

Abstract
Over the last two decades, beginning with the Avian Brain Nomenclature Forum in 2000, major revisions have been made to our understanding of the organization and nomenclature of the avian brain. However, there are still unresolved questions on avian pallial organization, particularly whether the cells above the vestigial ventricle represent distinct populations to those below it or similar populations. To test these two hypotheses, we profiled the transcriptomes of the major avian pallial subdivisions dorsal and ventral to the vestigial ventricle boundary using RNA sequencing and a new zebra finch genome assembly containing about 22,000 annotated, complete genes. We found that the transcriptomes of neural populations above and below the ventricle were remarkably similar. Each subdivision in dorsal pallium (Wulst) had a corresponding molecular counterpart in the ventral pallium (dorsal ventricular ridge). In turn, each corresponding subdivision exhibited shared gene co‐expression modules that contained gene sets enriched in functional specializations, such as anatomical structure development, synaptic transmission, signaling, and neurogenesis. These findings are more in line with the continuum hypothesis of avian brain subdivision organization above and below the vestigial ventricle space, with the pallium as a whole consisting of four major cell populations (intercalated pallium, mesopallium, hyper‐nidopallium, and arcopallium) instead of seven (hyperpallium apicale, interstitial hyperpallium apicale, intercalated hyperpallium, hyperpallium densocellare, mesopallium, nidopallium, and arcopallium). We suggest adopting a more streamlined hierarchical naming system that reflects the robust similarities in gene expression, neural connectivity motifs, and function. These findings have important implications for our understanding of overall vertebrate brain evolution.
Keywords: avian, brain evolution, pallium, RNA‐Seq, transcriptomics, WGNCA, RRIDs, SCR_014583, SCR_017036, SCR_001905, SCR_015954, SCR_010943, SCR_015687, SCR_021063, SCR_003092, SCR_012988, SCR_003302, SCR_018190, SCR_021061, SCR_021062, SCR_004277, SCR_000432
In 2013, our group examined the expression profiles of 50 genes to develop the continuum hypothesis of avian brain organization. It states that the subdivisions of the dorsal pallium develop continuously with those of the ventral pallium, resulting in a “partial mirror image” organization around the vestigial ventricle divide. However, these claims were challenged due to the small number of genes profiled. The present study uses RNA sequencing to profile the whole transcriptome (~20,000 genes) of the principal subdivisions of the avian telencephalon and confirms the remarkable molecularly similarities between the dorsal and ventral pallium. We recommend adopting a hierarchal nomenclature to reflect these robust molecular similarities.
1. INTRODUCTION
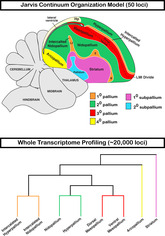
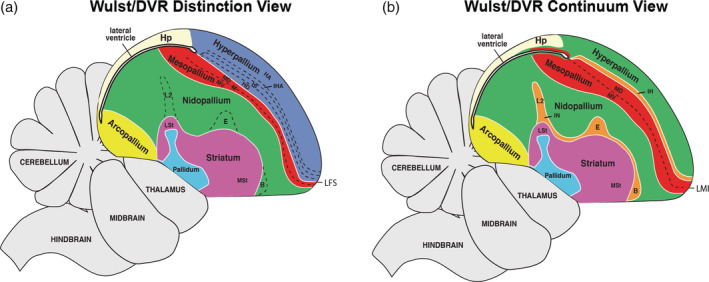
More than 120 years ago, some of the founders of comparative neurobiology proposed that the nonmammalian telencephalon consisted of mostly basal ganglia, homologous to the mammalian striatum and globus pallidus (Ariëns Kappers, 1922; Edinger, 1888; Edinger, 1908). This was the dominate view until the late 1960s, when the use of histochemical markers led to an alternative hypothesis that some of the striatal regions could instead be homologous to cell populations in the mammalian cortex (Karten, 1969). In the early 2000s, The Avian Brain Nomenclature Forum was formed, a consortium that evaluated the past century of findings and performed additional experiments, to develop a revised nomenclature. They concluded that the dorsal two‐thirds of the avian telencephalon was organized into distinct cell type subdivisions broadly homologous to the developing mammalian pallium, inclusive of the six‐layered cortex, claustrum, and pallial amygdala (Jarvis et al., 2005; Reiner, Perkel, Bruce, et al., 2004). As a result, the Forum, with support from the broader neuroscience community, developed a revised nomenclature that more accurately defined the organization of avian neural cell populations and their homologies with those of mammals and potentially other vertebrates. One important contribution of the Forum was to further define the relationship of cell populations around the prominent cell‐free lamina mesopallialis intermediate (LMI), the vestigial ventricle space that closes during avian embryonic development (Chen et al., 2013). Specifically, this new nomenclature considered the subdivisions dorsal to the vestigial ventricle as “hyperpallium” populations comprising the Wulst or dorsal cortex as distinct from those ventral to it (Reiner, Perkel, Mello, & Jarvis, 2004). These ventral pallium subdivisions were named the mesopallium, nidopallium (along with intercalated nidopallium [IN]), and arcopallium, comprising the dorsal ventricular ridge (DVR; Figure 1(a)). We term this view here as the “distinction hypothesis,” as it views the Wulst and DVR as neural populations broadly distinct from one another (Reiner, Perkel, Bruce, et al., 2004; Reiner, Perkel, Mello, & Jarvis, 2004).
FIGURE 1.
Two competing hypotheses on avian brain organization. (a) Dorsal and ventral pallium distinction hypothesis. This view treats the Wulst (blue) and dorsal ventricular ridge (DVR; red, green, yellow) above and below the vestigial ventricle divide as distinct sets of cell populations. The different subdivisions of the Wulst (separated by dotted lines) were given the hyperpallium prefix. The different subdivisions of the DVR were named mesopallium, nidopallium, and arcopallium. (b) Dorsal and ventral pallium continuum hypothesis. Each subdivision above the vestigial ventricle is colored according to gene expression similarity (n = 50) to cell populations below this divide. The lamina frontralis superior (LFS) (white line in a) or lamina mesopallialis intermediate (LMI) (dashed line in b) is the remaining vestigial lateral ventricle that has become condensed in adults but connected with the more posterior lateral open ventricle space shown below the hippocampus (Hp). Figure modified from Jarvis et al. (2013)
Despite this advance in our knowledge of these cell population relationships, a new model proposed nearly a decade later sparked a new debate in our understanding of avian telencephalon organization. Jarvis et al. (2013) and Chen et al. (2013) analyzed the mRNA in situ hybridization patterns of 50 marker genes in both coronal and sagittal planes of the adult and developing avian telencephalon. The findings suggested that what the Forum had defined as different hyperpallium populations above the vestigial ventricle (Figure 1(a), blue) exhibited shared gene expression profiles with cell populations below it (Figure 1(b), red, green, and orange). Combined with histological, functional, and connectivity evidence, they proposed a model of cell type continuities around the developing ventricular space. Gene expression patterns from the adult and developing avian telencephalon suggested that these continuous cell populations proliferate and simultaneously wrap around the ventricle, which closes prior to hatching to form an anatomical separation between the dorsal and ventral pallium regions. We term this view here as the “continuum hypothesis.”
Despite these advances, the continuum hypothesis or “partial mirror image” view of avian brain organization above and below the lamina divide has been met with some criticism. Some raised concern over the limited number and potentially biased selection of the genes sampled (Montiel & Molnár, 2013). Other concurrent gene expression studies appeared to support the Forum's distinction of cell populations above and below the vestigial ventricle divide (Belgard et al., 2013; Dugas‐Ford et al., 2012; Montiel & Molnár, 2013; Suzuki & Hirata, 2014). One study found that the developmental expression profile of the NR4A2 receptor only marked the mesopallium below the vestigial ventricle in birds, providing support to the Wulst/DVR distinction model (Puelles et al., 2016; Watson & Puelles, 2017). To try to reconcile these conflicting conclusions, another study proposed a hybrid hypothesis, which calls for the reclassification of hyperpallium densocellare (HD, Figure 1(a)) as dorsal mesopallium (MD, Figure 1(b)) while maintaining distinction between the nidopallium and the rest of the hyperpallium (Wullimann, 2017). However, this view did not address the proposed relationship of the intercalated sensory pallium associated with the hyperpallium and nidopallium. In contrast, the dorsal and ventral mesopallium relationship around the vestigial ventricle has since been supported with RNA‐Seq transcriptome profiling of embryonic chicken brains (Briscoe et al., 2018), but data from additional regions is necessary to further test these similarities. The ongoing debate over the precise organization of the avian brain makes it difficult to perform comparative and functional analyses within the avian brain, across vertebrate lineages, and between studies that rely different models (Briscoe & Ragsdale, 2018; Lovell et al., 2020; Puelles et al., 2016).
Here, we attempt to further resolve these hypotheses (Figure 1) by performing RNA‐Seq transcriptome profiling on the main avian pallium populations in question, using the zebra finch (Taeniopygia guttata), a songbird. The zebra finch belongs to the Neoaves clade, which makes up 95% of extant living bird species (Jarvis et al., 2014). Our comparative expression profiling of over 22,000 genes supports the continuum hypothesis of shared relationships of pallial populations below and above the lateral ventricle, resolves discrepancies with the prior literature, and reveals functional specializations specific to each combination of populations.
2. MATERIALS AND METHODS
2.1. Animals subjects
Tissue samples were collected from four adult male zebra finches (~1–6 years old, Table S1). Animals were individually housed overnight in sound isolation chambers. Birds were euthanized in the dark 2 h before the lights normally come on, by rapid decapitation within 1–2 min of handling, to limit activity‐dependent gene expression changes in the brain (Feenders et al., 2008; Wada et al., 2006; Whitney et al., 2014). The brains were extracted, bisected sagittally, and each hemisphere frozen in Tissue‐Tek OCT (Sakura, #4583) in a cryomold on dry ice. The entire procedure was performed within 5 min to reduce influence of activity‐dependent genes and preserve RNA integrity.
2.2. Laser capture microscopy and RNA‐Seq libraries/sequencing
One hemisphere/bird was sectioned on a cryostat at 12 μM and mounted on PEN membrane slides for laser capture microscopy (LCM, Arcturus XT). In brief, one slide containing reference sections from each slide series was stained with Cresyl violet to aid in subdivision identification. PEN slides from alternating series containing the sections of interest were individually dehydrated in serial alcohol baths from 50 to 100% and visualized under brightfield on the LCM (Arcturus Slide Prep Protocol #2). In addition to using the adjacent Cresyl violet stained section, axon bundles were visible in the LCM sections and used to help identify brain subdivisions. The region of interest was selected using a touch screen monitor and stylus pen and then laser dissected using the “Cut‐and‐Capture” method (Arcturus Instrument User Guide). First, a specialized cap with a microplastic film (Macro Caps: LCM0211) was placed on the tissue, then an infrared laser was used to melt the microplastic film to “capture” the tissue on the cap, and finally an ultraviolet laser was used to “cut” the region of interest from the larger tissue section. All tissue samples were dissected in under 30 min/slide to ensure high RNA integrity. RNA quality for all samples was measured by the RNA Integrity Number (RIN, range 1–10) provided by the Agilent RNA 6000 Pico kit run on the Bioanalyzer (Cat: 5067‐1513). We required a minimum RIN of 6 (mean = 7.1) before processing for sequencing. RNA was isolated from each sample using the Picopure RNA Isolation kit (Ref: KIT0204) and stored at −80° until all samples were collected. Samples were randomized across batches (n = 4) to minimize batch effects. However, some samples, specifically the IN and hyperpallium, were collected several years apart in sections from the same animals, so additional analyses were performed to test and normalize for any potential batch effects. Samples were randomized into batches and cDNA was generated using the SMART‐Seq Ultra Low‐Input RNA kit for sequencing (Clonetech, Ref: 634891). Each sample library was prepped using the NEBNext Ultra II DNA Library Prep Kit (Cat: E7645L) and dual‐indexed for sequencing using the NEBNext Multiplex Oligos for Illumina Set 2 (Cat: E6442S). RNA Sequencing of pair‐end, 150 bp reads was conducted on the NextSeq 500 system from Illumina.
2.3. Sequencing data processing and quality control
Quality of all raw sequence reads were verified using FastQC (v0.11.5, RRID:SCR_014583), trimming off low‐quality (<QV30) and adapter sequences using fastq‐mcf (v1.05). Transcript levels for each gene in each brain region were quantified using Salmon (v0.14.1, RRID:SCR_017036), a pseudoalignment software designed for fast quantification of annotated transcripts from RNA‐Seq data (Patro et al., 2017). The annotation used for quantification was generated using the newly assembled, high‐quality, long‐read‐based, Vertebrate Genome Project (VGP) zebra finch genome (bTaeGut1_v1, RefSeq Accession: GCF_003957565.1). A final gene × sample expression matrix was used as input for all downstream analyses. The empirical cumulative probability distributions were calculated for each of the two collection groups using the “ecdf” function in R (RRID:SCR_001905) and were plotted against each other in a P–P plot. The relative log expression (RLE) plot was generated using the “plotRLE” function from the scater package (v1.12.2, RRID:SCR_015954) in R (v4.0.2, RRID:SCR_000432). Housekeeping genes were empirically determined based on their expression variation, with any gene with a coefficient of variance (CV) of zero across all samples taken as housekeeping. The scater package was used to explore the effects of unwanted variations from known sources. Any significant sources of unwanted variation, like individual bird replicates, were accounted for in all downstream analyses either by inclusion as a term in linear models or direct correction using the “removeBatchEffect” function (Ritchie et al., 2015) from limma (v3.40.6, RRID:SCR_010943).
2.4. Principal component analysis
The unnormalized gene × sample expression matrix was supplied to DESeq2 (v1.24.0, RRID:SCR_015687) for differential expression testing between brain subdivisions. In order to visualize principal sources of variation in the data, the gene × sample expression matrix was normalized using the variance stabilizing transformation method from the DESeq2 package in R (Love et al., 2014). This method produces normalized data on a log2 scale with respect to library size, with the goal of removing the dependence of the variance on the mean (Huber et al., 2002). Such dependence on the mean can lead to high values on the logarithmic scale for lowly expressed genes which are frequent in expression data sets. Following normalization, we performed principal component analysis (PCA) and visualized the first two principal components (PCs) with DEseq2, applying the default setting limiting the analysis to the top 500 most variable genes. We verified the variance explained by this subset of genes most strongly correlated with the biological variable of interest using the scater package, indicating it is an appropriate threshold. Additional PCs were obtained using the “prcomp” function in R. Both functions center the data around the mean and calculate the covariance matrix, while “prcomp” computes additional PCs equal to the number of biological samples in the experiment. We also plotted the percent variance explained by each PC. We repeated this procedure for all samples (n = 36) and a subset of samples hypothesized to be similar in the dorsal and ventral pallium (n = 28).
2.5. Molecular anatomical cluster analysis of differentially expressed genes
For differential expression testing, a linear model was constructed on the gene × sample expression matrix without normalization with the design “~ subdivision + individual”. This allowed for modeling of the observed expression variance as a function of neural subdivision while controlling for the individual replicate effect. From this model, each subdivision was contrasted to all others in a pairwise manner. Tests conducted within each pairwise comparison were subjected to independent filtering from DESeq2, which filters out tests that have little or no chance of showing significant evidence, resulting in increased power with the same Type 1 error rate (Love et al., 2014). Genes were considered differentially expressed if they passed multiple test corrections (Benjamini–Hochberg; q < .05). A dissimilarity matrix was generated from the total number of differentially expressed genes (DEGs; i.e., degree of difference) for each comparison and results clustered using the “hclust” (method = “average”) function from R. A union set of all DEGs was taken and a similar clustering procedure was performed on the normalized counts to determined degree of shared expression between all samples for the genes with the strongest biological signal. Bootstrap resampling was also performed using 1000 iterations of all DEGs using the pvclust package in R (v2.2‐0; RRID:SCR_021063). Importantly, there are frequent improvements to the zebra finch assembly/annotation, so currently uncharacterized genes (LOC IDs) may be annotated with gene symbols following the publication of this manuscript. All genes from this analysis (Table S2) can be searched in NCBI's Genome Data Viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/; RRID:SCR_003092) by selecting the organism (zebra finch) and assembly version used in this study (bTaeGut1_v1.p, RefSeq Accession: GCF_003957565.1) and the most up‐to‐date records will be displayed.
2.6. Validations using in situ hybridization
A total of 64 genes with zebra finch brain in situ hybridization data were selected to test for concordance and validation of the differential expression tests results from our RNA‐Seq data. The in situ hybridization data were selected from a variety of sources (Wada et al., 2004; Kubikova et al., 2010; Jarvis et al., 2013; Chen et al., 2013; Pfenning et al., 2014; Whitney et al., 2014, Lovell et al., 2020 RRID:SCR_012988) based on high resolution images. Each gene was scored by an independent, blind observer as either a positive or negative marker for all subdivisions profiled in the present study. Pairwise brain subdivision comparisons were examined that were most relevant for testing between the two brain organization hypotheses. In each comparison, genes were scored as either: (1) true positive (TP)—specialized expression in the in situ is in agreement with the RNA‐Seq results; (2) true negative (TN)—absence of specialized expression in the in situ is in agreement with the differential expression results from RNA‐Seq, (3) false positive (FP)—differential expression testing failed to detect visible difference observed in the in situ; and (4) false negative (FN)—in situ expression profile does not match the results of differential expression. With these values, we defined an accuracy measure for each brain subdivision comparison as (TP + TN)/(TP + TN + FP + FN).
2.7. Weighted gene co‐expression network construction
Gene networks were assessed following best practices for weighted gene network co‐expression (WGCNA) analysis using the WGCNA package (v1.69; RRID:SCR_003302) in R (Langfelder & Horvath, 2008). The overall steps of WGCNA are to: (1) define a weighted correlation matrix of gene–gene similarities; (2) group similar gene clusters into distinct co‐expression modules; (3) correlate gene modules to biological features of interest; and (4) define “hub” genes that drive the desired phenotype. In order to construct the weighted co‐expression matrix, all samples were first screened for outlier genes using the “goodSamplegenes” function. This resulted in 20,822 stably expressed genes identified across all samples. A Pearson correlation adjacency matrix was then constructed for these genes across all input samples, raising each correlation to an empirically determined soft power exponent, depending on the network (all samples = 8, excluding intercalated samples = 6). Raising each correlation to a power helps to highlight disparity between strong and weak correlations which is helpful for thousands of correlations. Importantly, all networks were unsigned, which does not allow for both positive and negative correlations.
2.8. Identification of gene modules in specific brain subdivisions
The weighted adjacency matrix was then converted into a topological overlap matrix (TOM), which is a pairwise similarity measure of genes (nodes) in the network. Genes with a high TOM exhibit shared expression profiles. The TOM was then converted into a dissimilarity matrix (1‐TOM) to highlight differences between genes and allow for dendrogram clustering using the “hclust” function in R. Clustered genes exhibiting shared expression patterns were grouped into modules and each assigned individual color and number IDs (0‐n) to aid in identification. The minimum module size was set to 100 genes in order to reduce obtaining single sample modules specific to an individual/background noise. Module eigengenes (MEs) were calculated for each module by taking the first PC of the expression data for all assigned module genes. The ME allowed the expression patterns of all genes in a given module to be summarized to a single statistic and was useful for correlation with brain subdivisions. Each sample was binary coded as either belonging to a unique brain region or combination of regions depending on hypothesis, and significantly associated modules were determined by Pearson correlation with each module's eigengene using the “corPvalueStudent” function from the WGNCA package. p‐Values were corrected for multiple tests (method = “fdr”) using the “p.adj” function in R. Modules were considered highly significantly correlated for each brain subdivision if they exhibited an r 2 value >.90 and a q value <.001. Gene ontology analysis was conducted for modules significantly associated to subdivisions to assess functional enrichment, with a Homo sapiens background and custom expression background (mean expression <10/all samples) using gProfileR2 (v0.1.9; RRID:SCR_018190).
2.9. Hub gene identification for subdivision modules
To identify potential drivers of the shared expression of module genes, we looked for hub genes in each subdivision‐associated module. Here, Hub genes are defined as genes whose expression exhibits strong correlation with a given brain region and high connectivity with other genes (nodes) in a given module network. Two metrics were used to determine significant hub genes for each brain subdivision module. First, gene significance (GS, strength of correlation to a region) was defined as the Pearson correlation of each gene expression value to each brain subdivision. p‐Values were calculated for each correlation and corrected for multiple tests as described above. Second, module membership (MM, strength of connectivity in the module) was determined using the Pearson correlation of each gene's expression value with the eigengene vector for each module. Hub genes were defined by their significant correlation with a brain subdivision (absolute value of GS > 0.8) and strong connectivity with other module genes (absolute value of MM > 0.8). Similar thresholds have been utilized by other groups for identification of hub genes (Hilliard et al., 2012; Li et al., 2020; Liao et al., 2020). Networks of the top 50 interconnected hub genes were visualized using VizANT (v1.0; RRID:SCR_021061). Uncharacterized genes (LOC IDs) were replaced with other aliases whenever possible using the rentrez package (v1.2.2; RRID:SCR_021062).
3. RESULTS
3.1. Collecting basal transcriptome levels
Our goal was to determine the relationship between various subdivisions of the avian telencephalon according to the distinction and continuum hypotheses of brain organization (Figure 1) by measuring their transcriptomes at baseline. In order to compare different brain regions across different individuals, we needed to collect brain tissue from animals under carefully controlled conditions in order limit any confounding variables. Since up to 10% of the transcribed genome can be regulated in an activity‐dependent manner, with different cell populations having different sets of regulated genes in different brain regions controlled by different behaviors (Jarvis et al., 2013; Whitney et al., 2014), we designed our experiment to reduce these confounding variables. Male zebra finches were kept alone in sound isolation chambers overnight to keep gene expression levels at steady state and only males were used to prevent any differences in sex chromosome expressed genes to confound our analyses (Agate et al., 2003). Sagittal sections were processed in order to reduce section as a variable, as more brain subdivisions are captured together in the sagittal plane relative to the more commonly cut coronal plane.
To test both hypotheses (Figure 1), we targeted nine regions for transcriptomic profiling. These include seven regions from the dorsal and ventral pallium subdivisions on either side of the vestigial ventricle, and two control regions (arcopallium and striatum) agreed upon by both views (Figure 2). For the sake of simplicity, we use the Jarvis et al. (2013) terminology throughout the present study, but provide translation of names across studies in Table 1 and in the text when needed. A list of region abbreviations used in this study is provided in Figure 2. A series of sections was stained with Cresyl violet to help identify brain subdivisions. Adjacent sections were processed for laser capture microdissection (LCM), using bright‐field microscopy to help further identity brain subdivision boundaries, and aid in avoiding accidentally contaminating samples with adjacent brain subdivisions (Figure 2).
FIGURE 2.
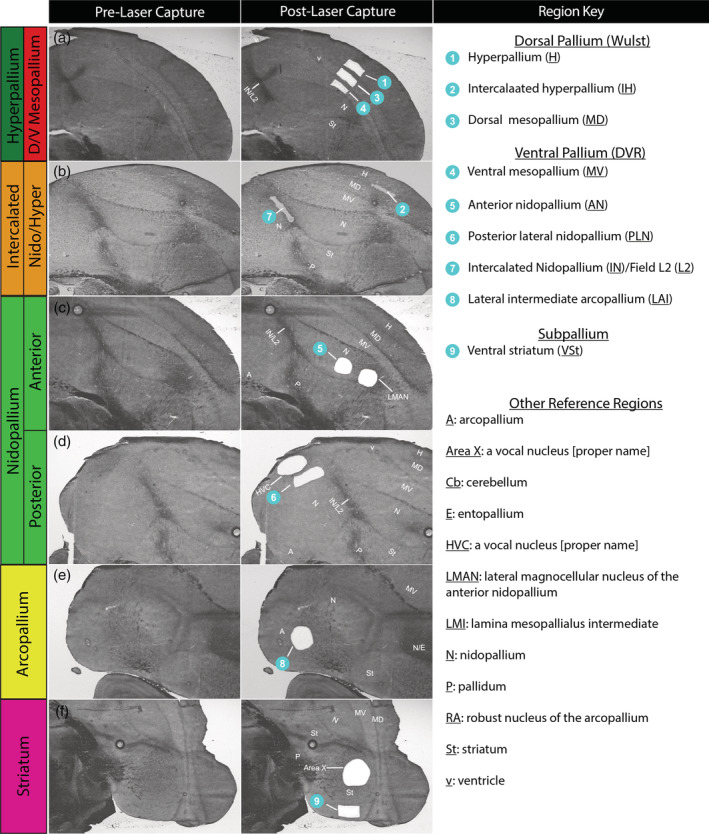
Example images of laser capture microscopy (LCM) dissections. (a–e) Bright‐field sections showing the brain regions profiled before (left column) and after (center column) LCM dissections. Dissected regions are numbered (blue circles). Abbreviations for all relevant brain regions according to the continuum hypothesis (Figure 1(b), Table 1) are provided (right column). Some sections have additional dissections of song nuclei as part of another study in progress. Darker brain regions are due to increased myelination, some of which separate brain subdivisions via axon tracts
TABLE 1.
Comparison of brain region nomenclature and connectivity motifs. List of subdivision terminology according to two competing hypotheses for all subdivisions profiled in the current study. Subdivisions are color‐coded based on their relationships according to each hypothesis in Figure 1. The 2004 view is based on Reiner, Perkel, Bruce, et al., 2004; Reiner, Perkel, Mello, & Jarvis, 2004 and Jarvis et al. (2005), while the 2013 view used in the present study is based on Jarvis et al. (2013) and Chen et al. (2013)
| Names 2004 (Figure 1(a)) | Names 2013 and this study (Figure 1(b)) |
|---|---|
| Hyperpallium apicale (HA) | Hyperpallium (H) |
| Interstitial hyperpallium apicale (IHA) | Intercalated hyperpallium (IH) |
| Hyperpallium densocellulare (HD) | Dorsal mesopallium (MD) |
| Mesopallium (M) | Ventral mesopallium (MV) |
| Field L2 (L2) | Intercalated nidopallium (IN) |
| Niodpallium (N) | Nidopallium (N) |
| Arcopallium (A) | Arcopallium (A) |
| Striatum (St) | Striatum (St) |
Specifically, from the dorsal pallium (e.g., Wulst) we captured: (1) a secondary visual region of the hyperpallium (H; also known as hyperpallium apicale; Figure 2(a)); (2) a primary visual region of the underlying intercalated hyperpallium (IH; also known as interstitial hyperpallium apicale; Figure 2(b)); and (3) a secondary visual region of the dorsal mesopallium (MD; also known as HD; Figure 2(a)). From the ventral pallium (e.g., DVR), we captured: (4) a motor region of the ventral mesopallium (MV; also known as mesopallium; Figure 2(a)); (5 and 6) anterior motor (AN) and posterior lateral auditory‐motor (PLN) regions of the nidopallium (Figure 2(c,d)); and (7) the Field L2 auditory portion of the IN (also known as L2; Figure 2(b)). We isolated two regions of the nidopallium to test for diversity within an accepted brain subdivision. We also isolated regions from two other unique subdivisions accepted by both hypotheses. These include: (8) a motor portion of the lateral intermediate arcopallium (LAI; Figure 2(e)); and (9) a motor portion of the ventral striatum (Figure 2(f)). The visual, auditory, and motor functional designations are based on stimulus‐ and movement‐regulated immediate early gene activation, electrophysiological activity, neural connectivity, and/or lesions studies (Feenders et al., 2008; Jarvis et al., 2013; Kelley & Nottebohm, 1979; Mandelblat‐Cerf et al., 2014; Shimizu & Hodos, 1989; Shimizu & Karten, 1993). The dissected motor regions of the nidopallium, arcopallium, and striatum were adjacent to the song nuclei of those brain subdivisions (Figure 2(c,d,f)). We could not find a fourth “interstitial” hyperpallial apicale region between the formally named HD and hyperpallium apicale (Figures 1(a) and 2(a,b)), consistent with our previous findings in zebra finches and other avian species (Jarvis et al., 2013).
RNA‐Seq expression profiling was performed on these samples, with a sequencing depth of ~20 million reads per sample. These reads were aligned to a new more complete and more error‐free 2019 zebra finch genome assembly from the VGP, containing 22,186 annotated genes, of which 17,438 are protein coding (Rhie et al., 2020). We were able to map nearly 98% of reads to transcripts from this new VGP assembly, maximizing the power of our analysis, compared to 87% to the old Sanger‐based assembly (Rhie et al., 2020; Warren et al., 2010). Importantly, 91% of the reads mapped to unique loci (21,617 genes, ≥ than one read in a sample) in the 2019 assembly (~84% in 2010 assembly), which were used for all downstream analyses.
3.2. Quality control and impact of bird specific transcriptome patterns
We first tested whether there were any batch affects or other covariate influences on the gene expression patterns across all samples. Although the intercalated pallium samples were collected from the same individuals as the other samples, they were collected at separate times, potentially introducing unwatched batch effects. However, we did not detect any major batch effects between these collection groups using a variety of approaches. For example, the cumulative probability distributions P–P plot of the normalized expression counts for the intercalated samples (collection Group 2) relative to the rest of the samples (collection Group 1) were nearly identical (Figure 3(a)), indicating no systematic shift between collection groups. A plot of the RLE of all genes in each sample indicated no evidence of a systematic shift in global expression in any sample or collection group (Figure 3(b)). The CV of house‐keeping genes (those with a CV = 0) showed about 81% of these genes in the first collection group had stable expression in the second collection group, further supporting their consistency following normalization regardless of batch (Figure 3(c)). Results presented further below suggest that the 19% difference in the housekeeping genes is biologically driven.
FIGURE 3.
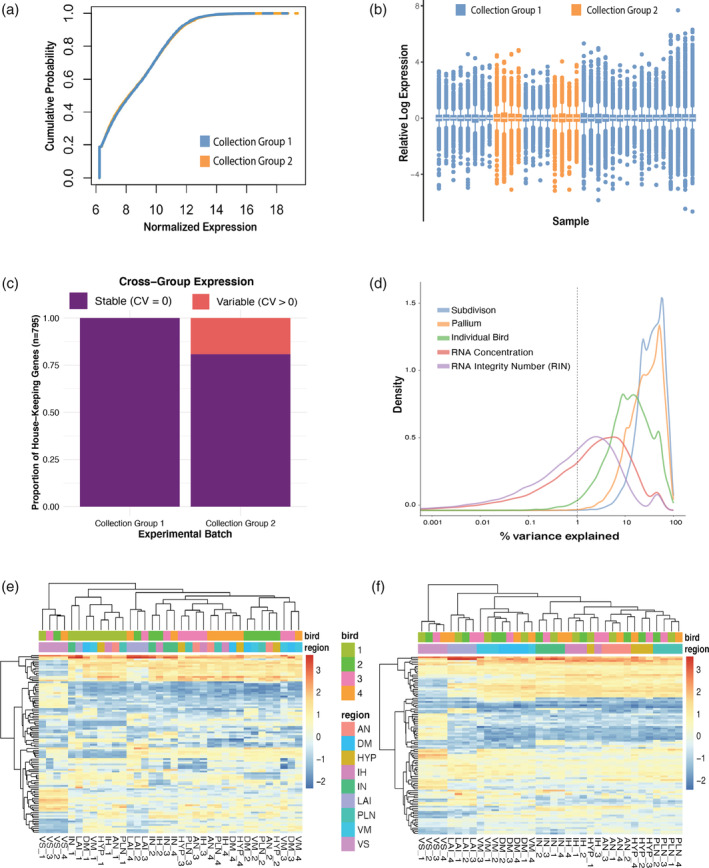
Minimal batch effects detected across collection groups, while individual bird is a strong source of unwanted variation. (a) P–P plot of normalized expression from collection Group 1 (all nonintercalated samples) versus collection Group 2 (intercalated samples). The distributions look nearly identical. (b) Relative log expression plot of all samples colored by collection group. There is no evidence of a systematic shift in global expression in any sample. (c) Proportion of stable house‐keeping genes (coefficient of variance [CV] = 0) across the collection groups. (d) Density plot of variance explained by individual variables. Note brain subdivisions (blue line) and broader pallium subdivisions (orange line) together explain most of the variance seen in the data, individual bird (green line) accounts for a nontrivial amount of variance, and RNA concentration and RNA Integrity Number (RIN) have little effect. (e) Heatmap of normalized expression of top 100 most variable genes. Regions cluster mostly by bird (top color bar) rather than brain subdivisions regions (bottom color bar). (f) After accounting for the bird effect as a covariate, these same genes exhibit robust clustering by brain region (bottom color bar)
We maximized the variance explained by our biological variables of interest (subdivision and pallium) by modeling and removing sources of unwanted variation. We found that brain subdivision, including broader pallium subdivisions, explained the vast majority (>90%) of the variance distribution (Figure 3(d)). However, there was a strong individual bird effect which peaked around 10% of variance, and a weaker one with ~5% of variance associated with RNA concentration and quality (Figure 3(d)). Despite the strong explanation by brain subdivision, hierarchical clustering of expression levels of the top 100 most variable genes in the data clustered samples more by individual bird than by brain region (Figure 3(e)). Removing this individual covariate effect from the data, resulted in robust clustering by brain region (Figure 3(f)), highlighting the importance of controlling for individual animal variation before conducting downstream analyses. These individual bird differences might have been driven by age or some other tightly correlated cofactor like genotype, but it is not easily determined with only four individuals. Nevertheless, this bird effect was accounted for in all analyses in this study, either by direct removal from the normalized expression matrix (PCA and WGCNA) or by inclusion as a term in the linear model for differential expression testing.
3.3. PCA and clustering of Wulst and DVR brain regions
First, we investigated the clustering patterns of all samples to see which regions, if any, shared similar transcriptomic profiles. We performed a PCA of the top 500 most variable genes in the bird‐normalized expression matrix for all samples. When including all regions, the first PC was explained by large differences between the striatal and pallial regions for all birds, while the second PC was explained by differences among pallial regions, with the arcopallium being the most distinct, followed by the intercalated pallial regions (Figure 4(a)). The remaining pallial regions (dorsal/ventral mesopallium, hyperpallium, nidopallium, and intercalated pallium) all grouped closely together. To further explore this tight clustering, we conducted an additional PCA on this subset of the data surrounding the vestigial ventricle. The Wulst samples above the vestigial ventricle (hyperpallium, IH, and dorsal mesopallium) did not form distinct clusters from the DVR samples below it (nidopallium, IN, and ventral mesopallium; Figure 4(b)). Instead, the samples from each subdivision in the Wulst clustered with one subdivision each in the DVR: hyperpallium with both anterior and posterior nidopallium; IH with IN; and dorsal mesopallium with ventral mesopallium (Figure 4(b)). This clustering pattern is most consistent with the broad pallial naming scheme provided by Jarvis et al. (2013).
FIGURE 4.
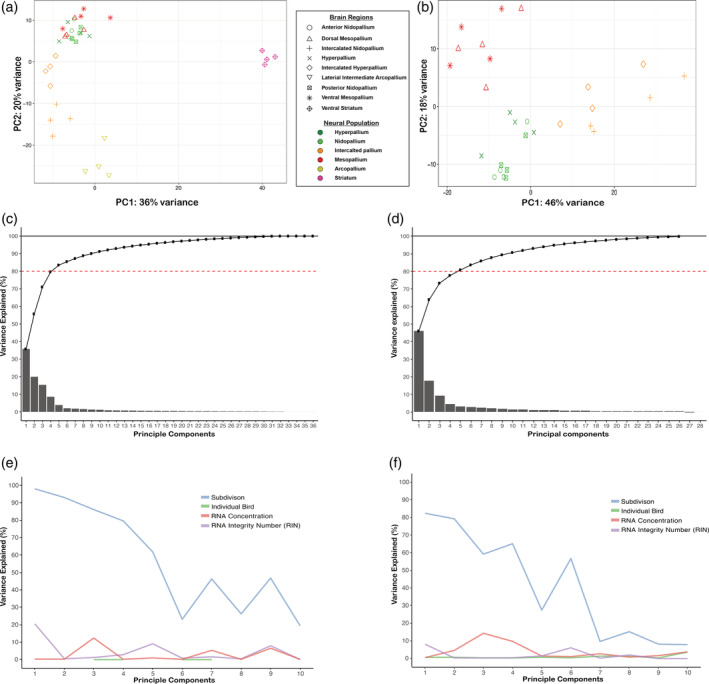
Principal component analysis (PCA) supports view of similar populations in Wulst and dorsal ventricular ridge (DVR). PCA plot using the top 500 most variable genes plotted for all samples (a) and a subset of samples excluding the arcopallium and striatum (b). Each point represents measurements from one bird. The symbol legend treats the brain regions above and below the vestigial ventricle as different in the context of the distinction hypothesis. The color treats them as similar in the context of the continuum hypothesis. Note that samples above the vestigial ventricle are not distinct but cluster with samples from below it. (c) The cumulative percent variance explained for all principal components identified (n = 36, consistent with the number of biological samples). (d) The cumulative percent variance explained from the subset of samples in (b) (n = 28). In both cases, the first four principal components (PCs) explain ~80% of the observed variance (dotted red line) and 100% of the variance is explained as PC = n is reached (solid black line). (e) Percent variance explained in each PC by main biological variables and other covariates for all samples. (f) A similar plot as (e) but for the subset of regions in (b). Brain subdivision is strongly associated with the primary PCs in both analyses
Since the first two PCs in each PCA captured a combined variance of 56% (Figure 4(a)) and 64% (Figure 4(b)), respectively, we wondered what contributed to the additional variance. When we plotted the percent variance explained by each PC, the first four PCs explained ~80% of the variance observed in the expression date for both analyses (Figure 4(c,d)). These four PCs were all best explained by brain subdivision differences, with smaller contributions (0–20%) from technical variables like RNA quality and RNA concentration, and no influence of differences between individual birds (Figure 4(e,f)). These findings indicate that the observed variance in the PCAs is primarily biological.
3.4. Shared molecular profiles between Wulst and DVR brain regions
To more comprehensively assess relationships in the molecular specializations of each brain region sampled, we next performed pairwise differential expression analysis on all measured genes (n = 21,617) for each subdivision. Results were hierarchically clustered based on the total number of genes with significant differences following multiple test corrections (false discovery rate < 0.5). In order to use total number of differentially expressed markers as an indicator of magnitude of difference or similarity, we first aimed to establish a baseline of expected molecular difference within a widely accepted brain subdivision. We tested two regions of the anterior and posterior nidopallium and found they differed by only 37 genes, ~0.1% of genes tested after DESeq2 independent filtering (Figure 5(a)), setting a threshold for when to consider if two regions could belong to the same brain subdivision. Given this threshold, we looked at the regions above and below the vestigial ventricle (Figure 5(a)). We found that the dorsal mesopallium (also known as HD in Figure 1(a)) above the vestigial ventricle was the most similar to the ventral mesopallium (also known as mesopallium) below it, differing by only four genes or about 0.01% of the genes tested for differential expression. These genes were Myelin Basic Protein (MBP), FSTL1, NINJ2, and DAPK2. The hyperpallium (also known as hyperpallium apicale) above the vestigial ventricle was most similar to the anterior and posterior nidopallium below it, differing by only 35 and 50 genes, respectively (0.1 and 0.2% of the genes tested; Figure 5(a)). Remarkably, the anterior nidopallium was more similar to the hyperpallium than it was to the posterior nidopallium, suggesting that two areas within the nidopallium are more different from each other than one of them is to a region of the hyperpallium. The IH (also known as IH apicale in Figure 1(a)) in the Wulst was the most similar to the IN (Field L2) in the DVR, but differing by many more genes (n = 532, 2.6% of genes tested; Figure 5(a)). Despite this greater difference, by comparison the IH and IN are four and six times more different than the brain subdivisions that they have long been considered to be associated with, the hyperpallium (n = 2097 genes; 10.4%) and nidopallium (n = 3230 genes; 16.1%; Figure 5(a)), respectively. These differences between the intercalated pallium regions with the nidopallium and hyperpallium are in the range of the number of genes that differ between well‐established brain subdivisions (~3000–6000 genes; ~15–30%), including between pallial and striatal regions (Figure 5(a)).
FIGURE 5.
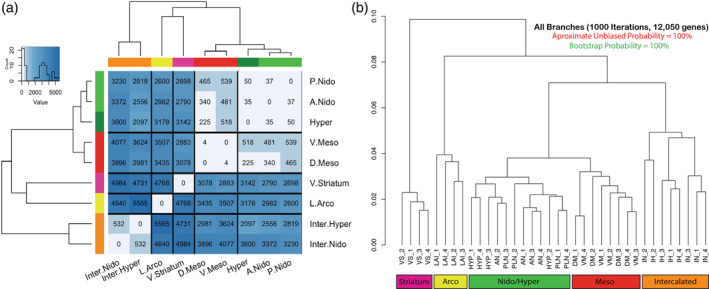
Molecular relationships between brain subdivisions. (a) Dissimilarity heatmap with cluster dendrograms of all differentially expressed genes in pairwise analyses of all brain subdivisions profiled. Heatmap is colored according to number of genes that are significantly differentially expressed at a false discovery rate (FDR) < 0.05. The leaves of the tree are colored and labeled according to the continuum hypothesis, which reflects their clustering patterns. (b) Hierarchical clustering with approximate unbiased (au, red) and bootstrap probability (bp, green) p‐values for all differentially expressed genes (n = 12,050) across all samples. All approximate unbiased (au, red) and bootstrap values were 100% for all branches. See Figure 2 for brain region abbreviation list; numbers next to the abbreviations are individual birds
While this clustering approach details the extent of total molecular differences, it does not reflect shared character of specializations, that is, regions that have the same genes specialized in the same/opposite directions. In order to visualize brain region relationships for the genes with the most biological signal, we took the union set of all statistically significant, DEGs (n = 12,050) and performed hierarchical clustering of the expression values with bootstrap sampling of all genes in all brain regions, keeping each sample from each bird independent (Figure 5(b)). This phylo‐gene expression tree showed a remarkably similar topology as that using only 50 genes sampled in Jarvis et al. (2013). The striatum clustered away from the other pallium samples, with the arcopallium being the most distinct of the pallial regions. The remaining pallial samples clustered together in a pattern that supports the hypothesized continuous relationships, with 100% bootstrap probability support in all branches. Further, the nidopallium/hyperpallium and mesopallium regions formed a super cluster, revealing higher order relationships. In both types of analyses, we did not observe any clustering pattern that supported grouping the hyperpallium subdivisions of the Wulst (Figure 1(a)) as more similar to each other. Rather, the gene expression clustering via PCA (Figure 4(a,b)), differential gene expression (Figure 5(a)), and phylogenetic bootstrapping similarities (Figure 5(b)) were antithetical to the distinction hypothesis.
3.5. Validations by in situ hybridization
To test the validity of our RNA‐Seq findings and determine if the expression profiles we discovered are characteristic of the brain subdivisions, we analyzed available in situ hybridization profiles of 64 genes from various studies (Chen et al., 2013; Jarvis et al., 2013; Kubikova et al., 2010; Pfenning et al., 2014; Wada et al., 2004; Whitney et al., 2014) and the zebra finch expression atlas (Lovell et al., 2020) which had clear expression profiles and high‐quality data (Table S2). This included searching for available in situs of all genes with significant differential RNA‐Seq expression in the Wulst and DVR populations above and below the vestigial ventricle. We scored each of these gene's patterns in pairwise differential expression results as TP, TN, FP, or FN, and calculated accuracy:
These findings show that RNA‐Seq accuracy was very high and is in concordance with in situ markers for these brain subdivisions (Table 2). The accuracy of the differential expression to markers for the two mesopallium regions was 100% (Table 2). The accuracy between the hyperpallium and anterior nidopallium expression was ~97%, further supporting the similarity observed between these regions. The accuracy between the intercalated pallium and the posterior nidopallium and hyperpallium regions in which they reside were ~86 and 90% respectively, while the accuracy between the two intercalated regions was 95%. By comparison, the accuracy of expression between the well‐established arcopallium versus striatum subdivisions was 89%. These findings indicate that the RNA‐Seq gene expression comparisons between more similar brain subdivisions have higher accuracy according to our in situ hybridization analyses.
TABLE 2.
Concordance of differential expression analyses with known control genes. True positive, false positive, true negative, and false negative values and the percent accuracy from those values are listed for 64 genes, between 10 brain subdivision comparisons that test the distinction versus continuum hypotheses relationships. All RNA‐Seq and in situ hybridization comparisons examined had a percent accuracy between 84 and 100%, adding confidence to the differential gene expression and hierarchical clustering results (Figure 5)
| Region 1 | Region 2 | True positive | False positive | True negative | False negative | Percent accuracy |
|---|---|---|---|---|---|---|
| MD | MV | 1 | 0 | 63 | 0 | 100.0 |
| H | AN | 5 | 1 | 57 | 1 | 96.9 |
| AN | PLN | 3 | 0 | 59 | 2 | 96.9 |
| A | PLN | 34 | 2 | 28 | 0 | 96.9 |
| IH | IN | 10 | 2 | 51 | 1 | 95.3 |
| IN | PLN | 27 | 4 | 31 | 2 | 90.6 |
| PLN | MV | 20 | 1 | 37 | 6 | 89.1 |
| A | St | 45 | 6 | 12 | 1 | 89.1 |
| IH | H | 11 | 4 | 44 | 5 | 85.9 |
| H | MD | 14 | 1 | 40 | 9 | 84.4 |
Abbreviations: A, arcopallium; AN, anterior nidopallium; H, hyperpallium; IH, intercalated hyperpallium; IN, intercalated nidopallium; MD, dorsal mesopallium; MV, ventral mesopallium; PLN, posterior lateral nidopallium; St, striatum.
Example validated genes of shared specialized expression between the Wulst and DVR regions included SATB2 (Special AT‐Rich Sequence‐Binding Protein 2) and CHRNA3 (Cholinergic Receptor Nicotinic Alpha 3 Subunit), both upregulated equally throughout the dorsal mesopallium and ventral mesopallium relative to other pallial regions (Figure 6(a,b)). KCTD12 (Potassium Channel Tetramerization Domain Containing 12) was equally upregulated throughout the hyperpallium and nidopallium, and not in the mesopallium regions (Figure 6(c)). SLC4A4 (Solute Carrier Family 4 Member 4), involved in the regulation of bicarbonate secretion and absorption, and intracellular pH, was confirmed with upregulation specific to the intercalated pallial regions (Figure 6(d)).
FIGURE 6.
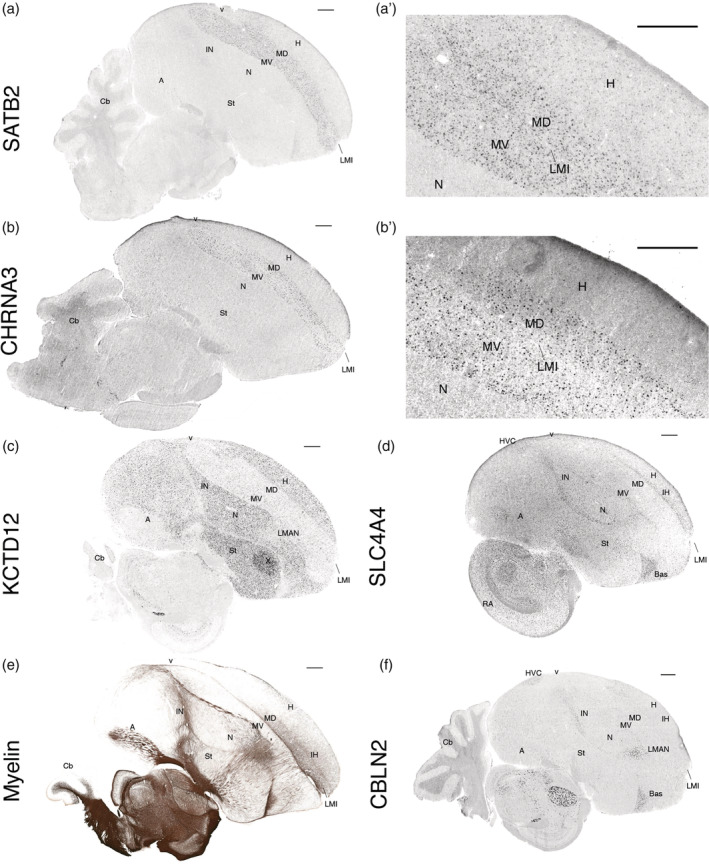
In situ hybridizations and myelin staining confirm RNA‐Seq profiles and reveal full anatomical expression patterns. (a) SATB2, a mesopallium upregulated gene; (a′) higher power image showing that it also has sparse hyperpallium expression, not seen in the nidopallium. (b) CHRNA3, a mesopallium upregulated gene; (b′) higher power image showing that it does not have sparse expression in the hyperpallium. (c) KCTD12, a nidopallium and hyperpallium upregulated gene. (d) SLC4A4, an intercalated pallium upregulated gene. (e) Myelin stain correlating with increased expression of MBP in the anterior dorsal mesopallium (MD) relative to the ventral mesopallium (MV). (f) CBLN2 is upregulated in the intercalated nidopallium but not in the intercalated hyperpallium. All in situ hybridization images are from the Zebra Finch Expression Brain Atlas (Lovell et al., 2020; RRID:SCR_012988) and downloaded as of August 2020. The myelin image is from the digital atlas of the Zebra Finch Brain Architecture Project (Karten et al., 2013; RRID:SCR_004277). All scale bars are 1 mm
Of the few rare genes that exhibited differential expression between the shared Wulst and DVR regions, we validated several. We observed that myelin, whose major component is MBP, was notably higher in the dorsal mesopallium relative to the ventral mesopallium, consistent with the RNA‐Seq expression data (Figure 6(e), Table S2). The reason for this difference appeared to be a higher number of myelinated fibers coursing medial‐laterally in the anterior half of the dorsal mesopallium. Notably, one of the other three genes with increased expression in dorsal mesopallium relative to ventral mesopallium, NINJ2 (Ninjurin 2), has been shown to be differentially expressed in adult myelinating oligodendrocytes in comparative high‐throughput microarray screens of mouse cortical cell types (Noroozi et al., 2019), further suggesting the principal difference in the mesopallium regions surrounding the ventricle is myelin based. SATB2 was also one of the top five of the 36 genes with differential expression between the nidopallium and hyperpallium, and the gene with the highest expression difference in the hyperpallium relative to the nidopallium (Table S2). The in situ hybridization revealed that the reason for this difference was higher expression in sparsely labeled cells throughout the hyperpallium not found in the nidopallium (Figure 6(a′)). This is similar to the pattern observed previously (Jarvis et al., 2013) with SCUBE1 (Signal Peptide, CUB Domain, EGF‐Like Domain Containing 1), which apparently was not strong enough to rise to the level of significance in the RNA‐Seq data after multiple test corrections. We also noticed a sparse hyperpallium expression pattern with NR4A2 (Nuclear Receptor Subfamily 4 Group A Member 2), which occurs as a result of activity‐dependent induction as shown in our companion study (Biegler et al., 2021). These findings hint at a sparse cell type unique to the hyperpallium relative to the nidopallium. However, most mesopallium markers did not show similarly labeled sparse cells in the hyperpallium at baseline (Figure 6(b′)). The top ranked gene with higher expression in the nidopallium relative to the hyperpallium was NR2F2 (Nuclear Receptor Subfamily 2 Group F Member 2, also known as COUPTFII), which had been previously identified as a nidopallium marker relative to the hyperpallium (Jarvis et al., 2013). The CBLN2 (Cerebellin 2 Precursor) in situ pattern confirmed its higher expression in intercalated nidopallium regions relative to IH regions (Figure 6(f)), the 57th such ranked gene (Table S2). We note that in searching through many in situ hybridization profiles, it was difficult to find constitutively expressed genes that differed between the two mesopallium regions and between the nidopallium and hyperpallium regions, consistent with the RNA‐Seq findings.
3.6. Functional gene networks in specific avian telencephalic populations
The results from the pairwise differential expression analysis highlight the shared expression profiles between the dorsal and ventral pallium subdivisions (Figure 5(a)). Investigating co‐expression networks of these genes could offer insights into whether the subdivisions above and below the vestigial ventricle also exhibit a functional gene network similarity, or if there are functional network distinctions between each subdivision regardless of shared expression profiles. To test for this possibility, we performed whole gene co‐expression network analysis (WGCNA) treating all samples independently. WGCNA finds patterns of co‐expression across all genes in the dataset and defines clusters of genes that fluctuate together, termed gene modules. These expression modules can then be associated to one or more brain regions and often confer functional significance (Langfelder & Horvath, 2008; Oldham et al., 2006). If the distinction hypothesis is correct, we would expect to find distinct gene modules for each of the proposed unique subdivisions. However, the presence of gene modules that significantly correlate with subdivisions above and below the vestigial ventricle would be strong evidence in favor of the continuum hypothesis.
We noted instability in gene MM when the intercalated pallial modules were included, potentially due to greater divergence between the two populations (Figure 5(a)), and thus we performed network analyses with and without the intercalated pallium regions included. For each network, we first constructed our co‐expression networks by conducting pairwise expression correlations between all genes, raising each to an empirically determined soft power, which is helpful to highlight the disparity between strong and weak correlations in large genomic datasets (Figure 7(a,c)). We then defined highly correlated genes into discrete modules with unique color/numeric IDs, each with a criterion of a 100‐gene minimum in order to avoid small modules driven by single samples and to obtain the most robust findings (Figure 7(b,d)). We summarized each module by their eigengenes (first PC of expression from all genes in the module) and tested for significant correlation of these values to one or more brain subdivisions with respect to each hypothesis (Figure 8). In other words, we tested to see if all replicates of any given subdivision were associated with positive eigengene values for all modules. Finally, we tested the functional enrichments of any subdivision‐specific modules using Gene Ontology analysis (Figure 9).
FIGURE 7.
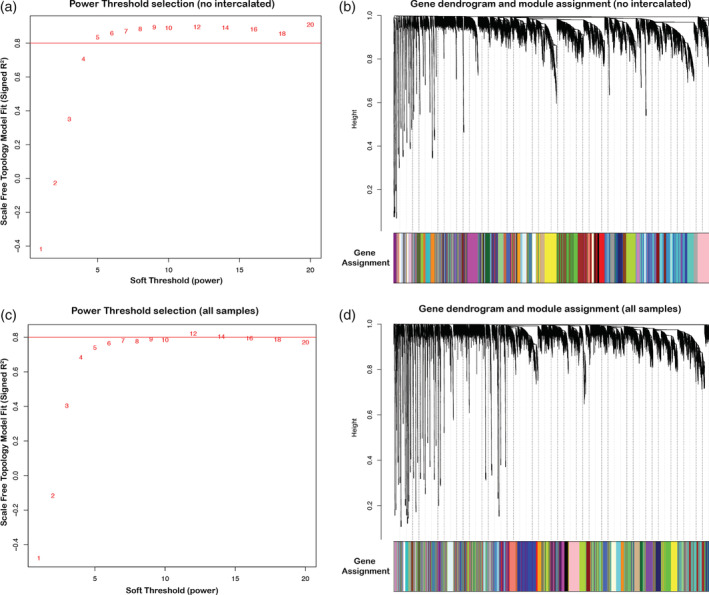
Soft power threshold and gene dendrogram for gene co‐expression networks. (a) For the network excluding the intercalated regions, a soft power (6) was selected to maximize mean connectivity between genes (at least 80%). (b) Gene co‐expression network dendrogram drawn from the soft power threshold in (a), resulting in 47 unique modules (colors). (c) For the network including the intercalated regions, a soft power (8) was selected to maximize mean connectivity between genes (at least 80%). (d) Gene co‐expression network dendrogram drawn from the soft power threshold in (c), resulting in 38 unique modules
FIGURE 8.
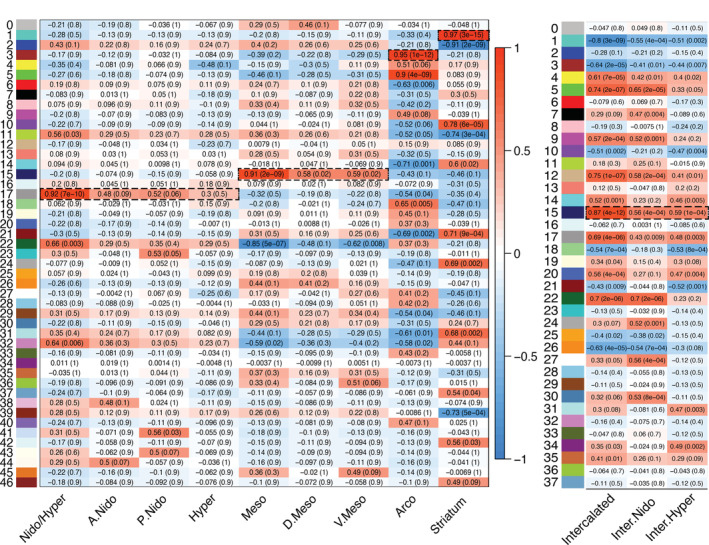
Statistical correlation results of all gene module co‐expression networks. (a) Module eigengene vectors and subdivision correlations with intercalated pallium regions excluded. The 47 subnetworks in the gene expression data are identified with unique module number and color (left), as in Figure 7(b). Entries show Pearson correlation and associated corrected q‐value (parenthesis), testing for statistical relationships between each module eigengene to a unique subdivision or combination of subdivisions. Color scale indicates strength of positive or negative correlation. Dashed boxed regions highlight strong (r 2 > .9) and highly significant (p < .0001) correlations. (b) Module eigengene vectors and subdivision correlations with intercalated pallium regions included. The 38 subnetworks in the gene expression data are identified with unique module number and color (left), as in Figure 7(d); only results for the intercalated regions are shown. Dashed boxed regions highlight strong (r 2 ~ .9) and highly significant (p < .0001) correlations
FIGURE 9.
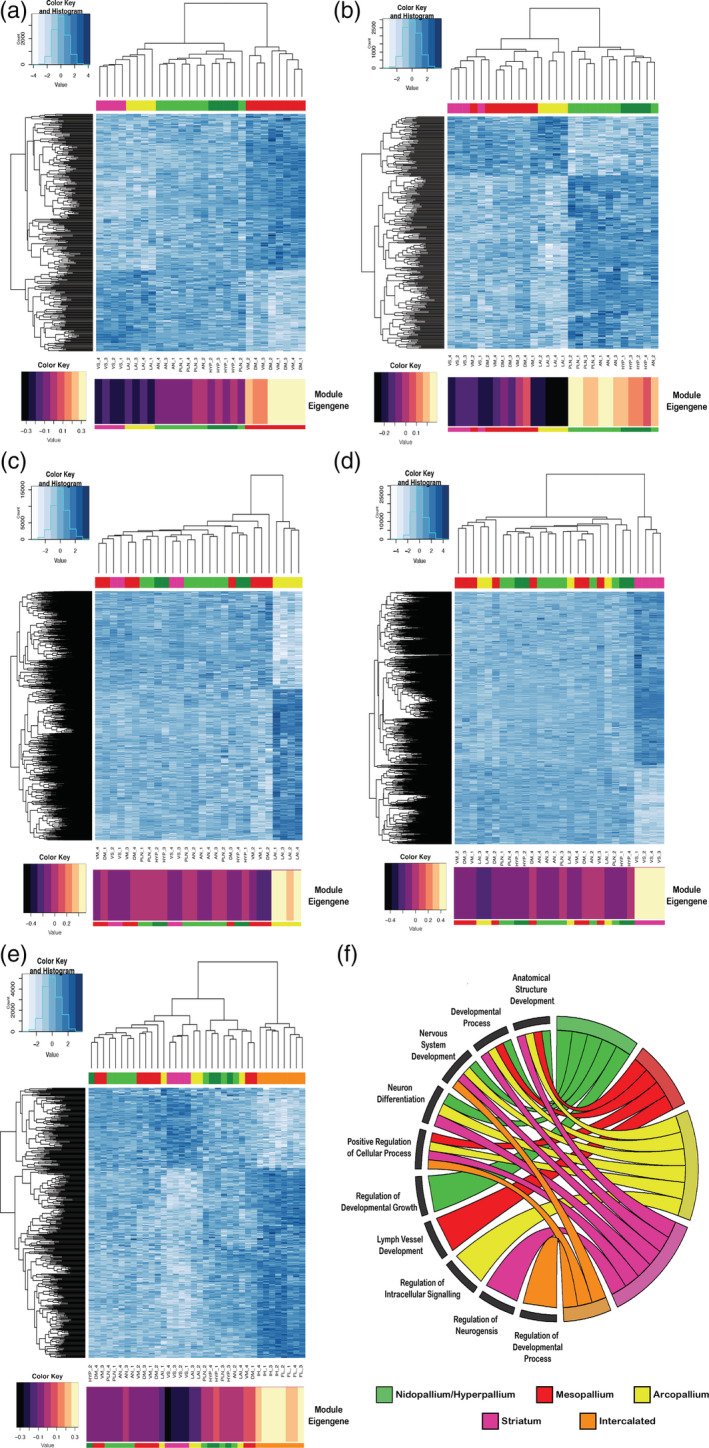
Gene expression module profiles specific to each major avian telencephalic subdivision and combinations of subdivisions profiled. (a–e) Heatmaps of gene expression for subdivision‐specific modules. All module specific genes (rows) are plotted; degree of blue indicates level of median scaled gene expression for all module genes. To the left is the gene expression tree dendrogram; at the top is the brain region dendrogram relationships for each sample color coded by pallial identity. At the bottom is module eigengene value for each subdivision module; the larger the number (or lighter color), the stronger the relationship of that sample to the module eigengene. (a) Mesopallium‐specific (MV, MD; red) module (15, n Gene = 363). There is a strong anticorrelation between this module's genes and the arcopallium and striatum. (b) Nidopallium (PLN, AN; light green) and hyperpallium (HYP; dark green) specific module (17, n Gene = 335). (c) Arcopallium (LAI) specific module (3, n Gene = 1501). (d) Striatum (VS; maroon) specific module (1, n Gene = 2239). (e) Intercalated‐specific (IH, IN; orange) specific module (15, n Gene = 442). (f) Chord diagram of significant GO terms for each neural subdivision module. Each subdivision module contains specific functional enrichments (bottom left quadrant), as well as substantial overlap in function for nervous system development and neuron differentiation (top left quadrant). A list of the most significant GO terms can be found in Table 3, with a complete list in Table S3(a–d) and Table S4(e)
Without the intercalated pallium regions, we selected a soft power (6), as it was the smallest power that allowed for maximum mean connectivity between genes (~85%, Figure 7(a)). With this network, we found a total of 47 modules (Figure 7(b)). Among these 47 modules, we found five with highly significant positive correlations (r 2 > .9, q < .0001) between their eigengenes and distinct brain subdivisions (Figure 8(a)). They included a mesopallium‐specific module of both dorsal and ventral regions (Module 15; Figure 8(a)). This mesopallium module consisted of 363 genes (Figure 9(a)), and was highly specialized for functions in lymph vessel development and anatomical structure development (Figure 9(f)). Another was a nidopallium/hyperpallium‐specific module (Module 17; Figure 8(a)), consisting of 335 genes (Figure 9(b)), with functional enrichments in regulation of development growth and anatomical structure development (Figure 9(f)). There were two arcopallium‐specific modules (Modules 3 and 5; Figure 8(a)), consisting of 1501 (Figure 9(c)) and 1205 genes, with distinguishing functional specializations of anatomical structure development and regulation of intracellular signaling, respectively (Figure 9(f)). Finally, we found a striatum‐specific module (Module 1; Figure 8(a)), consisting of 2239 genes (Figure 9(d)), and with a distinguishing specialization for neurogenesis and nervous system development (Figure 9(f)).
When including the intercalated regions in the WGNCA, we selected a higher soft power (8) to achieve maximum connectivity (~80%) and obtained fewer modules (n = 38, Figure 7(c,d)). Of these, there was one additional module with a highly significant correlation with the intercalated pallial regions (Module 15, r 2 = .87, q < .0001; Figure 8(b)), consisting of ~1700 genes (Figure 9(e)), with a distinguishing functional specialization for regulation of developmental processes (Figure 9(f)). Intriguingly, there were five development‐specific enrichments shared between most subdivision‐specific modules. These shared module enrichment ontologies were for anatomical structure development; developmental process; neuron system development; neuron development; and positive regulation of cellular process (Figure 9(f); Table 3). Importantly, these similar functional modules were composed of mostly nonoverlapping gene sets for each brain subdivision (Tables S3 and S4), suggesting these shared functional enrichments are achieved with unique sets of genes that could be critical for the development of each pallium's anatomical distinction.
TABLE 3.
Top 5 gene ontology terms and p‐values for the subdivision‐specific modules. All subdivision‐specific modules are enriched for either anatomical structure or nervous system development, but with different genes, suggesting these regions utilize different genes for the same functions to distinguish themselves from each other during development
| Subdivision | GO term | P‐value |
|---|---|---|
| Nidopallium/hyperpallium | Neuron development | 1.50e‐03 |
| Nervous system development | 1.75e‐03 | |
| System development | 1.79e‐03 | |
| Multicellular organism development | 1.83e‐03 | |
| Anatomical structure development | 3.49e‐03 | |
| Mesopallium | Lymph vessel development | 4.27e‐05 |
| Regulation of system process | 8.88e‐05 | |
| Multicellular organismal process | 5.31e‐04 | |
| Regulation of multicellular organismal process | 8.64e‐04 | |
| Lymphangiogenesis | 2.22e‐03 | |
| Arcopallium | System development | 4.63e‐12 |
| Anatomical structure development | 5.98e‐12 | |
| Multicellular organism development | 2.40e‐11 | |
| Developmental process | 8.58e‐11 | |
| Anatomical structure morphogenesis | 1.15e‐10 | |
| Striatum | Regulation of nervous system development | 8.91e‐09 |
| Developmental process | 1.25e‐08 | |
| Generation of neurons | 3.25e‐08 | |
| Anatomical structure development | 3.37e‐08 | |
| Neurogenesis | 4.71e‐08 | |
| Intercalated | Positive regulation of nervous system development | 6.82e‐04 |
| Positive regulation of cellular process | 2.50e‐03 | |
| Positive regulation of biological process | 8.60e‐03 | |
| Cellular component organization | 8.95e‐03 | |
| Regulation of multicellular organismal development | 9.19e‐03 |
Importantly, with or without the intercalated pallium samples included, there were no modules where the dorsal/ventral mesopallium, the nidopallium/hyperpallium, or the intercalated regions exhibited strong separate gene network correlations (at r 2 = ~.9, p < .0001; Figure 8(a,b)). These individual regions had weaker correlations (r 2 = .4–.6, p < .05), but they were much stronger when grouped together (Figure 8(a,b)). The hyperpallium on its own had no significant positive correlation at all (p > .1) and one weakly significant negative correlation (r 2 = .48, p = .01; meaning absence of this subnetwork; Figure 8(a)), indicating that the hyperpallium could not be distinguished from the nidopallium in terms of gene functional networks in this analysis. The higher correlations in the combined regions cannot be explained by higher sample numbers alone, because if the regions above and below the vestigial ventricle were significantly different, the correlations would be weakened, not strengthened, by combining them. The strengthening demonstrates shared functional molecular properties.
We also noted some intriguing higher order relationships among some brain subdivision‐specific modules. The mesopallium‐specific module had an inverse gene expression relationship of the same interacting genes in the arcopallium and striatum (negative correlations in Module 15 in Figure 8(a); expression profile in Figure 9(a)). A similar finding was seen for the nidopallium/hyperpallium Module 17 relative to the arcopallium (Figures 8(a) and 9(b)). In contrast, the gene modules exhibiting strong correlated expression with the arcopallium (Module 3) and striatum (Module 1) did not exhibit anticorrelation (reversed expression) with the other subdivisions. (Figures 8(a) and 9(c,d)). This suggests that there are broad programs of gene regulation that can be turned up in one brain subdivision and turned down in another.
3.7. Hub genes reveal key molecules involved in avian subdivision organization
Each gene module contains genes with co‐regulated expression, but some of these genes exhibit higher connectivity than others. Seo et al. (2009) proposed that genes with high connectivity in a regulatory module, known as hub genes, are promising candidates for master regulators of the module‐specific co‐expression. Indeed, studies have shown that hub genes, specifically transcription factors, are critical components of specialized expression networks in disease states like cancer (Castro et al., 2016; Fletcher et al., 2013) and developmental processes like cell differentiation in yeast (Borneman et al., 2006). Identifying hub genes in the brain subdivision‐specific co‐expression modules could offer important insights into essential genetic players for establishing the similar populations around the vestigial ventricle. To determine hub genes for each subdivision, we used two criteria. First, we calculated each gene's connectivity to all other genes in its module in order to measure the strength of its MM. Second, we calculated the strength of the correlation with each gene's expression to the subdivision of interest as a measure of GS. The GS can be a positive or negative value depending on the gene's expression relative to all other subdivisions, where one gene's downregulation might be just as important as another gene's upregulation. By selecting the upper‐most values of each measure as a threshold (absolute value of MM > 0.80 and an absolute value of GS > 0.80), we were able to define the most important genes for each subdivision‐specific module.
We found significant associated hub genes specific to each brain subdivision (Table S5) and visualized the top 50 in network diagrams (Figure 10(a–e)). Each subdivision had several hub gene transcription factors with various ranges of downstream target genes, which could be strong candidates for regulating the other genes within the module. For example, the mesopallium module exhibited moderate interconnectivity (median n = 7) between its top 50 hub genes. One of the most densely connected hub genes (individual n = 28) was the SATB2 transcription factor (Figures 6(a) and 10(a)), which is known to be expressed in the superficial layers of the mammalian cortex, and controls the expression of genes involved in intracortical pyramidal neuron connectivity (Alcamo et al., 2008; Cera et al., 2019). This suggests that SATB2 may help specialize the dorsal and ventral portions of the mesopallium in a similar manner. The nido‐hyperpallium module also exhibited moderate connectivity between its top 50 hub genes (median n = 7), and was similarly defined by strong connectivity (individual n = 16) of the DACT2 transcription factor (Figure 10(b)), which regulates intracellular signaling during development (Schubert et al., 2014). Two other hub genes in this module were the axon guidance genes SEMA6A (individual n = 6) and EPHA8 (individual n = 10), suggesting they may help establish the shared intratelencephalic connectivity motifs of the nidopallium and hyperpallium (Jarvis et al., 2013). The intercalated pallium (median n = 6.5) had GRIN2A ionotropic glutamate receptor and the doublecortin kinase DCLK1 specific to the IN (Figure 10(c)), suggesting these genes may control the specialized signaling pathways for this region. The arcopallium module exhibited many more connections between hub genes (median n = 24). Two of the most interconnected genes in the arcopallium (Figure 10(d)) are well‐known transcription factors LHX9 (individual n = 25) and ETV1 (also known as ER81, individual n = 39) previously studied in the avian brain (Jarvis et al., 2013). These two genes are expressed in deep layer cortical projection neurons and the pallial amygdala of mammals (Abellán et al., 2013; Dugas‐Ford et al., 2012). Likewise, the top hub genes for the striatum exhibited strong interconnectivity (median n = 25), and included well‐known dopamine receptors in that brain region (D1A, D1B; also known as DRD1 and DRD5, respectively). The FOXP2 transcription factor was found in the top 75 hub genes (Figure 10(e)), which has been shown to be critical for proper striatum specialization and function (Haesler et al., 2007; Kubikova et al., 2014; Teramitsu et al., 2010). Importantly, while uncharacterized genes (LOC IDs) were replaced with functional aliases whenever possible (see Methods), each subdivision‐specific hub network was composed of some genes of unknown function, many of which are ncRNAs (22–82% in top 50 hubs), highlighting need for further investigations into the roles of these genes in the differentiation of neural subdivisions. Overall, these analyses demonstrate that the molecular functions of the Wulst subdivisions are informative for the DVR subdivisions, and vice versa.
FIGURE 10.
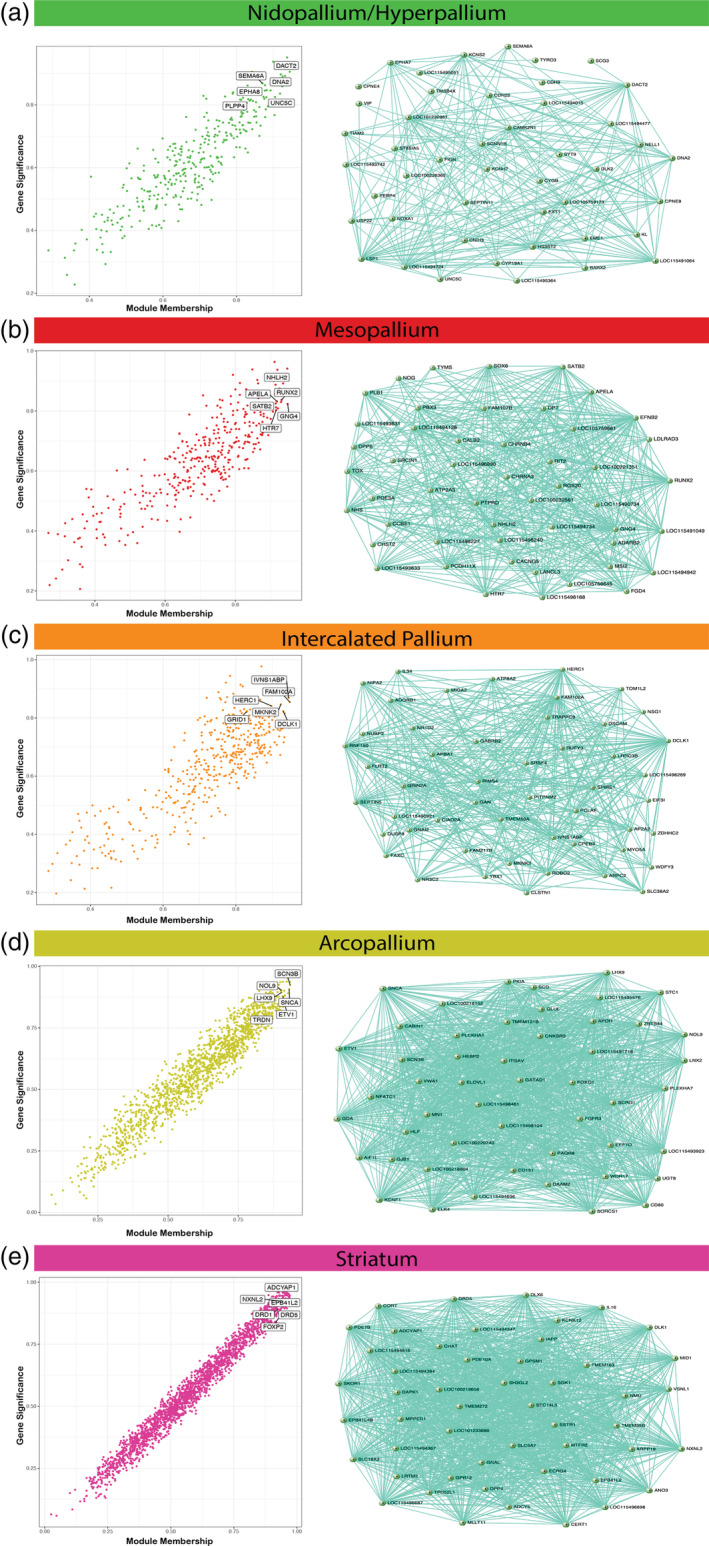
Hub gene identification and top 50 hub gene networks reveal best candidates for subdivision specific expression regulation. (a–e) Correlation between module membership (MM) and gene significance (GS) for the (a) dorsal/ventral mesopallium, (b) nidopallium/hyperpallium, (c) intercalated pallium, (d) arcopallium, and (e) striatum modules. Highlighted are example hub genes with MM > 0.80 and GS > 0.80. The top 50 hub genes and their connections are visualized adjacent to each subdivision‐specific module correlation plot. The genes with the highest connectivity offer prime candidates for key regulators of module expression for each subdivision. A full list of hub genes for each subdivision‐specific module can be found in Table S5
4. DISCUSSION
In the present study, we examined the entire annotated transcriptome of the major cell populations of the adult avian pallium and striatum at a well‐controlled behavioral baseline. We found that for each major population above the vestigial ventricle (i.e., LMI) divide there is a corresponding population below it. The dorsal/ventral mesopallium regions are the most similar, followed by the hyperpallium and nidopallium, while the intercalated pallial regions are the most divergent. These results are inconsistent with the distinction hypothesis, which states that the dorsal pallium constituting the Wulst is a “hyperpallium cluster” distinct in character and cell types from the ventral pallium constituting the DVR. Rather, these findings are more consistent with the continuum hypothesis of avian dorsal and ventral pallium organization (Figure 11), in which the six previous distinctly‐named pallial cell populations (hyperpallium apicale, interstitial hyperpallium apicale, HD, mesopallium, nidopallium, Field L2) are really three continuous cell populations (nido‐hyperpallium, mesopallium, and intercalated pallium) that wrap around the vestigial ventricle (Chen et al., 2013; Jarvis et al., 2013).
FIGURE 11.
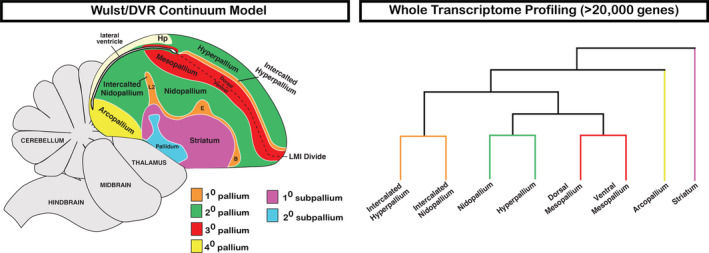
Summary of avian telencephalon transcriptomic profiling. Our results demonstrate robust clustering between subdivisions of the avian Wulst and DVR, most consistent with the continuum model of avian brain organization [Color figure can be viewed at wileyonlinelibrary.com]
We believe there are several key reasons why our findings and interpretations differ from some past individual gene expression and broader transcriptome studies (Belgard et al., 2013; Montiel et al., 2016; Montiel & Molnár, 2013; Puelles et al., 2016; Watson & Puelles, 2017). First, we used a set of in situ hybridization gene expression profiles, Nissl staining, and myelin staining (Chen et al., 2013; Jarvis et al., 2013; Karten et al., 2013) to helped guide our understanding of brain population boundaries and thereby our dissections for RNA‐Seq analyses. Second, we performed LCMs on thin 12 μm sections, as opposed to more gross dissections on thick sections, which allowed for easier identification of anatomical boundaries via fiber tracts. Both of these considerations helped increase our regional accuracy and to reduce cross contamination between brain subdivisions. However, we acknowledge that each subdivision contains heterogeneous expression profiles (e.g., anterior vs. posterior nidopallium), and such precise dissections may limit the generalization of claims to entire subdivisions as a whole. Nevertheless, the in situ hybridization results thus far indicate that most genes identified as differentially expressed in the RNA‐Seq data are representative of entire subdivisions. Finally, we kept animals in quiet control conditions in order to establish a consistent baseline of gene expression across animals. This is in contrast to freely behaving animals, where up to 10% of the genes in the genome can be regulated across different cell types within a forebrain circuit (Whitney et al., 2014). Even with this control of animal state, we still found a robust individual animal effect which we could fortunately normalize out due in part by a well‐balanced experimental design. Such unwanted individual variation can have a strong impact on the results of gene expression experiments and should be taken into consideration whenever possible. We believe these careful controls lend greater confidence to the results of our experiments.
One example where we see the impact of these key differences in experimental design is the NR4A2 transcription factor. Puelles et al. (2016) used this gene's expression pattern to argue that the continuum hypothesis was not plausible, because only the ventral mesopallium exhibited NR4A2 expression with no label above the LMI lamina divide. However, our companion study found that NR4A2 is an activity‐dependent gene, whose expression changes in different brain regions according to different sensory stimuli or behaviors, including in both ventral and dorsal mesopallium (Biegler et al., 2021). In the present study, we found that NR4A2 is a hub gene in the arcopallium‐specific module with faint expression in the mesopallium at baseline, highlighting the importance of utilizing animals with well‐controlled behavior states for interpretations of gene markers of cell types. Future studies on finer delineations within each subdivision, such as within the arcopallium (Mello et al., 2019), could help narrow down which cell types utilize this gene at rest or during behavior.
Previous studies from our group examined the expression profiles of 50 genes in the adult and a subset of 16 in the developing avian telencephalon and noted the transcriptomic similarity between the dorsal and ventral pallium populations (Chen et al., 2013; Jarvis et al., 2013). While this was an informative achievement at the time, the limited number of genes caused some to question the representative claims about the relationships of these neural populations (Montiel & Molnár, 2013). The present study uses a non‐biased examination of the entire annotated transcriptome (~20,000 genes), greater than 400% more genes from our previous studies, and confirms the remarkable transcriptomic similarity between dorsal and ventral pallium subdivisions. The hyperpallium and nidopallium differed by only 35–50 genes (~0.2%) depending on the portion sampled, and the mesopallium regions surrounding the vestigial ventricle differed by only four genes (0.01%). We are now confident that whole transcriptome analysis yields strong evidence for a robust similarity between dorsal and ventral pallium populations in the adult zebra finch telencephalon.
The shared gene co‐expression modules between similar neural populations above and below the vestigial ventricle contribute to a broader understanding of avian brain subdivision functions. Since all the shared subdivision‐specific modules contain nonoverlapping gene sets involved in anatomical structure and nervous system development, these genes represent candidates for establishing the principal neural connectivity and structure differences observed between subdivisions. For example, the nidopallium/hyperpallium hub transcription factor DACT2 is a promising candidate for establishing the shard intertelencephalic connections observed in these subdivisions (Jarvis et al., 2013), as its paralog DACT1 has been shown to mediate dendritic outgrowth of excitatory neurons in mammalian CNS (Okerlund et al., 2010). The prominently studied axon guidance receptors SEMA6A and EPHA8 also found in the nidopallium/hyperpallium module suggest a role for these receptors in establishing the shared connectivity motifs in these regions. Further studies are needed to elucidate the precise ligands utilized by each receptor.
There is also an important evolutionary implication of shared co‐expression modules between groups of avian neural subdivisions, as these may be the principal signatures of homology. Wagner (2014) proposed the concept of Character Identity Networks (ChINs), which are highly conserved core gene regulatory networks that provide a mechanism for diversity of homologous characteristics. For example, the Hox gene network is a ChIN that gives rise to the diverse body plans across vertebrate and invertebrate species (Mallo & Alonso, 2013). Importantly, ChINs work to regulate “realizer genes” which allow for phenotypic diversity from a homologous regulatory network. The presence of shared co‐expression networks, particularly hub genes, between a group of subdivisions (e.g., nidopallium/hyperpallium) is strong evidence for a homologous ChIN between these two brain subdivisions. This network would work to define broad character, but can allow for the phenotypic diversity of each region through a set of realizer genes (i.e., 35–50 DEGs). Such a similar network architecture is difficult to evolve convergently and is more often taken as evidence of shared functional networks in homologous brain regions (Oldham et al., 2006). Furthermore, the ChIN concept helps explain the interesting phenomenon of inverse expression profiles between subdivision‐specific modules. These gene networks might be “toggled” on or off during development to give rise to different neural populations in the avian brain. Further studies investigating the trajectory of these subdivision‐specific modules during development, as well as functional manipulation of key DEGs, are necessary to further test this idea.
While this work was conducted in zebra finch, other studies in chickens have supported the similarity of the avian Wulst and DVR cell types in terms of gene expression and neural connectivity. Briscoe et al. (2018) performed RNA‐Seq on a subset of chicken brain regions we profiled here in the zebra finch and found that the dorsal and ventral mesopallium contain similar cell types. Further, they found that this shared cell type was most similar to the intertelencephalic neurons (IT) of the mammalian cortex. They noted that the transcription factor SATB2 was a marker for these IT cells in mammals and birds and is likely an important regulator of genes in this cell type. Our findings suggest that SATB2 is a centralized hub gene in the mesopallium‐specific module that defines both dorsal and ventral regions, suggesting the principal cell type driving the signal in our bulk data are these IT‐like neurons. We also note that the in situ patterns of some of the genes (e.g., FOXP1, dopamine receptors, ER81, RORB, COUPTFII) we examined in the zebra finch, have also been examined in chicken with similar results (e.g.,Dugas‐Ford et al., 2012; Jarvis et al., 2013). As chickens belong to an entirely separate branch of the avian phylogenetic tree (Jarvis et al., 2014), these findings suggest that the major similarities above and below the vestigial ventricle are not limited to a local branch of the Neoaves. Putting these results together, it suggests that dorsal/ventral pallium similarities are likely a result of common cell types in these populations, and this shared architecture is likely present throughout the avian lineage. However, these neural populations are not homogenous, and understanding the cellular diversity within established subdivisions across species is necessary for defining evolutionary relationships across the lamina divide.
In contrast to these findings in birds, a study utilizing single cell RNA‐Seq of turtle pallium (Tosches et al., 2018) did not find such an overlap in gene expression between the turtle DVR and dorsal cortex (considered homologous to avian Wulst). One explanation for this discrepancy could lie in the cell type relationships in the brain of the reptilian last common ancestor. The DVR in birds and non‐avian reptiles is organized similarly, whereas the dorsal pallium (also known as Wulst) is thicker in birds compared to the thinly layered non‐avian reptilian dorsal pallium (Dugas‐Ford et al., 2012). Turtles diverged from other reptiles that gave rise to birds and crocodiles about 250 million years ago, offering ample time for divergent evolution of these neural populations. The turtle dorsal cortex neurons may have diverged in their expression from the turtle DVR neurons during this time, resulting in more distinct cell populations around the ventricle consistent with Tosches et al. (2018). Conversely, the avian and crocodile pallium would have retained the Wulst/DVR expression similarities, which is consistent with this study and crocodile in situ hybridization data for a small set of genes highlighting this relationship (Briscoe et al., 2018). This would suggest that the avian and crocodile pallium organization represents homology of cell types around the vestigial ventricle and is more reflective of the reptilian common ancestor. Alternatively, if the converse relationship is true, it would suggest the turtle Wulst/DVR organization is more reflective of the reptilian common ancestor, and the observed expression similarities of these populations in avian and crocodilian species is an example of convergence. However, we believe the dramatic similarity in expression between Wulst/DVR observed in this study is more parsimonious with shared homology than large‐scale, transcriptome‐wide convergence. Furthermore, the presence of shared co‐expression modules between subdivisions around the LMI suggest that these brain subdivisions share large‐scale organization in gene expression networks that is difficult to through convergence (Oldham et al., 2006; Wagner, 2014). Whole transcriptome and open chromatic analyses of carefully dissected lizard neural subdivisions, as well as developmental studies in all reptilian species, would provide the critical evidence necessary to distinguish between these two alternatives.
The evolutionary relationships between the avian dorsal and ventral pallium and the mammalian cortex are still a topic of intense debate. At a minimum, the present study suggests that when comparing birds relative to other non‐reptile vertebrates any evolution‐based hypotheses using gene expression profiling should consider the pallial populations above and below the vestigial ventricle together. The IN has been proposed to be homologous to Layer 4 thalamic recipient neurons of the mammalian cortex (Dugas‐Ford et al., 2012; Jarvis et al., 2013), and our findings indicate that this parallel would also apply to the avian IH (Jarvis et al., 2005). The nidopallium and ventral mesopallium populations have been proposed to be homologous to cell types in the upper Layers 2 and/or 3 of the mammalian cortex, respectively (Wang et al., 2010). If so, then the hyperpallium and dorsal mesopallium populations of birds may also be considered homologous to cell types predominately found in these mammalian cortical layers. A similar logic applies to the hypotheses that propose that the avian ventral pallial regions are homologous to the mammalian claustrum and amygdala (Puelles et al., 2016). In light of the present work, it will be difficult to justify the claim that the avian dorsal pallial regions alone are homologous to the six‐layered cortex separate from the claustrum and amygdala.
Consistent with this shared consideration, past studies have found a columnar organization in both avian dorsal and ventral pallial regions that encompass the three populations around the vestigial ventricle highlighted in this study (Feenders et al., 2008; Jarvis et al., 2013; Medina & Reiner, 2000; Wang et al., 2010). Furthermore, a recent study that examined neural connectivity in the pigeon brain found local microcircuits in the Wulst and DVR on either side of vestigial ventricle, reminiscent of mammalian cortical column architecture (Stacho et al., 2020). Despite this study having defined the brain regions according to the distinction hypothesis, our current study is nonetheless consistent with their overall view that the subdivisions on either side of the lamina contain similar canonical microcircuits generated by similar cell types. A key difference in our conclusions is the claim that the hyperpallium output population of the Wulst microcircuit is analogous to Layer 5 of the mammalian cortex (Stacho et al., 2020; Wild, 1997; Wild & Williams, 2000). If true, one might expect a similar gene expression profile of the hyperpallium output neurons with the arcopallium, the primary output population of the DVR microcircuit that is also proposed to be like Layer 5 neurons. But that is not what we observed. Instead, the hyperpallium profile was remarkably similar to the nidopallium, notably distinct from the arcopallium. However, one unifying view of both ideas is that while the primary cell type of the avian hyperpallium is nidopallium‐like, there are distinct cell types within the hyperpallium that resemble the arcopallium for some genes, like NR4A2, but these are too sparse to be detected as the primary signal in the present bulk RNA sequencing analysis. Indeed, such a sparse hyperpallium cell population with distinct gene expression profiles has been observed in this study and others (Jarvis et al., 2013, Biegler et al., 2021). This cell type might be the extratelencephalic neurons observed and could arise through modulation of the hyperpallium/nidopallium co‐expression module found in this study. However, other genes showing sparse expression in the hyperpallium, like SATB2, are not enriched in the arcopallium and challenge this idea. Alternatively, a recent study reported a sparse population of nidopallium projection neurons with extratelencephalic targets (Wild, 2017), further highlighting the potential cell type similarities between the hyperpallium and nidopallium subdivisions. Further molecular profiling using single cell/nuclei RNA sequencing technology is necessary to test these hypotheses and aid in our understanding of the evolutionary relationships of these cell types across vertebrates.
The present study is more consistent with the continuum hypothesis, which states that the dorsal and ventral pallium populations are continuous with one another around the vestigial ventral (Jarvis et al., 2013). A potential mechanism for this hypothesized cell type continuity was proposed by Chen et al. (2013), which involved tangential migration of cell types around the developing ventricle space, ultimately giving rise to the adjacent dorsal and ventral pallium populations upon occlusion. At that time, the literature had supported the presence of both radial and tangential migration of excitatory neurons in the developing avian telencephalon (Métin et al., 2007; Striedter & Keefer, 2000). However, since then the Molnar group has conducted more targeted embryonic developmental fate mapping studies to test for tangential migration and concluded that only radial migration occurs in the avian dorsal and ventral pallium domains away from the ventricle (García‐Moreno et al., 2018). It is important to note, however, that their embryonic injections did not encompass the SATB2‐labeled dorsal mesopallium identified here and in Chen et al. (2013), suggesting further studies are needed to truly test this hypothesis for the development of the mesopallium. Bruguier et al. (2020) also performed fate mapping of cells in the ventral mesopallium to test for tangential migration to other pallial regions. They found no migration of ventral mesopallium cells towards the dorsal pallium but did find evidence of tangential migration to the underlying nidopallium. While these findings were noted as preliminary, they offer evidence that tangential migration can play a role in the development of the avian telencephalon. Nevertheless, if radial migration were the sole trajectory of pallial derived neurons, then one possible mechanism to explain our findings in adults is that developing cells migrate radially from the ventricle in a similar pattern above and below the ventricle space. Ultimately, systematic lineage tracing experiments, paired with in situs for the shared expression markers found in this study, would provide more definitive evidence to test whether there is a physical continuum for the developmental origins of similar subdivisions above and below the ventricle.
Regardless of the evolution and developmental origin of the brain regions, we believe that the shared transcriptomes and molecular functions found in this study, combined with the shared neural connectivity motifs and developmental origins (Chen et al., 2013; Jarvis et al., 2013; Stacho et al., 2020), are evidence of an important functional relationship that should be recognized by a common naming scheme. Should more evidence emerge to support this view, we recommend future studies consider adopting a hierarchal subdivision naming system, as first proposed in the Jarvis et al.'s (2013) and Chen et al.'s (2013) studies (Figure 11). The IH and IN (L2, entopallium, and basorostralis) would be called the dorsal and ventral primary (1°) pallium, respectively, since they are the first recipient population of thalamic sensory input into the telencephalon. The hyperpallium and nidopallium would be called the dorsal and ventral secondary (2°) pallium, respectively, as they receive their main extra‐telencephalic input via of the 1° pallium regions. The dorsal and ventral mesopallium would be called the dorsal and ventral tertiary (3°) pallium, respectively, as they receive their main extra‐telencephalic input via the 2° pallium. The arcopallium would be called the quaternary (4°) pallium, as it contains cell populations that are the main output of the telencephalon. We believe this naming system is most reflective of the present knowledge of shared neural connectivity, developmental timelines, and gene expression patterns observed in the dorsal and ventral avian pallium.
In conclusion, the highly similar molecular makeup of the populations above and below the vestigial ventricle necessitate shared functions, helping to inform our understanding of avian brain organization and allowing for new interpretations and translations of findings between brain subdivisions and vertebrate species.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Gregory Gedman: Performed experiments and analyses and co‐wrote the article. Bettina Haase: Generated the RNA‐Seq libraries. Gillian Durieux: Helped with LCM dissections and processing primary pallium regions. Matthew T. Biegler: Provided in situ hybridization analysis. Olivier Fedrigo: Consulted on experimental design and supervised RNA sequencing. Erich D. Jarvis: Supervised the study and co‐wrote the article.
5.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/cne.25159.
Supporting information
Table S1. Sample_Metadata
Table S2. DEG_Lists_with_Validations 2
Table S3. Module_Assignments_and_GO_Enrichments_without_Intercalated_Samples
Table S4. Module_Assignments_and_GO_Enrichments_with_Intercalated_Samples
Table S5. Hub_Genes_Subdivision_Modules
ACKNOWLEDGMENTS
The authors thank Samara Brown for editing and constructive feedback on the manuscript, and Thomas Carroll of the Rockefeller University Bioinformatics Core Facility for his advice on several analyses. This project was funded by the Howard Hughes Medical Institute (OSU1013377) and Rockefeller University start‐up funds to E. D. J., and National Science Graduate Research Fellowship 2015202850 to G. G.
Gedman G, Haase B, Durieux G, Biegler MT, Fedrigo O, Jarvis ED. As above, so below: Whole transcriptome profiling demonstrates strong molecular similarities between avian dorsal and ventral pallial subdivisions. J Comp Neurol. 2021;529:3222–3246. 10.1002/cne.25159
Funding information Howard Hughes Medical Institute, Grant/Award Number: OSU1013377; National Science Foundation Graduate Research Fellowship, Grant/Award Number: 2015202850
Contributor Information
Gregory Gedman, Email: ggedman@rockefeller.edu.
Erich D. Jarvis, Email: ejarvis@rockefeller.edu.
DATA AVAILABILITY STATEMENT
All RNA sequencing data supporting the findings of this study are deposited in the NCBI SRA archives under the BioProject ID: PRJNA678351.
REFERENCES
- Abellán, A. , Desfilis, E. , & Medina, L. (2013). The olfactory amygdala in amniotes: An evo‐devo approach. Anatomical Record (Hoboken), 296(9), 1317–1332. [DOI] [PubMed] [Google Scholar]
- Agate, R. J. , Grisham, W. , Wade, J. , Mann, S. , Wingfield, J. , Schanen, C. , Palotie, A. , & Arnold, A. P. (2003). Neural, not gonadal, origin of brain sex differences in a gynandromorphic finch. Proceedings of the National Academy of Sciences of the United States of America, 100(8), 4873–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcamo, E. A. , Chirivella, L. , Dautzenberg, M. , Dobreva, G. , Fariñas, I. , Grosschedl, R. , & McConnell, S. K. (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron, 57(3), 364–377. [DOI] [PubMed] [Google Scholar]
- Ariëns Kappers, C. (1922). The ontogenetic development of the corpus striatum in birds and a comparison with mammals and man. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 26, 135–158. [Google Scholar]
- Belgard, T. G. , Montiel, J. F. , Wang, W. Z. , García‐Moreno, F. , Margulies, E. H. , Ponting, C. P. , & Molnár, Z. (2013). Adult pallium transcriptomes surprise in not reflecting predicted homologies across diverse chicken and mouse pallial sectors. Proceedings of the National Academy of Sciences of the United States of America, 110(32), 13150–13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegler, M. , Cantin, L. , Scarano, D. , & Jarvis, E. (2021). Controlling for activity‐dependent genes and behavioral states is critical for determining brain relationships within and across species. The Journal of Comparative Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman, A. R. , Leigh‐Bell, J. A. , Yu, H. , Bertone, P. , Gerstein, M. , & Snyder, M. (2006). Target hub proteins serve as master regulators of development in yeast. Genes & Development, 20(4), 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe, S. D. , Albertin, C. B. , Rowell, J. J. , & Ragsdale, C. W. (2018). Neocortical association cell types in the forebrain of birds and alligators. Current Biology, 28(5), 686–696.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe, S. D. , & Ragsdale, C. W. (2018). Homology, neocortex, and the evolution of developmental mechanisms. Science, 362(6411), 190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruguier, H. , Suarez, R. , Manger, P. , Hoerder‐Suabedissen, A. , Shelton, A. M. , Oliver, D. K. , Packer, A. M. , Ferran, J. L. , García‐Moreno, F. , Puelles, L. , & Molnár, Z. (2020). In search of common developmental and evolutionary origin of the claustrum and subplate. The Journal of Comparative Neurology, 528, 2956–2977. [DOI] [PubMed] [Google Scholar]
- Castro, M. A. , de Santiago, I. , Campbell, T. M. , Vaughn, C. , Hickey, T. E. , Ross, E. , Tilley, W. D. , Markowetz, F. , Ponder, B. A. , & Meyer, K. B. (2016). Regulators of genetic risk of breast cancer identified by integrative network analysis. Nature Genetics, 48(1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cera, I. , Whitton, L. , Donohoe, G. , Morris, D. W. , Dechant, G. , & Apostolova, G. (2019). Genes encoding SATB2‐interacting proteins in adult cerebral cortex contribute to human cognitive ability. PLoS Genetics, 15(2), e1007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. C. , Winkler, C. M. , Pfenning, A. R. , & Jarvis, E. D. (2013). Molecular profiling of the developing avian telencephalon: Regional timing and brain subdivision continuities. Journal of Comparative Neurology, 521(16), 3666–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas‐Ford, J. , Rowell, J. J. , & Ragsdale, C. W. (2012). Cell‐type homologies and the origins of the neocortex. Proceedings of the National Academy of Sciences of the United States of America, 109(42), 16974–16979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger, L. (1888) (Translation from German) Investigations on the comparative anatomy of the brain. Vol. 1. The forebrain. Vol. 2. The interbrain. First Theil: The interbrain of the shark fish and the amphibian. Vol. 3. New studies of the forebrain of reptiles. Vol. 4. Studies of the interbrain of reptiles. Vol. 5. Investigations of the forebrain of birds in collaboration with Dr. A. Wallenberg in Danzig and Dr. G. M. Holmes in London. (5 vols). Frankfurt/Main: Moritz Diesterweg. Special castings from the papers of the Senckenbergi nature‐searching society.
- Edinger, L. (1908). The relations of comparative anatomy to comparative psychology. Comparative Neurology and Psychology, 18(5), 437–457. [Google Scholar]
- Feenders, G. , Liedvogel, M. , Rivas, M. , Zapka, M. , Horita, H. , Hara, E. , Wada, K. , Mouritsen, H. , & Jarvis, E. D. (2008). Molecular mapping of movement‐associated areas in the avian brain: A motor theory for vocal learning origin. PLoS One, 3(3), e1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, M. N. , Castro, M. A. , Wang, X. , de Santiago, I. , OReilly, M. , Chin, S. F. , Rueda, O. M. , Caldas, C. , Ponder, B. A. , Markowetz, F. , & Meyer, K. B. (2013). Master regulators of FGFR2 signalling and breast cancer risk. Nature Communications, 4, 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Moreno, F. , Anderton, E. , Jankowska, M. , Begbie, J. , Encinas, J. M. , Irimia, M. , & Molnár, Z. (2018). Absence of tangentially migrating glutamatergic neurons in the developing avian brain. Cell Reports, 22(1), 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesler, S. , Rochefort, C. , Georgi, B. , Licznerski, P. , Osten, P. , & Scharff, C. (2007). Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus area X. PLoS Biology, 5(12), e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard, A. T. , Miller, J. E. , Fraley, E. R. , Horvath, S. , & White, S. A. (2012). Molecular microcircuitry underlies functional specification in a basal ganglia circuit dedicated to vocal learning. Neuron, 73(3), 537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, W. , von Heydebreck, A. , Sültmann, H. , Poustka, A. , & Vingron, M. (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics (Oxford, England), 18(Suppl 1), S96–S104. [DOI] [PubMed] [Google Scholar]
- Jarvis, E. D. , Gunturkun, O. , Bruce, L. , Csillag, A. , Karten, H. , Kuenzel, W. , Medina, L. , Paxinos, G. , Perkel, D. J. , Shimizu, T. , Striedter, G. , Wild, J. M. , Ball, G. F. , Dugas‐Ford, J. , Durand, S. E. , Hough, G. E. , Husband, S. , Kubikova, L. , Lee, D. W. , … Butler, A. B. (2005). Avian brains and a new understanding of vertebrate brain evolution. Nature Reviews. Neuroscience, 6(2), 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, E. D. , Mirarab, S. , Aberer, A. J. , Li, B. , Houde, P. , Li, C. , Ho, S. Y. , Faircloth, B. C. , Nabholz, B. , Howard, J. T. , Suh, A. , Weber, C. C. , da Fonseca, R. R. , Li, J. , Zhang, F. , Li, H. , Zhou, L. , Narula, N. , Liu, L. , … Zhang, G. (2014). Whole‐genome analyses resolve early branches in the tree of life of modern birds. Science, 346(6215), 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, E. D. , Yu, J. , Rivas, M. V. , Horita, H. , Feenders, G. , Whitney, O. , Jarvis, S. C. , Jarvis, E. R. , Kubikova, L. , Puck, A. E. , Siang‐Bakshi, C. , Martin, S. , McElroy, M. , Hara, E. , Howard, J. , Pfenning, A. , Mouritsen, H. , Chen, C. C. , & Wada, K. (2013). Global view of the functional molecular organization of the avian cerebrum: Mirror images and functional columns. The Journal of Comparative Neurology, 521(16), 3614–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten, H. J. (1969). The organization of the avian telencephalon and some speculations on the phylogeny of the amniote telencephalon. In Pertras J. (Ed.), Comparative and evolutionary aspects of the vertebrate central nervous system (pp. 164–179). New York Academy of Sciences. [Google Scholar]
- Karten, H. J. , Brzozowska‐Prechtl, A. , Lovell, P. V. , Tang, D. D. , Mello, C. V. , Wang, H. , & Mitra, P. P. (2013). Digital atlas of the zebra finch (Taeniopygia guttata) brain: A high‐resolution photo atlas. The Journal of Comparative Neurology, 521(16), 3702–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, D. , & Nottebohm, F. (1979). Projections of a telencephalic auditory nucleus‐field L‐in the canary. The Journal of Comparative Neurology, 183(3), 455–469. [DOI] [PubMed] [Google Scholar]
- Kubikova, L. , Bosikova, E. , Cvikova, M. , Lukacova, K. , Scharff, C. , & Jarvis, E. D. (2014). Basal ganglia function, stuttering, sequencing, and repair in adult songbirds. Scientific Reports, 4, 6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubikova, L. , Wada, K. , & Jarvis, E. D. (2010). Dopamine receptors in a songbird brain. The Journal of Comparative Neurology, 518(6), 741–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. , & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Wang, L. , Wu, Y. , Yuan, Z. , & Zhou, J. (2020). Weighted gene co‐expression network analysis to identify key modules and hub genes associated with atrial fibrillation. International Journal of Molecular Medicine, 45, 401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Wang, Y. , Cheng, M. , Huang, C. , & Fan, X. (2020). Weighted gene coexpression network analysis of features that control cancer stem cells reveals prognostic biomarkers in lung adenocarcinoma. Frontiers in Genetics, 11, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell, P. V. , Wirthlin, M. , Kaser, T. , Buckner, A. A. , Carleton, J. B. , Snider, B. R. , McHugh, A. K. , Tolpygo, A. , Mitra, P. P. , & Mello, C. V. (2020). ZEBrA: Zebra finch expression brain atlas—A resource for comparative molecular neuroanatomy and brain evolution studies. The Journal of Comparative Neurology, 528(12), 2099–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo, M. , & Alonso, C. R. (2013). The regulation of Hox gene expression during animal development. Development, 140(19), 3951–3963. [DOI] [PubMed] [Google Scholar]
- Mandelblat‐Cerf, Y. , Las, L. , Denisenko, N. , & Fee, M. S. (2014). A role for descending auditory cortical projections in songbird vocal learning. eLife, 3, e02152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, L. , & Reiner, A. (2000). Do birds possess homologues of mammalian primary visual, somatosensory and motor cortices? Trends in Neurosciences, 23(1), 1–12. [DOI] [PubMed] [Google Scholar]
- Mello, C. V. , Kaser, T. , Buckner, A. A. , Wirthlin, M. , & Lovell, P. V. (2019). Molecular architecture of the zebra finch arcopallium. The Journal of Comparative Neurology, 527(15), 2512–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métin, C. , Alvarez, C. , Moudoux, D. , Vitalis, T. , Pieau, C. , & Molnár, Z. (2007). Conserved pattern of tangential neuronal migration during forebrain development. Development, 134(15), 2815–2827. [DOI] [PubMed] [Google Scholar]
- Montiel, J. F. , & Molnár, Z. (2013). The impact of gene expression analysis on evolving views of avian brain organization. The Journal of Comparative Neurology, 521(16), 3604–3613. [DOI] [PubMed] [Google Scholar]
- Montiel, J. F. , Vasistha, N. A. , Garcia‐Moreno, F. , & Molnár, Z. (2016). From sauropsids to mammals and back: New approaches to comparative cortical development. The Journal of Comparative Neurology, 524(3), 630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noroozi, R. , Azari, I. , Taheri, M. , Omrani, M. D. , & Ghafouri‐Fard, S. (2019). A single nucleotide polymorphism within Ninjurin 2 is associated with risk of multiple sclerosis. Metabolic Brain Disease, 34(5), 1415–1419. [DOI] [PubMed] [Google Scholar]
- Okerlund, N. D. , Kivimäe, S. , Tong, C. K. , Peng, I. F. , Ullian, E. M. , & Cheyette, B. N. (2010). Dact1 is a postsynaptic protein required for dendrite, spine, and excitatory synapse development in the mouse forebrain. The Journal of Neuroscience, 30(12), 4362–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham, M. C. , Horvath, S. , & Geschwind, D. H. (2006). Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proceedings of the National Academy of Sciences of the United States of America, 103(47), 17973–17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , Love, M. I. , Irizarry, R. A. , & Kingsford, C. (2017). Salmon provides fast and bias‐aware quantification of transcript expression. Nature Methods, 14(4), 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenning, A. R. , Hara, E. , Whitney, O. , Rivas, M. V. , Wang, R. , Roulhac, P. L. , Howard, J. T. , Wirthlin, M. , Lovell, P. V. , Ganapathy, G. , Mouncastle, J. , Moseley, M. A. , Thompson, J. W. , Soderblom, E. J. , Iriki, A. , Kato, M. , Gilbert, M. T. , Zhang, G. , Bakken, T. , … Jarvis, E. D. (2014). Convergent transcriptional specializations in the brains of humans and song‐learning birds. Science, 346(6215), 1256846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles, L. , Ayad, A. , Alonso, A. , Sandoval, J. E. , MartÍnez‐de‐la‐Torre, M. , Medina, L. , & Ferran, J. L. (2016). Selective early expression of the orphan nuclear receptor Nr4a2 identifies the claustrum homolog in the avian mesopallium: Impact on sauropsidian/mammalian pallium comparisons. The Journal of Comparative Neurology, 524(3), 665–703. [DOI] [PubMed] [Google Scholar]
- Reiner, A. , Perkel, D. , Bruce, L. , Butler, A. , Csillag, A. , Kuenzel, W. , Medina, L. , Paxinos, G. , Shimizu, T. , Striedter, G. , Wild, M. , Ball, G. , Durand, S. , Gunturkun, O. , Lee, D. , Mello, C. , Powers, A. , White, S. , Hough, G. , … Jarvis, E. (2004). Revised nomenclature for avian telencephalon and some related brainstem nuclei (vol 473, pg 377, 2004). Journal of Comparative Neurology, 475(2), 288–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner, A. , Perkel, D. J. , Mello, C. V. , & Jarvis, E. D. (2004). Songbirds and the revised avian brain nomenclature. Annals of the New York Academy of Sciences, 1016, 77–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie, A. , McCarthy, S. A. , Fedrigo, O. , Damas, J. , Formenti, G. , Koren, S. , Uliano‐Silva, M. , Chow, W. , Fungtammasan, A. , Gedman, G. L. , Cantin, L. J. , Thibaud‐Nissen, F. , Haggerty, L. , Lee, C. , Ko, B. J. , Kim, J. , Bista, I. , Smith, M. , Haase, B. , … Jarvis, E. D. (2020). Towards complete and error‐free genome assemblies of all vertebrate species. bioRxiv, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, M. E. , Phipson, B. , Wu, D. , Hu, Y. , Law, C. W. , Shi, W. , & Smyth, G. K. (2015). limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Research, 43(7), 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, F. R. , Sobreira, D. R. , Janousek, R. G. , Alvares, L. E. , & Dietrich, S. (2014). Dact genes are chordate specific regulators at the intersection of Wnt and Tgf‐β signaling pathways. BMC Evolutionary Biology, 14, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, C. H. , Kim, J.‐R. , Kim, M.‐S. , & Cho, K.‐H. (2009). Hub genes with positive feedbacks function as master switches in developmental gene regulatory networks. Bioinformatics, 25(15), 1898–1904. [DOI] [PubMed] [Google Scholar]
- Shimizu, T. , & Hodos, W. (1989). Reversal learning in pigeons: Effects of selective lesions of the Wulst. Behavioral Neuroscience, 103(2), 262–272. [DOI] [PubMed] [Google Scholar]
- Shimizu, T. , & Karten, H. J. (1993). The avian visual system and the evolution of the neocortex. In Zeigler H. P. & Bischof H. J. (Eds.), Vision, brain, and behavior in birds (pp. 103–114). MIT. [Google Scholar]
- Stacho, M. , Herold, C. , Rook, N. , Wagner, H. , Axer, M. , Amunts, K. , & Güntürkün, O. (2020). A cortex‐like canonical circuit in the avian forebrain. Science, 369(6511), eabc5534. [DOI] [PubMed] [Google Scholar]
- Striedter, G. F. , & Keefer, B. P. (2000). Cell migration and aggregation in the developing telencephalon: Pulse‐labeling chick embryos with bromodeoxyuridine. The Journal of Neuroscience, 20(21), 8021–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, I. K. , & Hirata, T. (2014). A common developmental plan for neocortical gene‐expressing neurons in the pallium of the domestic chicken Gallus gallus domesticus and the Chinese softshell turtle Pelodiscus sinensis . Frontiers in Neuroanatomy, 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramitsu, I. , Poopatanapong, A. , Torrisi, S. , & White, S. A. (2010). 'Striatal FoxP2 is actively regulated during songbird sensorimotor learning. PLoS One, 5(1), e8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosches, M. A. , Yamawaki, T. M. , Naumann, R. K. , Jacobi, A. A. , Tushev, G. , & Laurent, G. (2018). Evolution of pallium, hippocampus, and cortical cell types revealed by single‐cell transcriptomics in reptiles. Science, 360(6391), 881–888. [DOI] [PubMed] [Google Scholar]
- Wada, K. , Howard, J. T. , McConnell, P. , Whitney, O. , Lints, T. , Rivas, M. V. , Horita, H. , Patterson, M. A. , White, S. A. , Scharff, C. , Haesler, S. , Zhao, S. , Sakaguchi, H. , Hagiwara, M. , Shiraki, T. , Hirozane‐Kishikawa, T. , Skene, P. , Hayashizaki, Y. , Carninci, P. , & Jarvis, E. D. (2006). A molecular neuroethological approach for identifying and characterizing a cascade of behaviorally regulated genes. Proceedings of the National Academy of Sciences of the United States of America, 103(41), 15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada, K. , Sakaguchi, H. , Jarvis, E. D. , & Hagiwara, M. (2004). Differential expression of glutamate receptors in avian neural pathways for learned vocalization. The Journal of Comparative Neurology, 476(1), 44–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, G. (2014). Homology, genes, and evolutionary innovation. Princeton University Press. [Google Scholar]
- Wang, Y. , Brzozowska‐Prechtl, A. , & Karten, H. J. (2010). Laminar and columnar auditory cortex in avian brain. Proceedings of the National Academy of Sciences of the United States of America, 107(28), 12676–12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, W. C. , Clayton, D. F. , Ellegren, H. , Arnold, A. P. , Hillier, L. W. , Kunstner, A. , Searle, S. , White, S. , Vilella, A. J. , Fairley, S. , Heger, A. , Kong, L. , Ponting, C. P. , Jarvis, E. D. , Mello, C. V. , Minx, P. , Lovell, P. , Velho, T. A. , Ferris, M. , … Wilson, R. K. (2010). The genome of a songbird. Nature, 464(7289), 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, C. , & Puelles, L. (2017). Developmental gene expression in the mouse clarifies the organization of the claustrum and related endopiriform nuclei. The Journal of Comparative Neurology, 525(6), 1499–1508. [DOI] [PubMed] [Google Scholar]
- Whitney, O. , Pfenning, A. R. , Howard, J. T. , Blatti, C. A. , Liu, F. , Ward, J. M. , Wang, R. , Audet, J. N. , Kellis, M. , Mukherjee, S. , Sinha, S. , Hartemink, A. J. , West, A. E. , & Jarvis, E. D. (2014). Core and region‐enriched networks of behaviorally regulated genes and the singing genome. Science, 346(6215), 1256780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild, J. M. (1997). The avian somatosensory system: the pathway from wing to Wulst in a passerine (Chloris chloris). Brain research, 759(1), 122–134. [DOI] [PubMed] [Google Scholar]
- Wild, J. M. (2017). The ventromedial hypothalamic nucleus in the zebra finch (Taeniopygia guttata): Afferent and efferent projections in relation to the control of reproductive behavior. The Journal of Comparative Neurology, 525(12), 2657–2676. [DOI] [PubMed] [Google Scholar]
- Wild, J. M. , & Williams, M. N. (2000). Rostral Wulst in passerine birds. I. Origin, course, and terminations of an avian pyramidal tract. The Journal of Comparative Neurology, 416(4), 429–450. [PubMed] [Google Scholar]
- Wullimann, M. F. (2017). Should we redefine the classic lateral pallium? The Journal of Comparative Neurology, 525(6), 1509–1501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sample_Metadata
Table S2. DEG_Lists_with_Validations 2
Table S3. Module_Assignments_and_GO_Enrichments_without_Intercalated_Samples
Table S4. Module_Assignments_and_GO_Enrichments_with_Intercalated_Samples
Table S5. Hub_Genes_Subdivision_Modules
Data Availability Statement
All RNA sequencing data supporting the findings of this study are deposited in the NCBI SRA archives under the BioProject ID: PRJNA678351.