

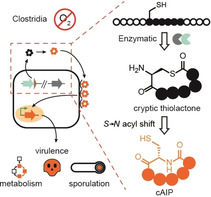
Abstract
Clostridia coordinate many important processes such as toxin production, infection, and survival by density‐dependent communication (quorum sensing) using autoinducing peptides (AIPs). Although clostridial AIPs have been proposed to be (thio)lactone‐containing peptides, their true structures remain elusive. Here, we report the genome‐guided discovery of an AIP that controls endospore formation in Ruminiclostridium cellulolyticum. Through a combination of chemical synthesis and chemical complementation assays with a mutant strain, we reveal that the genuine chemical mediator is a homodetic cyclopeptide (cAIP). Kinetic analyses indicate that the mature cAIP is produced via a cryptic thiolactone intermediate that undergoes a rapid S→N acyl shift, in a manner similar to intramolecular native chemical ligation (NCL). Finally, by implementing a chemical probe in a targeted screen, we show that this novel enzyme‐primed, intramolecular NCL is a widespread feature of clostridial AIP biosynthesis.
Keywords: Clostridia, cyclopeptide, native chemical ligation, quorum sensing, RiPP
The genome‐guided discovery of an autoinducing peptide (AIP) that controls endospore formation in Ruminiclostridium cellulolyticum is reported. A combination of chemical synthesis and chemical complementation assays with a mutant strain reveals that the genuine chemical mediator is a homodetic cyclopeptide (cAIP).
Introduction
The Clostridia are a heterogeneous class of anaerobic bacteria that play major roles in human and animal health, industrial biotechnology, and the environment. [1] There is growing evidence that Clostridia coordinate a broad range of crucial group behaviors that affect their physiology and pathogenicity by sensing the accumulation of secreted peptidic signals that regulate gene expression. [2] Since the concentration of these chemical mediators correlates with cell density (quorum sensing, QS), gene expression can be synchronized with specific environmental conditions and growth phases. [3] An impressive number of genetic studies have revealed that the accessory gene regulator (Agr) system is central to QS in diverse Clostridia.[ 2a , 2b , 2c , 2d , 2e , 2f , 2g , 2h , 2i , 2j , 2k , 2l , 2m ] For example, Agr‐based QS is linked to the virulence potential of the infamous human pathogens Clostridium difficile[ 2a , 2c ] and Clostridium perfringens, [2m] and regulates toxin production in C. difficile,[ 2a , 2b ] C. perfringens[ 2d , 2e , 2f , 2g , 2h , 2i , 2j ] and Clostridium botulinum. [2k] Furthermore, Agr‐mediated QS is implicated in the regulation of clostridial sporulation[ 2f , 2k , 2l ] as well as granulose production in the industrially important solvent producer Clostridium acetobutylicum. [2l]
The QS signals derived from the Agr system are called autoinducing peptides (AIPs) and belong to the ribosomally synthesized and post‐translationally modified peptide (RiPP) class of natural products. [4] The mechanistic details of AIP biosynthesis and signaling were deciphered in pathogenic Staphylococcus spp., [5] with the prototypical Agr system consisting of four components: a gene‐encoded precursor peptide (AgrD), an integral membrane macrocyclase (AgrB), and a sensor kinase (AgrC) and response regulator (AgrA) pair that transduce the signal (Figure 1 A). [6] AgrD is comprised of three regions: an N‐terminal leader peptide that aids in membrane localization, [7] a core peptide that encodes the QS signal, [8] and a C‐terminal follower peptide [9] that is recognized by AgrB. The biosynthesis of the mature QS signal from this unmodified precursor peptide occurs over two steps (Figure 1 B). First, the AgrB transmembrane cysteine protease[ 8 , 9 , 10 ] removes the follower peptide and catalyzes intramolecular thiolactone bond formation between a conserved cysteine in the core peptide and the liberated C‐terminus. [11] The leader peptide is then removed by an extracellular peptidase, [12] leaving the thiolactone macrocycle with an N‐terminal extension (NTE) of varying length (Figure 1 A and Figure 1 B).[ 5a , 13 ] In rare cases, cyclization involves a serine residue, resulting in the formation of a lactone macrocycle.[ 5c , 14 ] Akin to other QS systems, AIP production and sensing are mutually enhancing, leading to a positive‐feedback autoinduction circuit. [5a]
Figure 1.
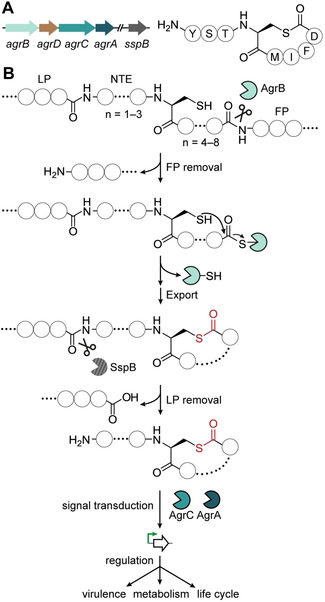
The prototypical Agr system of Staphylococcus aureus: A) Representation of an S. aureus AIP biosynthetic gene cluster showing genes encoding the integral membrane macrocyclase (AgrB), the precursor peptide (AgrD), the cognate two‐component transduction system (AgrC‐AgrA), as well as the distal gene encoding the membrane protease (SspB). Also shown is an example of a mature staphylococcal thiolactone‐containing AIP. B) Overview of the biosynthetic pathway to staphylococcal thiolactone‐containing AIPs. The AIP is derived from an internal fragment of the ribosomally synthesized precursor peptide AgrD. First, AgrB catalyzes C‐terminal proteolysis of the follower peptide (FP) and subsequent macrocyclization, followed by secretion. Then, the N‐terminal leader peptide (LP) is removed by SspB, leaving an N‐terminal extension (NTE) of varying length. The mature AIP, which varies among strains, consists of a five‐membered thiolactone ring with a 2–4 residue NTE. In response to binding of the AIP, the sensor kinase AgrC is autophosphorylated and the signal is transduced by the response regulator AgrA leading to global alterations in gene expression.
By analogy to their staphylococcal counterparts, it has been assumed that clostridial AIPs generally possess thiolactone moieties.[ 2a , 2b , 2h , 2j , 2l ] Considering the prevalence[ 2l , 15 ] and essential roles of Agr signaling circuits in Clostridia,[ 2a , 2b , 2c , 2d , 2e , 2f , 2g , 2h , 2i , 2j , 2k , 2l , 2m ] this assumption should be substantiated. Until now, the chemical structures of clostridial AIPs have been inferred from indirect experimental evidence such as signal degradation, [2b] and by chemical complementation of mutant phenotypes with synthetic AIPs designed from bioinformatics‐based structure predictions.[ 2j , 2l ] Here, we report the first complete structural assignment of a clostridial AIP and provide compelling evidence that the previously suggested structures are transient precursors of the true signaling molecules. Our findings not only prompt a re‐examination of some previously reported AIP structures, but also reveal a novel pathway to cyclic RiPPs by natural native chemical ligation (NCL).
Results and Discussion
Genomics‐guided discovery of an AIP from a cellulolytic, antibiotic‐producing anaerobe
Ruminiclostridium cellulolyticum DSM 5812 has been extensively studied for its ability to degrade cellulose [16] and to produce the DNA‐gyrase inhibiting antibiotic closthioamide, [17] but there has been no report on AIP‐mediated QS in this genetically tractable strain. [18] Indeed, AIP‐dependent signaling pathways have not been studied in any Ruminiclostridium species to date. We therefore chose R. cellulolyticum as a starting point for the characterization of clostridial AIPs. To gain insight into the potential of R. cellulolyticum to produce AIPs, we mined its genome for genes encoding members of the AgrB protein family (PF04 647). We identified four homologs of the AgrB peptidase gene (Table S1). Next, we scanned the genome region surrounding each candidate agrB for genes tentatively encoding a AIP precursor peptide (AgrD) and detected a candidate agrD in all four loci (Table S1 and Figure S1). In theory, the four agrBD gene pairs could enable R. cellulolyticum to produce four different AIPs (Rc‐AIP1‐4) (Figure S1). This finding is unusual since previous bioinformatic analyses detected just one or two AIP gene clusters in members of the Clostridia.[ 2b , 2c , 2l , 15 ]
The core peptide sequences of each possible R. cellulolyticum AIP were predicted by aligning Rc‐AgrD1‐4 to AgrD homologues for which the corresponding products have been structurally characterized (Figure S2). Based on previous synthetic studies involving complementation of mutant phenotypes in various Clostridia[ 2j , 2l ] and a preliminary MS‐based detection of an AIP from Hungateiclostridium thermocellum, [19] we predicted that the AIPs from R. cellulolyticum would not have NTEs, i.e. they should present a free N‐terminus at the ultimate cysteine residue.
In order to assess the production of AIPs by R. cellulolyticum, the strain was cultivated, the culture was extracted with ethyl acetate, and the extract was analyzed by HPLC‐HRMS. The metabolite profile was searched for the molecular ion masses calculated for each macrocyclic AIP (Rc‐AIP1‐4) without an NTE. No signals were detected that could account for the Rc‐AIP2‐4 predictions, regardless of NTE length (0 to 4 residues). However, we detected a metabolite with an exact mass consistent with the prediction for mature Rc‐AIP1 devoid of an NTE (calc. m/z 873.3389 [M+H]+; found m/z 873.3398) [M+H]+) (Figure 2 A and Figure 2 B). We also checked the metabolite profile for the exact masses of Rc‐AIP1 bearing NTEs of up to four residues; however, no corresponding signals were found (Figure S3). HRMS2‐based fragmentation of the ion corresponding to the Rc‐AIP1 candidate resulted in a pattern in which all observed fragment masses were in agreement with a macrocyclic peptide of the predicted amino acid sequence (CWFWSY) (Figure S4). Notably, Rc‐AIP1 is only the second example of a native clostridial AIP detected to date and, in both cases, the signaling molecules lack an NTE. [19]
Figure 2.
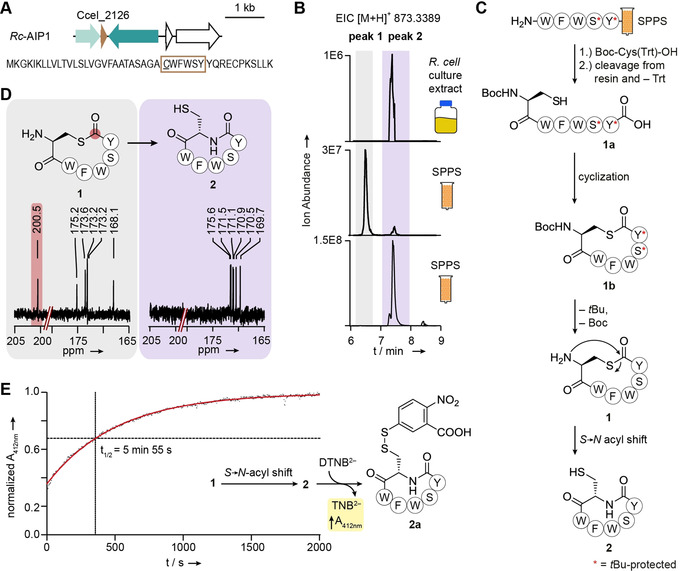
Ruminiclostridium cellulolyticum produces a homodetic cyclopeptide (cAIP) derived from an instable thiolactone: A) Representation of the Rc‐AIP1 biosynthetic gene cluster. The predicted core peptide sequence is surrounded by a brown box, with the cysteine residue at the putative cyclization position underlined. B) Extracted ion chromatogram (EIC) showing the detection of the mass corresponding to Rc‐AIP1 in the metabolite profile of R. cellulolyticum and the synthetic references of 1 and 2 (see Figure S7 for the mass spectra). SSPS, solid‐phase peptide synthesis. C) Synthetic route to synthetic references 1 and 2. The asterisk indicates the tBu‐protection of a hydroxyl group. D) 13C‐NMR spectra recorded for synthetic references 1 and 2, with a signal (highlighted in red) in the characteristic region for a thiolactone in the case of 1 (see Figure S6 and Figure S10 for full spectra). E) Determination of the stability of 1 by reaction of the spontaneously formed free thiol in 2 with Ellman's reagent. The increase in absorbance at 412 nm, resulting from the free anion (highlighted in yellow), was measured over time and the half‐life (t 1/2) of 1 was calculated based on the reaction rate. The graph shows a representative example (see Figure S11 for complete data set).
Rc‐AIP1 is a homodetic cyclopeptide derived from a thiolactone‐containing intermediate
The possibility of spontaneous S→N acyl migrations occurring in AIPs with thiolactones formed between the C‐ and N‐termini was only recently posited. [20] The identification of Rc‐AIP1, in which an NTE is absent, provided us with an opportunity to investigate this prospect. We attempted to isolate Rc‐AIP1 from R. cellulolyticum, but this proved challenging due to low production titers. As an alternative approach, we synthesized a reference compound for comparison to Rc‐AIP1. Specifically, we employed Fmoc‐based solid‐phase peptide synthesis (SPPS, for more experimental details see SI) to prepare the linear tBu‐protected peptide, WFWSY. The N‐terminal amino acid (Cys) was coupled to the linear protected peptide as a S‐Trt‐ and N‐Boc‐protected version by using ethyl cyanohydroxyiminoacetate (Oxyma), (1‐cyano‐2‐ethoxy‐2‐oxoethylidenaminooxy)‐dimethylamino‐morpholino‐carbenium hexafluorophosphate (COMU), and diisopropylethylamine (DIPEA) as coupling reagents. The Trt group was removed simultaneously as the Boc‐ and tBu‐protected peptide was liberated from the resin by treatment with hexafluoroisopropanol (HFIP) and purified by preparative HPLC. The obtained thiol peptide acid 1 a was cyclized with 1‐hydroxybenzotriazole hydrate (HOBt) and N‐(3‐dimethylaminopropyl)‐N′‐ethylcarbodiimide hydrochloride (EDCI) as coupling reagents, and DIPEA and 4‐dimethylaminopyridine (DMAP) as bases in DMF, [2j] to yield the corresponding thiolactone 1 b (Figure 2 C and Figure S5). After removal of the tBu‐based protecting groups with trifluoroacetic acid (TFA), the labile thiolactone 1, which could be handled under strictly acidic conditions only, was purified by preparative HPLC (3.4 % overall yield starting from SPPS). The characteristic 13C‐NMR signal at ≈200 ppm confirmed the successful formation of the thiolactone‐containing peptide 1 (Figure 2 D and Figure S6).
Immediately after deprotection, we subjected the synthetic thiolactone 1 to HPLC‐HRMS analysis in order to compare its retention time, accurate mass, and fragmentation patterns with those of Rc‐AIP1 from R. cellulolyticum culture extracts. We observed a divergence in the retention times of 1 (peak 1) and Rc‐AIP1 (Figure 2 B and Figure S7). However, we noted that a minor congener (peak 2) in the reference chromatogram had the same retention time as Rc‐AIP1. We also found that both compounds show the same m/z values and very similar HRMS2 fragmentation patterns, yet differ in their stabilities to the collision energies used (Figure S8). These observations led us to conclude that 1 is unstable and undergoes an S→N acyl shift to form the corresponding homodetic cyclopeptide 2 with a more stable amide bond (Figure 2 C).
To scrutinize this conjecture, we used HPLC‐HRMS to monitor the stability of thiolactone 1 in MeOH. We noted that 1 was indeed unstable, since the corresponding peak decreased upon formation of a new peak that came to predominate in the HPLC‐HRMS profile (Figure S9). In order to obtain a synthetic reference, we incubated 1 in 50 mM 3‐(N‐morpholino)propanesulfonic acid (MOPS) at pH 7 until peak 2 became the major signal in the chromatogram. The corresponding compound 2 was purified by preparative HPLC and its lactam structure was confirmed by NMR analyses (Figure 2 D and Figure S10). Notably, the retention time of 2 proved to be identical to that of the natural product (Rc‐AIP1) (Figure 2 B and Figure S7). Consequently, Rc‐AIP1 must exclusively possess amide bonds and hence deviates from the hitherto assumed (thio)lactone‐containing architecture of AIPs. In light of this difference, we designate Rc‐AIP1 as a “cAIP” to emphasize that it is a homodetic cyclopeptide.
To exclude the possibility that lactam 2 arose in the R. cellulolyticum metabolite profile as an artifact of the extraction procedure, we freshly prepared thiolactone 1 and, immediately after deprotection, determined its rearrangement kinetics under conditions mimicking those of the cultivation (at 37 °C in 50 mM MOPS, pH 7.1). As the conversion of 1 to 2 results in the formation of a thiol, the transformation was monitored by using a spectrophotometer and a thiol detection assay based on Ellman's reagent (Figure 2 E). [21] We determined an average half‐life of 5.4±0.6 min for 1 (Figure 2 E and Figure S11), indicating that the NTE‐free thiolactone‐containing AIP would not persist over the estimated generation time (6 h) of R. cellulolyticum determined under similar cultivation conditions. [22] We propose that by the time the threshold concentration needed to trigger a QS‐dependent response would be reached, the spontaneously formed 2 would dominate over 1. We therefore propose that 2 is the native structure of Rc‐AIP1.
Rc‐AIP1 controls endospore formation in Ruminiclostridium cellulolyticum
Our elucidation of the structure of Rc‐AIP1 revealed an unprecedented structural deviation for an AIP family member, but the question remained as to whether the homodetic cyclopeptide 2 acts as a competent signal. If so, it should be sensed by R. cellulolyticum and elicit a response. However, no physiological role has been reported for an R. cellulolyticum AIP. To this end, we created a null mutant by using CRISPR‐nCas9 genome editing [18] to incorporate a nonsense mutation in Rc‐agrD1 (Ccel_2126), the gene encoding Rc‐AIP1 (Figure 3 A and Figure S12). As expected, no signal for Rc‐AIP1 could be detected in the metabolite profile of R. cellulolyticum ΔagrD1 (Figure 3 B). This biosynthetic deficiency could be complemented by re‐introducing an intact copy of Rc‐agrD1 on a plasmid (Figure 3 B).
Figure 3.
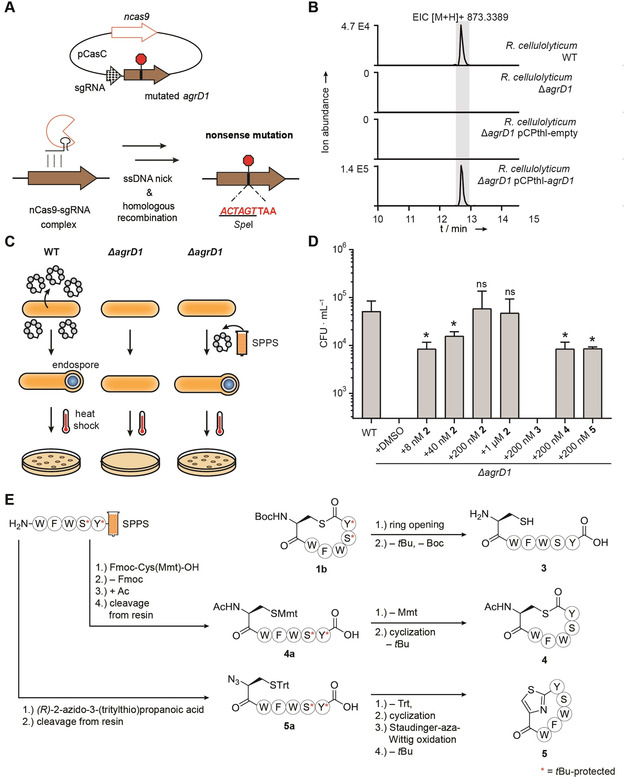
Control of endospore formation in Ruminiclostridium cellulolyticum by the cAIP Rc‐AIP1 and evaluation of synthetic surrogates: A) Scheme depicting the incorporation of a nonsense mutation into Rc‐agrD1 (Ccel_2126) of R. cellulolyticum to generate the null mutant R. cellulolyticum ΔagrD1. The simultaneous introduction of a SpeI recognition site enabled the differentiation of successfully edited R. cellulolyticum from the wild type by restriction digest of colony PCR products. B) Extracted ion chromatogram (EIC) showing the presence or absence of Rc‐AIP1 in the metabolite profiles of R. cellulolyticum wild type (WT), R. cellulolyticum ΔagrD1, R. cellulolyticum ΔagrD1 pCPthl‐empty (vector control) and R. cellulolyticum ΔagrD1 pCPthl‐agrD1 (complemented mutant). C) Overview of the endospore formation assay. SSPS, solid‐phase peptide synthesis. D) Quantification of endospore formation (colony forming units per milliliter, CFU ⋅ mL−1) by heat‐treated cultures of R. cellulolyticum wild type, R. cellulolyticum ΔagrD1, and R. cellulolyticum ΔagrD1 supplemented with the listed concentrations of 2, 3, 4 and 5. The results depicted are derived from at least three independent experiments. The colony counts for R. cellulolyticum ΔagrD1 + DMSO (vehicle control) and R. cellulolyticum ΔagrD1 + 200 nM 3 were below the quantification limit (300 CFU ⋅ mL−1) of the assay. For the other assay conditions, the displayed values are the mean colony counts (CFU ⋅ mL−1) with error bars showing the standard deviation. All values were compared to that of the wild type (ns, not significant; *, p≤0.05). DMSO, dimethyl sulfoxide. E) Synthetic route to compounds 3–5. The linear sequences for 4 and 5 were produced in the same manner as for 1 and 2 (see Figure 2 C). The asterisk indicates the tBu‐protection of a hydroxyl group.
Since Agr‐based QS has been implicated in the initiation of sporulation in Clostridia,[ 2f , 2k , 2l ] we assessed the sporulation efficiency of R. cellulolyticum ΔagrD1 (Figure 3 C). We found that the number of heat‐resistant endospores formed by R. cellulolyticum ΔagrD1 was indeed reduced by greater than two orders of magnitude compared to the wild‐type strain (Figure 3 D). Exogenous supplementation of R. cellulolyticum ΔagrD1 with 2 could rescue the spore formation deficiency in a concentration‐dependent manner (Figure 3 D). Importantly, 2 restored wild‐type levels of sporulation at 200 nM, a concentration considered physiologically relevant [23] and at least an order of magnitude lower than previously reported for clostridial Agr‐dependent phenotypes.[ 2j , 2l ] Combined with the inherent instability of 1 over a biologically relevant timescale, these chemical complementation experiments corroborate that 2 is an authentic signaling molecule involved in the orchestration of endospore formation in R. cellulolyticum.
Synthetic surrogates reveal the importance of a macrocycle to Rc‐AIP1 recognition
The rapid transformation of the thiolactone 1 into lactam 2 under culturing conditions precludes a direct comparison of their potencies in the endospore formation assay. In order to gain preliminary insights into the structure–activity relationship of Rc‐AIP1, we prepared three analogues: a linear derivative 3, an N‐terminally acetylated derivative 4, and a derivative 5 containing a peptidomimetic thiazole moiety at the macrocyclization site (Figure 3 E and Figure S5). N‐acetylated analogue 4 features an increased ring size compared to the naturally occurring lactam 2, but preserves the thiolactone moiety since it cannot undergo an S→N acyl shift. Thiazole‐containing analogue 5 has the same ring size as 2, but the risk of disulfide formation is eliminated. Moreover, it represents a stabilized analogue of the cAIP intermediate thiolactone 1 and is not prone to hydrolysis. Heterocyclic motifs are present in many natural products and their incorporation in cyclic peptides has led to molecules with improved properties. [24]
In brief, the linear peptide 3 was obtained by hydrolysis of the protected, SPPS‐derived thiolactone 1 b with aqueous NaOH, followed by TFA treatment to remove the protecting groups (0.8 % overall yield starting from SPPS) We prepared the N‐acetylated analogue 4 from the corresponding N‐acetylated, fully‐protected linear peptide 4 a, which was assembled on solid support. Treatment of 4 a with TFA and Et3SiH yielded the ω‐mercapto carboxylic acid, which was transformed into the thiolactone by using benzotriazole‐1‐yl‐oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and DIPEA. [25] Global cleavage of the protecting groups was performed under acidic conditions to give thiolactone 4 (8.4 % overall yield starting from SPPS). The thiazole analogue 5 was synthesized from the fully protected linear peptide 5 a, which was assembled by SPPS in analogy to 1 a and 4 a, but with (R)‐2‐azido‐3‐(tritylthio)propanoic acid replacing the protected cysteine. [25] Upon deprotection, the ω‐mercapto carboxylic acid was cyclized to 5 by using PyBOP as a coupling reagent and DIPEA as a base. The heterocyclization was promoted by means of a Staudinger‐aza‐Wittig sequence [25c] by using PPh3 in 2,6‐lutidine. The resulting thiazoline was oxidized by using 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) and bromotrichloromethane. Subsequently the protecting groups were cleaved by TFA/Et3SiH/H2O (16 % overall yield starting from SPPS). [26]
With these structural analogues at hand, we tested their potencies in the endospore formation bioassay with R. cellulolyticum ΔagrD1. In stark contrast to the response to 2, supplementation with 200 nM of the linear peptide 3 was indistinguishable from the phenotype of the untreated mutant (Figure 3 D). It is likely that the conformation needed for effective binding of Rc‐AIP1 to its cognate receptor is favored by its cyclic nature compared to the conformationally more flexible linear peptide. Both macrocyclic analogues 4 and 5 were active in the endospore formation assay, albeit less so than 2. Specifically, both 4 and 5 were only capable of partially complementing the sporulation deficient phenotype of R. cellulolyticum ΔagrD1 at 200 nM, a level at which 2 completely restored sporulation. As well as further supporting that a macrocyclic nature is important for recognition, the partial functionality of analogues 4 and 5 indicate that there is some variation tolerated in terms of linkage moiety and ring size of synthetic surrogates. However, the relative impact of these structural variations on biological function is difficult to distinguish. More elaborate structure‐activity studies will be necessary to precisely evaluate the structural features that are crucial for Rc‐AIP1 to adopt the conformation required for optimal interaction with its receptor.
Clostridial AIPs commonly undergo intramolecular S→N rearrangements
Prompted by the discovery that Rc‐AIP1 is a potent QS signal, we wondered if the products of other clostridial Agr clusters might be homodetic cyclopeptides. We therefore mined the genomes of diverse Clostridia spanning four genera for genes encoding homologous Agr systems (Figure S13A and Table S1). From this bioinformatic analysis, we selected four strains from two genera for cultivation and extraction. After performing HPLC‐HRMS analyses of the culture extracts, we scrutinized the metabolite profiles for signals consistent with the relevant mature AIPs having NTEs of up to four residues. We identified a number of candidates, all of which lacked an NTE. The sequences of the candidate AIPs were verified by HRMS2‐based fragmentation (Figure S4 and Figure S14–18). Our detection of the AIP produced by C. acetobutylicum confirms the prediction that it is a 6‐membered macrocyclic peptide without an NTE. [2l]
Since the absence of an NTE is a prerequisite for the S→N acyl shift to occur, we presumed that the newly detected AIPs should be subject to NCL‐like rearrangement to form cAIPs. However, such a connectivity difference would again be indistinguishable by HRMS2. Although it has been proposed that the AIPs of C. acetobutylicum [2l] and C. perfringens [2j] would not have NTEs based on chemical complementation experiments, the resulting possibility of rearrangement was not explored. Our detection of numerous NTE‐free AIPs allowed us to investigate whether the S→N acyl shift observed for Rc‐AIP1 is a more general phenomenon guided by the innate chemical reactivity of the α‐amino thioester.
In order to circumvent the need for synthetic references to finalize the structures for all candidate AIPs, we developed a method that could distinguish between a thiolactone or lactam moiety (Figure S19). We employed N‐benzylmaleimide to chemoselectively label the free thiols that would only be present in the lactam forms and exploited the fact that cAIPs would be resistant to basic hydrolysis (Figure 4 A). The method was validated by way of synthetic standards (Figure S20–23) and by showing that Rc‐AIP1 in a crude extract behaved as expected for a cAIP (Figure S24). When applied in a screen of the crude extracts containing candidate AIPs, a thiol adduct was detected in all (Figure 4 B–E), indicating that the cysteine side chain of each AIP had indeed been liberated by an S→N acyl shift. In accordance with the results obtained from N‐benzylmaleimide labeling, no signals corresponding to the linearized forms were detected upon NaOH treatment of the extracts (Figure 4 B–E). Hence, we conclude that, like Rc‐AIP1, these NTE‐free AIPs from Clostridia are homodetic cyclopeptides and should be classified as cAIPs.
Figure 4.
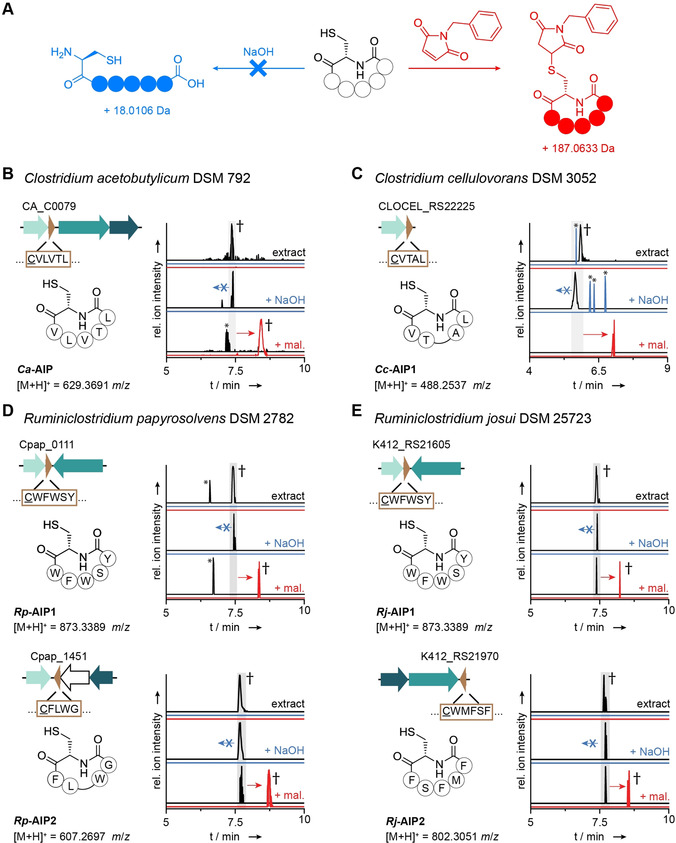
Identification of cAIPs in extracts of diverse Clostridia: A) General scheme depicting the reaction of a cAIP with N‐benzylmaleimide resulting in the formation of a thiol adduct (red) and the absence of hydrolysis by NaOH (blue). B–E) After treatment with N‐benzylmaleimide (+ mal) and NaOH, the AIPs detected in extracts of the named Clostridia behaved in a manner characteristic of homodetic peptides (cAIPs) (Figure 4 A). Specifically, new species corresponding to the mal‐adducts were detected (red), whereas no hydrolytic ring‐openings were observed (blue). EICs are shown at either a 1.0 ppm window (panel B) or a 1.5 ppm window (panels C–E). † Further analysis by HRMS2 experiments (untreated: Figure S4 and Figure S14–18, + mal: Figure S25–28). * Unrelated peaks detected in the crude extract.
A biosynthetic route to cyclopeptides involving enzyme‐primed native peptide ligation
Two in vivo routes for the formation of the cAIPs may be conceived (Figure 5 A). By analogy to the staphylococcal Agr system, AgrB should cleave the follower peptide of AgrD and catalyze the formation of a thiolactone‐containing macrocycle, which is then exported. Concomitant proteolytic cleavage of the leader peptide would liberate a free N‐terminus that can undergo a spontaneous S→N acyl shift (route (i)). Alternatively, the leader peptide may be removed before the core peptide is processed by AgrB. The resulting free amine would then liberate the AgrB‐bound intermediate by directly forming the amide bond (route (ii)), mirroring other RiPP maturation routes (e.g. cyanobactins). [27] Whereas there is no direct proof of a cryptic thiolactone intermediate, multiple lines of evidence support route (i) as the more probable pathway. Firstly, a cysteine residue at the cyclization position is indispensable for the formation of a cryptic thiolactone in route (i), whereas any amino acid would be suitable for route (ii). We and others noted that the cysteine residue is a conserved feature of clostridial AgrD peptides (Figure S13B).[ 2b , 2g , 2k , 2l , 15 ] Secondly, in characterized Agr systems the leader peptide is removed after the thiolactone‐containing peptide is secreted from the cell. [5a] Route (ii) would be inconsistent with these data.
Figure 5.
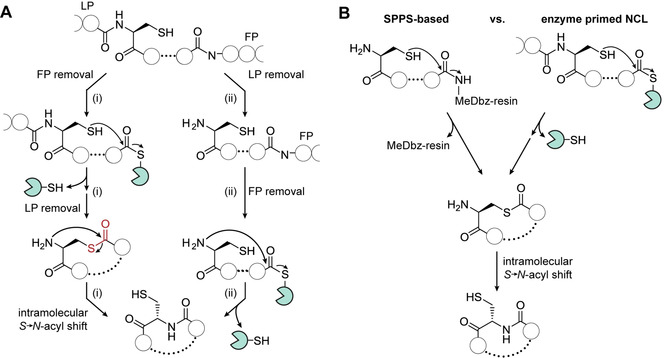
Model for cAIP biosynthesis: A) Two possible routes for in vivo cAIP formation involving either (i) a native chemical ligation (NCL)‐like S→N shift or (ii) the direct formation of the lactam. FP, follower peptide; LP leader peptide. B) Comparison of of solid‐phase peptide synthesis (SPPS)‐based and enzyme‐primed NCL strategies for cyclopeptide production. In chemical synthesis, direct attack of a free thiol leads to self‐cleaving from the 3‐amino‐4‐(methylamino)benzoic acid (MeDbz)‐linked resin yielding the thiolactone. In cAIP biosynthesis, the same product is produced by the attack of a free thiol to the thioester. The intermediary thiolactones undergo intramolecular S→N acyl shifts to give the corresponding cyclopeptides.
Taken together, these findings reveal a specialized biosynthetic pathway to cAIPs in Clostridia. By cleaving the follower peptide, AgrB generates a thioester‐bound peptide and promotes an entropy‐driven cyclization reaction to release the corresponding thiolactone. Liberation of the amine by cleavage of the NTE region sets the stage for a consecutive, maturing S→N acyl shift. This mechanism is strikingly similar to NCL strategies employed for the synthesis of (cyclo)peptides (Figure 5 B). [28] In particular, the AgrB‐primed pathway mirrors intramolecular variants of NCL that permit the on‐resin synthesis of cyclopeptides. [29] The reactivities and mechanisms are identical in both the synthetic and biosynthetic routes, yet the synthetic peptide is linked to a bead, whereas the cAIP precursor is tethered to an enzyme. Spontaneous S→N acyl migrations have been reported in protein splicing pathways, [30] as well as in a recently discovered compensatory pathway for glutathione biosynthesis in bacteria lacking the GshA glutathione ligase. [31] By drawing on chemical logic, it had been suspected that the thiolactone formed from an unprotected N‐terminal cysteine in a subset of AIPs could rearrange to yield an amide bond.[ 20 , 32 ] Our work describing the intramolecular, enzyme‐primed NCL route to clostridial cAIPs now provides support for this proposal.
Notably, the propensity of thioesters to undergo exchange reactions and S→N acyl shifts was recently exploited in a discovery strategy for canonical thiolactone‐possessing AIPs produced in culture.[ 20 , 32 ] Efforts to apply the NCL‐based technique to identify the NTE‐free AIP of the pathogen Listeria monocytogenes [33] were unsuccessful. [20] On the basis of our findings, we propose, as did the authors, that the mature AIP presents as the lactam form, meaning it would be incompatible with chemoselective trapping by NCL. We speculate that the NTE‐free AIP of the human‐associated Lactobacillus plantarum, [34] an important anaerobe used for food fermentation, could also be a cAIP. The fate of the lactone in NTE‐free AIPs cyclized through a serine residue, such as that of H. thermocellum, remains to be clarified. [19]
Finally, it should be highlighted that this previously overlooked natural NCL‐like cyclopeptide formation adds to the known pathways for RiPP maturation. Previously, ATP‐dependent and protease‐dependent avenues have been reported for cyclopeptide biosynthesis in diverse RiPP families,[ 27 , 35 ] with a maturation enzyme installing the lactam bond in each case. The mechanism for cAIP macrolactamization described herein markedly differs from these routes and represents a novelty in the RiPP biosynthetic toolbox.
Conclusion
Many genetic studies have indicated that Agr‐derived peptides mediate important processes in Clostridia. However, the true nature of the implicated signaling molecules has remained uncertain. In this study, we provide the first rigorously determined structure of a clostridial AIP. On the basis of chemical synthesis and meticulous analysis, we ascertained that the signals detected in clostridial cultures do not correspond to thiolactones; they are homodetic cyclopeptides or cAIPs that result from enzyme‐primed S→N acyl shifts. In the case of a model clostridial cAIP, Rc‐AIP1 produced by R. cellulolyticum, kinetic analyses exclude the possibililty that the thiolactone precursor could play a significant biological role, which is also supported by chemical complementation experiments.
Until now, it was believed that cyclization through a thioester or ester was a conserved feature of AIPs.[ 4 , 5c ] In expanding the chemical diversity of the AIP family to include cAIPs, this discovery highlights the importance of rigorous chemical analyses and synthesis, and serves as a cautionary tale regarding cursory bioinformatics‐based assumptions about the structures of natural products. Moreover, the structures and pathways described herein are an unexpected divergence from the canonical AIP biosynthetic routes and prompt a revision of the clostridial AIPs and their formation. This novel route to cyclic RiPPs via cryptic thiolactone intermediates may be considered as the natural equivalent of an intramolecular NCL. From a translational point of view, identification of the cAIP structures opens new possibilities to control anaerobic QS‐regulated processes using synthetic surrogates in biotechnology and remediation. Providing the homodetic cyclopeptide architecture is conserved in the products of the Agr systems implicated in the virulence of pathogenic Clostridia,[ 2a , 2b , 2c , 2d , 2e , 2f , 2g , 2h , 2i , 2j , 2k , 2m ] this knowledge could also assist current research on QS antagonists as therapeutics. [36]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank A. Perner for MS measurements, H. Heinecke for NMR measurements, and H. Büttner for providing culture extracts. Financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 1127/2 ChemBioSys‐239748522 (to H.D.A. and C.H.), Leibniz Award (to C.H.), and Cluster of Excellence “Balance of the Microverse” (to R.F., H.D.A. and C.H.), the Alexander von Humboldt Foundation (postdoctoral fellowship to K.L.D.), and the Carl Zeiss Foundation (predoctoral fellowship to A.O.) is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
E. M. Molloy, M. Dell, V. G. Hänsch, K. L. Dunbar, R. Feldmann, A. Oberheide, L. Seyfarth, J. Kumpfmüller, T. Horch, H.-D. Arndt, C. Hertweck, Angew. Chem. Int. Ed. 2021, 60, 10670.
References
- 1. Cruz-Morales P., Orellana C. A., Moutafis G., Moonen G., Rincon G., Nielsen L. K., Marcellin E., Genome Biol. Evol. 2019, 11, 2035–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Darkoh C., Odo C., DuPont H. L., mBio 2016, 7, e01237-16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Darkoh C., DuPont H. L., Norris S. J., Kaplan H. B., mBio 2015, 6, e02569-14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Martin M. J., Clare S., Goulding D., Faulds-Pain A., Barquist L., Browne H. P., Pettit L., Dougan G., Lawley T. D., Wren B. W., J. Bacteriol. 2013, 195, 3672–3681; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Chen J., McClane B. A., Infect. Immun. 2012, 80, 3008–3017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Chen J., Rood J. I., McClane B. A., mBio 2011, 2, e00275-11; [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2f. Li J., Chen J., Vidal J. E., McClane B. A., Infect. Immun. 2011, 79, 2451–2459; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2g. Ohtani K., Yuan Y., Hassan S., Wang R., Wang Y., Shimizu T., J. Bacteriol. 2009, 191, 3919–3927; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Vidal J. E., Ma M., Saputo J., Garcia J., Uzal F. A., McClane B. A., Mol. Microbiol. 2012, 83, 179–194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2i. Vidal J. E., Chen J., Li J., McClane B. A., PLoS One 2009, 4, e6232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2j. Ma M., Li J., McClane B. A., J. Bacteriol. 2015, 197, 1807–1818; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2k. Cooksley C. M., Davis I. J., Winzer K., Chan W. C., Peck M. W., Minton N. P., Appl. Environ. Microbiol. 2010, 76, 4448–4460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2l. Steiner E., Scott J., Minton N. P., Winzer K., Appl. Environ. Microbiol. 2012, 78, 1113–1122; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2m. Navarro M. A., Li J., Beingesser J., McClane B. A., Uzal F. A., mSphere 2020, 5, e00500-20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2n. Kosaka T., Nakayama S., Nakaya K., Yoshino S., Furukawa K., Biosci. Biotechnol. Biochem. 2007, 71, 58–68; [DOI] [PubMed] [Google Scholar]
- 2o. Feng J., Zong W., Wang P., Zhang Z. T., Gu Y., Dougherty M., Borovok I., Wang Y., Biotechnol. Biofuels 2020, 13, 84; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2p. Kotte A. K., Severn O., Bean Z., Schwarz K., Minton N. P., Winzer K., Microbiology 2020, 166, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Waters C. M., Bassler B. L., Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346; [DOI] [PubMed] [Google Scholar]
- 3b. Williams P., Winzer K., Chan W. C., Cámara M., Philos. Trans. R. Soc. London Ser. B 2007, 362, 1119–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., Dorrestein P. C., Entian K. D., Fischbach M. A., Garavelli J. S., Göransson U., Gruber C. W., Haft D. H., Hemscheidt T. K., Hertweck C., Hill C., Horswill A. R., Jaspars M., Kelly W. L., Klinman J. P., Kuipers O. P., Link A. J., Liu W., Marahiel M. A., Mitchell D. A., Moll G. N., Moore B. S., Müller R., Nair S. K., Nes I. F., Norris G. E., Olivera B. M., Onaka H., Patchett M. L., Piel J., Reaney M. J., Rebuffat S., Ross R. P., Sahl H. G., Schmidt E. W., Selsted M. E., Severinov K., Shen B., Sivonen K., Smith L., Stein T., Süssmuth R. D., Tagg J. R., Tang G. L., Truman A. W., Vederas J. C., Walsh C. T., Walton J. D., Wenzel S. C., Willey J. M., van der Donk W. A., Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Novick R. P., Geisinger E., Annu. Rev. Genet. 2008, 42, 541–564; [DOI] [PubMed] [Google Scholar]
- 5b. Monnet V., Juillard V., Gardan R., Crit. Rev. Microbiol. 2016, 42, 339–351; [DOI] [PubMed] [Google Scholar]
- 5c. Thoendel M., Kavanaugh J. S., Flack C. E., Horswill A. R., Chem. Rev. 2011, 111, 117–151; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Vasquez J. K., West K. H. J., Yang T., Polaske T. J., Cornilescu G., Tonelli M., Blackwell H. E., J. Am. Chem. Soc. 2020, 142, 750–761; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Yang T., Tal-Gan Y., Paharik A. E., Horswill A. R., Blackwell H. E., ACS Chem. Biol. 2016, 11, 1982–1991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Tal-Gan Y., Ivancic M., Cornilescu G., Blackwell H. E., Org. Biomol. Chem. 2016, 14, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Queck S. Y., Jameson-Lee M., Villaruz A. E., Bach T. H., Khan B. A., Sturdevant D. E., Ricklefs S. M., Li M., Otto M., Mol. Cell 2008, 32, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang L., Lin J., Ji G., J. Biol. Chem. 2004, 279, 19448–19456. [DOI] [PubMed] [Google Scholar]
- 8. Ji G., Beavis R. C., Novick R. P., Proc. Natl. Acad. Sci. USA 1995, 92, 12055–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thoendel M., Horswill A. R., J. Biol. Chem. 2009, 284, 21828–21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Zhang L., Gray L., Novick R. P., Ji G., J. Biol. Chem. 2002, 277, 34736–34742; [DOI] [PubMed] [Google Scholar]
- 10b. Qiu R., Pei W., Zhang L., Lin J., Ji G., J. Biol. Chem. 2005, 280, 16695–16704. [DOI] [PubMed] [Google Scholar]
- 11. Wang B., Zhao A., Novick R. P., Muir T. W., Proc. Natl. Acad. Sci. USA 2015, 112, 10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kavanaugh J. S., Thoendel M., Horswill A. R., Mol. Microbiol. 2007, 65, 780–798. [DOI] [PubMed] [Google Scholar]
- 13. Otto M., Süssmuth R., Jung G., Götz F., FEBS Lett. 1998, 424, 89–94. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Nakayama J., Cao Y., Horii T., Sakuda S., Akkermans A. D., de Vos W. M., Nagasawa H., Mol. Microbiol. 2001, 41, 145–154; [DOI] [PubMed] [Google Scholar]
- 14b. Nakayama J., Chen S., Oyama N., Nishiguchi K., Azab E. A., Tanaka E., Kariyama R., Sonomoto K., J. Bacteriol. 2006, 188, 8321–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wuster A., Babu M. M., J. Bacteriol. 2008, 190, 743–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desvaux M., FEMS Microbiol. Rev. 2005, 29, 741–764. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lincke T., Behnken S., Ishida K., Roth M., Hertweck C., Angew. Chem. Int. Ed. 2010, 49, 2011–2013; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2055–2057; [Google Scholar]
- 17b. Chiriac A. I., Kloss F., Krämer J., Vuong C., Hertweck C., Sahl H. G., J. Antimicrob. Chemother. 2015, 70, 2576–2588; [DOI] [PubMed] [Google Scholar]
- 17c. Dunbar K. L., Dell M., Gude F., Hertweck C., Proc. Natl. Acad. Sci. USA 2020, 117, 8850–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Fedorova I., Arseniev A., Selkova P., Pobegalov G., Goryanin I., Vasileva A., Musharova O., Abramova M., Kazalov M., Zyubko T., Artamonova T., Artamonova D., Shmakov S., Khodorkovskii M., Severinov K., Nucleic Acids Res. 2020, 48, 2026–2034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Xu T., Li Y., Shi Z., Hemme C. L., Li Y., Zhu Y., Van Nostrand J. D., He Z., Zhou J., Appl. Environ. Microbiol. 2015, 81, 4423–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verbeke T. J., Giannone R. J., Klingeman D. M., Engle N. L., Rydzak T., Guss A. M., Tschaplinski T. J., Brown S. D., Hettich R. L., Elkins J. G., Sci. Rep. 2017, 7, 43355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gless B. H., Bojer M. S., Peng P., Baldry M., Ingmer H., Olsen C. A., Nat. Chem. 2019, 11, 463–469. [DOI] [PubMed] [Google Scholar]
- 21. Riddles P. W., Blakeley R. L., Zerner B., Anal. Biochem. 1979, 94, 75–81. [DOI] [PubMed] [Google Scholar]
- 22. Jennert K. C. B., Tardif C., Young D. I., Young M., Microbiology 2000, 146 Pt 12, 3071–3080. [DOI] [PubMed] [Google Scholar]
- 23. MDowell P., Affas Z., Reynolds C., Holden M. T., Wood S. J., Saint S., Cockayne A., Hill P. J., Dodd C. E., Bycroft B. W., Chan W. C., Williams P., Mol. Microbiol. 2001, 41, 503–512. [DOI] [PubMed] [Google Scholar]
- 24. Smolyar I. V., Yudin A. K., Nenajdenko V. G., Chem. Rev. 2019, 119, 10032–10240. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Oberheide A., Pflanze S., Stallforth P., Arndt H.-D., Org. Lett. 2019, 21, 729–732; [DOI] [PubMed] [Google Scholar]
- 25b. Schwenk S., Ronco C., Oberheide A., Arndt H.-D., Eur. J. Org. Chem. 2016, 4795–4799; [Google Scholar]
- 25c. Riedrich M., Harkal S., Arndt H.-D., Angew. Chem. Int. Ed. 2007, 46, 2701–2703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2755–2758. [Google Scholar]
- 26. Phillips A. J., Uto Y., Wipf P., Reno M. J., Williams D. R., Org. Lett. 2000, 2, 1165–1168. [DOI] [PubMed] [Google Scholar]
- 27. Ongpipattanakul C., Nair S. K., Biochemistry 2018, 57, 3201–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Burke H. M., McSweeney L., Scanlan E. M., Nat. Commun. 2017, 8, 15655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. White C. J., Yudin A. K., Nat. Chem. 2011, 3, 509–524; [DOI] [PubMed] [Google Scholar]
- 28c. Tam J. P., Wong C. T., J. Biol. Chem. 2012, 287, 27020–27025; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d. Agouridas V., El Mahdi O., Diemer V., Cargoët M., Monbaliu J.-C. M., Melnyk O., Chem. Rev. 2019, 119, 7328–7443; [DOI] [PubMed] [Google Scholar]
- 28e. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Tulla-Puche J., Barany G., J. Org. Chem. 2004, 69, 4101–4107; [DOI] [PubMed] [Google Scholar]
- 29b. Serra G., Posada L., Hojo H., Chem. Commun. 2020, 56, 956–959; [DOI] [PubMed] [Google Scholar]
- 29c. Liu C.-F., Tam J. P., J. Am. Chem. Soc. 1994, 116, 4149–4153. [Google Scholar]
- 30. Paulus H., Annu. Rev. Biochem. 2000, 69, 447–496. [DOI] [PubMed] [Google Scholar]
- 31. Veeravalli K., Boyd D., Iverson B. L., Beckwith J., Georgiou G., Nat. Chem. Biol. 2011, 7, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McBrayer D. N., Tal-Gan Y., Nat. Chem. 2019, 11, 398–399. [DOI] [PubMed] [Google Scholar]
- 33. Zetzmann M., Sanchez-Kopper A., Waidmann M. S., Blombach B., Riedel C. U., Front. Microbiol. 2016, 7, 989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sturme M. H., Nakayama J., Molenaar D., Murakami Y., Kunugi R., Fujii T., Vaughan E. E., Kleerebezem M., de Vos W. M., J. Bacteriol. 2005, 187, 5224–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montalbán-López M., Scott T. A., Ramesh S., Rahman I. R., van Heel A. J., Viel J. H., Bandarian V., Dittmann E., Genilloud O., Goto Y., Grande Burgos M. J., Hill C., Kim S., Koehnke J., Latham J. A., Link A. J., Martínez B., Nair S. K., Nicolet Y., Rebuffat S., Sahl H. G., Sareen D., Schmidt E. W., Schmitt L., Severinov K., Süssmuth R. D., Truman A. W., Wang H., Weng J. K., van Wezel G. P., Zhang Q., Zhong J., Piel J., Mitchell D. A., Kuipers O. P., van der Donk W. A., Nat. Prod. Rep. 2021, 38, 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. LaSarre B., Federle M. J., Microbiol. Mol. Biol. Rev. 2013, 77, 73–111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Salam A. M., Quave C. L., mSphere 2018, 3, e00500-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36c. Tal-Gan Y., Stacy D. M., Foegen M. K., Koenig D. W., Blackwell H. E., J. Am. Chem. Soc. 2013, 135, 7869–7882; [DOI] [PubMed] [Google Scholar]
- 36d. Vasquez J. K., Blackwell H. E., ACS Infect. Dis. 2019, 5, 484–492; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36e. Tal-Gan Y., Ivancic M., Cornilescu G., Yang T., Blackwell H. E., Angew. Chem. Int. Ed. 2016, 55, 8913–8917; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9059–9063; [Google Scholar]
- 36f. Vasquez J. K., Tal-Gan Y., Cornilescu G., Tyler K. A., Blackwell H. E., ChemBioChem 2017, 18, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary