

Abstract
Objective
The aim was to demonstrate that continuous s.c. infusion of a soluble levodopa (LD)/carbidopa (CD) phosphate prodrug combination effectively delivers stable LD exposure via a minimally invasive and convenient mode and has the potential to treat Parkinson's disease (PD) patients who are not well controlled on oral medication.
Methods
Foslevodopa and foscarbidopa were prepared and the equilibrium solubility and chemical stability examined in aqueous media with different values of pH. Solutions of foslevodopa/foscarbidopa (ratios ranging from 4:1 to 20:1) were prepared by dissolving pH‐adjusted lyophilized materials in water and infused s.c. in healthy volunteers for ≤72 hours. Frequent blood samples were collected to measure LD and CD exposure, and safety was monitored throughout the study.
Results
Foslevodopa/foscarbidopa (ABBV‐951) demonstrates high water solubility and excellent chemical stability near physiological pH, enabling continuous s.c. infusion therapy. After s.c. infusion, a stable LD pharmacokinetic (PK) profile was maintained for ≤72 hours, and the infusion was well tolerated.
Interpretation
Preparation of foslevodopa and foscarbidopa enables preclinical and clinical PK, safety, and tolerability studies in support of their advancement for the treatment of PD. In phase 1 clinical trials, foslevodopa/foscarbidopa demonstrates consistent and stable LD plasma exposure, supporting further studies of this treatment as a potentially transformational option for those suffering from PD. ANN NEUROL 2021;90:52–61
Globally, Parkinson's disease (PD) affects >6,000,000 people and has the fastest rate of growth among neurological disorders, the leading source of worldwide disability. 1 Our understanding of the complex interplay of genetic and environmental factors involved in the development of PD 2 has advanced substantially 3 over the 2 centuries since James Parkinson's Essay on the Shaking Palsy 4 ; however, the primary pathological hallmark of the disease is still dopamine (DA) deficiency within the basal ganglia, leading to death of dopaminergic neurons in the substantia nigra pars compacta. 5 Despite significant efforts to fulfill the substantial clinical need to identify disease‐modifying agents, 6 chronic DA replacement therapy remains the most effective method to treat the tremor, bradykinesia, and rigidity motor symptoms of PD. 7
Levodopa (LD) remains the gold standard in DA replacement therapy. 7 , 8 , 9 The mechanism of action for LD as DA replacement therapy in the central nervous system involves the enzymatic decarboxylation of LD (a DA prodrug) by central dopa decarboxylase (Fig 1A). Without a peripheral dopa decarboxylase inhibitor, minimal LD crosses the blood–brain barrier owing to premature decarboxylation; thus, high LD doses are required for PD therapy, leading to side effects. The development of the dopa decarboxylase inhibitor, carbidopa (CD), 10 was a key breakthrough in LD treatment and led to the approval of oral LD/CD combination therapy in 1975. In early stage PD, oral LD/CD is effective at managing symptoms (see Fig 1B) when there is tolerance of fluctuating brain DA concentrations. 7 Later in disease progression, higher and more frequent doses are required, eventually leading to dyskinesias (involuntary exaggerated movements) at high DA concentrations and increased “off” time at low DA concentrations. Efforts to remove the peaks and troughs from oral LD/CD therapy and deliver a consistent and optimal DA level led to the development of levodopa/carbidopa intestinal gel (Duodopa). Levodopa/carbidopa intestinal gel decreases the “off” time of advanced PD patients by an average of 1.9 hours per day 11 but requires surgery for administration. Nevertheless, clinical experience with levodopa/carbidopa intestinal gel demonstrated that sustained LD concentrations of ≤5μg/ml should be effective in treating the vast majority of patients with advanced PD, 12 , 13 an important observation guiding our efforts to develop improved LD therapeutic approaches.
FIGURE 1.
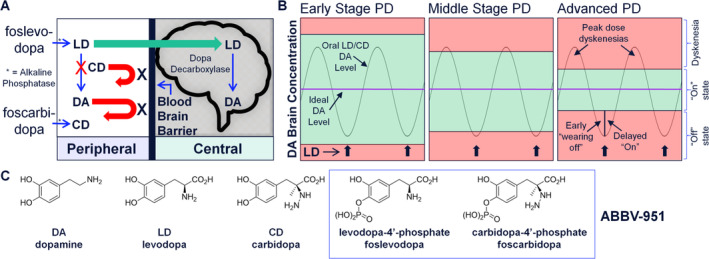
Foslevodopa and foscarbidopa are designed and prepared as highly water‐soluble prodrugs to enable continuous s.c. infusion therapy. (A) Mechanism of action for foslevodopa/foscarbidopa leading to increased brain DA levels. (B) Stages of PD, with representation of ideal DA level to achieve optimal therapeutic benefit. (C) Structures of DA, LD, CD, foslevodopa, and foscarbidopa. CD = carbidopa; DA = dopamine; LD = levodopa; PD = Parkinson's disease. [Color figure can be viewed at www.annalsofneurology.org]
Despite >50 years of patient experience with LD, a product that sustains high and personalized LD plasma exposure via a minimally invasive and convenient mode of delivery to treat PD does not exist. 9 To enable potential continuous s.c. infusion therapy with a highly concentrated liquid formulation of LD/CD near physiological pH, the phosphate prodrugs foslevodopa and foscarbidopa were considered (Fig 1C). No selective LD monophosphate synthesis has been reported previously, and there was no precedent for CD phosphates; therefore, synthetic routes to access foslevodopa and foscarbidopa were developed. 14
This work describes preclinical solubility, stability, and pharmacokinetic (PK) data and the results of a phase 1 first‐in‐human clinical study in healthy volunteers to support advancement of foslevodopa and foscarbidopa into PD patient clinical trials. There are several ongoing and completed phase 1 and phase 3 clinical trials in PD patients to evaluate the PK, safety, and efficacy of foslevodopa and foscarbidopa. Details of the additional clinical trials can be found at clinicaltrials.gov (NCT03033498, NCT03374917, NCT03781167, NCT04379050, and NCT04380142).
Methods
Solubility Methods for Prodrugs
Approximately 2g of solids was added to 5ml of aqueous solution in a 20ml scintillation vial. The suspension was kept in a 25°C water bath while stirring for 2 hours after pH stabilization. The suspension was filtered (Acrodisc 4mm syringe filter, 0.45μm polytetrafluoroethylene membrane; Pall Life Sciences, Cortland, NY, USA). The filtrate was diluted appropriately and analyzed by high‐performance liquid chromatography (HPLC). The solid residue was examined by powder X‐ray diffractometry to ensure no phase change. Water, aqueous solutions with different concentrations of acid or base were used to achieve different values of pH.
Solubility Methods for LD and CD
Suspensions were prepared by adding sufficient solids to 1ml of aqueous buffer. They were tumbled end to end for 3 days in a water bath at 25°C. After equilibration, the samples were filtered (Acrodisc 4mm syringe filter, 0.45 μm polytetrafluoroethylene membrane; Pall Life Sciences), diluted, and analyzed by HPLC. The solid residue was examined by powder X‐ray diffractometry to ensure no phase change. Different aqueous buffers were used to achieve the range of pH values for the profile.
Preparation of Formulation
In the preclinical studies, fresh foslevodopa/foscarbidopa formulations were used, dissolved in water and titrated with strong base to the target pH immediately before dosing. In the clinical study, lyophilized formulations were used at foslevodopa‐to‐foscarbidopa ratios of 4:1, 10:1, and 20:1. Foslevodopa and foscarbidopa were dissolved in water and titrated with strong base to the target pH. The solution was then lyophilized in glass vials and stoppered under vacuum. To evaluate the liquid stability of foslevodopa and foscarbidopa, liquid formulations at a ratio of 20:1 were prepared at different pH values and stoppered in low‐oxygen conditions in glass vials.
Assay Method
Assay values and the identification of foslevodopa and foscarbidopa for all samples were performed using reversed‐phase HPLC equipped with an ultraviolet detector. The samples were analyzed using gradient elution. Concentrations were determined using relative areas of the peaks in the samples compared with standard solutions.
Preclinical Pharmacokinetics
To establish preclinical proof of concept and to enable estimation of human PK, the preclinical pharmacokinetic profiles of foscarbidopa, foslevodopa, CD, and LD were characterized in rats, Göttingen minipigs, dogs, and monkeys after doses delivered by i.v. bolus, s.c. bolus, or infusion. The use of animals in this study followed the guidelines provided in the NRC Guide for the Care and Use of Laboratory Animals (1996) and the Animal Welfare Act (National Research Council, 1986). The use of animals in this study was approved by the AbbVie Institutional Animal Care and Use Committee and was conducted in an Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility.
Three animals were included in each dose group. Plasma samples were quantified by HPLC–tandem mass spectrometry after protein precipitation with 5% trichloroacetic acid in water to separate the compounds of interest from plasma. Pharmacokinetic parameters were determined using noncompartmental methods. The area under the plasma concentration–time curve from time 0 to t hours (time of the last measurable plasma concentration) after dosing (AUC0–t) was calculated using the linear trapezoidal rule for the plasma concentration–time profiles. The residual area extrapolated to infinity, determined as the final measured serum concentration (C t) divided by the terminal elimination rate constant (β), was added to AUC0–t to produce the total area under the curve (AUC0–∞ [area under the concentration time curve from time 0 to infinity]). Clearance (CL) was calculated by dividing the administered dose by AUC0–∞.
Clinical Study Design
A clinical trial in healthy human volunteers was designed to assess the PK of LD and CD, in addition to the safety following different foslevodopa/foscarbidopa infusion regimens. Additionally, the study was designed to assess the PK of LD and CD following oral doses of LD/CD to use as a comparator to the PK following the foslevodopa/foscarbidopa infusion. The study had a total of 3 groups of healthy volunteers, for which the dosing, number of subjects, and the PK sampling schedule are described in Table 1. Group 1 consisted of foslevodopa/foscarbidopa or placebo infusion in period 1 and oral LD/CD dosing (a single 100/25mg LD/CD immediate‐release tablet every 5 hours for 3 doses in total) in period 2. Group 2 consisted of foslevodopa/foscarbidopa or placebo infusion, whereby 3 different ratios of foslevodopa to foscarbidopa were delivered in a randomized crossover: 4:1, 10:1, and 20:1. Group 3 consisted of a foslevodopa/foscarbidopa loading dose infusion followed by a continuous infusion for a total infusion period of 72 hours. All foslevodopa/foscarbidopa infusions were delivered s.c. to the abdomen by an ambulatory infusion pump (model no. 871303U; B Braun Medical Inc., Evanston, IL, USA).
TABLE 1.
Outline of Clinical Study Design
| Group | Period | n | Treatment | PK Sampling |
|---|---|---|---|---|
| 1 | 1 | 6 | 360/90mg foslevodopa/foscarbidopa infused over 24 hours | Before priming of catheter, before infusion (0 hours), and at 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 24.5, 25, 26, 28, 30, 36, and 48 hours after the start of infusion |
| 2 | Placebo | |||
| 2 | 8 | Three separate oral doses of 100/25mg LD/CD each (for a total of 300/75mg LD/CD) | Before first dose (0 hours) and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, before second dose (5 hours), 5.25, 5.5, 5.75, 6, 6.5, 7, 8, 9, before third dose (10 hours), 10.25, 10.5, 10.75, 11, 11.5, 12, 13, 14, 15, 19, and 24 hours after initial dosing | |
| 2 | 1 | 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | Before priming of catheter, before infusion (0 hours), and at 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 26, and 28 hours after the start of infusion |
| 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 2 | Placebo | |||
| 2 | 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | ||
| 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 2 | Placebo | |||
| 3 | 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 h | ||
| 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/64mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/32mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 1 | 640/160mg foslevodopa/foscarbidopa CSCI infused over 16 hours | |||
| 2 | Placebo | |||
| 3 | 1 | 6 | 200/10mg foslevodopa/foscarbidopa infused over 5 minutes | Before priming of catheter, before infusion (0 hours), and at 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, 48, 52, 56, 60, 64, 68, 72, 72.5, 73, 74, 76, 78, 84, and 96 hours after the start of infusion |
| 2 | 6 | 960/48mg foslevodopa/foscarbidopa per 24 hours infused over 71 hours 55 minutes |
CD = carbidopa; CSCI = continuous s.c. infusion; LD = levodopa; PK = pharmacokinetic.
All procedures performed in this study were in accordance with the ethical standards of the Declaration of Helsinki and its later amendments or comparable ethical standards. The study protocol was approved by the local institutional review board, and written informed consent was obtained from each subject before any study‐related procedures were performed. Men and women aged 45–75 years with a body mass index between 18 and 30kg/m2 and in general good health were eligible to enroll in the study. Subjects were excluded if they tested positive for human immunodeficiency virus or hepatitis A, B, or C, or had alanine aminotransferase or aspartate aminotransferase concentrations >1.5× the upper limit of normal, or if they had contraindications to LD (eg, narrow‐angle glaucoma, pheochromocytoma, Cushing's syndrome) or a history of orthostatic hypotension. Subjects must not have used or consumed any of the following before study drug administration: tobacco or nicotine products within 6 months; another investigational product within 6 weeks; a drug by injection within 30 days; over‐the‐counter or prescription medications, vitamins, or herbal supplements within 2 weeks; or grapefruit, grapefruit products, Seville oranges, star fruit, or alcohol within 72 hours. Subjects were instructed not to sunbathe, use tanning salons, hot tubs, saunas, steam baths, spas, heat lamps, heated water beds, or to apply heat sources of any kind (such as heating pads or electric blankets) for 7 days before study drug administration.
A total of 22 subjects were enrolled in the study: 8 in Group 1, 8 in Group 2, and 6 in Group 3. Serial plasma PK samples were collected by venipuncture to assay for LD following the oral LD/CD dosing and the foslevodopa/foscarbidopa dosing. Human plasma with K2EDTA treated with sodium metabisulfite and disodium hydrogen arsenate was used to prepare the calibration standards, with a curve range of 9.99 to 10,000ng/ml for LD and 9.29 to 10,000ng/ml for CD. The accuracy (percentage bias) at the lower limit of quantitation was between −1.0 and 0.1% for LD and between −0.6 and 0.2% for CD. The accuracy of the quality control samples prepared at concentrations distributed throughout the calibration curve range was between −7.8 and 3.0%, and the precision (coefficient of variation) was ≤13.8% for LD. The accuracy of the quality control samples prepared at concentrations distributed throughout the calibration curve range was between −5.4 and 2.8%, and the precision (coefficient of variation) was ≤14.6% for CD. All PK parameters were determined using noncompartmental methods with SAS® version 9.2 (SAS Institute, Cary, NC). LD‐to‐CD exposure ratios were calculated from the PK parameter output as the LD area under the plasma concentration–time curve (AUC) divided by the CD AUC, where the AUC was from time 0 to the last observed time point.
Safety and tolerability were assessed throughout the study. All adverse events reported were collected from the time of dosing until 30 days after discontinuation of study drug administration. In addition, serious adverse events were collected from the time the subject signed the study‐specific informed consent. In addition to spontaneous reports by the subjects and observations by the investigator, adverse events were monitored by measurements on vital signs, physical examination, electrocardiogram, clinical laboratory test assessments, and Columbia‐Suicide Severity Rating Scale. Adverse events were coded using the Medical Dictionary for Regulatory Activities (https://www.meddra.org/). The number and percentage of subjects having treatment‐emergent adverse events were tabulated by primary System Organ Class and preferred term.
Results
Preclinical Characterization
Preclinical data were obtained to establish the potential for foslevodopa and foscarbidopa to treat PD via continuous s.c. infusion. Attributes of a viable prodrug candidate included high aqueous solubility, acceptable prodrug chemical stability, and PK proof of concept. These data were then used to project foslevodopa and foscarbidopa human PK.
Aqueous Solubility
Solubility values were determined for LD, CD, and their corresponding phosphate prodrugs (foslevodopa and foscarbidopa; Fig 2). For LD and CD, the pH–solubility profiles cover pH 2 to 10. For their phosphate prodrugs, however, the profiles are limited to pH 0 to 3.5 owing to their extremely high solubility at higher pH. The symbols represent experimental determinations, and dotted lines represent fitting against the Henderson–Hasselbalch equation that describes ionization of acids and bases at different values of pH. At a physiological pH of 7.4, the pH of the intended s.c. solution formulation, LD and CD solubility values are relatively low, at 3.95 and 2.75mg/ml respectively. In contrast, the solubility values of the phosphate prodrugs are high. Extrapolation of the pH–solubility profiles to pH 7.4 indicates that their solubility values are >1g/ml. Thus, the solubility of these phosphate prodrugs enabled a concentrated solution formulation to be developed for s.c. administration.
FIGURE 2.
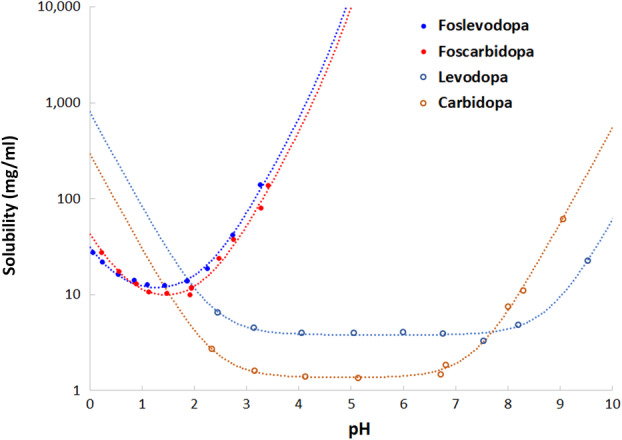
pH–solubility profiles of levodopa, carbidopa, foslevodopa, and foscarbidopa drug substance at 25°C. [Color figure can be viewed at www.annalsofneurology.org]
Chemical Stability
Given that both LD and CD are known to be susceptible to oxidative or decarboxylative degradation, 15 , 16 the long‐term stability of solutions of foslevodopa and foscarbidopa was assessed. In oxygen‐protected conditions, foslevodopa and foscarbidopa were observed to be stable (<2% change in foslevodopa and foscarbidopa assay values) for >1 year in solution at high concentrations (eg, 240/12 and 360/18mg/ml) across a wide pH range (6.5–9.2) including the physiological pH of 7.4.
Preclinical Proof of Concept
Preclinical proof of concept was established in rats, in which continuous s.c. administration of a 4:1 ratio of foslevodopa to foscarbidopa delivers concentrations of LD that would be effective in the large majority of advanced PD patients (1.5–4.5μg/ml LD). The continuous s.c. infusion was well tolerated throughout the study. Studies in rats and pigs exploring a range of formulation ratios of foslevodopa to foscarbidopa established that therapeutically relevant LD levels are attainable over a broad range of ratios from 4:1 to 15:1.
Projection of Human PK
To enable estimation of human PK, the prodrugs were co‐administered to preclinical species at 2 different dose ratios: 4:1 and 10:1 (foslevodopa:foscarbidopa). The clearance of the prodrugs across species was high (≥2l/h/kg). In addition, LD and CD PK were determined in rats, dogs, and monkeys after administration of LD or CD separately and after co‐administration. Without co‐administration, LD clearance is high across species: rats (4.2l/h/kg), monkeys (3.7l/h/kg), and humans (0.8l/h/kg). With co‐administration of CD, the reduction in LD clearance was somewhat greater for the 4:1 dose ratio (69 and 68% in rats and monkeys, respectively) compared with the 10:1 dose ratio (50 and 65% in rats and monkeys, respectively). CD clearance was not affected by LD co‐administration. These data were used to estimate a human in vivo half maximal inhibitory concentration (IC50) of 44ng/ml CD for inhibition of LD clearance, through inhibition of the enzyme amino acid decarboxylase in the periphery. The fraction of a human foslevodopa dose metabolized by amino acid decarboxylase was estimated as the mean (0.7) of the values observed in rats (0.8) and monkeys (0.6).
Foslevodopa/foscarbidopa dose rates of 140/35mg/h (4:1 dose ratio) or 140/14mg/h (10:1 dose ratio) by continuous s.c. infusion were estimated to achieve and maintain the steady‐state LD target plasma concentration of 3000ng/ml (Fig 3). A dose rate of 140/7mg/h (20:1 dose ratio) was also projected to achieve and maintain the target LD plasma concentration. However, this ratio was projected to reduce the estimated steady‐state CD concentration to 192ng/ml, below the target plasma concentration CD. For foslevodopa/foscarbidopa dose ratios of 4:1, 10:1, and 20:1, the projected steady‐state levodopa/carbidopa exposure ratios were 4, 8, and 16, respectively.
FIGURE 3.
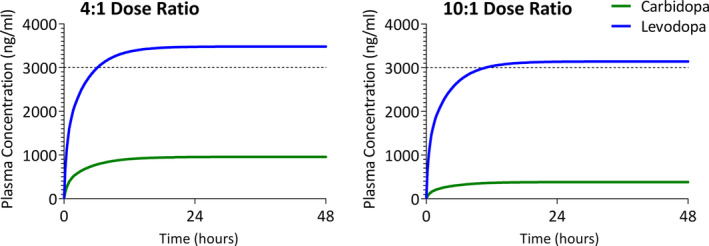
Estimated levodopa and carbidopa plasma concentration–time profiles following continuous s.c. infusions of foslevodopa/foscarbidopa. [Color figure can be viewed at www.annalsofneurology.org]
Clinical Assessment
All 22 subjects from the study were enrolled, and 21 of 22 subjects received the intended regimen of foslevodopa/foscarbidopa, placebo infusion, or oral LD/CD. One subject from Group 3 who was assigned to the foslevodopa/foscarbidopa regimen received the bolus dosing rate for 12 minutes rather than the protocol‐specified 5 minutes, resulting in a bolus dose 2.4 times higher than desired. The subject was not administered the total 72 hour infusion in order not to receive more than the planned dose. This subject was excluded from the PK analysis.
Results for Group 1, Period 1 show that after foslevodopa/foscarbidopa infusion, LD and CD exposures increase until reaching a steady state at approximately 16 hours (Fig 4A). After oral LD/CD dosing in Group 1, Period 2, the LD and CD PK fluctuations were higher than with the foslevodopa/foscarbidopa regimen (see Fig 4E). Additionally, results from Group 1 showed that the LD:CD AUC0–∞ exposure ratio (ER) after the foslevodopa/foscarbidopa infusion results in a lower‐than‐expected ratio (higher CD exposure than expected) compared with preclinical data and the oral LD/CD dosing regimen. Owing to the higher‐than‐expected CD exposure, Group 2 explored a range of foslevodopa/foscarbidopa ratios (4:1, 10:1, and 20:1) to identify the ratio that would provide the desired CD exposure.
FIGURE 4.
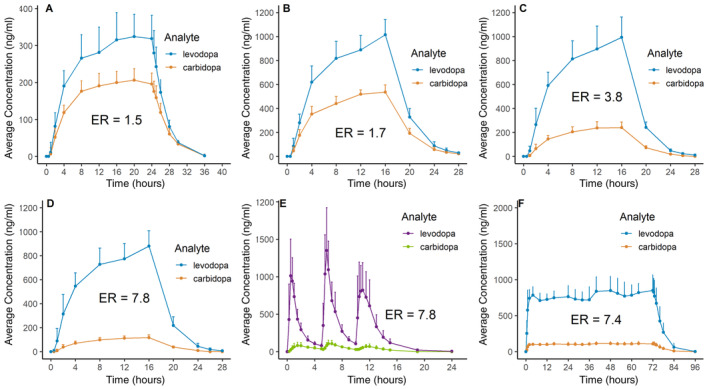
Clinical pharmacokinetic results. (A) Group 1, Period 1, foslevodopa‐to‐foscarbidopa dosing ratio of 4:1. (B) Group 2, foslevodopa‐to‐foscarbidopa dosing ratio of 4:1. (C) Group 2, foslevodopa‐to‐foscarbidopa dosing ratio of 10:1. (D) Group 2, foslevodopa‐to‐foscarbidopa dosing ratio of 20:1. (E) Group 1, Period 2, oral levodopa‐to‐carbidopa dosing ratio of 4:1. (F) Group 3, foslevodopa‐to‐foscarbidopa dosing ratio of 20:1, 72 hour infusion. ER = levodopa‐to‐carbidopa exposure ratio based on AUC0–∞. [Color figure can be viewed at www.annalsofneurology.org]
A gradual increase in both LD and CD exposure throughout the duration of infusion was observed in Group 2, similar to the foslevodopa/foscarbidopa PK profile from Group 1, Period 1. The 3 PK profiles of the different foslevodopa/foscarbidopa ratios show that CD exposure decreased with decreasing foscarbidopa doses going from the 4:1 to 20:1 foslevodopa‐to‐foscarbidopa ratios, while the LD exposure remained similar for the different ratios (Fig 4B–D). To assess the overall similarity of exposure between the different foslevodopa‐to‐foscarbidopa ratios and oral LD/CD dosing from Group 1, the AUC0–∞ ERs were calculated and shown with each PK plot (see Fig 4).
Foslevodopa/foscarbidopa had an ER most similar to oral LD/CD (4:1) when dosed at a 20:1 ratio. Foslevodopa/foscarbidopa dosing from Group 3 used the 20:1 ratio for dosing. Results from Group 3 (foslevodopa/foscarbidopa loading dose followed by continuous foslevodopa/foscarbidopa infusion) indicated that LD exposure appeared to be stable once a steady state was achieved at approximately 2 hours and was maintained for the remainder of the 72 hour infusion period (Fig 4F). The variability in LD exposure appeared to be higher with oral LD/CD (see Fig 4E) in comparison to foslevodopa/foscarbidopa infusion from Group 3 (see Fig 4F).
The foslevodopa/foscarbidopa PK profile indicated rapid distribution of active LD in the systemic circulation after s.c. infusion of foslevodopa. The average foslevodopa/foscarbidopa LD degree of fluctuation for Group 3 ([maximum concentration minus minimum concentration]/average concentration) from 2 to 72 hours was 0.41 (range 0.21–0.43), and from 2 to 16 hours it was 0.20 (range 0.10–0.42). For comparison, the average degree of fluctuation after oral LD/CD administration (mean ± SD) was 4.1 ± 1.4 (range 2.74–7.24).
For subjects taking oral LD/CD, no treatment‐emergent adverse events were assessed by the investigator as having a reasonable possibility of being related to the study drug. One subject reported enteritis that was moderate in severity and was determined by the investigator as having no reasonable possibility of being related to study drug administration. For subjects taking foslevodopa/foscarbidopa, the infusion was generally well tolerated. All adverse events were mild in severity and did not cause any discontinuations from the study. Out of the 8 subjects in Group 2, 1 subject who received study drug reported skin irritation from electrocardiogram electrodes that was mild in severity, and 2 subjects who received placebo reported 3 treatment‐emergent adverse events (1 subject reported subconjunctival hemorrhage and a contusion; 1 subject reported diarrhea) that were mild in severity. Four subjects from Group 3 reported mild infusion site pain within 30 minutes after initiation of the infusion with an average duration of around 30 minutes, and 1 subject had an adverse event of edema at the infusion site with a duration of approximately 19 hours. There were no reports of pain or discomfort for the remaining 72 hours of the study.
Discussion
A successful continuous s.c. infusion prodrug will require 3 fundamental features: (1) high aqueous solubility, (2) excellent chemical stability, and (3) highly efficient conversion of the water‐soluble salt or prodrug to the active pharmaceutical ingredient. A phosphate prodrug approach was selected to deliver these objectives. Challenging nonselective methods for foslevodopa synthesis provided small quantities for initial assessment, but no carbidopa phosphate prodrug had been reported, and the methods reported for levodopa failed with carbidopa owing to stability challenges. Therefore, regioselective syntheses based on asymmetric hydrogenation and asymmetric hydrazination were developed to prepare foslevodopa and foscarbidopa, respectively, and a stable trihydrate solid form of foscarbidopa was identified to facilitate its isolation and storage. 14
Initial preclinical data on foslevodopa/foscarbidopa demonstrated that these prodrugs had exceptional aqueous solubility at physiological pH (>1g/ml at pH 7.4) and very good chemical stability (<2% decomposition in solution for 1 year). On the basis of these attractive physiochemical properties, subsequent preclinical evaluation in rats, dogs, and minipigs demonstrated PK proof of concept that continuous s.c. administration at 4:1 and 15:1 ratios can deliver concentrations of LD that would be effective in advanced PD patients (>5μg/ml at steady state). In addition, these PK proof‐of‐concept studies were also used to model clinical doses and enable initiation of foslevodopa/foscarbidopa clinical studies.
Foslevodopa/foscarbidopa was infused s.c. for ≤72 hours to a total of 18 healthy volunteers. The prodrugs converted to active LD and CD, and stable exposures were observed once a steady state was achieved. In Group 1 and Group 2, foslevodopa/foscarbidopa was infused without a loading dose, and this resulted in a steady state being achieved approximately 12 to 16 hours after the infusion started. When treating patients with PD, it is desirable to achieve a steady state quickly in order to control motor symptoms. Therefore, Group 3 received a loading dose of foslevodopa/foscarbidopa followed by a continuous infusion, and this resulted in an LD steady state within 2 hours. The stable LD PK profile following foslevodopa/foscarbidopa infusion is consistent with being able to maintain LD exposure within a narrow therapeutic window for PD patients and is consistent with the LD PK profile previously shown with levodopa/carbidopa intestinal gel. 13
Results from Group 1 showed that the CD exposure following the foslevodopa/foscarbidopa infusion was higher than expected based on preclinical foslevodopa/foscarbidopa PK data and PK data following oral LD/CD dosing, suggesting that foscarbidopa delivered by s.c. administration has a higher bioavailability than CD administered orally. Based on results from Group 1, Group 2 was designed with a range of formulations to determine a foslevodopa‐to‐foscarbidopa dosing ratio, where the desirable dosing ratio was defined as having a LD:CD ER similar to oral LD/CD dosed at the 4:1 ratio. Results from Group 2 showed that the 20:1 foslevodopa‐to‐foscarbidopa dosing ratio resulted in an LD:CD ER similar to oral LD/CD dosed at the 4:1 ratio. Therefore, a 20:1 foslevodopa‐to‐foscarbidopa ratio was considered the appropriate dosing ratio.
The degree of fluctuation for plasma LD exposures is an important parameter for assessing whether a treatment can maintain exposure within a narrow therapeutic window for PD patients (Fig 1B). The mean (±SD) degree of fluctuation of LD exposure for the subjects who received foslevodopa/foscarbidopa for 72 hours in Group 3 was 0.24 ± 0.12 over the 2 to 16 hour period and 0.37 ± 0.10 for the 2 to 72 hour period. Foslevodopa/foscarbidopa infusion in Group 3 showed a lower degree of fluctuation of LD than that of oral administration in the present study (4.1 ± 1.4 [mean ± SD]), previous oral LD/CD administration in healthy elderly volunteers (4.3 ± 0.9),17 and PD patients (3.2 ± 1.3). 18 A previous phase 1 study, in which levodopa/carbidopa intestinal gel was infused directly into the jejunum reported a degree of fluctuation of 0.52 ± 0.20 (mean ± SD) over the 2 to 16 hour period; however, that study was run in PD patients rather than healthy volunteers. 19 The degree of fluctuation after foslevodopa/foscarbidopa infusion in PD patients will be evaluated in a separate study (NCT03033498).
Overall, the oral LD/CD dosing and foslevodopa/foscarbidopa infusion were generally well tolerated, with no adverse events causing discontinuation from the study. No pattern was evident with regard to the nature or frequency of treatment‐emergent adverse events after continuous s.c. infusion or bolus injection of ABBV‐951 compared with subjects who received placebo. All the adverse events reported by subjects from Group 1 and Group 2 were mild or moderate in severity and were determined by the investigator to have no reasonable possibility of being related to study drug administration. Four of the 6 subjects from Group 3 reported mild infusion site pain within 30 minutes after initiation of the infusion, with no reports of pain or discomfort for the remaining 72 hours of the study. Given that pain was reported only ≤30 minutes after infusion initiation, an association between pain/discomfort and the loading dose (which was delivered at a higher infusion rate than the 72 hour continuous infusion) was suggested. Foslevodopa/foscarbidopa is intended to be a 24 hour treatment for advanced PD. Therefore, a loading dose will be needed only for initiation of treatment or to resume treatment after an interruption of infusion. Although no human data are available to exclude the possibility of tolerance to ABBV‐951 after its continuous, 24 hour per day infusion, anecdotal data from ongoing studies in patients with PD receiving stable rates of foslevodopa, in addition to data from 24 hour per day intestinal delivery of levodopa, with or without reducing the nighttime infusion rate, 20 , 21 , 22 , 23 do not seem to report an increase in such risk.
The first studies in healthy volunteers were conducted using the B. Braun syringe pump, an ambulatory infusion system frequently used in laboratory experiments but whose physical characteristics (dimensions, operability, etc.) make it unsuitable for daily use in clinical settings. The efficacy of foslevodopa/foscarbidopa relies on its conversion to the active moieties of LD and CD when delivered at rates that can meet individual patient needs, and infusion pumps are deployed to achieve those LD plasma concentrations within the personalized therapeutic window. Pumps with the technical specifications required to deliver foslevodopa/foscarbidopa are commercially available and have been considered or used during the development of this therapy. However, a parallel plan to develop a more user‐friendly, small, and portable delivery system, targeted to be used by patients with PD daily in their home and that fulfills the same technical requirements, is ongoing.
In conclusion, with this manuscript we introduce foslevodopa/foscarbidopa (ABBV‐951), a highly concentrated (20:1) solution of levodopa/carbidopa prodrugs for s.c. delivery. Its stable liquid formulation at near neutral pH enabled the preclinical characterization and the beginning of the clinical testing with phase 1 studies. This paper also illustrates the therapeutic potential of ABBV‐951 by means of continuous s.c. infusion via a portable pump. ABBV‐951 monotherapy might prove to be beneficial in controlling both daytime and nighttime levodopa‐responsive PD symptoms, improving convenience over currently available therapeutic alternatives (reduced pill burden, no surgery needed, no sleep interruption for those patients taking oral levodopa overnight). We showed that a loading dose preceding the start of the continuous infusion helps quickly to achieve and sustain steady‐state LD concentrations that are clinically relevant and can be personalized to each patient's need, although more studies are warranted to refine its tolerability profile. Our vision is that by giving the possibility of a loading dose via the pump rather than via oral formulations, together with the ability to administer extra doses or change the infusion rate during the day to respond to specific clinical needs, ABBV‐951 might offer a more convenient approach to the management of PD symptoms, motor fluctuations, and dyskinesia. More studies are ongoing and in planning to generate the necessary data to support this vision.
Author Contributions
M.R., E.V., E.M.M., F.J., X.L., G.G.Z.Z., D.S., R.A.C., B.E., W.L., and P.R.K. contributed to the conception and design of the study. M.R., E.V., E.M.M., X.L., G.G.Z.Z., P.T.M., D.S., R.A.C., B.E., and M.F.F. acquired and analyzed the data. M.R., E.V., E.M.M., X.L., G.G.Z.Z., R.A.C., B.E., M.F.F., and P.R.K. drafted a significant portion of the manuscript or figures.
Potential Conflicts of Interest
M.R., E.A.V., E.M.M., X.L., G.G.Z.Z., P.T.M., D.S., R.A.C., B.P.E., W.L., M.F.F., and P.R.K.: AbbVie employees and may hold stock or options. F.J.: AbbVie employee at the time this work was conducted and may hold stock or options.
Acknowledgments
This study was sponsored by AbbVie, Inc. AbbVie provided review and approval of the study design and this publication. Wesley Wayman, PhD, an employee of AbbVie, collated the manuscript and provided copyediting support.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), in addition to other information (eg, protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html
References
- 1. Dorsey ER, Elbaz A, Nichols E, et al. Global, regional, and national burden of Parkinson's disease, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol 2018;17:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896–912. [DOI] [PubMed] [Google Scholar]
- 3. del Rey NL, Quiroga‐Varela A, Garbayo E, et al. Advances in Parkinson's disease: 200 years later. Front Neuroanat 2018;12:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 2002;14:223–236. [DOI] [PubMed] [Google Scholar]
- 5. Parent M, Parent A. Substantia nigra and Parkinson's disease: a brief history of their long and intimate relationship. Can J Neurol Sci 2010;37:313–319. [DOI] [PubMed] [Google Scholar]
- 6. Elkouzi A, Vedam‐Mai V, Eisinger RS, Okun MS. Emerging therapies in Parkinson disease ‐ repurposed drugs and new approaches. Nat Rev Neurol 2019;15:204–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA 2020;323:548–560. [DOI] [PubMed] [Google Scholar]
- 8. LeWitt PA, Fahn S. Levodopa therapy for Parkinson disease: a look backward and forward. Neurology 2016;86:S3–S12. [DOI] [PubMed] [Google Scholar]
- 9. Poewe W, Antonini A. Novel formulations and modes of delivery of levodopa. Mov Disord 2015;30:114–120. [DOI] [PubMed] [Google Scholar]
- 10. Sletzinger M, Chemerda JM, Bollinger FW. Potent decarboxylase inhibitors. Analogs of methyldopa. J Med Chem 1963;6:101–103. [DOI] [PubMed] [Google Scholar]
- 11. Fernandez HH, Standaert DG, Hauser RA, et al. Levodopa‐carbidopa intestinal gel in advanced Parkinson's disease: final 12‐month, open‐label results. Mov Disord 2015;30:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang HY, Chen X, Jiang J, et al. Evaluating a physiologically based pharmacokinetic model for predicting the pharmacokinetics of midazolam in Chinese after oral administration. Acta Pharmacol Sin 2016;37:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Othman AA, Rosebraugh M, Chatamra K, et al. Levodopa‐carbidopa intestinal gel pharmacokinetics: lower variability than oral levodopa‐carbidopa. J Parkinsons Dis 2017;7:275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cardinal‐David B, Chan V, Dempah K, et al. Carbidopa and L‐dopa prodrugs and methods of use. US patent application US 2016/0106765Α1. 2016.
- 15. Mondal S, Thampi A, Puranik M. Kinetics of melanin polymerization during enzymatic and nonenzymatic oxidation. J Phys Chem B 2018;122:2047–2063. [DOI] [PubMed] [Google Scholar]
- 16. Gasowska‐Bajger B, Frackowiak‐Wojtasek B, Koj S, et al. Oxidation of carbidopa by tyrosinase and its effect on murine melanoma. Bioorg Med Chem Lett 2009;19:3507–3510. [DOI] [PubMed] [Google Scholar]
- 17. Yeh KC, August TF, Bush DF, et al. Pharmacokinetics and bioavailability of Sinemet CR: a summary of human studies. Neurology 1989;39:25–38. [PubMed] [Google Scholar]
- 18. Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson's disease. Mov Disord 2011;26:2246–2252. [DOI] [PubMed] [Google Scholar]
- 19. Nyholm D, Odin P, Johansson A, et al. Pharmacokinetics of levodopa, carbidopa, and 3‐O‐methyldopa following 16‐hour jejunal infusion of levodopa‐carbidopa intestinal gel in advanced Parkinson's disease patients. AAPS J 2013;15:316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ricciardi L, Bove F, Espay KJ, et al. 24‐hour infusion of levodopa/carbidopa intestinal gel for nocturnal akinesia in advanced Parkinson's disease. Mov Disord 2016;31:597–598. [DOI] [PubMed] [Google Scholar]
- 21. Chang FC, Tsui DS, Mahant N, et al. 24 h levodopa‐carbidopa intestinal gel may reduce falls and “unresponsive” freezing of gait in Parkinson's disease. Parkinsonism Relat Disord 2015;21:317–320. [DOI] [PubMed] [Google Scholar]
- 22. Cruse B, Morales‐Briceño H, Chang FCF, et al. 24‐hour levodopa‐carbidopa intestinal gel may reduce troublesome dyskinesia in advanced Parkinson's disease. NPJ Parkinson's Dis 2018;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nyholm D, Adnan M, Senek M. Real‐life use of levodopa/Carbidopa intestinal gel in Parkinson's disease according to analysis of pump data. J Parkinsons Dis 2020;10:1529–1534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), in addition to other information (eg, protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html