

Abstract
A synthetic pathway for the synthesis of novel anionic sandwich complexes with a cyclo‐E3 (E=P, As) ligand as an end deck was developed giving [Cp′′′Co(η3‐E3)]− (Cp′′′=1,2,4‐tri‐tert‐butylcyclopentadienyl, E=P ([5]), As ([6])) in good yields suitable for further reactivity studies. In the reaction with the chlorophosphanes R2PCl (R=Ph, Cy, tBu), neutral complexes with a disubstituted cyclo‐E3P (E=P, As) ligand in [Cp′′′Co(η3‐E3PR2)] (E=P (7 a–c), As (9 a–c)) were obtained. These compounds can be partially or completely converted into complexes with a cyclo‐E3 (E=P, As) ligand with an exocyclic {PR2} unit in [Cp′′′Co(η2:η1‐E3PR2)] (E=P (8 a–c), As (10 a–c)). Additionally, the insertion of the chlorosilylene [LSiCl] (L=(tBuN)2CPh) into the cyclo‐E3 ligand of [5] and [6] was achieved and the novel heteroatomic complexes [Cp′′′Co(η3‐E3SiL)] (E=P (11), As (12)) could be isolated. The reaction pathway was elucidated by DFT calculations.
Keywords: electrophile, heterocycle, polypnictogen, rearrangement, ring expansion
The transfer of an intact cyclo‐E3 ligand (E=P, As) from the nickel complex [Cp′′′Ni(η3‐E3)] to the anionic cobalt complex [Cp′′′Co(η3‐E3)]− was achieved, which was afterwards quenched with main group electrophiles such as chlorophosphanes (R2PCl). That way, the E3 cycle can be expanded to a substituted four‐membered ring, which undergoes further rearrangement processes by ring contractions to a cyclo‐E3 ligand with an exocyclic {PR2} unit.
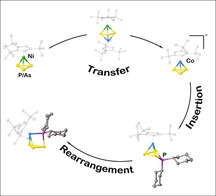
Introduction
During the last decades, the conversion of white phosphorus and yellow arsenic in the coordination sphere of transition metal complexes has been widely investigated yielding a plethora of polypnictogen (En) complexes. [1] While neutral complexes with cyclic En ligands are well known, anionic derivatives are rather limited for E=P, but completely unknown for E=As. Two principal routes for the synthesis of the P derivatives have been reported. One is the reduction of a suitable halogen‐containing precursor in the presence of white phosphorus. Cummins et al. reported the first derivative [Na(thf)3][(DippO)3Nb(η3‐P3)] (A, Scheme 1) obtained that way in 2009. [2] [K(18‐c‐6)][CpArFe(η4‐P4)] (B, Scheme 1) reported by Wolf et al. was also obtained by the reduction of the related bromo dimer [(CpArFe)2(μ,η1:η1‐Br)2] in the presence of P4. [3] A similar pathway is the reaction of an isolated anionic precursor with P4, for example, in the reaction of the cobaltate [K(18‐c‐6)(thf)1.5][(PHDI)Co(η4‐1,5‐cod)] with P4 yielding [K(18‐c‐6)][(PHDI)Co(η4‐P4)] (C, Scheme 1). [4] Another route is the abstraction of P atoms from neutral precursors, for example, by the reaction of [(PHDI)Co(η4‐P5R2)] (D) with cyanides under formation of [K(18‐c‐6)][(PHDI)Co(CN)(η3‐P3)] (E, Scheme 1). [4] A drawback of the mentioned procedures is the partially low yield synthesis and that they can hardly be scaled up in most of the cases. In contrast, we recently showed that one P atom of [M(η4‐P4)] (M=Cp′′′Co (F), Cp′′Ta(CO)2 (G); Cp′′′=1,2,4‐tri‐tert‐butylcyclopentadienyl) can be abstracted by MeNHC to yield [(MeNHC)2P][Cp′′′Co(η3‐P3)] (H, Scheme 1) and [(MeNHC)2P][Cp′′Ta(CO)2(η3‐P3)] (I, Scheme 1) in very good yields.[ 5 , 6 ] However, the laborious synthesis of the starting materials F and G represents a slight drawback in this approach. The anion [Cp′′′Co(η3‐P3)]− was also observed by the reduction of F with alkali metals in the presence of crown ether or as a side product in the functionalization of F with main group nucleophiles. [7] Figueroa et al. could show that a cyclo‐P4 ligand in molybdenum complexes, for example, in [(CNArDipp2)MoI2(CO)(η4‐P4)] (J), stays intact upon reduction under formation of the dianionic complex [K2(db‐18‐c‐6)][(CNArDipp2)Mo(CO)(η4‐P4)] (K, Scheme 1). [8] The subsequent chemistry of these anionic complexes is only barely investigated. Compound C can be quenched with chlorophosphanes to form neutral complexes with a disubstituted five‐membered P5R2 ligand. [4] Compound A can be reacted with electrophiles, for example, AsCl3, to give the landmark access to AsP3 [2] or with the neutral phosphinidene Cp*P{W(CO)5}2 to form the anionic complex [Na(18‐c‐6)(thf)2][(DippO)3Nb(μ3,η3:η1:η1‐P4Cp*){W(CO)5}2]. [9] The first reaction can be considered as a transfer of a cyclo‐P3 unit to an As atom after reduction of AsCl3. There is only one other example reported where a complete homoatomic cyclo‐En ligand is transferred, namely a cyclo‐E5 ligand from [Cp*Fe(η5‐E5)] (E=P, As) to Ru and Os in the reaction with [Cp*M(CO)2]2 (M=Ru, Os) yielding [Cp*M(η5‐E5)] (M=Ru, Os; E=P, As). [10] Moreover, Di Vaira et al. were able to transfer cyclo‐E2S (E=P, As) units from cobalt to rhodium centers. [11] A subsequent investigation of the reactivity of the cyclo‐P3 complexes E, H and I has not been reported yet due to their limited accessibility. The anion H containing a cyclo‐P3 ligand might be an interesting starting material for quenching reactions with main group electrophiles. Although the yield of 79 % is quite high, the laborious and resource‐consuming synthesis of F is a limiting factor. [5] Additionally, the salt elimination in reactions with, for example, chlorophosphanes, is less favored if the [(MeNHC)2P]+ cation is present compared to alkali metal ions. Therefore, the question arises as to whether it is possible to synthesize [Cp′′′Co(η3‐P3)]− in a more efficient way and with an alkali metal as counterion. Moreover, the access to compounds of the heavier homologue arsenic to receive first anionic cyclo‐As3 ligand complexes is still a challenge. Thus, we took pains to synthesize the As analogue complex of A via the route described by Cummins et al. for the P derivative, [2] however, without success. In search of different approaches, the idea came up to combine the features of transfer reactions and reductive reaction conditions. Hence, [Cp′′′Ni(η3‐E3)] (E=P (1), As(2)) seems to be a suitable starting material including a cyclo‐E3 ligand which is accessible in gram scale.[ 12 , 13 ] [Cp′′′Ni(η3‐E3)] (E=P, As) can be reacted with the cobalt toluene complex [(Cp′′′Co)2(μ,η4:η4‐C7H8)] to yield the heterobimetallic triple‐decker complexes [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (3), As (4)) in gram scale.[ 12 , 14 ] The investigation of their redox chemistry reveals an interesting fact, namely that in the monocations [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)][FAl] (E=P, As; [FAl]=FAl{OC6F10(C6F5)}3) the {Cp′′′M} fragments are not strongly bound to the E3 ligand and a rearrangement process takes place under formation of the homometallic species [(Cp′′′Co)2(μ,η3:η3‐E3)][FAl] and [(Cp′′′Ni)2(μ,η3:η3‐E3)][FAl]. [12] On the other hand, the reduction of 3 and 4 yields the monoanionic triple‐decker complexes [K(thf)n][(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P, As), which retain their heterobimetallic character. Herein we report the reaction of 3 and 4 with an excess of potassium leading to [Cp′′′Co(η3‐E3)]− (E=P ([5]), As ([6])) in high yields, the latter being the first anionic cyclo‐As3 ligand complex, and their reactivity towards chlorophosphanes and chlorosilylene to obtain first cyclo‐E4 heteroelement ligands with P/As/Si sequences.
Scheme 1.
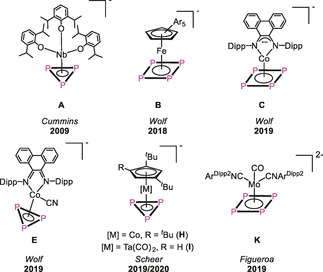
Selected examples of anionic complexes bearing cyclo‐Pn ligand as an end deck.
Results and Discussion
The reduction of 3 and 4 with an excess of elemental potassium (Scheme 2) leads to [K(thf)n][Cp′′′Co(η3‐E3)] (E=P, n=0.7 (5); E=As, n=0.8 (6)), KCp′′′ and a black precipitate. [15] After workup, a 1:0.8 mixture of KCp′′′ and [5] and a 1:0.7 mixture of KCp′′′ and [6] can be obtained as a red and a green solid in yields of 69 and 63 %, respectively. The content of thf was determined by 1H NMR spectroscopy. Addition of 18‐c‐6 and recrystallization from a concentrated thf solution of [5]/[6] layered with n‐hexane at room temperature yields large colorless single crystals of [K(18‐c‐6)]Cp′′′ (cf. SI) and red and green crystals of [K(18‐c‐6)][Cp′′′Co(η3‐E3)] (E=P (5), As (6)), respectively, which can be separated under the microscope giving analytically pure [K(18‐c‐6)][5] and [K(18‐c‐6)][6] in isolated yields of 23 and 15 %, respectively. Since the anion [Cp′′′Co(η3‐P3)]− was already reported with [(MeNHC)2P]+ as a counterion, [5] only the structure of the novel arsenic derivative [6] will be discussed in the following. The structure in the solid state (Figure 1) reveals a sandwich complex with a cyclo‐As3 ligand as an end deck. The As−As distances are between 2.3876(4) and 2.3969(4) Å and in the range between single and double bonds,[ 16 , 17 ] which is in agreement with the calculated Wiberg Bond Indices (WBIs) of 1.07 for all As−As bonds. The novel anion [6] is isoelectronic to the neutral nickel complex [CpRNi(η3‐As3)] (CpR=C5 iPr4H, [18] Cp′′′ [12] ) and represents the first anionic complex with a cyclo‐As3 ligand as end deck. As already mentioned, a few subsequent reactions of anionic complexes with a cyclo‐E3 (E=P, As) ligand were only investigated for the phosphorus niobium complex A reported by Cummins.[ 2 , 9 , 19 ]
Scheme 2.
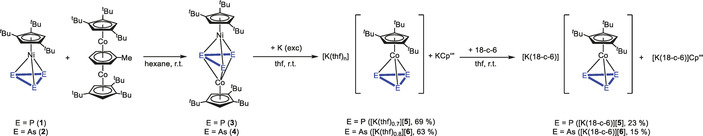
Reaction of 1/2 with [(Cp′′′Co)2(μ,η4:η4‐C7H8)] to the heterobimetallic triple‐decker complexes 3/4; reduction to [K(thf)0.7][5]/[K(thf)0.8][6] and complexation with 18‐c‐6 (Cp′′′=1,2,4‐tri‐tert‐butylcyclopentadienyl).
Figure 1.
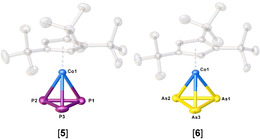
Molecular structure of the anions [5] and [6] in the solid state. [31] Counterions, solvent molecules and hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn with 50 % probability level.
Due to the much higher yields of [K(thf)0.7][5] and [K(thf)0.8][6] and the fact that KCp′′′ does not disturb much the reaction with chlorine‐containing electrophiles, mixtures of [K(thf)0.7][5] and [K(thf)0.8][6] with KCp′′′, respectively, were used for the subsequent reactivity investigations.
Firstly, the reactivity of [K(thf)0.7][5] towards the chlorophosphanes R2PCl (R=Ph, Cy, tBu) was studied. The reaction of [K(thf)0.7][5] with an excess of R2PCl in thf at −80 °C leads to an immediate color change from dark red to brown in all cases. After warming to room temperature, the solvent was removed in vacuo and the residue extracted with n‐pentane. After filtration from a large amount of white solid (KCp′′′+KCl), the solvent was removed in vacuo and NMR spectra in [D8]toluene were recorded. The 31P{1H} NMR spectra each reveal the complete conversion of [5] and the formation of two types of compounds [Cp′′′Co(η3‐P4R2)] (R=Ph (7 a), Cy (7 b), tBu (7 c)) and [Cp′′′Co(η2:η1‐P4R2)] (R=Ph (8 a), Cy (8 b), tBu (8 c)) in ratios of 1:0.2 (7 a:8 a), 1:0.01 (7 b:8 b) and 1:0.01 (7 c:8 c), respectively (Scheme 3). For R=Ph, instead of unused Ph2PCl, small amounts of Ph3P, Ph4P2 and (PhP)5 can be detected, while for R=Cy, tBu unused R2PCl can be detected in the 31P{1H} NMR spectra. For R=tBu, Cy, a column‐chromatographic workup was conducted. For R=tBu, clean 7 c was the only obtained fraction, while for R=Cy, a first weak red fraction of 8 b and a second brown fraction of 7 b could be separated. Finally, single crystals of 7 b, 7 c and 8 a (details cf. SI) could be obtained.
Scheme 3.
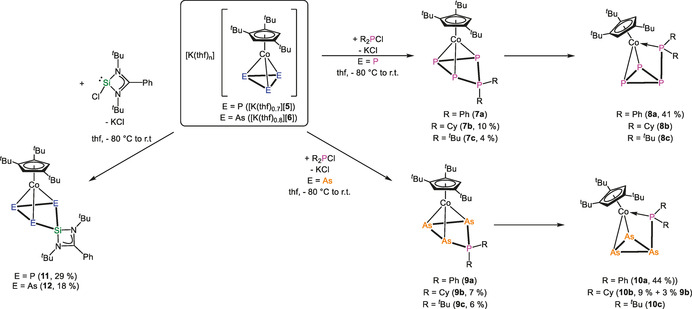
Reaction of [K(thf)0.7][5] and [K(thf)0.8][6] with R2PCl (R=Ph, Cy, tBu) and [(tBuN)2CPhSiCl]. The yields correspond to the isolated crystalline material.
The molecular structure of 7 b/7 c (Figure 2) reveals a neutral complex with a disubstituted cyclo‐P4R2 ligand, while P1 is bent out of the plane (P2‐P3‐P4) by 29° (7 b) and 33° (7 c) and three P atoms (P2‐P4) remain coordinated to Co1 (Figure 2). There are two shorter P−P distances (P1−P2 2.1583(5) Å (7 b), 2.1810(8) Å (7 c) and P1−P4 2.1544(5) Å (7 b), 2.1756(8) Å (7 c)) and two slightly longer P−P distances within the P3 unit coordinated to cobalt (P2−P3 2.2021(6) Å (7 b), 2.2023(8) Å (7 c) and P3−P4 2.2029(6) Å (7 b), 2.2017(8) Å (7 c). All P−P bonds are in the range of single bonds, [17] underlined by WBIs in the range of 0.99 and 1.04. The molecular structure of 8 a in the solid state reveals a cyclic P3 ligand with an exocyclic {PPh2} unit which is η1 coordinated to the Co atom together with one side of the cyclo‐P3 unit in η2 fashion, respectively. The phosphorus ligand shows two shorter P−P distances (P1−P2 2.1774(15) Å, P3−P4 2.1316(17) Å) and two longer ones (P2−P3 2.2309(15) Å, P2−P4 2.2372(15) Å). The P3−P4 distance lies between a single and a double bond, indicated by a WBI of 1.14, while all other P−P distances are in the range of single bonds (WBIs of 0.95–0.96).
Figure 2.
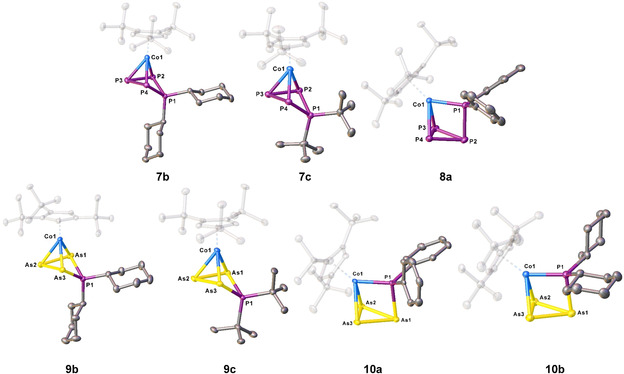
Molecular structure of 7 b, 7 c, 8 a, 9 b, 9 c, 10 a and 10 b in the solid state. [31] Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn with 50 % probability level.
Moreover, the reactivity of the arsenic derivative [6] towards chlorophosphanes was investigated. The reaction of [K(thf)0.8][6] with R2PCl (Ph, Cy, tBu) in thf at −80 °C leads to an immediate color change from dark green to brown. After workup, NMR spectra in [D8]toluene of these reactions were recorded. The reaction with Ph2PCl is again less selective. The 31P{1H} NMR spectrum shows the formation of [Cp′′′Co(η3‐As3PPh2)] (9 a), [Cp′′′Co(η2:η1‐As3PPh2)] (10 a) in a ratio of 1:3.5 beside Ph3P, Ph4P2 and (PhP)5, while the 1H NMR spectrum additionally shows signals for [(Cp′′′Co)2(μ,η2:η2‐As2)2]. For R=Cy, only the two products [Cp′′′Co(η3‐As3PCy2)] (9 b) and [Cp′′′Co(η2:η1‐As3PCy2)] (10 b) are detected in the 31P{1H} NMR spectrum in a ratio of 8.5:1. For R=tBu [Cp′′′Co(η3‐As3PtBu2)], (9 c) can be detected as the major product beside traces (1:0.01) of [Cp′′′Co(η2:η1‐As3PtBu2)] (10 c).
The reaction mixtures were purified by column chromatography yielding analytically clean 9 b, 9 c and 10 a, with 10 b, however, being contaminated with 9 b. During this workup, no fraction of 9 a and 10 c, respectively, could be obtained, probably due to a complete conversion of 9 a to 10 a caused by the reaction time and by chromatographic workup. Moreover, 10 c was only present in very small amounts in the reaction mixture.
After crystallization, 9 b could be isolated in 7 %, 9 c in 6 %, 10 a in 44 % and 10 b in 12 % (mixture of 9 b:10 b 1:3, cf. Scheme 3) yields. The molecular structures of 9 b and 9 c in the solid state (Figure 2) each reveal unprecedented four‐membered cycles consisting of three As atoms and one P atom, with all As atoms being coordinated to Co1. The PR2 unit deviates from the plane (As1‐A2‐As3) by 33° (9 b) and 32° (9 c). The P−As bonds (P1−As1 2.2967(5) Å (9 b), 2.3052(8) Å (9 c) and P1−As3 2.3002(5) Å (9 b), 2.3113(7) Å (9 c)) are in the range of single bonds, indicated by WBIs of 0.95 (9 b) and 0.94 (9 c). The As−As bonds (As1−As2 2.4381(3) Å (9 b), 2.4404(4) Å (9 c) and As2−As3 2.4264(3) Å (9 b), 2.4196(5) Å (9 c)) are also in the range of slightly shortened single bonds [17] underlined by WBIs of 1.01/1.02 (9 b) and 1.01/1.02 (9 c). The molecular structure of 10 a/10 b reveals a cyclo‐As3 ligand with an exocyclic {PR2} unit. One side of the As3 unit coordinates in an η2 fashion and the {PR2} unit in an η1 fashion to the cobalt atom. The P1−As1 distance is 2.3111(8) Å (10 a)and 2.2991(11) Å (10 b) (WBI of 0.94 and 0.97), respectively. The As3 unit shows two As−As distances in the range of single bonds (As1−As2 2.4699(5) Å (10 a), 2.4731(6) Å (10 b), As1−As3 2.4644(5) Å (10 a), 2.4717(7) Å (10 b); WBIs of 0.94/0.93 (10 a) and 0.94/0.94(10 b)) and one shorter distance (As2−As3 2.3531(5) Å (10 a), 2.3496(6) Å(10 b)), which is between a single and double bond,[ 16 , 17 ] indicated by WBI of 1.12 and 1.10. To obtain further insight into how these reactions proceed and whether it is possible to transfer the four‐membered compounds (7 a–c, 9 a–c) into the species with the cyclo‐E3 ligand containing an exocyclic {PR2} unit (8 a–c, 10 a–c), systematic NMR investigations were conducted. For each system (except [6]+Ph2PCl), one sample of the crude reaction mixture (after extraction with n‐pentane and filtration) in [D8]toluene was kept at room temperature for several days and a second sample in [D8]toluene was tempered at 60 °C and NMR spectra were recorded from time to time (see Figure 3 and SI for further details). For the reaction of [5] with Ph2PCl, the 31P{1H} NMR spectrum measured directly after the reaction shows the presence of 7 a and 8 a in a ratio of 5:1. After 4 days at room temperature, an inverted ratio of 0.5:1 is observed and after nine days, 7 a cannot be detected at all. Keeping the sample at 60 °C, after one hour, a ratio of 1.1:1 is observed and after 14 h, 7 a cannot be detected anymore. For the reaction of [5] with Cy2PCl, the 31P{1H} NMR spectrum measured directly after the reaction shows 7 b and 8 b in a ratio of 86:1. After four days at room temperature, the ratio changed to 49:1 and after nine days to 30:1. Keeping the sample at 60 °C, 7 b can be successively transferred into 8 b, for example, ratios of 32:1 after one hour and of 0.75:1 after 86 hours were observed (Figure 3). Directly after the reaction of [5] with tBu2PCl, the presence of 7 c and 8 c in a ratio of 86:1 is observed in the 31P{1H} NMR spectrum. After four days, the ratio changed to 24:1 and after nine days to 22:1. Tempering of the sample at 60 °C leads after one hour to a ratio of 17:1, which does not change further upon longer thermolysis times (after 14 hours still the same ratio). The behavior of the reaction of the arsenic compound [6] is quite similar (cf. SI).
Figure 3.
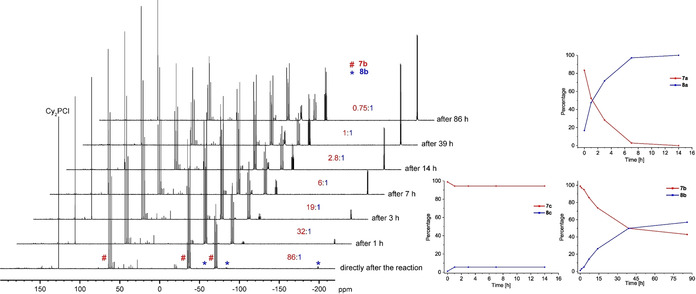
31P{1H} NMR spectra of the reaction of [5] with Cy2PCl directly after the reaction and after thermolysis at 60 °C (left) and plotted percentage ratios of products after thermolysis of the crude mixtures from the reactions of [5] with R2PCl (R=Ph, Cy, tBu) at 60 °C (right).
In order to elucidate the reaction pathway and clarify the outcome of the reactions that are so strongly dependent on the substituents on the chlorophosphanes, DFT calculations were performed for the model reaction of [CpHCo(η3‐E3)]− (E=P, As; E‐1) with the phosphenium ion PMe2 + (E‐2). The optimization of the starting material, products, intermediates and transition states was performed at the BP86/def2‐SVP level of theory and the energies were calculated at the B3LYP/def2‐TZVP level of theory including solvent effects and dispersion correction D3BJ. [20] The complete pathway was calculated only for the methyl‐substituted phosphenium ion (PMe2 +), while for the other substituents (Ph, Cy, tBu) only the products [CpHCo(η3‐E3PR2)] (P‐1), [CpHCo(η2:η1‐E3PR2)] (P‐2) and the highest transition state TS‐2 between P‐1 and P‐2 were optimized. A combined summary of the essential steps is depicted in Figure 4 (for the phosphorus species; for the arsenic species cf. SI). The first step displays the addition of the {PMe2} unit at the cyclo‐E3 ligand in E‐1 leading to the intermediate Int‐1. Over the transition state TS‐1 with a barrier of 33 (P) and 10 kJ mol−1 (As), respectively, the products P‐1 are formed. The energy of P‐1 was set arbitrarily to 0. It has to be noted that for E=P, an additional intermediate and transition state could be modeled, representing a rotation of the {PMe2} unit along the P−P bond before the optimization leads to P‐1 (cf. SI). The {PMe2} unit migrates to one E atom over the transition state TS‐2, which is 126 (P) and 138 kJ mol−1 (As), respectively, higher in energy and the products P‐2 are formed. Again, for E=P, further intermediates and transition states could be modeled which are all lower in energy than TS‐2 and mainly represent rotation processes of the {PMe2} unit (cf. SI). Compared to P‐1, the final products P‐2 are lower in energy by 28 and 39 kJ mol−1 for E=P and As, respectively, and the conversion of P‐1 into P‐2 is exothermic overall. For the other substituents on the electrophile (Ph, Cy, tBu), the formation of all six products P‐2 is exothermic related to their respective P‐1. The cyclohexyl‐substituted species are less favored (−26 (P) and −30 kJ mol−1 (As)), followed by the tert‐butyl‐substituted ones (−33 (P) and −35 kJ mol−1 (As)) and the phenyl‐substituted ones which are the most favored ones (−36 (P) and −47 kJ mol−1 (As)). [21] Nevertheless, the highest transition state (TS‐2) between P‐1 and P‐2 determines how easy the conversion of P‐1 into P‐2 can take place. The highest energy shows the tert‐butyl derivative (170 (P) and 176 kJ mol−1 (As)), followed by the cyclohexyl derivative (141 (P) and 145 kJ mol−1 (As)) while the phenyl‐substituted derivative shows the lowest activation barrier (126 (P) and 128 kJ mol−1 (As)). The results are in good agreement with the experimental observations from the reactions of the Cp′′′ derivatives [5] and [6] with R2PCl (Ph, Cy, tBu). The conversion of the phenyl‐substituted species 7 a and 9 a into 8 a and 10 a proceeds much faster at room temperature and for short thermolysis times and only 8 a and 10 a could be isolated finally. The cyclohexyl derivatives 7 b and 9 b can be partially converted at room temperature only after a long time or after long thermolysis times into 8 b and 10 b, while no full conversion was observed within the investigated time range. For the tert‐butyl derivatives 7 c and 9 c, only traces of 8 c and 10 c can be detected after a long time at room temperature or after thermolysis.
Figure 4.
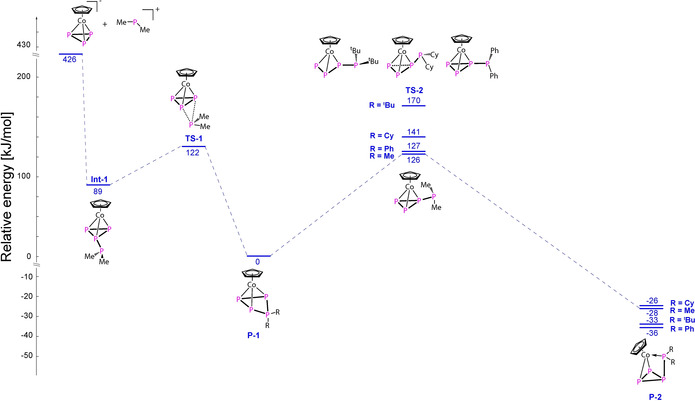
Calculated reaction mechanism for the reaction of CpHCoE3 − (E‐1) with Me2P+ (E‐2) at the B3LYP‐D3BJ/def2‐TZVP level of theory (for details see SI). Solvent effects were included via the polarizable continuum model (CPM). Energy values are referenced to P‐1. Additional intermediates and transition states are omitted for clarity (cf. SI). The relative energy of the transition states for [CpHCoP4R2] (R=tBu, Cy, Ph; TS‐2) and the final products P‐2 are also given.
An insertion of a chlorophosphane or an in situ generated phosphenium ion starting from a chlorophosphane into a cyclo‐Pn ligand was only reported for the reaction of the anionic cobalt complex [K(18‐c‐6)][(PHDI)Co(η4‐P4)] yielding [(PHDI)Co(η4‐P5R2)][ 4 , 22 ] reported by Wolf et al. and by ourselves for the reaction with the neutral nickel complex [Cp′′′Ni(η3‐P3)] yielding cationic species of the type [Cp′′′Ni(η3‐P4R2)]+. [23] The latter complexes are isoelectronic and isostructural to 7 a–c but represent the final products of their reactions and no further rearrangements to species along the lines of 8 a–c were found, which are formed after an unprecedented ring contraction reaction. A similar structural motif of the latter complexes was already reported for the cationic rhodium complex [(triphos)Rh(η2:η1‐P4RR′)]+ with overall P−P distances closely related to those observed for 8 a. [24] Note that, for [Cp*Fe(η4‐P5{GeL}2)] (L=(tBuN)2CPh), a migration of one exocyclic germylene fragment towards the Cp*Fe unit was observed. [25] The mixed phosphorus and arsenic complexes 9 a–c and 10 a–c expand the series of rare examples of complexes comprising mixed polypnictogen ligands, namely, EP3 (E=As, Sb), [2] [Cp′′′Co(μ3,η4:η1:η1‐AsP4Cp*){W(CO)5}2], [9] [(Cp′′′Fe)2(μ,η4:η4‐PxAsy)] (x+y=4), [Cp′′′Fe(η5‐PxAsy)] (x+y=5) [26] and [{CpRMo(CO)2}2(μ,η2:η2‐EE′)] (E/E′=P, As, Sb, Bi) [27] were reported.
In order to investigate if this reactivity pattern of [5] and [6] can be extended to other halogen‐containing species, the reaction of [K(thf)0.7][5] and [K(thf)0.8][6] with the chlorosilylene [LSiCl] (L=(tBuN)2CPh) (Scheme 3) was studied. The reactions were conducted at −80 °C in thf, while a color change was first observed upon warming to room temperature form dark red to brown and from dark green to brown‐green, respectively. After workup, [Cp′′′Co(η3‐E3SiL)] (L=(tBuN)2CPh, E=P (11), As (12)) could be isolated in crystalline yields of 29 and 18 %, respectively. Crystals suitable for X‐ray single crystal structure analysis could be obtained from a concentrated solution of 11 in o‐difluorobenzene layered with n‐pentane at −30 °C and from a concentrated solution of 12 in a mixture of n‐pentane and toluene at −30 °C. The molecular structures in the solid state (Figure 5) reveal that the silylene has been inserted into the cyclo‐E3 ligand and novel four‐membered rings consisting of three pnictogen atoms and one Si atom are formed. All three E atoms are coordinated to Co1, while Si1 is bent out of the plane by 30° (11/12). The Si1−E1 distance amounts to 2.1682(17) Å (11) and 2.2853(14) Å (12), Si1−E3 distance to 2.1696(18) Å (11) and 2.2775(14) Å (12), the E1−E2 bond to 2.2283(19) Å (11) and 2.4417(8) Å (12), and the E2−E3 bond to 2.2032(18) Å (11) and 2.4243(8) Å (12). All mentioned distances are in the range between single and double bonds, confirmed by the calculated WBIs in the range of 1.01 and 1.08 (11) and 0.98 and 1.09 (12), respectively. Compound 11 and 12 represent the first complexes including a four‐membered cycle consisting of three pnictogen atoms and one silicon atom and add to the row of pnictogen silicon heterocycles, for example, [(LSi)2P2] [28] reported by the groups of H. W. Roesky and Driess in 2011 and its heavier analogue [(LSi)2As2] reported 2016 by our group. [29] The latter synthetic approach gave also access to the inorganic benzene derivatives [(LSi)3E3] (E=P, As). [29] P. Roesky et al. recently showed that the chlorosilylene [LSiCl] (L=(tBuN)2CPh) can be used for a substitution of one P atom in [Cp*Fe(η5‐P5)] to form [Cp*Fe(η4‐P4SiL)]. [30] However, compound 12 represents the first example where an AsnSiL heterocycle acts as a ligand for a transition metal complex.
Figure 5.
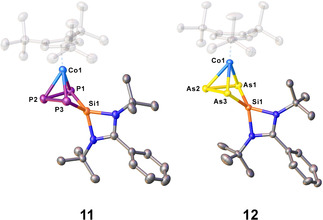
Molecular structure of 11 and 12 in the solid state. [31] Hydrogen atoms and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn with 50 % probability level.
The 31P{1H} NMR of 11 in [D8]thf shows a doublet of doublets centered at δ=−66.6 ppm and two overlapping doublets centered at δ=−75.0 ppm with an integral ratio of 1:1:1 with 1 JPP coupling constants of 213 and 262 Hz. The signal at −75.0 ppm additionally reveals silicon satellites with a 1 JPSi coupling constant of 109 Hz. The related 29Si{1H} NMR spectrum shows a triplet of doublet centered at δ=−24.6 ppm with a 1 JPSi coupling constant of 109 Hz and a 2 JPSi coupling constant of 12 Hz. The 29Si{1H} NMR spectrum of 12 in C6D6 shows one singlet centered at δ=−51.6 ppm.
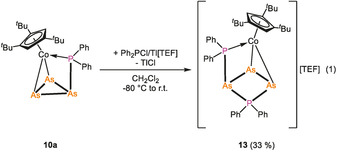
As a proof of principle and to check whether the complexes with a cyclo‐E3 ligand and the exocyclic {PR2} unit can be further functionalized, [Cp′′′Co(η2:η1‐As3PPh2)] (10 a) was reacted with the in situ generated phosphenium ion PPh2 + [halide abstraction from Ph2PCl with Tl[TEF], ([TEF]=[Al{OC(CF3)3}4], cf. Eq. 1). The 31P{1H} NMR spectrum of the reaction solution in CD2Cl2 reveals the formation of [Cp′′′Co(η2:η1‐As3P2Ph4)][TEF] (13), beside starting material and small amounts of decomposition products. After workup and crystallization, 13 could be isolated in 33 % crystalline yield.
Single crystals of 13 suitable for X‐ray single‐crystal structure analysis could be obtained from a concentrated solution in CH2Cl2 layered with n‐pentane at room temperature. The molecular structure (Figure 6) reveals that the PPh2 + has been inserted into the cyclo‐As3 ligand of 10 a. One As−As edge of the As3PPh2 cycle coordinates in η2 fashion to the cobalt atom together with the exocyclic {PPh2} unit in η1 fashion. The P2 atom deviates from the plane of the arsenic atoms by approx. 21° (fold angle As1‐As2‐As3‐P2). All E−E distances are in the range of single bonds, only the As1−As2 bond is slightly elongated (2.5758(5) Å). The 1H NMR spectrum of 13 in CD2Cl2 shows five signals for the Cp′′′ ligand indicating that the rotation of the Cp′′′ is hindered (usually, three signals are observed). The 31P{1H} spectrum in CD2Cl2 shows two doublets centered at δ=2.4 and −27.1 ppm with an 2 JPP coupling constant of 41 Hz.
Figure 6.
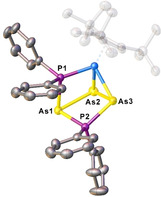
Molecular structure of the cation in 13. [31] Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn with 50 % probability level.
Conclusion
We have shown that it is possible to obtain access to the anionic complex [Cp′′′Co(η3‐P3)]− [5] in a much easier preparative way and in reasonable preparative scale for further reactivity studies. In addition, the approach could be successfully extended to the analogous arsenic species [Cp′′′Co(η3‐As3)]− [6], representing the first anionic compound with a cyclo‐As3 ligand as end deck. Both compounds could be electrophilically quenched with chlorophosphanes to yield compounds with a disubstituted cyclo‐E3PR2 ligand in [Cp′′′Co(η3‐E3PR2)] (E=P (7 a–c), As (9 a–c)) in a first step. These complexes tend to undergo an unusual ring contraction reaction from a four‐membered cyclic ligand to complexes with an cyclo‐E3 ligand with an exocyclic {PR2} unit in [Cp′′′Co(η2:η1‐E3PR2)] (E=P (8 a–c), As (10 a–c)). This transformation proceeds at room temperature in solution or at elevated temperatures and is dependent on the substituents on the used chlorophosphane. Such behavior was not observed before for other complexes with a P4R2 ligand. Moreover, the complexes 9 a–c and 10 a–c add to the only scarcely known complexes containing mixed phosphorus and arsenic ligands, but here with As as major component. Therefore, the use of [6] presents a unique possibility to obtain As‐rich derivatives. The conversion processes could be monitored by NMR spectroscopy and the reaction pathway could be elucidated by DFT calculations. Furthermore, this synthetic concept was extended to other halogen‐containing main group compounds such as the chlorosilylene [LSiCl] (L=(tBuN)2CPh). That way, the first cyclic P3Si and unprecedented As3Si rings were formed in [Cp′′′Co(η3‐E3SiL)] (E=P (11), As (12)), contributing to the rare family of known pnictogen–silicon heterocycles. Furthermore, it could be shown that the complexes with a cyclo‐E3 ligand and an exocyclic {PR2} unit are still reactive and can be further functionalized. As a proof of principle, an in situ generated PPh2 + could be inserted into the cyclo‐As3 ligand in 10 a yielding the cationic complex 13 which contains a novel mixed As3(PPh2)2 ligand coordinating in an η2:η1 fashion. This shows that the initially formed products have big potential for subsequent reactivity.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the projects Sche 384/33‐2 and 384/36‐1. M.P. is grateful to the Fonds der Chemischen Industrie for a PhD fellowship and S.R. is grateful to the Studienstiftung des Deutschen Volkes for a PhD fellowship. Open access funding enabled and organized by Projekt DEAL.
M. Piesch, S. Reichl, M. Seidl, G. Balázs, M. Scheer, Angew. Chem. Int. Ed. 2021, 60, 15101.
Dedicated to Professor Matthias Driess on the occasion of his 60th birthday
Contributor Information
Dr. Martin Piesch, https://www.uni‐regensburg.de/chemie‐pharmazie/anorganische‐chemie‐scheer/
Prof. Dr. Manfred Scheer, Email: manfred.scheer@ur.de.
References
- 1.
- 1a. Hoidn C. M., Scott D. J., Wolf R., Chem. Eur. J. 2021, 27, 1886–1902; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Scalambra F., Peruzzini M., Romerosa A. in Advances in Organometallic Chemistry (Ed.: Pérez P. J.), Academic Press, San Diego, 2019, pp. 173–222; [Google Scholar]
- 1c. Caporali M., Gonsalvi L., Rossin A., Peruzzini M., Chem. Rev. 2010, 110, 4178–4235; [DOI] [PubMed] [Google Scholar]
- 1d. Seidl M., Balázs G., Scheer M., Chem. Rev. 2019, 119, 8406–8434. [DOI] [PubMed] [Google Scholar]
- 2. Cossairt B. M., Diawara M.-C., Cummins C. C., Science 2009, 323, 602. [DOI] [PubMed] [Google Scholar]
- 3. Chakraborty U., Leitl J., Mühldorf B., Bodensteiner M., Pelties S., Wolf R., Dalton Trans. 2018, 47, 3693–3697. [DOI] [PubMed] [Google Scholar]
- 4. Hoidn C. M., Maier T. M., Trabitsch K., Weigand J. J., Wolf R., Angew. Chem. Int. Ed. 2019, 58, 18931–18936; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19107–19112. [Google Scholar]
- 5. Piesch M., Reichl S., Seidl M., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2019, 58, 16563–16568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16716–16721. [Google Scholar]
- 6. Riedlberger F., Todisco S., Mastrorilli P., Timoshkin A. Y., Seidl M., Scheer M., Chem. Eur. J. 2020, 26, 16251–16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piesch M., Seidl M., Scheer M., Chem. Sci. 2020, 11, 6745–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mandla K. A., Neville M. L., Moore C. E., Rheingold A. L., Figueroa J. S., Angew. Chem. Int. Ed. 2019, 58, 15329–15333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15473–15477. [Google Scholar]
- 9. Piesch M., Seidl M., Stubenhofer M., Scheer M., Chem. Eur. J. 2019, 25, 6311–6316. [DOI] [PubMed] [Google Scholar]
- 10. Rink B., Scherer O. J., Wolmershäuser G., Chem. Ber. 1995, 128, 71–73. [Google Scholar]
- 11. Di Vaira M., Mani F., Moneti S., Peruzzini M., Sacconi L., Stoppioni P., Inorg. Chem. 1985, 24, 2230–2236. [Google Scholar]
- 12. Piesch M., Reichl S., Riesinger C., Seidl M., Balázs G., Scheer M., Chem. Eur. J. 2021, 27, 10.1002/chem.202100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mädl E., Balázs G., Peresypkina E. V., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 7702–7707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7833–7838. [Google Scholar]
- 14. Piesch M., Dielmann F., Reichl S., Scheer M., Chem. Eur. J. 2020, 26, 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The black residue of the reaction was dissolved in concentrated nitric acid, neutralized with KOH and filtered. The obtained green solid (Ni(OH)2) was dissolved in a concentrated ammonia solution. After addition of an alcoholic solution of diacetyl dioxime, a pink precipitate of the related nickel complex was obtained.
- 16. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 12770–12779. [DOI] [PubMed] [Google Scholar]
- 17. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186–197. [DOI] [PubMed] [Google Scholar]
- 18. Scherer O. J., Braun J., Walther P., Wolmershäuser G., Chem. Ber. 1992, 125, 2661–2665. [Google Scholar]
- 19.
- 19a. Velian A., Cossairt B. M., Cummins C. C., Dalton Trans. 2016, 45, 1891–1895; [DOI] [PubMed] [Google Scholar]
- 19b. Velian A., Cummins C. C., Chem. Sci. 2012, 3, 1003; [Google Scholar]
- 19c. Cossairt B. M., Cummins C. C., Angew. Chem. Int. Ed. 2010, 49, 1595–1598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1639–1642. [Google Scholar]
- 20.For the calculations, the in situ generated phosphenium ion was used as a model system instead of the obviously much more complicated reaction of the chlorophosphane with [CpHCoE3]− under salt elimination.
- 21.In some cases, the conversion of P-1 into P-2 does not proceed in one step over TS-2. Since TS-2 represents the highest barrier, no further intermediates and transition states were modeled.
- 22. Ziegler C. G. P., Maier T. M., Pelties S., Taube C., Hennersdorf F., Ehlers A. W., Weigand J. J., Wolf R., Chem. Sci. 2019, 10, 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riesinger C., Dütsch L., Balazs G., Bodensteiner M., Scheer M., Chem. Eur. J. 2020, 26, 17165–17170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barbaro P., Ienco A., Mealli C., Peruzzini M., Scherer O. J., Schmitt G., Vizza F., Wolmershäuser G., Chem. Eur. J. 2003, 9, 5195–5210. [DOI] [PubMed] [Google Scholar]
- 25. Yadav R., Goswami B., Simler T., Schoo C., Reichl S., Scheer M., Roesky P. W., Chem. Commun. 2020, 56, 10207–10210. [DOI] [PubMed] [Google Scholar]
- 26. Schwarzmaier C., Bodensteiner M., Timoshkin A. Y., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 290–293; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 295–299. [Google Scholar]
- 27. Dütsch L., Riesinger Ch., Balázs G., Scheer M., Chem. Eur. J. 2021, 27, 10.1002/chem.202100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Inoue S., Wang W., Präsang C., Asay M., Irran E., Driess M., J. Am. Chem. Soc. 2011, 133, 2868–2871; [DOI] [PubMed] [Google Scholar]
- 28b. Sen S. S., Khan S., Roesky H. W., Kratzert D., Meindl K., Henn J., Stalke D., Demers J.-P., Lange A., Angew. Chem. Int. Ed. 2011, 50, 2322–2325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2370–2373. [Google Scholar]
- 29. Seitz A. E., Eckhardt M., Erlebach A., Peresypkina E. V., Sierka M., Scheer M., J. Am. Chem. Soc. 2016, 138, 10433–10436. [DOI] [PubMed] [Google Scholar]
- 30. Yadav R., Simler T., Reichl S., Goswami B., Schoo C., Köppe R., Scheer M., Roesky P. W., J. Am. Chem. Soc. 2020, 142, 1190–1195. [DOI] [PubMed] [Google Scholar]
- 31. Deposition Numbers CCDC 2068508, 2068509, 2068510, 2068511, 2068512, 2068513, 2068514, 2068515, 2068516, 20685187, 2068518, and 2068519 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary