018, 140, 12040–12055. [DOI] [PubMed] [Google Scholar]
- 23. Kandasamy B., Vanhaecht S., Nkala M. F., Beelen T., Bassil B. S., Parac-Vogt T. N., Kortz U., Inorg. Chem. 2016, 55, 9204–9211. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Stracke J. J., Finke R. G., J. Am. Chem. Soc. 2011, 133, 14872–14875; [DOI] [PubMed] [Google Scholar]
- 24b. Stracke J. J., Finke R. G., ACS Catal. 2013, 3, 1209–1219; [Google Scholar]
- 24c. Vickers J. W., Lv H. J., Sumliner J. M., Zhu G. B., Luo Z., Musaev D. G., Geletii Y. V., Hill C. L., J. Am. Chem. Soc. 2013, 135, 14110–14118. [DOI] [PubMed] [Google Scholar]
- 25. Hara M., Waraksa C. C., Lean J. T., Lewis B. A., Mallouk T. E., J. Phys. Chem. A 2000, 104, 5275–5280. [Google Scholar]
- 26. Phillips M., J. Soc. Chem. Ind. 1950, 69, 282–284. [Google Scholar]
- 27. Paille G., Boulmier A., Bensaid A., Ha-Thi M.-H., Tran T.-T., Pino T., Marrot J., Rivière E., Hendon C. H., Oms O., Gomez-Mingot M., Fontecave M., Mellot-Draznieks C., Dolbecq A., Mialane P., Chem. Commun. 2019, 55, 4166–4169. [DOI] [PubMed] [Google Scholar]
- 28. Natali M., Orlandi M., Berardi S., Campagna S., Bonchio M., Sartorel A., Scandola F., Inorg. Chem. 2012, 51, 7324–7331. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. López X., Carbo J. J., Bo C., Poblet J. M., Chem. Soc. Rev. 2012, 41, 7537–7571; [DOI] [PubMed] [Google Scholar]
- 29b. López X., Fernández J. A., Poblet J. M., Dalton Trans. 2006, 1162–1167; [DOI] [PubMed] [Google Scholar]
- 29c. Solé-Daura A., Rodríguez-Fortea A., Poblet J. M., Robinson D., Hirst J. D., Carbó J. J., ACS Catal. 2020, 10, 13455–13467. [Google Scholar]
- 30. Haider A., Bassil B. S., Soriano-López J., Qasim H. M., Sáenz de Pipaón C., Ibrahim M., Dutta D., Koo Y.-S., Carbó J. J., Poblet J. M., Galán-Mascarós J. R., Kortz U., Inorg. Chem. 2019, 58, 11308–11316. [DOI] [PubMed] [Google Scholar]
- 31. Aprà E., Bylaska E. J., de Jong W. A., Govind N., Kowalski K., Straatsma T. P., Valiev M., van Dam H. J. J., Alexeev Y., Anchell J., Anisimov V., Aquino F. W., Atta-Fynn R., Autschbach J., Bauman N. P., et al., J. Chem. Phys. 2020, 152, 184102. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Lee C., Yang C., Parr R. G., Phys. Rev. B 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 32b. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 33. Hay P. J., Wadt W. R., J. Chem. Phys. 1985, 82, 270–283. [Google Scholar]
- 34. Ditchfield R., Hehre W., People J. A., J. Chem. Phys. 1971, 54, 724–728. [Google Scholar]
- 35.
- 35a. Klamt A, Schüürmann G., J. Chem. Soc. 1993, 2, 799–805; [Google Scholar]
- 35b. York D. M., Karplus M., J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary