

Abstract
Objective
Data on the magnitude of benefit of modern therapies for pulmonary arterial hypertension (PAH) in connective tissue disease (CTD)–associated PAH are limited. In this study, we performed meta‐analyses of randomized, controlled trials (RCTs) and registries to quantify the benefit of these modern therapies in patients with CTD‐PAH.
Methods
The PubMed and Embase databases were searched for articles reporting data from RCTs or registries published between January 1, 2000 and November 25, 2019. Eligibility criteria included multicenter studies with ≥30 CTD‐PAH patients. For an RCT to be included, the trial had to evaluate an approved PAH therapy, and long‐term risks of clinical morbidity and mortality or 6‐minute walk distance had to be reported. For a registry to be included, survival rates had to be reported. Random‐effects models were used to pool the data.
Results
Eleven RCTs (total of 4,329 patients; 1,267 with CTD‐PAH) and 19 registries (total of 9,739 patients; 4,008 with CTD‐PAH) were included. Investigational therapy resulted in a 36% reduction in the risk of clinical morbidity/mortality events both in the overall PAH population (hazard ratio [HR] 0.64, 95% confidence interval [95% CI] 0.54, 0.75; P < 0.001) and in CTD‐PAH patients (HR 0.64, 95% CI 0.51, 0.81; P < 0.001) as compared to control subjects. The survival rate was lower in CTD‐PAH patients compared to all PAH patients (survival rate 62%, 95% CI 57, 67% versus 72%, 95% CI 69, 75% at 3 years). The survival rate in CTD‐PAH patients treated primarily after 2010 was higher than that in CTD‐PAH patients treated before 2010 (survival rate 73%, 95% CI 62, 81% versus 65%, 95% CI 59, 71% at 3 years).
Conclusion
Modern therapy provides a similar reduction in morbidity/mortality risk in patients with CTD‐PAH when compared to the PAH population overall. Risk of death is higher in CTD‐PAH patients than in those with PAH overall, but survival has improved in the last 10 years, which may be related to increased screening and/or new treatment approaches. Early detection of PAH in patients with CTD and up‐front intensive treatment are warranted.
INTRODUCTION
Pulmonary arterial hypertension (PAH) leads to right ventricular dysfunction and failure, with a median survival of ~3 years from the time of diagnosis (1,2). Connective tissue disease (CTD)–associated PAH is historically associated with shortened survival compared to idiopathic PAH (IPAH) (3, 4, 5, 6). Early detection of PAH with established methods among patients with CTDs, such as those with systemic sclerosis (SSc) (7), and subsequent early treatment may improve survival outcomes (8). Rheumatologists are in a unique and critical position to identify these patients.
Availability of new and combination therapy approaches targeting multiple pathophysiologic pathways have led to improved outcomes in PAH (9, 10, 11, 12, 13, 14, 15, 16). However, trials of PAH therapies generally enroll patients with different etiologies of PAH, and trials devoted solely to those with CTD‐PAH are rare; therefore, the magnitude of treatment effect in CTD‐PAH is poorly defined, as these patients represent a subgroup in most trials, albeit a large one. Furthermore, data on whether new treatment approaches have resulted in improved survival in CTD‐PAH are lacking.
For the present study, we conducted 2 meta‐analyses. In one meta‐analysis, we analyzed randomized, controlled trials (RCTs) to evaluate the magnitude of benefit of US Food and Drug Administration (FDA)–approved PAH therapies in patients with CTD‐PAH. In the other meta‐analysis, we analyzed real‐world observational disease registries to compare survival outcomes between patients with CTD‐PAH and the overall PAH population, and between patients treated mostly before 2010 and those treated mostly after 2010. Compared to prior meta‐analyses that have evaluated outcomes in RCTs among patients with CTD‐PAH (17,18), our RCT meta‐analysis provides a more contemporary data set that includes modern agents and treatment paradigms, as well as a larger sample size. Our second meta‐analysis extends these findings by evaluating long‐term survival outcomes, an end point that is not typically included in RCTs because of their shorter duration. We also investigated survival over time, to determine whether the availability of newer therapies and treatment approaches has translated into improved survival in real‐world settings.
PATIENTS AND METHODS
Study design
These meta‐analyses were conducted in accordance with the Preferred Reporting Items for Systematic Review and Meta‐Analysis guidelines (19), with a modification suited to the rare disease state of PAH. Specifically, we conducted a comprehensive literature search, instead of a systematic review, to identify peer‐reviewed reports of RCTs evaluating new therapies and disease registries. We did not expand the search to databases beyond PubMed and Embase, nor did we examine reference lists and non‐database sources for additional information, because of the very low likelihood of this method yielding additional articles in this rare disease. The protocol was registered with the International Prospective Register of Systematic Reviews (PROSPERO registration no. CRD42020167119) (20).
Search strategy and selection criteria
PubMed and Embase (Elsevier) were searched for English‐only articles reporting data from RCTs or registries and published between January 1, 2000 and November 25, 2019. Specific parameters (search terms, Boolean operators, and filters) were applied. To identify RCTs, we searched in PubMed for the term “pulmonary arterial hypertension” in the title along with “AND (randomized OR randomised)” and restricted the search to human subjects; in Embase, we searched for “(‘pulmonary hypertension’/exp OR ‘pulmonary hypertension’)” and restricted the search to phase III or phase IV RCTs. To identify registries, we searched in PubMed for the term “pulmonary arterial hypertension” in the title along with “AND (registry OR observation OR consecutive OR multicenter OR multicentre)” and restricted the search to human subjects; in Embase, we searched for “pulmonary hypertension” in the title along with “AND (‘observational study’/exp OR ‘observational study’).”
RCTs and registries had to meet the following inclusion criteria. 1) The RCT or registry was conducted at multiple centers. 2) Enrolled patients were adult patients with World Health Organization (WHO) group 1 pulmonary hypertension (i.e., PAH) (21). 3) The RCT or registry included ≥30 patients with CTD‐PAH. 4) Publicly available CTD‐PAH–specific outcomes data were provided for the CTD‐PAH subgroup. 5) Enrollment of patients began in 2000 or later. 6) Long‐term incidence rates of clinical morbidity and/or mortality were reported (median enrollment time ≥6 months).
Only peer‐reviewed data were included. Additional inclusion criteria for RCTs were as follows: the RCT was a phase III or phase IV study; the evaluated PAH therapy had received current approval from the FDA; patients were exposed to the study drug for PAH treatment for at least 3 months; and one of the defined primary or secondary end points was time to clinical morbidity/mortality, time to clinical worsening, or 6‐minute walk distance (6MWD) measured 3–6 months from baseline.
To minimize the risk of bias in study selection, we utilized the above‐noted strict prespecified inclusion/exclusion criteria. This involved a detailed review of each study design, patient inclusion and exclusion criteria, and definition of study end points. Studies not meeting the prespecified criteria were excluded. In addition, at least 2 reviewers independently verified the studies that were to be included in the analyses, with any disagreements arbitrated by the lead author (DK) and senior author (VM).
Publications providing the same data from the same RCT or registry were removed. For multiple publications from a single study, the most recent publication containing data on the CTD‐PAH population was utilized. Data from all primary reports of RCTs were included in the analyses of all PAH patients and CTD‐PAH patients, unless more detailed information for CTD‐PAH patients were included in later post hoc analyses. When we extracted data from the post hoc analyses of the CTD‐PAH subgroup, we ensured that the number of patients in the CTD‐PAH subgroup and the statistical analysis method were consistent with that described in the primary report. If multiple registries were conducted in a single country, only studies that did not substantially overlap in enrollment period were included, to avoid capturing data from the same patient in multiple registries.
Data were extracted from RCT and registry publications separately by 2 team members (with medical, science, or statistical expertise) under the leadership of statisticians at Actelion Pharmaceuticals. Extracted data were verified by a third team member independently. In the event of a discrepancy, a statistician verified the data prior to the final statistical analysis, and one of the authors (JH) arbitrated any disagreements.
Data were extracted separately for patients with any of the PAH etiologies and for patients with CTD‐PAH. Baseline data extracted for both RCTs and registries were the 6MWD, age, sex, WHO functional class, and PAH etiology. Data extracted after baseline were the change in the 6MWD from baseline to between 3 and 6 months, number of clinical morbidity/mortality events, and hazard ratio (HR) (with 95% confidence interval [95% CI]) for the long‐term risk of morbidity/mortality in RCTs. In addition, data from the registries included the survival rates at 1, 2, and 3 years, as reported in the registry or as determined from Kaplan‐Meier survival curves using a graph digitizer.
Statistical analysis
The meta‐analysis of RCTs evaluated the effect of PAH therapies on time to clinical morbidity and/or mortality in all patients and in patients with CTD‐PAH, as well as the effect on the 6MWD measured between 3 and 6 months after initiation of study treatment. The components of the clinical morbidity/mortality end points varied among the studies (see Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract). The meta‐analysis of registries evaluated survival outcomes in all patients and in patients with CTD‐PAH. Analysis populations are defined in Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract.
To assess heterogeneity among studies, we calculated the I2 value associated with the fixed‐effects meta‐analysis models. These values indicated that most analyses using fixed‐effects models had high heterogeneity, whereas analyses using random‐effects meta‐analysis models had I2 values that were considered to be within the acceptable range of heterogeneity (I2 lower than 50%) (see Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract). Thus, we controlled for heterogeneity among studies by consistently using random‐effects meta‐analysis models to pool results, using inverse variance weighting followed by unweighting with application of a random‐effects variance component. The overall treatment effect estimate was calculated using the DerSimonian and Laird random‐effects method (22).
Time‐to‐event end points were estimated using the Kaplan‐Meier method. Survival rates at 1, 2, and 3 years in the registries were extracted from Kaplan‐Meier curves and were stratified by study period mostly before or after 2010, to assess the impact of newer treatment approaches. Outcomes were analyzed for the overall PAH population and stratified by disease etiology (all CTD‐PAH patients and CTD‐PAH subtypes [SSc or systemic lupus erythematosus (SLE) or IPAH]). Registries with ≥50% of the study period in 2010 or later were classified as the after‐2010 group.
Sensitivity analyses included analysis of treatment effect in RCTs in patients with IPAH compared to patients with CTD‐PAH. In the registries, analysis of survival rate in selected studies containing both CTD‐PAH and other etiologies was performed to confirm the historical difference between etiologies.
A forest plot showing the effect size and associated variability in each study, as well as the combined effect, was created to examine the consistency of results. If any outliers were apparent, the data extraction was verified from the original source and the units were confirmed to ensure that no unit conversion was necessary. If, after this, an outlier was detected, a sensitivity analysis removing the outlier could be conducted to assess the impact on the overall analysis. However, no such outliers were found in our analysis.
Statistical analyses were performed using Comprehensive Meta‐Analysis version 3 software (Biostat).
RESULTS
Study and patient characteristics
With regard to RCTs, a total of 801 articles were identified through our comprehensive search strategy (see Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract) and 11 studies were ultimately included in the meta‐analysis (as listed in Supplementary Table 4 on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract). Among those that met the criteria for one of the defined primary end points, 5 RCTs reported time to clinical morbidity/mortality events (12, 13, 14, 15, 16,23,24) (specifically defined in Supplementary Table 1 [http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract]), and 6 RCTs reported change in the 6MWD (10,11,25, 26, 27, 28, 29, 30). The 11 RCTs enrolled a total of 4,329 patients with PAH, including 1,267 patients with CTD‐PAH (29.3%). Each RCT evaluated the addition of a PAH‐specific therapy to a patient’s current care, so that patients were stratified according to whether they received no PAH‐specific treatment, monotherapy, or dual combination therapy.
With regard to observational registries, a total of 1,389 articles were identified through our search (see Supplementary Figure 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract) and 19 registries were ultimately included in the meta‐analysis (as listed in Supplementary Table 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract): in 9 of the registries, patients with PAH of all etiologies were enrolled (4,5,31, 32, 33, 34, 35, 36, 37), and in 10 registries, only patients with CTD‐PAH were enrolled (38, 39, 40, 41, 42, 43, 44, 45, 46, 47). The 19 registries enrolled 9,739 patients with PAH, including 4,008 patients with CTD‐PAH (41.2%).
At baseline both in the RCTs and in the registries, patients with CTD‐PAH were older and had a lower mean 6MWD compared to the overall PAH population. In RCTs, patients with PAH of any etiology had a mean age of 50 years, 78–79% were female, and 41–43% had WHO functional class I or II disease (Table 1). Patients with CTD‐PAH in the RCTs had a mean age of 55–56 years, compared to a mean age of 50 years among patients of all PAH etiologies, and had a mean 6MWD of 337–339 meters, compared to a mean 6MWD of 355–357 meters among patients of all PAH etiologies (see Supplementary Tables 6 and 7 on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract).
Table 1.
Baseline characteristics of all patients with PAH in the randomized, controlled trials*
| Study (ref.) | Investigational treatment group | Control group | ||||||
|---|---|---|---|---|---|---|---|---|
| No. of patients |
Age, mean ± SD years |
Female, % | WHO functional class I–II, % | No. of subjects |
Age, mean ± SD years |
Female, % | WHO functional class I–II, % | |
| AMBITION (12) | 253 | 55 ± 14 | 74 | 30 | 247 | 54 ± 15 | 81 | 32 |
| GRIPHON (15) | 574 | 48 ± 15 | 80 | 48 | 582 | 48 ± 16 | 80 | 45 |
| SERAPHIN (14) | 242 | 45 ± 15 | 80 | 50 | 250 | 47 ± 17 | 74 | 52 |
| PHIRST (10) | 79 | 53 ± 15 | 75 | 35 | 82 | 55 ± 15 | 79 | 29 |
| ARIES‐1 (28) | 67 | 49 ± 16 | 79 | 36 | 67 | 48 ± 16 | 88 | 37 |
| ARIES‐2 (28) | 63 | 50 ± 16 | 81 | 46 | 65 | 51 ± 14 | 68 | 40 |
| PATENT (11) | 254 | 51 ± 17 | 80 | 45 | 126 | 51 ± 17 | 78 | 51 |
| SUPER‐1 (9) | 71 | 48 ± 15 | 79 | 39 | 70 | 49 ± 17 | 81 | 47 |
| BREATHE‐1 (25) | 144 | 49 ± 16 | 79 | 0 | 69 | 47 ± 16 | 78 | 0 |
| COMPASS‐2 (13) | 159 | 53 ± 15 | 79 | 45 | 175 | 55 ± 16 | 73 | 39 |
| FREEDOM‐EV (16) | 346 | 46 ± 16 | 80 | 62 | 344 | 45 ± 15 | 78 | 70 |
| All studies | 2,252 | 50 ± 1.1† | 79 | 41 | 2,077 | 50 ± 1.2† | 78 | 43 |
PAH = pulmonary arterial hypertension; WHO = World Health Organization; AMBITION = Ambrisentan plus Tadalafil in PAH; GRIPHON = Prostacyclin Receptor Agonist (Prostaglandin I2) in PAH; SERAPHIN = Study with an Endothelin Receptor Antagonist in PAH to Improve Clinical Outcome; PHIRST = PAH and Response to Tadalafil; ARIES = Randomized, Double‐blind, Placebo‐controlled, Multicenter, Efficacy Study of Ambrisentan for PAH (1 and 2); PATENT = PAH Soluble Guanylate Cyclase–Stimulator Trial 1; SUPER‐1 = Sildenafil Use in PAH; BREATHE‐1 = Bosentan Randomized Trial of Endothelin Antagonist Therapy; COMPASS‐2 = Combination of Bosentan and Sildenafil Versus Sildenafil Monotherapy on PAH; FREEDOM‐EV = International, Multicenter, Randomized, Double‐blind, Placebo‐controlled Event‐driven Trial of Oral Treprostinil in Subjects with PAH.
Values are the estimated mean ± SEM from the random‐effects model.
In all 16 registries in which baseline characteristics were reported separately for the CTD‐PAH population, patients with CTD‐PAH had a mean age of 55 years, 87% were female, 30% had WHO functional class I or II disease, and the mean 6MWD was 327 meters (see Supplementary Table 8 on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract). In the 9 registries in which patients with all PAH etiologies were enrolled, the patients had a mean age of 51 years, 74% were female, 28% had WHO functional class I or II disease, and the mean 6MWD was 348 meters. Patients with CTD‐PAH in these 9 registries had a mean age of 56 years, 84% were female, 24% had WHO functional class I or II disease, and the mean 6MWD was 328 meters (Table 2). Baseline data from the registries for the CTD‐PAH subgroups treated before 2010 and those treated after 2010 are shown in Supplementary Table 9, and baseline data from the registries for the CTD‐PAH subgroups of SSc and SLE are shown in Supplementary Table 10 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract).
Table 2.
Baseline characteristics of the registry patients with PAH of any etiology and patients with CTD‐PAH*
| Registry (ref.) | All patients (n = 7,844) | Patients with CTD‐PAH (n = 2,113) | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
Age, mean ± SD years |
Female, % |
WHO functional class I–II, % |
6MWD, mean ± SD meters |
CTD, % |
Age, mean ± SD years |
Female, % |
WHO functional class I–II, % |
6MWD, mean ± SD meters |
|
| REHAP (31) | 45 ± 17 | 71 | 31 | 363 ± 120 | 18 | 54 ± 15 | 90 | 21 | 309 ± 115 |
| PAH‐QuERI (32) | 55 ± 16 | 77 | 47 | NR | 29 | NR | NR | NR | NR |
| COMPERA (5) | 64 ± 16 | 64 | 11 | 298 ± 126 | 22 | 66 ± 13 | 78 | 11 | 273 ± 130 |
| French PAH Network Registry (4) | 50 ± 15 | 66 | 25 | 329 ± 109 | 15 | 56 ± 15 | 80 | 26 | 315 ± 111 |
| REVEAL (33) | 50 ± 17 | 77 | NR | NR | 28 | NR | NR | NR | NR |
| Turkish registry (34) | 46 ± 17 | 77 | 21 | NR | 22 | NR | NR | NR | NR |
| Chinese Registry‐PAH (35) | 36 ± 15 | 76 | 46 | 390 ± 111 | 37 | 42 ± 14 | 85 | 45 | 384 ± 107 |
| BPR (36) | 59 ± 17 | 77 | 11 | NR | 42 | 62 ± 11 | 85 | 6 | NR |
| KORPAH (37) | 50 ± 17 | 78 | 53 | 363 ± 116 | 58 | 54 ± 17 | 85 | 63 | 358 ± 114 |
| All registries | 51 ± 2.7† | 74 | 28 | 348 ± 16.4† | 29 | 56 ± 3.3† | 84 | 24 | 328 ± 20.1† |
PAH = pulmonary arterial hypertension; CTD‐PAH = connective tissue disease–associated PAH; WHO = World Health Organization; 6MWD = 6‐minute walk distance; REHAP = Spanish Registry of PAH; PAH‐QuERI = PAH Quality Enhancement Research Initiative; NR = not reported; COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; REVEAL = Registry to Evaluate Early and Long‐term PAH Disease Management; BPR = Bosentan Patient Registry; KORPAH = Korean Registry of PAH.
Values are the estimated mean ± SEM from the random‐effects model.
Outcomes from RCTs
Among the 5 RCTs in which time to clinical morbidity/mortality events was reported as the primary end point, 3,172 patients were enrolled (941 with CTD‐PAH [30%]) (12, 13, 14, 15, 16,23,24). Additional PAH therapy resulted in a 36% reduction in the risk of morbidity/mortality events in the overall PAH population compared to that in control subjects (HR 0.64, 95% CI 0.54, 0.75; P < 0.001), and a 36% reduction in the risk of morbidity/mortality events in patients with CTD‐PAH compared to controls (HR 0.64, 95% CI 0.51, 0.81; P < 0.001) (Figure 1).
Figure 1.
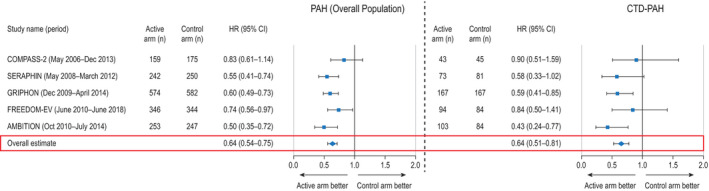
Time to clinical morbidity/mortality event for all patients with pulmonary arterial hypertension (PAH) (left) and patients with connective tissue disease (CTD)–associated PAH (right) in randomized, controlled trials that evaluated time to clinical morbidity/mortality event as a primary end point (5 trials). Results are depicted as forest plots, showing the hazard ratio (HR) with 95% confidence interval (95% CI) in the active treatment group relative to the control group. Overall HRs were estimated using random‐effects models. COMPASS‐2 = Combination of Bosentan and Sildenafil Versus Sildenafil Monotherapy on PAH; SERAPHIN = Study with an Endothelin Receptor Antagonist in PAH to Improve Clinical Outcome; GRIPHON = Prostacyclin Receptor Agonist (Prostaglandin I2) in PAH; FREEDOM‐EV = International, Multicenter, Randomized, Double‐blind, Placebo‐controlled Event‐driven Trial of Oral Treprostinil in Subjects with PAH; AMBITION = Ambrisentan plus Tadalafil in PAH.
In the overall PAH population, additional PAH therapy led to a placebo‐ or monotherapy‐corrected increase in the 6MWD (mean increase of 28.6 meters, 95% CI 19.2, 38.0; P < 0.001) (Figure 2A). In 8 RCTs (total of 2,874 patients; 882 with CTD‐PAH [31%]), this end point was reported according to CTD‐PAH etiology (9, 10, 11, 12,15,23, 24, 25, 26, 27, 28, 29, 30). Additional PAH therapy led to an increase in the 6MWD in the overall PAH population (mean increase of 34.6 meters, 95% CI 22.1, 47.1; P < 0.001) and in patients with CTD‐PAH (mean increase of 20.4 meters, 95% CI 10.9, 29.9; P < 0.001) (Figures 2B and C).
Figure 2.
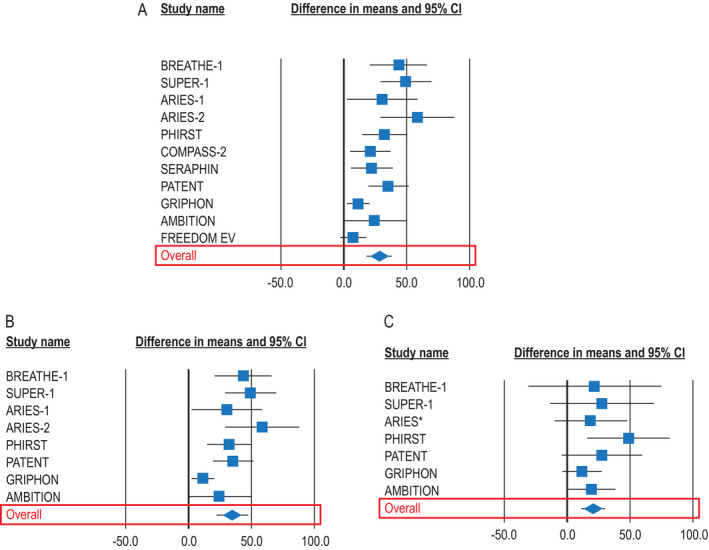
Change in the 6‐minute walk distance (6MWD) for all patients with pulmonary arterial hypertension (PAH) of any etiology in all randomized, controlled trials (RCTs) (11 trials) (A), for all patients in RCTs that reported 6MWD in patients with connective tissue disease (CTD)–associated PAH (8 trials) (B), and for patients with CTD‐PAH (8 trials) (C). Results are depicted as forest plots, showing the mean change in the 6MWD from baseline to between 3 and 6 months, with 95% confidence interval (95% CI). *Combined data from the Randomized, Double‐blind, Placebo‐controlled, Multicenter, Efficacy Study of Ambrisentan for PAH 1 and 2 (ARIES‐1 and ARIES‐2, respectively). BREATHE‐1 = Bosentan Randomized Trial of Endothelin Antagonist Therapy; SUPER‐1 = Sildenafil Use in PAH; PHIRST = PAH and Response to Tadalafil; COMPASS‐2 = Combination of Bosentan and Sildenafil Versus Sildenafil Monotherapy on PAH; SERAPHIN = Study with an Endothelin Receptor Antagonist in PAH to Improve Clinical Outcome; PATENT = PAH Soluble Guanylate Cyclase–Stimulator Trial 1; GRIPHON = Prostacyclin Receptor Agonist (Prostaglandin I2) in PAH; AMBITION = Ambrisentan plus Tadalafil in PAH; FREEDOM‐EV = International, Multicenter, Randomized, Double‐blind, Placebo‐controlled Event‐driven Trial of Oral Treprostinil in Subjects with PAH.
Sensitivity analyses were performed to compare outcomes between patients with CTD‐PAH and patients with IPAH among the subset of trials in which outcomes were reported separately in the IPAH subpopulation. Results from patients with IPAH trended similar to those in the overall PAH population (HR for risk of morbidity/mortality events 0.63, 95% CI 0.54, 0.73; P < 0.001).
Outcomes from registries
Among the 9 registries in which patients with PAH were included irrespective of etiology (4,5,31,32, 33, 34, 35, 36, 37), survival rates in the 2,113 patients with CTD‐PAH were lower than in the 7,829 patients in the overall PAH population (Figure 3A).
Figure 3.
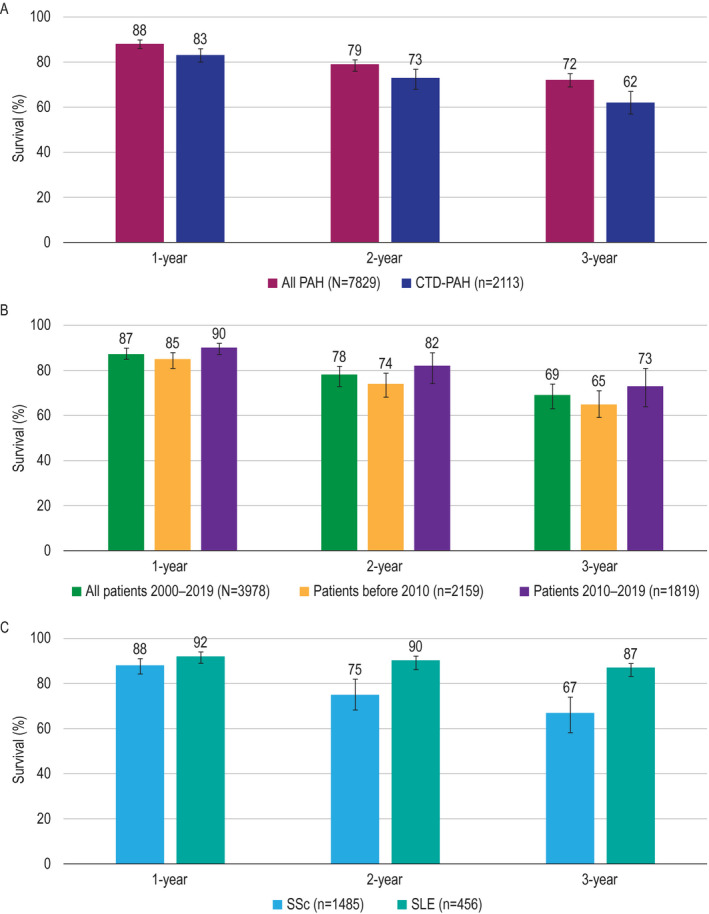
Survival estimates at 1 year, 2 years, and 3 years in all patients with pulmonary arterial hypertension (PAH) and patients with connective tissue disease (CTD)–associated PAH from the registries in which all PAH etiologies were included (9 registries) (A), in patients with CTD–associated PAH from all registries (19 registries) by enrollment period (B), and by CTD subtype from disease‐specific registries or registries that included disease‐specific outcomes (8 registries for systemic sclerosis [SSc] and 4 registries for systemic lupus erythematosus [SLE]) (C). Results are shown as the survival rates with 95% confidence intervals.
Among all CTD‐PAH patients with available data, including those from both the all‐PAH registries and the CTD‐PAH–specific registries (19 registries, 3,978 patients), survival rates in patients with CTD‐PAH in the registries in which treatment was received within ≥50% of the study period during or after 2010 (n = 1,819) were higher than in patients with CTD‐PAH in the registries in which treatment was received within ≥50% of the study period occurring before 2010 (n = 2,159) (Figure 3B).
Among all patients with CTD‐PAH, survival rates were lower for those with SSc (n = 1,485) (36,38,41,43,44,46,47) compared to those with SLE (n = 456) (39,40,42,45) (Figure 3C).
DISCUSSION
Our meta‐analysis of RCTs demonstrated that patients with CTD‐PAH derive a clinically significant benefit from currently available PAH therapies, which, in many patients, comprises the addition of a drug targeting a second or third pathway involved in the pathophysiology of PAH. Our meta‐analysis of registries showed that patients with CTD‐PAH have a higher risk of death than the overall PAH population; however, survival has improved among the CTD‐PAH population treated mostly in the last 10 years compared to earlier patient populations.
Two other relatively recent meta‐analyses have also evaluated the benefit of PAH‐specific therapy in patients with CTD‐PAH (17,18). Rhee and colleagues (17) evaluated individual patient data from 11 RCTs published between 2002 and 2013 (total of 2,762 patients; 827 with CTD‐PAH [30%]). Most of the trials (59% of patients) evaluated endothelin receptor antagonists (ERAs). Similar to our findings based on the 6MWD, patients with CTD‐PAH experienced less benefit than patients with IPAH. The mean placebo‐corrected treatment effect, measured as the change in 6MWD from baseline to 3 months, was 23.1 meters in patients with CTD‐PAH versus 40.4 meters in patients with IPAH (adjusted treatment effect difference −17.3 meters, 90% CI −31.3, −3.3; P for interaction = 0.043). We reported a similar placebo‐ or monotherapy‐corrected mean change in the 6MWD of 20.4 meters in patients with CTD‐PAH. Our reference population included patients with all PAH etiologies, which may explain the lower benefit (difference of 34.6 meters for the change in the 6MWD) observed in our study compared to that observed in patients with IPAH reported by Rhee and colleagues (17). However, earlier meta‐analyses of RCTs reporting change in the 6MWD are also conflicting, with one study showing a similar treatment benefit between patients with CTD‐PAH and those with PAH of all etiologies (48) and another showing no treatment benefit in patients with CTD‐PAH (49). Time to clinical worsening was not significantly prolonged among patients with CTD‐PAH in the meta‐analysis by Rhee and colleagues (17), as the odds ratio for the likelihood of a longer time to clinical worsening was 0.72 (95% CI 0.45, 1.16), whereas we demonstrated a reduction in the risk of a morbidity/mortality event with PAH‐specific therapy (HR 0.64, 95% CI 0.51, 0.80).
The difference between meta‐analyses may result from several factors. We believe that our analysis provides a more precise estimate of treatment effect because we applied more stringent statistical methods to pool the studies. Specifically, we measured the time to a clinical morbidity/mortality event by using the HR, which averages the treatment effect over the entire study period. Rhee and colleagues measured clinical worsening events using an odds ratio, which is affected by differences in study duration. Further, our study required a median study duration of ≥6 months to capture long‐term clinical morbidity and mortality (current standards to assess overall benefit), whereas ~50% of the trials included in the meta‐analysis by Rhee and colleagues (17) were of 12 weeks to 18 weeks in duration (i.e., a previous standard to assess PAH therapy efficacy). Finally, we used a more contemporary data set, which included trials of the most recently available PAH therapies, comprising oral ERAs, phosphodiesterase type 5 inhibitors, oral prostacyclin pathway agents, and riociguat, as well as more use of combination therapy. This data set, thus, more accurately reflects current treatment approaches. This approach also resulted in a larger patient population (1,267 CTD‐PAH patients from RCTs, compared to 827 patients in the analysis by Rhee and colleagues), which increases the precision of the statistical estimates.
Pan and colleagues (18) analyzed data extracted from 6 RCTs published between 2011 and 2017 (total of 3,262 patients; 963 with CTD‐PAH [30%]). These trials evaluated ERAs, tadalafil, selexipag, and riociguat. This meta‐analysis aimed to compare combination therapy to monotherapy; however, background therapy varied among studies and patients within the studies. Among 4 RCTs in the CTD‐PAH subset included in the analysis, additional PAH therapy led to a 27% reduction in relative risk for clinical worsening (pooled relative risk 0.73, 95% CI 0.60, 0.89; P = 0.002). These data are consistent with our finding of a 36% reduction in risk of a clinical morbidity/mortality event in the CTD‐PAH population. There were differences in methodology between the 2 analyses. In our study, for clinical relevance, only those treatment groups receiving FDA‐approved doses were analyzed. Additionally, our meta‐analysis included the more recently published FREEDOM‐EV trial (International, Multicenter, Randomized, Double‐blind, Placebo‐controlled Event‐driven Trial of Oral Treprostinil in Subjects with PAH) (16), the results of which were published after completion of the meta‐analysis by Pan and colleagues (18). Pan and colleagues also found no statistically significant benefit from additional therapy in terms of improvement in the 6MWD among patients with CTD‐PAH (mean change 21.38 meters, 95% CI −20.38, 63.14; P = 0.32). This end point was derived from 3 RCTs. Our meta‐analysis, which included 8 trials in which this end point was evaluated, demonstrated a similar benefit (measured numerically as change in the 6MWD) that was statistically significant in patients with CTD‐PAH (mean change 20.4 meters, 95% CI 10.9, 29.9; P < 0.001), perhaps reflecting greater statistical power due to increased sample size. Overall, compared to the meta‐analysis by Pan and colleagues (18), our study provides an expanded evaluation, including the FREEDOM‐EV trial, with an additional meta‐analysis of survival rates in registries, because data on long‐term survival outcomes and longitudinal analysis of survival outcomes over decades cannot be feasibly obtained from RCTs.
Patients with CTD‐PAH have a substantial risk of death; however, patients with CTD‐PAH who were treated within the last 10 years have numerically higher survival rates than those treated earlier. This difference may be related to increased screening for PAH, especially in those with SSc. Increased screening leads to earlier diagnosis, which provides the opportunity for earlier management (8) but also introduces lead‐time bias (50). If lead‐time bias is present, patients in later registries would be expected to be younger and to have less severe disease. Our analysis found that in the later registries, patients were older than those in the earlier registries (mean age 57 years versus 54 years), but had less severe disease (as defined by the proportion of patients with WHO functional class I or II disease) (40% versus 23%) and had a higher 6MWD (336 meters versus 321 meters) (see Supplementary Table 9, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract). Whether lead‐time bias is playing a substantial role in our results cannot be definitively determined from the current analysis.
The difference in survival over time also may reflect the availability of new treatment approaches. The improvement in survival is likely underestimated, since just 6 registries (32%) enrolled patients in 2015 or later, when all currently available treatments were in use and early combination therapy became more prevalent (5,34,39,41,46,47). More recent data are available from the United Kingdom Pulmonary Hypertension Audit (51). The most recent peer‐reviewed published data from this database (38) are included in our meta‐analysis; however, the latest report available (data from 2009–2019) is not included due to lack of peer review. Published data from 2001–2007 reported 1‐, 2‐, and 3‐year survival rates among patients with SSc‐associated PAH of 78%, 58%, and 47%, respectively. Corresponding survival rates from 2009–2019 were 81%, 61%, and 55%, respectively. These data corroborate the improved survival rates observed over time in our meta‐analysis. Consistent with clinical observations and published data (6,38,52), our meta‐analysis demonstrated that patients with SSc have worse survival rates than those with SLE. It should be noted, however, that patients with SSc in our analysis were older than those with SLE and appeared to have more severe disease, as indicated by fewer patients with WHO functional class I or II disease and a shorter 6MWD (see Supplementary Table 10, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract), which likely also contributed to their poorer survival. We were not able to use meta‐analysis to compare the treatment effect in RCTs between patients with SSc and those with SLE since only 2 RCTs provided sufficient data on patients with SSc (23,24) and only 1 provided sufficient data on patients with SLE (24).
Because all‐cause mortality was evaluated, we may have overestimated the incidence of death due to PAH among patients with CTD‐PAH. These patients were older and experienced a greater comorbidity burden compared to the overall PAH population. As such, these patients were possibly frailer and may have died from causes other than PAH. Although registries are subject to bias, these sources of long‐term data and larger sample size were deemed important to include in order to provide prolonged survival data unavailable from RCTs. Current guidelines now recommend combination therapy and more intensive therapy regardless of PAH etiology (53), and our meta‐analysis of registries provides evidence suggesting that the modern approach to treatment focuses on improving survival in CTD‐PAH. Nonetheless, survival remains lower for these patients, highlighting the need for continued research into the best treatment approaches and screening programs to promote early diagnosis and prompt management. Additional avenues for research to improve outcomes in this population include standardized reporting of comorbidities, which can substantially impact outcomes in CTD‐PAH as well as in PAH of other etiologies (54). Identification of comorbidities is further complicated by the lack of a consensus definition for significant interstitial lung disease in SSc. An additional area of focus should be standardized reporting of baseline risk profiles, since data suggest that patients with CTD‐PAH are at greater risk of death despite a less severe hemodynamic phenotype (5,55). Identification of clinically relevant changes in outcome measures, which may differ among PAH subtypes, would also be helpful. Finally, the era of personalized medicine may enable smaller study sizes and, ultimately, facilitate the discovery of treatment approaches that would yield a greater benefit within CTD‐PAH populations.
A strength of our meta‐analyses is the inclusion of only trials evaluating therapies that are approved for PAH treatment. By limiting the RCTs to only those with approved therapies, the results better reflect the benefit that can be observed in real‐world settings. In addition, our meta‐analysis of RCTs assessed the impact of current PAH treatments on morbidity and mortality, as endorsed by the 6th World Symposium on Pulmonary Hypertension (53). A limitation of our meta‐analysis of RCTs was that definitions of a clinical morbidity/mortality event (as listed in Supplementary Table 1 [http://onlinelibrary.wiley.com/doi/10.1002/art.41669/abstract]) varied to a limited extent across studies.
A limitation of our meta‐analysis of registries was the limited availability of studies that enrolled patients from 2015 onward, which would provide a survival estimate consistent with that observed in modern clinical practice. In all analyses, the overall PAH population to which we compared the CTD‐PAH population included patients with CTD‐PAH, because not all studies provided IPAH‐specific data. However, sensitivity analyses of RCTs that provided IPAH‐specific data demonstrated similar trends in patients with IPAH compared to those with CTD‐PAH as had been observed for the comparison between all PAH etiologies and CTD‐PAH.
An additional limitation is that it is unknown to what extent the treatment effect is influenced by different background therapies, potential variability in exposure to therapies, concomitant medications (such as immunosuppressants), as well as different proportions of newly and previously diagnosed patients in the study populations. Finally, the diagnosis of PAH was accepted on the basis of the criteria used in each study or registry; it is possible that underlying conditions, such as pulmonary veno‐occlusive disease and concomitant interstitial lung disease, were present and to differing degrees among the various studies. A limitation of our search methodology was that we did not search additional databases beyond PubMed and Embase. As noted, however, we do not expect any differences in outcomes as a result of this, given the parameters of our meta‐analyses, the rarity of this disease state, and the relatively small number of studies reporting data separately for the subset of patients with CTD‐PAH.
In conclusion, these complementary meta‐analyses of RCTs and observational disease registries demonstrated that with modern PAH treatments, patients with CTD‐PAH had a similar reduction in the risk of clinical morbidity and mortality events when compared to the overall PAH population. The improvement in the 6MWD in patients with CTD‐PAH appeared smaller than in those with other types of PAH, perhaps reflecting comorbidities (such as musculoskeletal involvement), independent of their cardiopulmonary capacity. Patients with CTD‐PAH have a higher risk of death than the overall PAH population; however, survival has improved among this subgroup treated in the last 10 years compared to earlier cohorts. Patients with SSc have worse survival rates than those with SLE. Given the high risk of mortality in these patients, early detection and up‐front aggressive treatment are warranted (56).
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Khanna had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Khanna, Zhao, Mathai, Coghlan, Shah, Hartney, McLaughlin.
Acquisition of data
Zhao, Shah, Hartney.
Analysis and interpretation of data
Khanna, Zhao, Saggar, Mathai, Chung, Coghlan, Shah, Hartney, McLaughlin.
ROLE OF THE STUDY SPONSOR
Funding for this analysis was provided by Actelion Pharmaceuticals US, Inc., a Janssen Pharmaceutical Company of Johnson & Johnson. Medical writing support was provided by Holly Strausbaugh, PhD, and Laura Evans, PharmD, on behalf of Twist Medical, LLC, which was funded by Actelion Pharmaceuticals. Data were collected and analyzed by some of the authors who are employees of Actelion Pharmaceuticals. All authors had full access to the data, contributed to data interpretation, contributed to manuscript writing, and had final responsibility for the decision to submit for publication. The publication of this article was not contingent upon approval by anyone from Actelion Pharmaceuticals except for authors who are employees of Actelion Pharmaceuticals, who, like all authors regardless of affiliation, are ethically required to approve of an article before it is submitted for publication.
Supporting information
Supplementary Material
Acknowledgments
The authors would like to thank Saling Huang, PhD, for consulting on the statistical analysis, and Kristine Jermakian for her contributions to the comprehensive review and data extraction.
Supported by Actelion Pharmaceuticals US, Inc.
Dr. Khanna has received consulting fees from Acceleron, Bristol Myers Squibb, Gilead, Genentech/Roche, GlaxoSmithKline, Mitsubishi Tanabe, and Sanofi Aventis/Genzyme (less than $10,000 each) and from Actelion, Bayer, Boehringer Ingelheim, Corbus, CSL Behring, and Horizon (more than $10,000 each) and holds stock in Eicos Sciences. Ms Zhao and Drs. Shah and Hartney are employees of Actelion and own stock or stock options in Johnson & Johnson, the parent company of Janssen Pharmaceuticals. Dr. Saggar has received consulting fees, speaking fees, and/or honoraria from Actelion, Gilead, and United Therapeutics (more than $10,000 each) and research support from those companies. Dr. Mathai has received consulting fees from Arena and Liquidia (less than $10,000 each) and from Actelion and United Therapeutics (more than $10,000 each). Dr. Chung has received consulting fees from Bristol Myers Squibb, Eicos Sciences, and Mitsubishi Tanabe (less than $10,000 each) and from Boehringer Ingelheim (more than $10,000), has received research support from Boehringer Ingelheim and United Therapeutics, and has served on a data safety monitoring board for Reata. Dr. Coghlan has received consulting fees, speaking fees, and/or honoraria from Bayer and GlaxoSmithKline (less than $10,000 each) and from Johnson & Johnson (more than $10,000) and research support from Johnson & Johnson. Dr. McLaughlin has received consulting fees from Altavant, CVC Caremark, CiVi Biopharma, and Gossamer Bio (less than $10,000 each) and from Actelion, Acceleron, Arena, Bayer, and United Therapeutics (more than $10,000 each) and research support from Acceleron, Actelion, Bayer, Reata, SoniVie, and United Therapeutics.
References
- 1. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- 2. D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991;115:343–9. [DOI] [PubMed] [Google Scholar]
- 3. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62:D34–41. [DOI] [PubMed] [Google Scholar]
- 4. Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J 2010;36:549–55. [DOI] [PubMed] [Google Scholar]
- 5. Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017;50:1700740. [DOI] [PubMed] [Google Scholar]
- 6. Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, et al. Characterization of connective tissue disease‐associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest 2010;138:1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Young A, Nagaraja V, Basilious M, Habib M, Townsend W, Gladue H, et al. Update of screening and diagnostic modalities for connective tissue disease‐associated pulmonary arterial hypertension. Semin Arthritis Rheum 2019;48:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Humbert M, Yaici A, de Groote P, Montani D, Sitbon O, Launay D, et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long‐term survival. Arthritis Rheum 2011;63:3522–30. [DOI] [PubMed] [Google Scholar]
- 9. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148–57. [DOI] [PubMed] [Google Scholar]
- 10. Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119:2894–903. [DOI] [PubMed] [Google Scholar]
- 11. Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330–40. [DOI] [PubMed] [Google Scholar]
- 12. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 2015;373:834–44. [DOI] [PubMed] [Google Scholar]
- 13. McLaughlin V, Channick RN, Ghofrani HA, Lemarié JC, Naeije R, Packer M, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J 2015;46:405–13. [DOI] [PubMed] [Google Scholar]
- 14. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809–18. [DOI] [PubMed] [Google Scholar]
- 15. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015;373:2522–33. [DOI] [PubMed] [Google Scholar]
- 16. White RJ, Jerjes‐Sanchez C, Bohns Meyer GM, Pulido T, Sepulveda P, Wang KY, et al. Combination therapy with oral treprostinil for pulmonary arterial hypertension: a double‐blind placebo‐controlled clinical trial. Am J Respir Crit Care Med 2020;201:707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of treatment response in idiopathic and connective tissue disease‐associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192:1111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pan J, Lei L, Zhao C. Comparison between the efficacy of combination therapy and monotherapy in connective tissue disease associated pulmonary arterial hypertension: a systematic review and meta‐analysis. Clin Exp Rheumatol 2018;36:1095–102. [PubMed] [Google Scholar]
- 19. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group . Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: the PRISMA statement. J Clin Epidemiol 2009;62:1006–12. [DOI] [PubMed] [Google Scholar]
- 20. National Institute for Health Research . PROSPERO: international prospective register of systematic reviews. URL: https://www.crd.york.ac.uk/prospero/.
- 21. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DerSimonian R, Laird N. Meta‐analysis in clinical trials. Control Clin Trials 1986;7:177–88. [DOI] [PubMed] [Google Scholar]
- 23. Coghlan JG, Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease‐associated pulmonary arterial hypertension (CTD‐PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017;76:1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galiè N, et al. Selexipag for the treatment of connective tissue disease‐associated pulmonary arterial hypertension. Eur Respir J 2017;50:1602493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896–903. [DOI] [PubMed] [Google Scholar]
- 26. Denton CP, Humbert M, Rubin L, Black CM. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open‐label extensions. Ann Rheum Dis 2006;65:1336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Badesch DB, Hill NS, Burgess G, Rubin LJ, Barst RJ, Galiè N, et al. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol 2007;34:2417–22. [PubMed] [Google Scholar]
- 28. Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double‐blind, placebo‐controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010–9. [DOI] [PubMed] [Google Scholar]
- 29. Badesch DB. Ambrisentan therapy for pulmonary arterial hypertension: a comparison by PAH etiology [abstract]. Chest 2007;132 Suppl:488b–9. [Google Scholar]
- 30. Galiè N, Denton CP, Dardi F, Manes A, Mazzanti G, Li B, et al. Tadalafil in idiopathic or heritable pulmonary arterial hypertension (PAH) compared to PAH associated with connective tissue disease. Int J Cardiol 2017;235:67–72. [DOI] [PubMed] [Google Scholar]
- 31. Escribano‐Subias P, Blanco I, López‐Meseguer M, Lopez‐Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J 2012;40:596–603. [DOI] [PubMed] [Google Scholar]
- 32. McLaughlin VV, Langer A, Tan M, Clements PJ, Oudiz RJ, Tapson VF, et al. Contemporary trends in the diagnosis and management of pulmonary arterial hypertension: an initiative to close the care gap. Chest 2013;143:324–32. [DOI] [PubMed] [Google Scholar]
- 33. Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long‐term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012;142:448–56. [DOI] [PubMed] [Google Scholar]
- 34. Yaylalı YT, Başarıcı I, Avcı BK, Meriç M, Sinan UY, Şenol H, et al. Risk assessment and survival of patients with pulmonary hypertension: multicenter experience in Turkey. Anatol J Cardiol 2019;21:322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang R, Dai LZ, Xie WP, Yu ZX, Wu BX, Pan L, et al. Survival of Chinese patients with pulmonary arterial hypertension in the modern treatment era. Chest 2011;140:301–9. [DOI] [PubMed] [Google Scholar]
- 36. Keogh A, Strange G, McNeil K, Williams TJ, Gabbay E, Proudman S, et al. The Bosentan Patient Registry: long‐term survival in pulmonary arterial hypertension. Intern Med J 2011;41:227–34. [DOI] [PubMed] [Google Scholar]
- 37. Chung WJ, Park YB, Jeon CH, Jung JW, Ko KP, Choi SJ, et al. Baseline characteristics of the Korean registry of pulmonary arterial hypertension. J Korean Med Sci 2015;30:1429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, et al. Connective tissue disease‐associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med 2009;179:151–7. [DOI] [PubMed] [Google Scholar]
- 39. Qian J, Li M, Zhang X, Wang Q, Zhao J, Tian Z, et al. Long‐term prognosis of patients with systemic lupus erythematosus‐associated pulmonary arterial hypertension: CSTAR‐PAH cohort study. Eur Respir J 2019;53:1800081. [DOI] [PubMed] [Google Scholar]
- 40. Kang KY, Jeon CH, Choi SJ, Yoon BY, Choi CB, Lee CH, et al. Survival and prognostic factors in patients with connective tissue disease‐associated pulmonary hypertension diagnosed by echocardiography: results from a Korean nationwide registry. Int J Rheum Dis 2017;20:1227–36. [DOI] [PubMed] [Google Scholar]
- 41. Kolstad KD, Li S, Steen V, Chung L, on behalf of the PHAROS Investigators . Long‐term outcomes in systemic sclerosis‐associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). Chest 2018;154:862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hao YJ, Jiang X, Zhou W, Wang Y, Gao L, Wang Y, et al. Connective tissue disease‐associated pulmonary arterial hypertension in Chinese patients. Eur Respir J 2014;44:963–72. [DOI] [PubMed] [Google Scholar]
- 43. Ngian GS, Stevens W, Prior D, Gabbay E, Roddy J, Tran A, et al. Predictors of mortality in connective tissue disease‐associated pulmonary arterial hypertension: a cohort study. Arthritis Res Ther 2012;14:R213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Launay D, Humbert M, Berezne A, Cottin V, Allanore Y, Couderc LJ, et al. Clinical characteristics and survival in systemic sclerosis‐related pulmonary hypertension associated with interstitial lung disease. Chest 2011;140:1016–24. [DOI] [PubMed] [Google Scholar]
- 45. Hachulla E, Jais X, Cinquetti G, Clerson P, Rottat L, Launay D, et al. Pulmonary arterial hypertension associated with systemic lupus erythematosus: results from the French pulmonary hypertension registry. Chest 2018;153:143–51. [DOI] [PubMed] [Google Scholar]
- 46. Morrisroe K, Stevens W, Huq M, Prior D, Sahhar J, Ngian GS, et al. Survival and quality of life in incident systemic sclerosis‐related pulmonary arterial hypertension. Arthritis Res Ther 2017;19:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weatherald J, Boucly A, Launay D, Cottin V, Prévot G, Bourlier D, et al. Haemodynamics and serial risk assessment in systemic sclerosis associated pulmonary arterial hypertension. Eur Respir J 2018;52:1800678. [DOI] [PubMed] [Google Scholar]
- 48. Kuwana M, Watanabe H, Matsuoka N, Sugiyama N. Pulmonary arterial hypertension associated with connective tissue disease: meta‐analysis of clinical trials. BMJ Open 2013;3:e003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Avouac J, Wipff J, Kahan A, Allanore Y. Effects of oral treatments on exercise capacity in systemic sclerosis related pulmonary arterial hypertension: a meta‐analysis of randomised controlled trials. Ann Rheum Dis 2008;67:808–14. [DOI] [PubMed] [Google Scholar]
- 50. Weatherald J, Montani D, Jevnikar M, Jaïs X, Savale L, Humbert M. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev 2019;28:190023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. NHS Digital . National audit of pulmonary hypertension: Great Britain, 2018‐19. October 24, 2019. URL: https://files.digital.nhs.uk/BA/4EF20E/NAPH%2010AR%20‐%20Main%20Report.pdf.
- 52. Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, et al. Characteristics and survival of anti–U1 RNP antibody–positive patients with connective tissue disease–associated pulmonary arterial hypertension. Arthritis Rheumatol 2016;68:484–93. [DOI] [PubMed] [Google Scholar]
- 53. Galiè N, Channick RN, Frantz RP, Grünig E, Jing ZC, Moiseeva O, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019;53:1801889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lewis RA, Thompson AA, Billings CG, Charalampopoulos A, Elliot CA, Hamilton N, et al. Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. Eur Respir J 2020;55:2000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peacock AJ, Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, et al. Idiopathic pulmonary arterial hypertension and co‐existing lung disease: is this a new phenotype? Pulm Circ 2020;10:2045894020914851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagaraja V, Cerinic MM, Furst DE, Kuwana M, Allanore Y, Denton CP, et al. Current and future outlook on disease modification and defining low disease activity in systemic sclerosis. Arthritis Rheumatol 2020;72:1049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material