

Abstract
Thoracic aortic aneurysm (TAA) develops silently and asymptomatically and is a major cause of mortality. TAA prevalence is greatly underestimated, it is usually diagnosed incidentally, and its treatment consists mainly of prophylactic surgery based on the aortic diameter. The lack of effective drugs and biological markers to identify and stratify TAAs by risk before visible symptoms results from scant knowledge of its pathophysiological mechanisms. Here we integrate the structural impairment affecting non‐syndromic non‐familial TAA with the main cellular and molecular changes described so far and consider how these changes are interconnected through specific pathways. The ultimate goal is to define much‐needed novel markers of TAA, and so the potential of previously identified molecules to aid in early diagnosis/prognosis is also discussed. © 2021 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: thoracic aortic aneurysm, bicuspid aortic valve, biomarkers, cellular phenotype, arterial remodeling, oxidative stress, clinical trials
The etiology of thoracic aortic aneurysms (TAAs)
A true arterial aneurysm is defined as local dilation of the vessel with at least 50% enlargement beyond the normal diameter [1], leading to a weakening of the vessel wall and promoting dissection or rupture. TAA is a silent and life‐threatening disease with an estimated incidence of at least 6–10 cases/100 000 patients/year. By location, ascending TAAs are the most common (60%), followed by descending TAAs (35%) [2]. When the aneurysm is not detected and an acute aortic event occurs, 21% of patients die before arriving at hospital, and of those who are alive on arrival, over 50% die in the following 48 h if they receive no intervention [3].
The etiology of TAA has not yet been fully described, but it is multifactorial and complex. TAA can develop alongside other organ system abnormalities, as in the case of syndromic TAA, representing 20% of all TAAs. If TAA is not associated with other systemic abnormalities, it is referred to as non‐syndromic TAA (Figure 1).
Figure 1.
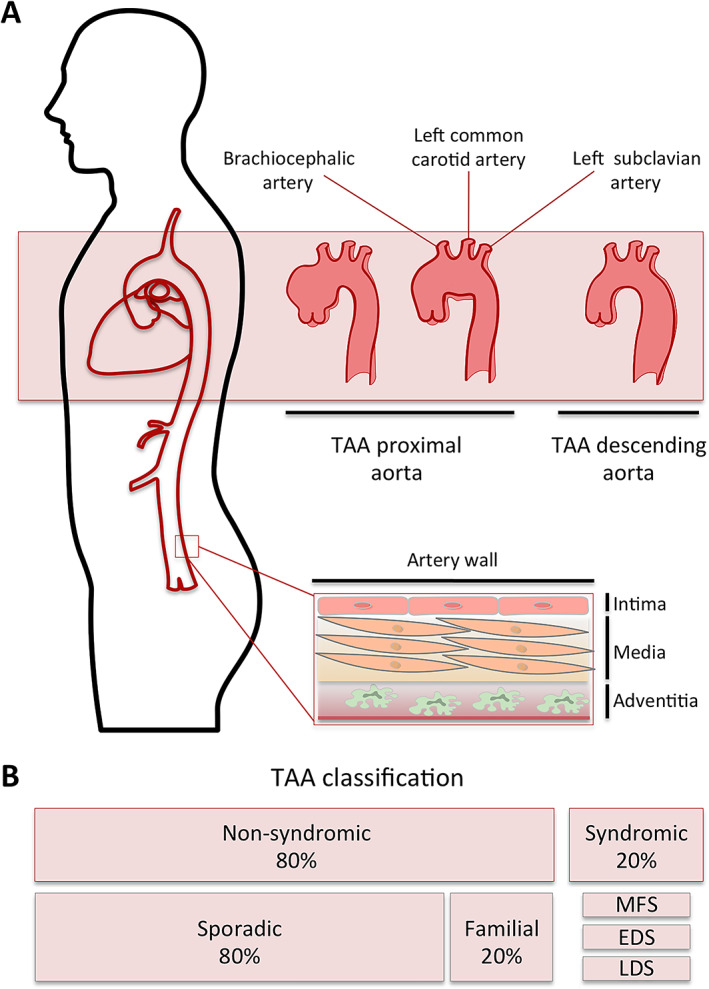
(A) TAA subtypes based on localization within the aortic tree. Within the thoracic aorta, three sections can be differentiated: the ascending aorta runs from the aortic valve in the left ventricle to the brachiocephalic trunk; the aortic arch starts at the second sternocostal joint and finishes at the level of the fourth vertebra, containing the head and neck vessels; and the descending thoracic aorta, which goes beyond the ligamentum arteriosum and ends at the level of the diaphragm. (B) TAA classification as syndromic and non‐syndromic. EDS, Ehlers‐Danlos syndrome; LDS, Loeys‐Dietz syndrome; MFS, Marfan syndrome.
Diagnosis and medical treatment of TAAs
Despite the rise in diagnoses due to the increase in imaging tests performed in recent years, the incidence and prevalence are probably underestimated, as TAA formation is slow, gradual, and painless, and patients remain asymptomatic. This results in 95% of all cases being undetected until an acute aortic event occurs, or the expansion leads to a compression of nearby anatomical structures. Thus, when symptoms appear, the aneurysm is usually very large and at risk of rupture, an event associated with high rates of mortality [4]. Oladokun et al [5] systematically reviewed the assessment of TAA growth and influencing risk factors, agreeing on aneurysm size, anatomical location, and the presence of Marfan's syndrome or a bicuspid aortic valve (BAV). Chronic dissections and chronic obstructive pulmonary disease were also implicated as risk factors. Sex is also an influencing factor of TAA growth, which is more than twice as fast in women than in men [6].
Currently, the only effective treatment for TAA patients is surgery. Prophylactic surgery is indicated when the aortic diameter exceeds 50 mm, although dilatation between 40 and 50 mm may also be clinically relevant depending on the nature and location of the aneurysm [2, 7]. However, there is widespread agreement regarding the lack of sensitivity and specificity of the aortic diameter as a proper estimator of dissection/rupture risk, which often causes patients to develop an aortic dissection or rupture with an aortic diameter below optimal threshold and leading to other patients undergoing surgery despite any real threat of dissection or rupture [8]. No specific drugs provide effective treatment for TAA. A systematic review on medication for TAA was carried out following the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) standards (see supplementary material, Figure S1). Improved long‐term outcomes were associated with statins intake [9], reducing adverse events (death, rupture, dissection), and surgical rate mainly in the ascending aorta and arch, and descending and thoracoabdominal aortic aneurysms, not in aortic root [10]. Additionally, a systematic review on clinical trials for TAA was carried out in ClinicalTrials.gov. Most of them aim to reduce complications from surgery and only one will investigate the pharmacological treatment effect on reducing the growth rate of TAAs and rupture [11]. Of 142 studies found, 15 included medications, which have been compiled in supplementary material, Table S1.
The pathogenic mechanisms of non‐syndromic TAA remain widely unclear, resulting in a lack of effective medication or specific TAA prevention programs beyond general guidelines for cardiovascular disease prevention [12, 13]. This review summarizes the main cellular and molecular changes taking place in non‐syndromic non‐familial TAA formation. The selection of studies was carried out in accordance with PRISMA standards (Figure 2). Among 4601 screened records, 305 were assessed for eligibility, excluding those studies dealing with only abdominal aortic aneurysm (AAA), syndromic TAA or familial TAA.
Figure 2.
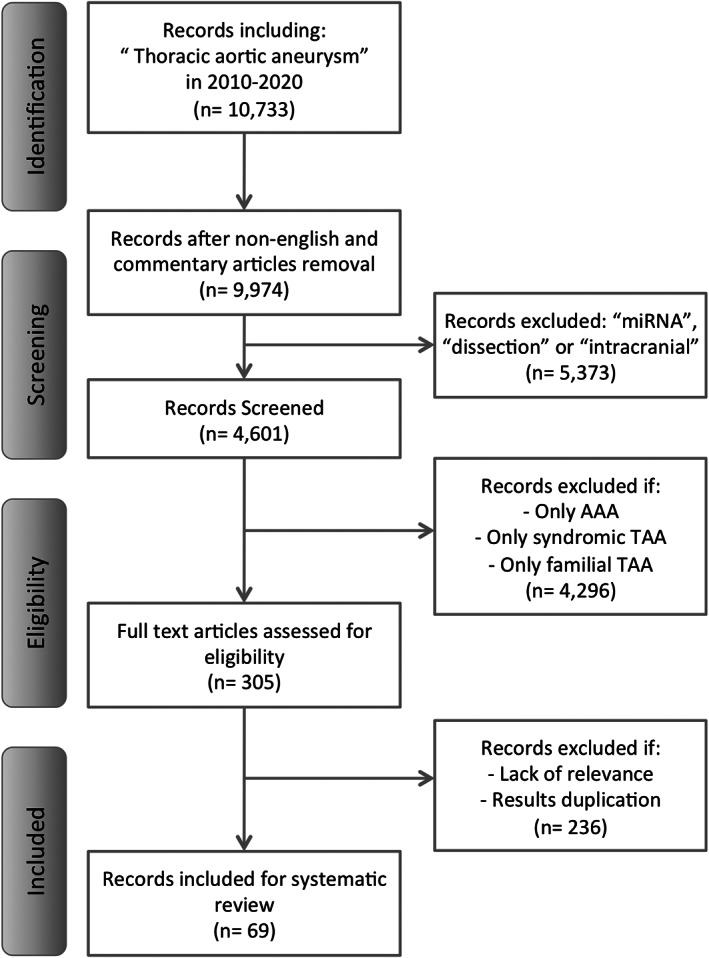
PRISMA flowchart.
Structural impairment and deregulated mechanisms in TAAs
Aortic structure and mechanical properties
The aortic wall is composed of three layers, i.e. the intima, the media, and the adventitia, essential in maintaining vessel homeostasis and mechanical integrity. During the dilatation process, crosstalk between cells, subcellular compartments, and the extracellular matrix (ECM) takes place, resulting in a network of interconnected changes artery‐wide.
The intima is a single layer of endothelial cells (ECs) that is in direct contact with the blood flow. The media layer is mainly composed of VSMCs and ECM, which confers biochemical and structural support and coordinate the contractile VSMCs and elastic tensions in response to mechanical stresses by blood flow. VSMCs are layered longitudinally alongside interspersed elastic fibers, forming elastin lamellae. VSMCs and elastin lamellae are anchored by focal adhesions, forming VSMC elastin‐contractile units, also reinforced by fibrillin, integrins, and collagen [14]. VSMCs are a major source of proteolytic enzymes and their inhibitors, such as matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family, serine/cysteine proteases, or alpha‐2‐macroglobulin, which maintain the ECM environment [15]. The ECM is the non‐cellular component of the aortic wall. It provides structural support and regulates cellular function and vessel remodeling through growth factors and cytokine delivery. ECM is composed of proteoglycans, glycoproteins, glycosaminoglycans, elastin, and collagen. Medial degeneration is marked by VSMC necrosis and disorganization, particularly around microvessels, and loss of elastin content and collagen fibers, making the aortic wall vulnerable to TAA [16].
The adventitia interacts with the nervous system and the vasa vasorum, acting as a channel for the entry of circulating cells and inflammatory mediators into the aortic wall. It is composed of fibroblasts embedded in a collagenous ECM. The adventitia may also be involved in disease evolution by promoting the development of neovessels toward the medial layer of TAAs. However, these neovessels were found to have defective endothelial junctions and lack mural cells, suggesting decreased EC interactions and subsequently high permeability, which leads to plasma protein accumulation in the aneurysmal wall [17, 18]. The vasa vasorum has been shown to be disrupted in human ascending TAA specimens, as evidence by aberrations in size, abundance, density, and wall thickness [19]. Additionally, the adventitial vasa vasorum has been found to harbor perivascular progenitor cells within the human adult thoracic aorta [20]. An important signaling pathway involved in TAA is NF‐kB, which upregulates adhesion molecule expression and triggers macrophage infiltration and inflammation in the adventitia and media [21].
Inflammation
When the endothelial barrier becomes destabilized and junctional permeability increases, T lymphocytes and macrophages are recruited, promoting vascular inflammation [22, 23]. Increased levels of 5‐hydroxyeicosatetraenoic acid (5‐HETE) were also found in the medial layer of TAAs [24], which could be a consequence of the enhanced activity displayed by 5‐lipoxygenase‐expressed macrophages in the adventitia [25]. Furthermore, adventitial inflammation with infiltration of lymphoplasmacytic cells has been detected in samples of sporadic TAAs, whereas there was no evidence of chronic inflammation in non‐aneurysmal patients [19]. During inflammation, angiogenesis can be promoted. Higher levels of pro‐ and anti‐angiogenic proteins, including angiopoietin‐1, angiopoietin‐2, fibroblast growth factor‐acidic, thrombospondin, plasminogen, and albumin were found in the medial layer of TAAs compared with healthy aortas. Particularly, these proteins were described in the areas of mucoid degeneration [26] (Figure 3, grey).
Figure 3.
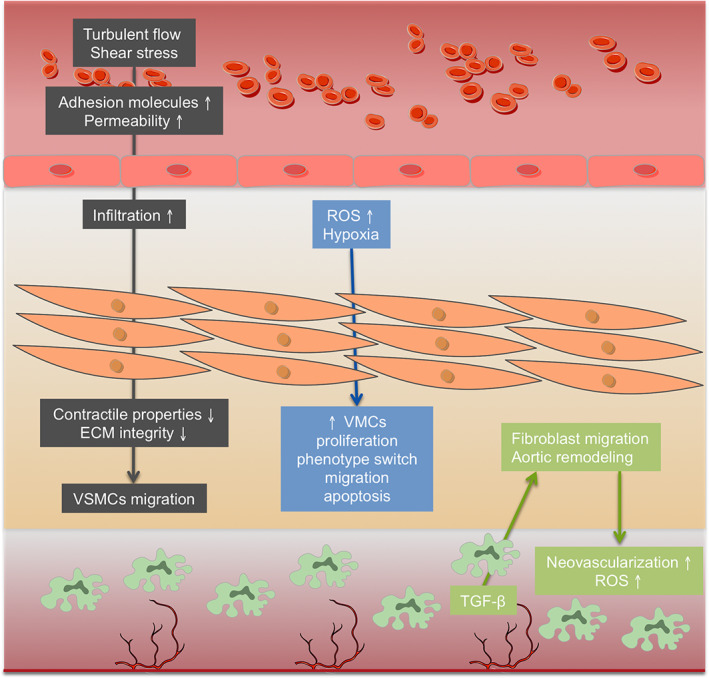
Main structural and cellular changes in TAA development. In grey, mechanical changes, permeability, and inflammation components; in blue, oxidative stress and hypoxia effects; in green, the role of adventitia and neovascularization.
Impaired mechanotransduction
VSMCs and ECs communicate through electrical coupling mechanisms and gap junctions to regulate aortic tone and diameter, and control blood pressure [27]. The sequential pathophysiology of aneurysm formation is still unknown, although dysfunction of ECs and altered mechanosensing in the aortic wall could be a first step. By means of mechanotransduction, ECs convert the mechanical stimuli caused by blood pressure into biochemical signals through the cytoskeleton [28]. ECs regulate these processes by activating mechanosensors, including the mechanosensory complex vascular endothelial‐cadherin (VE‐cadherin), vascular endothelial growth factor receptor 2 (VEGFR2), and platelet EC adhesion molecule (PECAM‐1) [29]. In particular, it has been shown that primary EC cultures from the aneurysmal wall of thoracic aorta present decreased levels of PECAM‐1, von Willebrand factor (vWF), and VE‐cadherin [18], which disrupt mechanotransduction and induce local proinflammatory activation [21] (Figure 3, grey).
Arterial wall remodeling
In TAA, there is a loss of VSMCs by apoptosis and a change in VSMC phenotype, impairing VSMCs' mechanotransduction ability and ECM remodeling [16, 30]. An EC–VSMCs co‐culture model described that altered flow in ECs can induce a synthetic/proliferative VSMC phenotype, leading to medial degeneration and TAA [31]. Increased angiotensin II (Ang‐II) signaling and TGF‐β production in TAA contribute to ECM degradation and facilitate VSMC migration to the wounded area [32]. The release of platelet‐derived growth factor, TGF‐β1, and insulin‐like growth factor‐1 is induced, also promoting VSMC migration, hypertrophy, and vascular remodeling [33]. The phenotypic switch in VSMCs can also be enhanced by a chronic hypoxic status in the media of TAAs [34], highlighted by the observed accumulation of the hypoxia marker GLUT1 within regions of elastin degeneration in aneurysmal media [19]. In addition, persistent tissue damage induces cystathionine‐γ‐lyase overexpression, which enhances H2S production and VSMC apoptosis in sporadic non‐syndromic TAAs, thus hindering any effective repair [35]. Additionally, the expression of α‐smooth muscle actin (α‐SMA), transgelin (SM22), and vimentin in VSMCs from ascending TAAs was found to be reduced, as was ECM synthesis [18].
With regard to lipids, decreased levels of ether‐type phosphatidylethanolamines were found within TAA arteries, whereas lysophosphatidylcholines and glycosylceramides were found to be increased, pointing to oxidative stress and intensified migratory activity of VSMCs [24]. An increase in sphingomyelins was also revealed, suggesting a repression of sphingomyelinase activity and the sphingomyelinase–ceramide pathway, which could lead to the inhibition of tissue regeneration, thereby favoring TAA progression [36] (Figure 3, blue).
In the adventitia, TGF‐β can induce fibroblasts to differentiate into myofibroblasts, which cause MMP production and matrix degradation and TAA [37, 38]. When the aortic wall is injured, adventitial fibroblasts migrate to the media and can be transformed into VSMCs, although showing lower expression of MMP‐3, α‐SMA, SM22, and Ki67 proteins in TAA, and thus highlighting defects in VSMC transformation and ECM remodeling [39]. Upregulated expression of monocyte chemoattractant protein‐1 (MCP‐1) was observed in primary human aortic adventitial fibroblast culture in response to Ang‐II, leading to fibroblast proliferation, adventitial thickening, and cytokine production [40] (Figure 3, green).
The renin–angiotensin system (RAS) and oxidative stress
One of the main signaling pathways altered in TAA is RAS. The turbulent blood flow in TAAs maintains RAS activation, which favors aneurysm formation [17, 41]. The high shear stress during TAA increases endothelium‐derived nitric oxide (eNO) production, an endothelial metabolite that is essential to maintain a healthy aortic wall [17]. Impaired regulation of eNO in TAA produces free oxygen radicals, favoring an oxidative stress status [42]. Increased reactive oxygen species (ROS) stimulated collagen degradation by MMP activation through a p22phox‐based NADP/NADPH oxidase in rat cardiac fibroblasts [43]. The increased levels of ROS detected in human ascending TAA tissues were correlated with medial degeneration and increased connective tissue growth factor, which modulates the synthetic/proliferative VSMC phenotype in the human and murine model of TAA based on Ang‐II infusion [44]. In a similar model, increased medial thickness, erythrocyte extravasation in the media, elastin fragmentation, and accumulation of CD45+ cells in the adventitia were observed [45].
Several lines of evidence also indicate that autophagy controls VSMC apoptosis in response to ROS generation and stimulates the transition of VSMCs into a proliferative/synthetic phenotype [46]. Although it is not fully known if autophagy plays a detrimental or a protective mechanism in TAA progression, it has been described as a dysregulated process in TAA, and high expression of osteopontin (OPN) in aortic aneurysms has been proven to promote autophagy in cultured VSMCs [47].
A molecular perspective of TAA
Elastin and collagen fragmentation
Elastic fibers supply compliance and resilience to the aortic wall and collagen plays an essential role in aortic stiffness and strength. Deregulated activity, such as that observed in TAA, degrades collagen and elastic fibers, causing irreversible breakdown of the elastic fiber network [48]. Degradation of elastin contributes to the production of elastokines or elastin‐derived peptides, which are elevated in aortic aneurysms [49].
Aortic samples from sporadic ascending TAAs showed that decreased collagens type I and III and increased collagens α1(XI) and V were linearly correlated with the size of the aneurysm [50]. In this context, cyclic mechanical stress constantly increases the synthesis, secretion, and assembly of tropocollagen into fibers via intensified Ang‐II activity and TGF‐β/Smad signaling [38]. In human non‐syndromic TAA samples, an increase in Smad2 activation was associated with disruption of elastic lamellae [51]. Furthermore, an enhancement of TGF‐β signaling and Smad2 activation was detected in the aortic wall of ascending TAA patients, whatever the etiology, with the resulting increase in TGF‐β1 and phosphorylated Smad2 protein levels [52] (Figure 4). This is in line with the described dysregulation of TGF‐β signaling in syndromic and familial non‐syndromic TAA. In Marfan syndrome, mutations in FBN1 cause overactivity of the TGF‐β signaling cascade with an increased accumulation of phosphorylated Smad2 [53]. In Loeys‐Dietz syndrome, mutations in TGF‐β receptors (TGFBR1 and TGFBR2) cause TGF‐β signaling upregulation (TGF‐β paradox) [38, 54]. In the same line, upregulated TGF‐β signaling has been related to mutations in ACTA2 and MYH11 genes in familial non‐syndromic TAAs [55, 56]. However, several mouse studies also support detrimental effects on aortic structure when TGF‐β or related signaling were inhibited, revealing a dimorphic role of TGF‐β signaling in the development of TAA [57].
Figure 4.
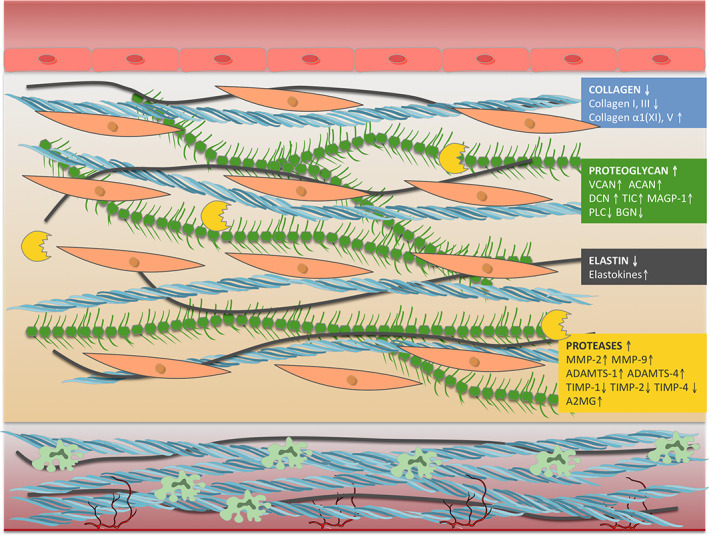
Schematic view of main molecular changes within the aortic structure in TAA.
Proteoglycans and glycoproteins
Proteoglycans provide mechanical support and mediate cellular signaling responses in the aortic wall. Versican was identified as the most upregulated ECM protein in a mouse model of aortic dilatation by Ang‐II infusion [58]. Similarly, an accumulation of versican together with an increase in aggrecan within the aortic intima was detected in ascending aortas from TAA patients [59]. The increase in total versican levels has been associated with upregulation of TGF‐β in TAA mice, which is known to induce the expression of different proteoglycans [38]. Decorin was focally increased in human ascending thoracic aortic samples compared with normal aortic wall, whereas biglycan was decreased overall [52]. Increased levels of decorin and microfibril‐associated glycoprotein I have been described in the media layer from sporadic non‐syndromic TAAs, leading to defective collagen and elastic fiber assembly and favoring TAA development [60]. Perlecan was also found to be downregulated in human aortic media of TAA patients [61]. In contrast, testican 2 levels were significantly increased in surgical media samples from non‐syndromic TAAs compared with control subjects [60].
Thrombospondins, tenascins, and fibronectin are some of the main glycoproteins of the ECM that play a role in TAA development. Fibrinogen, an essential glycoprotein precursor to fibrin, was found to be downregulated in aortic media from TAA patients [61] and associated with fibrin degradation products, such as D‐dimer, whose levels were elevated in TAA [3, 7] (Figure 4).
Proteases
MMPs, the ADAMTS family, and TIMPs are the main proteases and inhibitors present within the media that contribute to ECM environment maintenance. MMPs are in charge of cleavage and digestion of extracellular proteins such as elastin and collagen for wall repairment, remodeling and homeostasis, whereas the ADAMTS family is key to ECM remodeling by proteoglycan cleavage and procollagens and vWF processing. TIMPs can regulate MMPs and ADAMTS family activity to avoid over‐destruction. When TAA occurs, the combination of altered levels of MMPs, ADAMTS, and TIMPs together with biomechanical factors is thought to cause medial degeneration. A rat model of TAA induced by calcium chloride displayed significantly increased expressions of MMP‐2 and MMP‐9 in the intima and media [62]. A significantly higher expression of ADAMTS‐1 and ADAMTS‐4 proteins was found in TAA tissues compared with control aortic tissues [63]. Furthermore, in a mouse model of sporadic TAA induced by Ang‐II infusion, ADAMTS‐4 deficiency significantly reduced aortic diameter and aneurysm formation. Comparably, significant expression of ADAMTS‐4 was detected in aortic tissues from patients with sporadic ascending TAA [64]. The same induction by Ang‐II infusion in a mouse model lacking the catalytic domain of ADAMTS‐5 resulted in a notable increase in versican levels in those animals compared with control mice, suggesting that this protease is the key to versican regulation during aortic dilatation [58]. TIMPs inhibit MMP and ADAMTS activity and a significant reduction in TIMP‐1, TIMP‐2, and TIMP‐4 was detected in TAA aortas [65, 66], suggesting an over‐degradation of ECM products. Alpha‐2‐macroglobulin also inhibits MMPs in TAA. The expression of this acute‐phase protein was increased in TAA tissues compared with adjacent non‐aneurysmal tissues, playing a role in the inhibition and clearance of these active proteases [67] (Figure 4).
TAA and BAV
BAV is the most common congenital heart defect and approximately 40% of BAV individuals develop ascending aortic dilatation [17]; thus, BAV is considered to be an independent risk factor in TAA development. BAV anatomical variations may alter the normal outflow patterns in the ascending aorta, promoting aneurysms, although the specific mechanisms are unknown.
Research focused on patients with BAV has highlighted the implication of proteases, VSMCs, and fibroblasts. Particularly, a significant increase in MMP‐2 and a significant reduction for TIMP‐1 were found in TAA with BAV compared with TAA with tricuspid aortic valve (TAV) [65]. The activity of MMP‐2 and MMP‐9 were also measured in TAA associated with BAV and TAV, suggesting that the greater MMP‐2 and MMP‐9 activity found in aneurysms associated with BAV could explain the higher prevalence of TAA associated with BAV [68].
The expression of VSMC markers, such as α‐SMA, vimentin, calponin, transgelin, and OPN were found to be differentially altered between both aortic valves. Increased levels of calponin, α‐SMA, and SM22 were detected in BAV‐derived compared with TAV‐derived VSMCs [18, 69]. On the contrary, decreased levels of vimentin were found in VSMCs from BAV subjects [18]. Increased levels of OPN were found in aortic VSMCs from BAV patients compared with the TAV group [70]. In TAA samples, BAV subjects displayed increased endothelial nitric oxide synthase (eNOS) expression and activation compared with the TAV group [71] and, particularly in aortic media, elevated oxidative stress (increased superoxide anion production and lipid peroxidation) was observed [72]. Remarkably, higher infiltration of lymphocytes and plasma cells localized in small vessels of the vasa vasorum within the adventitia was found in TAV specimens when compared with BAV samples [19]. Higher levels of the heat shock protein HSP27 were described in dilated aortas of BAV and TAV patients compared with non‐dilated aortas [69]. In BAV patients, HSP27 was significantly lower compared with TAV patients, suggesting a decreased resistance to physical and chemical stress in BAV aortas and thus an increased risk of TAA development [73]. Plasma TGF‐β was found to be significantly increased in patients with TAV and aortic dilatation compared with non‐dilated aortas. When BAV is present, an increase in TGF‐β was also observed, although the difference failed to reach statistical significance [74].
These findings have led some authors to suggest that dilatation in patients with TAV involves inflammatory processes, whereas aortic aneurysm in patients with BAV may be the consequence of impaired repair capacity [69]. Alterations in VSMC markers point to a contractile prevalence of VSMCs in BAV patients, whereas cases of TAA associated with TAV present VSMCs with a synthetic phenotype. However, more studies should be carried out to confirm this, especially in light of the fact that multiple subtypes of VSMCs in TAA have recently been described [75].
Molecular markers: from basic science to the clinic
Currently, there is a lack of molecular diagnostic markers of TAA. In fact, there is extremely limited evidence of any biomarker. As a result, when TAA is known to exist, the risk of a fatal event is estimated based on the aortic diameter, despite its poor sensitivity and specificity. In particular, few studies point to specific alterations in biological fluids, which may serve to monitor progression, rupture, or postsurgical follow‐up. Supplementary material, Table S2 summarizes the main molecular changes reported so far. Most reported variations were identified in the aortic tissue, and a few studies focused on specific arterial layers (mainly media) or cell types (VSMCs, ECs). An analysis of biological networks revealed three main biological networks involved in TAA, shown in supplementary material, Figure S2: (A) connective tissue disorders, (B) tissue development, and (C) cellular movement. Approximately 11–14% of molecules included in the three networks are enzymes. Transcription regulators are mainly involved in (A) (8.5%), transmembrane receptors in (B) (14%) and growth factors in (C) (11%).
Plasma levels of stromal cell‐derived factor‐1α were positively correlated with aneurysm size [76]. Stromal cell‐derived factor‐1α has been linked to cardiac regeneration, tissue repair, and wound healing in different cardiovascular diseases, thus casting doubt on its specificity for TAA. Similarly, plasma TGF‐β was found to be altered in TAA [74]. However, the difficulties that inhibit accurate measurement of plasma levels owing to the possible interference of platelets has compromised its use as a progression marker [77]. Alpha‐2‐macroglobulin was decreased in the sera of TAA patients [78]. The four and a half LIM domains protein 1 (FHL1) was also increased in TAA patients independently of aortic valve configuration [79]. Plasma levels of the amino terminal pro‐peptide of type III procollagen were higher in patients with a significant increase in aneurysm size, during a follow‐up period of 1.6 ± 0.8 years, compared with those showing no or limited growth [80]. This biological marker of collagen metabolism is not specific for TAA, as it has also been involved in hepatic disease or other affections of the cardiovascular system.
Serum MMP‐9 levels were higher in TAA compared with control subjects, influenced by age and hypertension, and were positively associated with C‐reactive protein [81]. Decreased levels of MMP‐8, MMP‐9, and TIMP‐1 were detected in plasma samples of TAA patients, and TIMP‐2 was found to be decreased in BAV subjects [66]. Pro‐MMP‐2 was found to be the predominant isoform in the serum and tissue of TAA, irrespective of aortic valve configuration, whereas active MMP‐2 was not detectable in serum, suggesting that it may not be released into systemic circulation [82]. Despite their promising role in TAA diagnosis, these markers are involved in several processes that are not specific to TAA: MMP‐2 and MMP‐9 in AAA; MMP‐8, MMP‐9, TIMP‐1, and TIMP‐2 in cancer; TIMP‐2 and tumor necrosis factor receptor (TNFR) in renal disease; MMP‐8, TIMP‐2, and collagens in pulmonary disease; and alpha‐2‐macroglobulin, MMP‐9, TIMP‐1, or collagens in other cardiovascular pathologies. Some authors have pointed out that measuring the MMP and TIMP ratio could improve specificity [83]. Nevertheless, more research should be undertaken to confirm its utility.
Significantly higher amounts of fibrillin‐1 fragments were detected in the blood of aneurysmatic patients [84]. Collagen content was also higher in the blood of TAA patients, a hallmark of aortic elastic lamellae fragmentation [50, 79], which triggers an inflammatory response. Elevated levels of IL‐6 and its receptor, IL6R, were found together with TNFR1 and TNFR2 in blood samples of TAA patients. An increase in C‐reactive protein was also found in blood samples of TAA patients [85]. However, these are general markers of inflammation and cannot be considered specific TAA markers.
Concluding remarks and future perspectives
This review has presented and discussed a full array of mechanical, structural, cellular, and molecular changes affecting the aneurysmatic aorta in non‐syndromic TAA, including the biological pathways involved. Specific attention has been paid to alterations associated with BAV.
One of the main goals in TAA cardiovascular medicine is to identify specific changes in the aorta that can be monitored in a biological fluid with screening potential and diagnostic purposes. However, previous evidence underscores the lack of proper markers of risk estimation for TAA patients. Individual altered molecules cannot be used for this purpose, so combining molecular markers in panels could help to define a specific tool with clinical potential. Additional efforts should follow to explore the potential of complementary multi‐omics approaches to provide a deeper insight into the molecular content of blood and so identify minor molecules playing key roles in TAA development that may serve as markers or/and therapeutic targets.
Author contributions statement
GAL, MML and GAE conceived the idea. GAL and MML designed the study. AMB and AHJ performed the systematic review. AMB, AHJ, MML and GAL drafted the first version of the manuscript. All authors provided critical feedback, contributed to the final manuscript, and accepted the final version.
Supporting information
Figure S1. PRISMA flowchart for medication review in TAA
Figure S2. Pathway analysis reveals three main networks involved in TAA
Table S1. Clinical trials of medication for TAA
Table S2. Molecular variations identified in sporadic TAA
Acknowledgements
The authors acknowledge Oliver Shaw from Fundación Jiménez Díaz for English‐language editing. This work was supported by the Instituto de Salud Carlos III co‐supported by FEDER grants (PI20/01103) and CAM (PEJD‐2019‐PRE/BMD‐16992 and 2018‐T2/BMD‐11561).
No conflicts of interest were declared.
References
- 1. Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res 2019; 124: 588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuzmik GA, Sang AX, Elefteriades JA. Natural history of thoracic aortic aneurysms. J Vasc Surg 2012; 56: 565–571. [DOI] [PubMed] [Google Scholar]
- 3. Elefteriades JA, Farkas EA. Thoracic aortic aneurysm: clinically pertinent controversies and uncertainties. J Am Coll Cardiol 2010; 55: 841–857. [DOI] [PubMed] [Google Scholar]
- 4. Sidloff D, Choke E, Stather P, et al. Mortality from thoracic aortic diseases and associations with cardiovascular risk factors. Circulation 2014; 130: 2287–2294. [DOI] [PubMed] [Google Scholar]
- 5. Oladokun D, Patterson BO, Sobocinski J, et al. Systematic review of the growth rates and influencing factors in thoracic aortic aneurysms. Eur J Vasc Endovasc Surg 2016; 51: 674–681. [DOI] [PubMed] [Google Scholar]
- 6. Boczar KE, Cheung K, Boodhwani M, et al. Sex differences in thoracic aortic aneurysm growth. Hypertension 2019; 73: 190–196. [DOI] [PubMed] [Google Scholar]
- 7. Erbel R, Aboyans V, Boileau C, et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2873–2926. [DOI] [PubMed] [Google Scholar]
- 8. Rylski B, Beyersdorf F, Kari FA, et al. Acute type A aortic dissection extending beyond ascending aorta: limited or extensive distal repair. J Thorac Cardiovasc Surg 2014; 148: 949–954. [DOI] [PubMed] [Google Scholar]
- 9. Jovin IS, Duggal M, Ebisu K, et al. Comparison of the effect on long‐term outcomes in patients with thoracic aortic aneurysms of taking versus not taking a statin drug. Am J Cardiol 2012; 109: 1050–1054. [DOI] [PubMed] [Google Scholar]
- 10. Stein LH, Berger J, Tranquilli M, et al. Effect of statin drugs on thoracic aortic aneurysms. Am J Cardiol 2013; 112: 1240–1245. [DOI] [PubMed] [Google Scholar]
- 11. Spartalis M, Tzatzaki E, Iliopoulos DC, et al. Captopril versus atenolol to prevent expansion rate of thoracic aortic aneurysms: rationale and design. Future Cardiol 2021; 17: 189–195. [DOI] [PubMed] [Google Scholar]
- 12. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71: 2199–2269.29146533 [Google Scholar]
- 13. Williams B, Mancia G, Spiering W, et al. 2018 practice guidelines for the management of arterial hypertension of the European Society of Hypertension and the European Society of Cardiology: ESH/ESC Task Force for the Management of Arterial Hypertension. J Hypertens 2018; 36: 2284–2309. [DOI] [PubMed] [Google Scholar]
- 14. Ashkan K, Milewicz DM. Structure of the elastin‐contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol 2016; 32: 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Khalil RA. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol 2018; 81: 241–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Humphrey JD, Schwartz MA, Tellides G, et al. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res 2015; 116: 1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van de Pol V, Kurakula K, DeRuiter MC, et al. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: what is the role of endothelial cells? Front Physiol 2017; 8: 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malashicheva A, Kostina D, Kostina A, et al. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int J Vasc Med 2016; 2016: 3107879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Billaud M, Hill JC, Richards TD, et al. Medial hypoxia and adventitial vasa vasorum remodeling in human ascending aortic aneurysm. Front Cardiovasc Med 2018; 5: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Billaud M, Donnenberg VS, Ellis BW, et al. Classification and functional characterization of vasa vasorum‐associated perivascular progenitor cells in human aorta. Stem Cell Reports 2017; 9: 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saito T, Hasegawa Y, Ishigaki Y, et al. Importance of endothelial NF‐κB signalling in vascular remodelling and aortic aneurysm formation. Cardiovasc Res 2013; 97: 106–114. [DOI] [PubMed] [Google Scholar]
- 22. Dinesh NEH, Reinhardt DP. Inflammation in thoracic aortic aneurysms. Herz 2019; 44: 138–146. [DOI] [PubMed] [Google Scholar]
- 23. He R, Guo DC, Sun W, et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg 2008; 136: 922–929, 929.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saito K, Yagi H, Maekawa K, et al. Lipidomic signatures of aortic media from patients with atherosclerotic and nonatherosclerotic aneurysms. Sci Rep 2019; 9: 15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao L, Moos MP, Gräbner R, et al. The 5‐lipoxygenase pathway promotes pathogenesis of hyperlipidemia‐dependent aortic aneurysm. Nat Med 2004; 10: 966–973. [DOI] [PubMed] [Google Scholar]
- 26. Kessler K, Borges LF, Ho‐Tin‐Noé B, et al. Angiogenesis and remodelling in human thoracic aortic aneurysms. Cardiovasc Res 2014; 104: 147–159. [DOI] [PubMed] [Google Scholar]
- 27. Bacakova L, Travnickova M, Filova E, et al. The role of vascular smooth muscle cells in the physiology and pathophysiology of blood vessels. In Muscle Cell and Tissue ‐ Current Status of Research Field, Sakuma K (ed.). IntechOpen: London, 2018; 229–257. [Google Scholar]
- 28. Martino F, Perestrelo AR, Vinarský V, et al. Cellular mechanotransduction: from tension to function. Front Physiol 2018; 9: 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol Rev 2019; 99: 1467–1525. [DOI] [PubMed] [Google Scholar]
- 30. Mimler T, Nebert C, Eichmair E, et al. Extracellular matrix in ascending aortic aneurysms and dissections – what we learn from decellularization and scanning electron microscopy. PLoS One 2019; 14: e0213794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakamoto N, Kiuchi T, Sato M. Development of an endothelial‐smooth muscle cell coculture model using phenotype‐controlled smooth muscle cells. Ann Biomed Eng 2011; 39: 2750–2758. [DOI] [PubMed] [Google Scholar]
- 32. Petsophonsakul P, Furmanik M, Forsythe R, et al. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol 2019; 39: 1351–1368. [DOI] [PubMed] [Google Scholar]
- 33. Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol 2017; 219: 382–408. [DOI] [PubMed] [Google Scholar]
- 34. Liu K, Fang C, Shen Y, et al. Hypoxia‐inducible factor 1α induces phenotype switch of human aortic vascular smooth muscle cell through PI3K/AKT/AEG‐1 signaling. Oncotarget 2017; 8: 33343–33352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Chiarini A, Onorati F, Marconi M, et al. Studies on sporadic non‐syndromic thoracic aortic aneurysms: 1. Deregulation of Jagged/Notch 1 homeostasis and selection of synthetic/secretor phenotype smooth muscle cells. Eur J Prev Cardiol 2018; 25(1_suppl): 42–50. [DOI] [PubMed] [Google Scholar]
- 36. Doppler C, Arnhard K, Dumfarth J, et al. Metabolomic profiling of ascending thoracic aortic aneurysms and dissections ‐ implications for pathophysiology and biomarker discovery. PLoS One 2017; 12: e0176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forte A, Della Corte A, De Feo M, et al. Role of myofibroblasts in vascular remodelling: focus on restenosis and aneurysm. Cardiovasc Res 2010; 88: 395–405. [DOI] [PubMed] [Google Scholar]
- 38. Jones JA, Spinale FG, Ikonomidis JS. Transforming growth factor‐beta signaling in thoracic aortic aneurysm development: a paradox in pathogenesis. J Vasc Res 2009; 46: 119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suh JH, Yoon JS, Kim HW, et al. Adventitial fibroblast abnormality in thoracic aortic aneurysms and aortic dissections. Korean J Thorac Cardiovasc Surg 2011; 44: 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tieu BC, Ju X, Lee C, et al. Aortic adventitial fibroblasts participate in angiotensin‐induced vascular wall inflammation and remodeling. J Vasc Res 2011; 48: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu CH, Mohammadmoradi S, Chen JZ, et al. Renin‐angiotensin system and cardiovascular functions. Arterioscler Thromb Vasc Biol 2018; 38: e108–e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ejiri J, Inoue N, Tsukube T, et al. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res 2003; 59: 988–996. [DOI] [PubMed] [Google Scholar]
- 43. Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 2001; 280: C53–C60. [DOI] [PubMed] [Google Scholar]
- 44. Branchetti E, Poggio P, Sainger R, et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res 2013; 100: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rateri DL, Davis FM, Balakrishnan A, et al. Angiotensin II induces region‐specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol 2014; 184: 2586–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salabei JK, Hill BG. Autophagic regulation of smooth muscle cell biology. Redox Biol 2015; 4: 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng YH, Tian C, Meng Y, et al. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J Cell Physiol 2012; 227: 127–135. [DOI] [PubMed] [Google Scholar]
- 48. Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non‐matrix substrates. Cardiovasc Res 2006; 69: 614–624. [DOI] [PubMed] [Google Scholar]
- 49. Le Page A, Khalil A, Vermette P, et al. The role of elastin‐derived peptides in human physiology and diseases. Matrix Biol 2019; 84: 81–96. [DOI] [PubMed] [Google Scholar]
- 50. Toumpoulis IK, Oxford JT, Cowan DB, et al. Differential expression of collagen type V and XI α‐1 in human ascending thoracic aortic aneurysms. Ann Thorac Surg 2009; 88: 506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nagasawa A, Yoshimura K, Suzuki R, et al. Important role of the angiotensin II pathway in producing matrix metalloproteinase‐9 in human thoracic aortic aneurysms. J Surg Res 2013; 183: 472–477. [DOI] [PubMed] [Google Scholar]
- 52. Gomez D, Al Haj Zen A, Borges LF, et al. Syndromic and non‐syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 2009; 218: 131–142. [DOI] [PubMed] [Google Scholar]
- 53. Takeda N, Hara H, Fujiwara T, et al. TGF‐β signaling‐related genes and thoracic aortic aneurysms and dissections. Int J Mol Sci 2018; 19: 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luyckx I, Loeys BL. The genetic architecture of non‐syndromic thoracic aortic aneurysm. Heart 2015; 101: 1678–1684. [DOI] [PubMed] [Google Scholar]
- 55. Renard M, Callewaert B, Baetens M, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int J Cardiol 2013; 165: 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor‐β signaling and vascular smooth muscle cell contractility. Circ Res 2013; 113: 327–340. [DOI] [PubMed] [Google Scholar]
- 57. van Dorst DCH, de Wagenaar NP, van der Pluijm I, et al. Transforming growth factor‐β and the renin‐angiotensin system in syndromic thoracic aortic aneurysms: implications for treatment. Cardiovasc Drugs Ther 2020. 10.1007/s10557-020-07116-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fava M, Barallobre‐barreiro J, Mayr U, et al. Role of ADAMTS‐5 in aortic dilatation and extracellular matrix remodeling. Arterioscler Thromb Vasc Biol 2018; 38: 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cikach FS, Koch CD, Mead TJ, et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight 2018; 3: e97167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chiarini A, Onorati F, Marconi M, et al. Studies on sporadic non‐syndromic thoracic aortic aneurysms: II. Alterations of extra‐cellular matrix components and focal adhesion proteins. Eur J Prev Cardiol 2018; 25: 51–58. [DOI] [PubMed] [Google Scholar]
- 61. Serhatli M, Baysal K, Acilan C, et al. Proteomic study of the microdissected aortic media in human thoracic aortic aneurysms. J Proteome Res 2014; 13: 5071–5080. [DOI] [PubMed] [Google Scholar]
- 62. Geng L, Wang W, Chen Y, et al. Elevation of ADAM10, ADAM17, MMP‐2 and MMP‐9 expression with media degeneration features CaCl2‐induced thoracic aortic aneurysm in a rat model. Exp Mol Pathol 2010; 89: 72–81. [DOI] [PubMed] [Google Scholar]
- 63. Ren P, Zhang L, Xu G, et al. ADAMTS‐1 and ADAMTS‐4 levels are elevated in thoracic aortic aneurysms and dissections. Ann Thorac Surg 2013; 95: 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ren P, Hughes M, Krishnamoorthy S, et al. Critical role of ADAMTS‐4 in the development of sporadic aortic aneurysm and dissection in mice. Sci Rep 2017; 7: 12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rabkin SW. Differential expression of MMP‐2, MMP‐9 and TIMP proteins in thoracic aortic aneurysm – comparison with and without bicuspid aortic valve: a meta‐analysis. Vasa 2014; 43: 433–442. [DOI] [PubMed] [Google Scholar]
- 66. Ikonomidis JS, Ivey CR, Wheeler JB, et al. Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurysm disease. J Thorac Cardiovasc Surg 2013; 145: 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matsumoto K, Satoh K, Maniwa T, et al. Proteomic comparison between abdominal and thoracic aortic aneurysms. Int J Mol Med 2014; 33: 1035–1047. [DOI] [PubMed] [Google Scholar]
- 68. Boyum J, Fellinger EK, Schmoker JD, et al. Matrix metalloproteinase activity in thoracic aortic aneurysms associated with bicuspid and tricuspid aortic valves. J Thorac Cardiovasc Surg 2004; 127: 686–691. [DOI] [PubMed] [Google Scholar]
- 69. Kjellqvist S, Maleki S, Olsson T, et al. A combined proteomic and transcriptomic approach shows diverging molecular mechanisms in thoracic aortic aneurysm development in patients with tricuspid‐ and bicuspid aortic valve. Mol Cell Proteomics 2013; 12: 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stern C, Scharinger B, Tuerkcan A, et al. Strong signs for a weak wall in tricuspid aortic valve associated aneurysms and a role for osteopontin in bicuspid aortic valve associated aneurysms. Int J Mol Sci 2019; 20: 4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kotlarczyk MP, Billaud M, Green BR, et al. Regional disruptions in endothelial nitric oxide pathway associated with bicuspid aortic valve. Ann Thorac Surg 2016; 102: 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Billaud M, Phillippi JA, Kotlarczyk MP, et al. Elevated oxidative stress in the aortic media of patients with bicuspid aortic valve. J Thorac Cardiovasc Surg 2017; 154: 1756–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matt P, Fu Z, Carrel T, et al. Proteomic alterations in heat shock protein 27 and identification of phosphoproteins in ascending aortic aneurysm associated with bicuspid and tricuspid aortic valve. J Mol Cell Cardiol 2007; 43: 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rueda‐Martínez C, Lamas O, Carrasco‐Chinchilla F, et al. Increased blood levels of transforming growth factor β in patients with aortic dilatation. Interact Cardiovasc Thorac Surg 2017; 25: 571–574. [DOI] [PubMed] [Google Scholar]
- 75. Li Y, Ren P, Dawson A, et al. Single‐cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation 2020; 142: 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Parietti E, Pallandre JR, Deschaseaux F, et al. Presence of circulating endothelial progenitor cells and levels of stromal‐derived factor‐1α are associated with ascending aorta aneurysm size. Eur J Cardiothorac Surg 2011; 40: e6–e12. [DOI] [PubMed] [Google Scholar]
- 77. Fletcher AJ, Syed MBJ, Aitman TJ, et al. Inherited thoracic aortic disease: new insights and translational targets. Circulation 2020; 141: 1570–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Satoh K, Maniwa T, Oda T, et al. Proteomic profiling for the identification of serum diagnostic biomarkers for abdominal and thoracic aortic aneurysms. Proteome Sci 2013; 11: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Black KM, Masuzawa A, Hagberg RC, et al. Preliminary biomarkers for identification of human ascending thoracic aortic aneurysm. J Am Heart Assoc 2013; 2: e000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ormezzano O, Baguet JP, Thony F, et al. Aminoterminal propeptide of type III procollagen (PIIINP) is associated with ascending aortic aneurysm growth rate. Int J Cardiol 2010; 145: 379–380. [DOI] [PubMed] [Google Scholar]
- 81. Li T, Jiang B, Li X, et al. Serum matrix metalloproteinase‐9 is a valuable biomarker for identification of abdominal and thoracic aortic aneurysm: a case‐control study. BMC Cardiovasc Disord 2018; 18: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tscheuschler A, Meffert P, Beyersdorf F, et al. MMP‐2 isoforms in aortic tissue and serum of patients with ascending aortic aneurysms and aortic root aneurysms. PLoS One 2016; 11: e0164308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wilton E, Bland M, Thompson M, et al. Matrix metalloproteinase expression in the ascending aorta and aortic valve. Interact Cardiovasc Thorac Surg 2008; 7: 37–40. [DOI] [PubMed] [Google Scholar]
- 84. Marshall LM, Carlson EJ, O'Malley J, et al. Thoracic aortic aneurysm frequency and dissection are associated with fibrillin‐1 fragment concentrations in circulation. Circ Res 2013; 113: 1159–1168. [DOI] [PubMed] [Google Scholar]
- 85. Rabajdová M, Urban P, Špaková I, et al. Detection of pathological changes in the aorta during thoracic aortic aneurysm progression on molecular level. Dis Markers 2017; 2017: 9185934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PRISMA flowchart for medication review in TAA
Figure S2. Pathway analysis reveals three main networks involved in TAA
Table S1. Clinical trials of medication for TAA
Table S2. Molecular variations identified in sporadic TAA