

Abstract
n‐peri‐Acenes (n‐PAs) have gained interest as model systems of zigzag‐edged graphene nanoribbons for potential applications in nanoelectronics and spintronics. However, the synthesis of n‐PAs larger than peri‐tetracene remains challenging because of their intrinsic open‐shell character and high reactivity. Presented here is the synthesis of a hitherto unknown n‐PA, that is, peri‐heptacene (7‐PA), in which the reactive zigzag edges are kinetically protected with eight 4‐tBu‐C6H4 groups. The formation of 7‐PA is validated by high‐resolution mass spectrometry and in situ FT‐Raman spectroscopy. 7‐PA displays a narrow optical energy gap of 1.01 eV and exhibits persistent stability (t 1/2≈25 min) under inert conditions. Moreover, electron‐spin resonance measurements and theoretical studies reveal that 7‐PA exhibits an open‐shell feature and a significant tetraradical character. This strategy could be considered a modular approach for the construction of next‐generation (3 N+1)‐PAs (where N≥3).
Keywords: acenes, graphene, nanostructures, radicals, Scholl reaction
The synthesis of the hitherto unknown peri‐heptacene 7‐PA is demonstrated by a rational molecular design strategy in which the reactive zigzag edges are kinetically protected. The formation of 7‐PA is validated by high‐resolution mass spectrometry and in situ FT‐Raman spectroscopy. The open‐shell 7‐PA displays a narrow optical energy gap of 1.01 eV and exhibits a persistent stability (half‐life ≈25 min) under inert conditions.
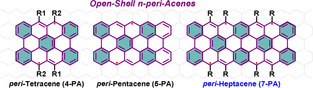
Bottom‐up synthesis of atomically precise nanographenes (NGs) with rich zigzag edges have attracted increasing attention due to their unique electronic structures associated with fascinating physicochemical properties. [1] These NGs are generally achieved from tailor‐made oligophenylene precursors based on solution‐mediated or surface‐assisted synthetic strategies. [2] The opto‐electronic and magnetic properties as well as their reactivities are critically dependent on their sizes, shapes, and edge topologies. [3] In particular, the laterally π‐extended zigzag‐edged NGs,[ 1a , 4 , 5 ] such as acenes and peri‐acenes, [6] are attractive candidates for carbon‐based spintronics due to their prominent open‐shell characters. In addition, they are also considered as ideal model compounds for exploring the spin‐polarized edge states of the elusive zigzag‐edged graphene nanoribbons (ZGNRs). [1a]
n‐peri‐Acenes (n‐PAs), comprised by two rows of peri fused linear acenes, can exhibit diradical or polyradical character upon the lateral π‐extension as a result of the gain of additional Clar's sextets in the open‐shell resonance form (Figure 1). [7] Recently, the in‐solution and on‐surface syntheses of peri‐tetracene (4‐PA) derivatives have been made independently by us [8] and Wu's group [9] showing a large diradical character (y 0=0.7) with a half‐life of ≈3 h in solution under ambient conditions. The next n‐PA homologue, i.e., peri‐pentacene (5‐PA, y 0=0.9), was synthesized in 2015 on Au (111) from 6,6′‐bipentacene precursor under ultrahigh vacuum conditions [10a] and its singlet open‐shell behavior was recently revealed by scanning tunneling spectroscopy. [10b] However, attempts to synthesize 5‐PA in solution have been unsuccessful so far due to the poor solubility of the fully planarized 5‐PA tetraketone precursor [11a] or the undesired Michael addition of the bispentacenequinone compound. [11b] Thereby, the synthetic access to n‐PAs beyond 4‐PA requires the development of more efficient synthetic strategies, which need to circumvent the difficulties associated with the low solubility and high reactivity of the targeted n‐PAs.
Figure 1.
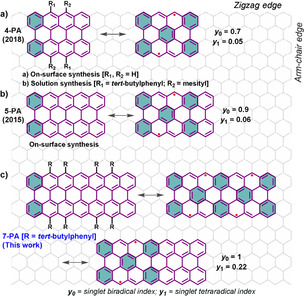
Clar's sextet (cyan‐colored benzenoid ring) depictions of 4‐PA, 5‐PA and 7‐PA, and the calculated (CASSCF (8,8)/6‐31G**) diradical (y 0) and tetraradical (y 1) indices. The substituents in 4‐PA and 7‐PA are omitted in the resonance structures for clarity.
In this work, we report the synthesis and in situ characterization of the unprecedented peri‐heptacene (7‐PA, N=2), which belongs to the family of (3 N+1)‐PAs, via an efficient six‐step synthetic strategy (Scheme 1) in solution. The key step of our strategy is the one‐pot reaction sequence where the 4‐(tert‐butylphenyl)ethynyl functionalized oligophenylene (1) undergoes iodine chloride‐mediated benzannulation and intramolecular cyclodehydrogenation reaction to afford the tetraiodo‐scaffold 2. The successive nucleophilic substitution and Friedel–Crafts cyclization reactions convert 2 to the key tetrahydro‐precursor 3, which successfully yields the targeted 7‐PA through oxidative dehydrogenation with 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ). The eight 4‐tert‐butylphenyl groups at the reactive zigzag‐edge peripheries of 7‐PA provide substantial steric protection, ensuring a reasonable stability with a half‐life (t 1/2) of ≈25 min in solution under inert conditions. The in situ formation of 7‐PA is firstly confirmed by high‐resolution (HR) matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) mass spectrometry. FT‐Raman experiments corroborate the in situ generation of 7‐PA from the precursor 3, where the resultant single prominent G band (1593 cm−1) and D bands (1331 and 1402 cm−1) are well supported by the simulated spectrum of 7‐PA. The 7‐PA displays a broad absorption band in the near‐infrared region with a maximum at 812 nm and an optical energy‐gap (ΔE g opt) of 1.01 eV. The theoretical calculation predicts that 7‐PA has a significant tetraradical character and the open‐shell character is supported by observing a broad signal with a g value of 2.003 in the electron spin resonance (ESR) spectrum. Moreover, the described synthetic strategy provides a possible guideline for accomplishing higher homologues of (3 N+1)‐PAs (N=3, 4, 5, etc.), where the steric protection of the zigzag edges can play a crucial role.
Scheme 1.
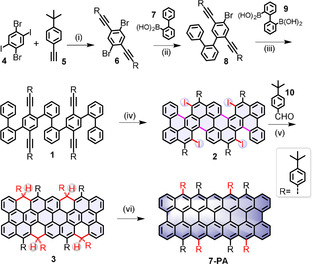
Synthesis of peri‐heptacene (7‐PA). Reagents and conditions: (i) Pd(PPh3)2Cl2, CuI, Et3N, THF, RT, 24 h, 87 %; (ii) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, 85 °C, 24 h, 50 %; (iii) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, 85 °C, 24 h, 41 %; (iv) ICl, dichloromethane, −78 °C, 3 h, 29 %; (v) 1. n‐BuLi, 10, toluene, −10 °C, 2 h; 2. BF3⋅OEt2, dichloromethane, 25 °C, 30 min, 40 % (over two steps); (vi) DDQ, toluene, 25 °C, 10 min.
To realize the synthesis of 7‐PA with sufficient steric protection as well as adequate solubility, we conceived the synthetic route as illustrated in Scheme 1. Firstly, the 4,4′‐((2,5‐dibromo‐1,4‐phenylene)bis(ethyne‐2,1‐diyl))bis(tert‐butylbenzene) (6) was synthesized via Sonagashira coupling of 1,4‐dibromo‐2,5‐diiodobenzene (4) and 1‐(tert‐butyl)‐4‐ethynylbenzene (5) in 87 % yield. Subsequently, compound 6 was reacted with one equiv (1,1′‐biphenyl)‐2‐ylboronic acid (7) by Suzuki coupling to afford 4‐bromo‐2,5‐bis((4‐tert‐butylphenyl)ethynyl)‐1,1′:2′,1′′‐terphenyl (8) in 50 % yield. After that, a two‐fold Suzuki coupling of 8 with (1,1′‐biphenyl)‐2,2′‐diyldiboronic acid (9) [12] gave compound 1 in 41 % yield. Next, we carried out the ICl‐induced benzannulation reaction of 1 in dichloromethane at −78 °C. Interestingly, the benzannulation and intramolecular cyclodehydrogenation occurred simultaneously, resulting in tetraiodo‐scaffold 2 with a yield of 29 % via a one‐pot reaction. Treatment of 2 with n‐butyllithium followed by the addition of 4‐(tert‐butyl)benzaldehyde (10) generated the corresponding tetraol compound, which was further subjected to an intramolecular Friedel–Crafts cyclization promoted by BF3⋅OEt2 to provide the tetrahydro‐precursor 3 as configurational isomers [13] in 40 % yield. All of the intermediates were purified, and fully characterized by HR MALDI‐TOF mass spectrometry and NMR (1H and 13C) spectroscopy (see Supporting Information (SI)).
To accomplish the targeted 7‐PA, the tetrahydro‐precursor 3 was dehydrogenated with slight excess of DDQ (≈2.5 equiv) in anhydrous toluene for 10 min at room temperature (RT). HR MALDI‐TOF mass analysis of the reaction mixture at positive mode revealed that the intense signal at m/z=1802.9496 of the precursor 3 ([M +], calcd for C140H122 is 1802.9547) disappeared and a new signal emerged at m/z=1798.9257 (Figure 2 a), which is fully consistent with the expected molecular mass of 7‐PA ([M +], calcd for C140H118 is 1798.9234), validating the successful formation of the targeted 7‐PA. Notably, the isotopic distribution observed for the obtained 7‐PA is in agreement with the simulated pattern for C140H118. Due to the open‐shell character (see below) and high reactivity of 7‐PA, conventional structural characterization by NMR spectroscopy and single‐crystal structural analysis were not possible. Therefore, we turned to Raman spectroscopy that has emerged as a unique tool to characterize the molecular structures of NGs in combination with density functional theory (DFT) simulations. [14] Thin films of precursor 3 and the in situ generated 7‐PA were analyzed with excitation at 1064 nm. It is evident in Figure 2 b that the spectra of 3 and 7‐PA differ due to their marked change of π‐conjugation. To foster the vibrational assignment of the Raman spectra of 3 and 7‐PA, we carried out DFT calculations [15] (Figure 2 c) on the respective molecular models (see SI for details). For 3, the experimental Raman bands at 1288, 1576 and 1597 cm−1 are assigned to the nuclear displacement patterns computed for the D (1272 cm−1), G1 (1598 cm−1) and G2 (1622 cm−1) modes (Figure 2 d), respectively. The inspection of the D mode of 3 highlights collective ring‐breathing displacements occurring in the planar π‐conjugated region of the molecule, similar to previous observations in related graphene nanostructures. [16] Such D mode of the graphene core is also found to couple with in‐plane bending modes of the aromatic edge C−H groups and with the CC stretching displacements of the lateral phenyl substituents. The G1 and G2 bands are due to collective ring stretching modes, coupled with in‐plane bending modes of the aromatic C−H groups at the graphene edges. In addition, the manifold of G lines observed for 3 can be ascribed to the presence of edge C(sp 3)‐H groups, which disrupts the aromaticity of the central core. As supported by DFT calculations, such G manifold is absent in the case of 7‐PA, where the four sp 3 hybridized carbons are transformed into sp 2 carbons upon the aromatization reaction of 3. The 7‐PA thin film exhibits prominent Raman bands at 1331, 1402 and 1593 cm−1 (Figure 2 b), and these bands are successfully assigned to the D1 (1298, 1301 cm−1), D2 (1428 cm−1) and G (1604 cm−1) modes computed by DFT (Figure 2 e). The comparison between the simulated and the experimental Raman spectrum of 7‐PA highlights solid‐state intermolecular interaction effects that are not accounted for by the gas‐phase DFT model (see SI for details). The D modes involve collective CC stretching displacements of the graphene core that result in a characteristic ring‐stretching pattern. The D1 mode is coupled to the bending of the aromatic C−H groups located on the graphene edge and to the CC stretching of the lateral phenyl moieties (similar to the case of 3). Besides, the D2 mode of 7‐PA involves an alternated CC stretching displacement pattern largely localized at the center of the graphene core. Finally, the G band of 7‐PA is attributed to collective CC stretching displacements of the graphene core that occur transversally with respect to the principal molecular axis. Thus, the FT‐Raman observations are fully rationalized by the respective theoretical simulations, and hence the formation of 7‐PA is validated.
Figure 2.
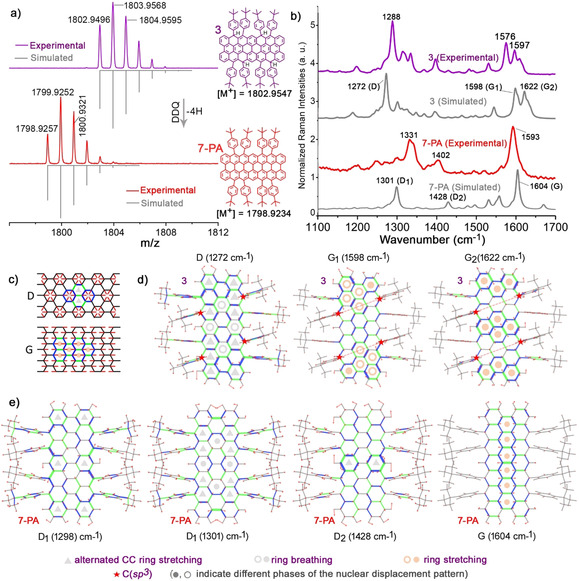
a) HR MALDI‐TOF mass spectra of 3 and 7‐PA in dithranol matrix. The 7‐PA was in situ generated by mixing toluene solution of 3 (8×10−4 M) with DDQ. b) Experimental and simulated Raman spectra of precursor 3 and 7‐PA. c) Representation of the D and G nuclear displacement patterns of graphene molecules. d) The normal modes of 3 corresponding to the D and G Raman transitions. e) The D and G Raman transitions of 7‐PA. Red arrows indicate nuclear displacements, whereas green and blue segments indicate stretching and shrinking bonds, respectively.
We further monitored the reaction between precursor 3 and DDQ using UV‐vis‐NIR spectroscopy. As presented in Figure 3, the 3 (3×10−5 M) in toluene shows strong absorption bands centered at 381, 403, 551, and 596 nm. Upon the addition of DDQ (≈2.5 equiv) under inert conditions, the bright pink colored solution of 3 instantaneously turned to pale brown (Figure 3). The resultant solution shows the evolution of clear vibronic structures, with an intense absorption peak at 812 nm along with four weak bands centered at 733, 924, 1050 and 1160 nm, in the near‐infrared region. The color change and the accompanied spectral changes can be ascribed to the in situ formation of 7‐PA. Based on this result, the optical energy gap (ΔE g opt) was estimated to be 1.01 eV from the lowest absorption onset, [17] which is notably smaller than that of 4‐PA (ΔE g opt=1.10 eV). [8] The in situ generated 7‐PA was persistent in solution under inert condition with a half‐life time of ≈25 min, as shown in Figure 3. The time‐dependent UV‐vis‐NIR measurement revealed that the intensity of the peak at 812 nm diminished with the concomitant increase of the peaks at 671 and 741 nm. Next, electron spin resonance (ESR) spectroscopy was used to examine the reaction between 3 and DDQ (2 equiv) at RT. The open‐shell character of 7‐PA is supported by observing a broad signal (linewidth=5.3 G) with a g value of 2.003 in the ESR spectrum under argon atmosphere (Figure S4 in the SI), while both precursor 3 and DDQ are ESR silent.
Figure 3.
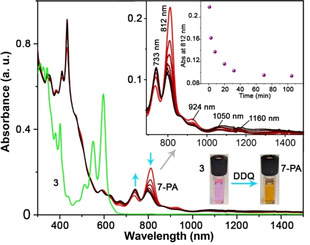
UV‐vis‐NIR spectra of the tetrahydro‐precursor 3 (green) (3×10−5 M) and the time‐dependent absorption changes of the 7‐PA (red) in dry toluene at RT under Ar (in the absence of light). The insets show the magnified view of NIR region and the absorbance (at 812 nm) of the 7‐PA (under argon) at different time intervals. The 7‐PA was in situ generated by mixing toluene solution of 3 with 2.5 equiv DDQ.
To gain deeper insight into the electronic structure of 7‐PA in contrast to 4‐PA and 5‐PA, quantum chemical simulations were performed using the Gaussian 09 package (Figure 4). [15] The geometries of 4‐PA, 5‐PA and 7‐PA were relaxed at the UCAM‐B3LYP/6–311G** level of theory. The diradical character y 0 and the tetraradical character y 1 were calculated using CASSCF(8,8)/6‐31G**, according to the occupation numbers of the lowest unoccupied natural orbitals (LUNO and LUNO+1). [18] 7‐PA exhibits an open‐shell singlet ground state like 4‐PA (y 0=0.7; y 1=0.05) and 5‐PA (y 0=0.9; y 1=0.06), but the diradical and tetraradical indices (y 0=1.0; y 1=0.22) are substantially higher. The densities of the unpaired electrons of 4‐PA, 5‐PA and 7‐PA are majorly located at the opposing zigzag peripheries. This delocalization is extending along the zigzag edges with increasing the molecular size (Figure 4 a). The calculated ΔE S‐T value (Figure S7) of 4‐PA is slightly smaller than that of 5‐PA and thereafter it gradually increases for the higher homologues such as 7‐PA. The molecular conformation of the open‐shell singlet ground state of 7‐PA is depicted in Figure 4 b, which shows a twisted backbone due to the steric bulkiness of the side groups. However, compared to the pristine planar 7‐PA molecule (Figure S8), the open‐shell characteristics are only slightly affected. Nucleus‐independent chemical shift (NICS) [19] and anisotropy of the induced current density (ACID) [20] calculations (Figure 4 c) were conducted at the UB3LYP/6‐31G** level of theory to analyze the aromaticity of 7‐PA. The center (A, B) and the corner (C, D) benzenoid rings are more aromatic than other benzenoid rings as indicated by their large negative NICS(1) values. The ACID plot of the 7‐PA displays a clockwise diatropic ring current of the π‐electrons along the periphery, and hence it is globally aromatic in nature. [9]
Figure 4.
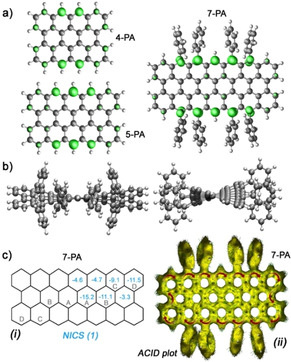
a) Calculated (UCAM‐B3LYP/6–311G**) density of the unpaired electrons of the singlet diradical forms of 4‐PA, 5‐PA and 7‐PA in gas‐phase. b) Side views of the relaxed structure of 7‐PA (to reduce the calculation time tert‐butyl groups on the phenyl rings are ignored). c) Calculated (UB3LYP/6–311G**) NICS(1) and ACID plots of 7‐PA.
In summary, we have successfully extended the boundary of the synthesis of n‐PAs up to 7‐PA. The in situ generated 7‐PA was remarkably persistent (t 1/2≈25 min) in solution under inert conditions, and thereby facilitated its structural confirmation by HR‐MALDI TOF mass and in situ FT‐Raman measurements. Moreover, its pronounced open‐shell character (y 0=1.0; y 1=0.22) and narrow optical energy‐gap (1.01 eV) were further unraveled by ESR and UV‐vis‐NIR measurements as well as quantum chemical simulations. We envisage that the synthetic strategy described here can provide tremendous stimulation towards the synthesis of the family of (3 N+1)‐PAs or even the zigzag‐edged graphene nanoribbons, which are attractive for the future development of carbon‐based spintronics and quantum devices. [21]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was financially supported by the EU Graphene Flagship (Graphene Core 3, 881603), ERC Consolidator Grant (T2DCP, 819698), the Center for Advancing Electronics Dresden (cfaed) and DFG‐NSFC Joint Sino‐German Research Project (EnhanceNano, No. 391979941), as well as the DFG‐SNSF Joint Switzerland‐German Research Project (EnhanTopo, No. 429265950). We thank Mr. Federico Lombardi and Prof. Dr. Lapo Bogani for helpful discussions. We also thank Dr. Paola Moretti (Politecnico di Milano, Italy) for the technical support. The authors acknowledge the use of computational facilities at the Center for information services and high‐performance computing (ZIH) at TU Dresden. J. Liu is grateful for the startup funding from The University of Hong Kong. G.S. and M.T acknowledge funding by the Italian Ministry of Education, Universities and Research (MIUR) through the PRIN 2017 program (Project No. 2017PJ5XXX “MAGIC DUST”). Open access funding enabled and organized by Projekt DEAL.
M. R. Ajayakumar, J. Ma, A. Lucotti, K. S. Schellhammer, G. Serra, E. Dmitrieva, M. Rosenkranz, H. Komber, J. Liu, F. Ortmann, M. Tommasini, X. Feng, Angew. Chem. Int. Ed. 2021, 60, 13853.
Contributor Information
Dr. Ji Ma, Email: ji.ma@tu-dresden.de.
Prof. Dr. Xinliang Feng, Email: xinliang.feng@tu-dresden.de.
References
- 1.
- 1a. Ruffieux P., Wang S., Yang B., Sánchez-Sánchez C., Liu J., Dienel T., Talirz L., Shinde P., Pignedoli C. A., Passerone D., Dumslaff T., Feng X., Müllen K., Fasel R., Nature 2016, 531, 489–492; [DOI] [PubMed] [Google Scholar]
- 1b. Wang X.-Y., Narita A., Müllen K., Nat. Rev. Chem. 2018, 2, 0100; [Google Scholar]
- 1c. Liu J., Feng X., Angew. Chem. Int. Ed. 2020, 59, 23386–23401; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23591–23607. [Google Scholar]
- 2.
- 2a. Moreno C., Vilas-Varela M., Kretz B., Garcia-Lekue A., Costache M. V., Paradinas M., Panighel M., Ceballos G., Valenzuela S. O., Peña D., Mugarza A., Science 2018, 360, 199–203; [DOI] [PubMed] [Google Scholar]
- 2b. Chen L., Hernandez Y., Feng X., Müllen K., Angew. Chem. Int. Ed. 2012, 51, 7640–7654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7758–7773. [Google Scholar]
- 3.
- 3a. From Polyphenylenes to Nanographenes and Graphene Nanoribbons (Eds.: Müllen K., Feng X.), Springer, Heidelberg, 2017; [Google Scholar]
- 3b. Ai Q., Jarolimek K., Mazza S., Anthony J. E., Risko C., Chem. Mater. 2018, 30, 947–957; [Google Scholar]
- 3c. Mishra S., Beyer D., Berger R., Liu J., Gröning O., Urgel J. I., Müllen K., Ruffieux P., Feng X., Fasel R., J. Am. Chem. Soc. 2020, 142, 1147–1152; [DOI] [PubMed] [Google Scholar]
- 3d. Fujii S., Enoki T., Acc. Chem. Res. 2013, 46, 2202–2210. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Son Y.-W., Cohen M. L., Louie S. G., Nature 2006, 444, 347–349; [DOI] [PubMed] [Google Scholar]
- 4b. Jiang D.-e., Sumpter B. G., Dai S., J. Chem. Phys. 2007, 127, 124703; [DOI] [PubMed] [Google Scholar]
- 4c. Xu Y., Wang B.-J., Ke S.-H., Yang W., Alzahrani A. Z., J. Chem. Phys. 2012, 137, 104107. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Zuzak R., Dorel R., Kolmer M., Szymonski M., Godlewski S., Echavarren A. M., Angew. Chem. Int. Ed. 2018, 57, 10500–10505; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10660–10665; [Google Scholar]
- 5b. Krüger J., García F., Eisenhut F., Skidin D., Alonso J. M., Guitián E., Pérez D., Cuniberti G., Moresco F., Peña D., Angew. Chem. Int. Ed. 2017, 56, 11945–11948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12107–12110; [Google Scholar]
- 5c. Kaur I., Jazdzyk M., Stein N. N., Prusevich P., Miller G. P., J. Am. Chem. Soc. 2010, 132, 1261–1263; [DOI] [PubMed] [Google Scholar]
- 5d. Purushothaman B., Bruzek M., Parkin S. R., Miller A. F., Anthony J. E., Angew. Chem. Int. Ed. 2011, 50, 7013; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7151. [Google Scholar]
- 6.
- 6a. Ye Q., Chi C., Chem. Mater. 2014, 26, 4046–4056; [Google Scholar]
- 6b. Sun Z., Ye Q., Chi C., Wu J., Chem. Soc. Rev. 2012, 41, 7857–7889; [DOI] [PubMed] [Google Scholar]
- 6c. Sun Z., Zeng Z., Wu J., Acc. Chem. Res. 2014, 47, 2582–2259; [DOI] [PubMed] [Google Scholar]
- 6d. Jiang D.-e., Dai S., Chem. Phys. Lett. 2008, 466, 72–75; [Google Scholar]
- 6e. Moscardó F., San-Fabián E., Chem. Phys. Lett. 2009, 480, 26–30; [Google Scholar]
- 6f. Konishi A., Kubo T., Top. Curr. Chem. 2017, 375, 83; [DOI] [PubMed] [Google Scholar]
- 6g. Malrieu J.-P., Trinquier G., J. Phys. Chem. A 2016, 120, 9564–9578; [DOI] [PubMed] [Google Scholar]
- 6h. Trinquier G., Malrieu J.-P., J. Phys. Chem. A 2018, 122, 1088–1103. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Clar E., The Aromatic Sextet, Wiley-Interscience, London, 1972; [Google Scholar]
- 7b. Clar E., Polycyclic Hydrocarbons, Academic Press, London, 1964; [Google Scholar]
- 7c. Bendikov M., Duong H. M., Starkey K., Houk K. N., Carter E. A., Wudl F., J. Am. Chem. Soc. 2004, 126, 7416–7417. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Ajayakumar M. R., Fu Y., Ma J., Hennersdorf F., Komber H., Weigand J. J., Alfonsov A., Popov A. A., Berger R., Liu J., Müllen K., Feng X., J. Am. Chem. Soc. 2018, 140, 6240–6244; [DOI] [PubMed] [Google Scholar]
- 8b. Ajayakumar M. R., Fu Y., Liu F., Komber H., Tkachova V., Xu C., Zhou S., Popov A. A., Liu J., Feng X., Chem. Eur. J. 2020, 26, 7497–7750; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Mishra S., Lohr T. G., Pignedoli C. A., Liu J., Berger R., Urgel J., Müllen K., Feng X., Ruffieux P., Fasel R., ACS Nano 2018, 12, 11917–11927. [DOI] [PubMed] [Google Scholar]
- 9. Ni Y., Gopalakrishna T. Y., Phan H., Herng T. S., Wu S., Han Y., Ding J., Wu J., Angew. Chem. Int. Ed. 2018, 57, 9697–9701; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9845–9849. [Google Scholar]
- 10.
- 10a. Rogers C., Chen C., Pedramrazi Z., Omrani A. A., Tsai H.-Z., Jung H. S., Lin S., Crommie M. F., Fischer F. R., Angew. Chem. Int. Ed. 2015, 54, 15143–15146; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15358–15361; [Google Scholar]
- 10b. Sánchez-Grande A., Urgel J. I., Veis L., Edalatmanesh S., Santos J., Lauwaet K., Mutombo P., Gallego J. M., Brabec J., Beran P., Nachtigallová D., Miranda R., Martín N., Jelínek P., Écija D., J. Phys. Chem. Lett. 2021, 12, 330–336. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Zöphel L., Berger R., Gao P., Enkelmann V., Baumgarten M., Wagner M., Müllen K., Chem. Eur. J. 2013, 19, 17821–17826; [DOI] [PubMed] [Google Scholar]
- 11b. Zhang X., Li J., Qu H., Chi C., Wu J., Org. Lett. 2010, 12, 3946–3949. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. IJpeij E. G., Beijer F. H., Arts H. J., Newton C., de Vries J. G., Gruter G.-J. M., J. Org. Chem. 2002, 67, 169–176; [DOI] [PubMed] [Google Scholar]
- 12b. Chai G.-L., Han J.-W., Wong H. N. C., Synthesis 2017, 49, 181–187. [Google Scholar]
- 13.
- 13a. Gu Y., Wu X., Gopalakrishna T. Y., Phan H., Wu J., Angew. Chem. Int. Ed. 2018, 57, 6541–6545; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6651–6655; [Google Scholar]
- 13b. Gu Y., Tullimilli Y. G., Feng J., Phan H., Zeng W., Wu J., Chem. Commun. 2019, 55, 5567–5570. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Castiglioni C., Tommasini M., Zerbi G., Philos. Trans. R. Soc. London Ser. A 2004, 362, 2425–2459; [DOI] [PubMed] [Google Scholar]
- 14b. Tommasini M., Castiglioni C., Zerbi G., Phys. Chem. Chem. Phys. 2009, 11, 10185–10194. [DOI] [PubMed] [Google Scholar]
- 15.Gaussian 09, Revision D.01.
- 16.
- 16a. Maghsoumi A., Narita A., Dong R., Feng X., Castiglioni C., Müllen K., Tommasini M., Phys. Chem. Chem. Phys. 2016, 18, 11869–11878; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Dumslaff T., Yang B., Maghsoumi A., Velpula G., Mali K. S., Castiglioni C., Feyter S. D., Tommasini M., Narita A., Feng X., Müllen K., J. Am. Chem. Soc. 2016, 138, 4726–4729. [DOI] [PubMed] [Google Scholar]
- 17. Rudebusch G. E., Zafra J. L., Jorner K., Fukuda K., Marshall J. L., Arrechea-Marcos I., Espejo G. L., Ortiz R. P., Gómez-García C. J., Zakharov L. N., Nakano M., Ottosson H., Casado J., Haley M. M., Nat. Chem. 2016, 8, 753–759. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Yamaguchi K., Self-consistent field: theory and applications (Eds.: Carbó R., Klobukowski M.), Elsevier, Amsterdam, 1990, pp. 727–823; [Google Scholar]
- 18b. Yamanaka S., Okumura M., Nakano M., Yamaguchi K., J. Mol. Struct. 1994, 310, 205–218. [Google Scholar]
- 19. Schleyer P. v. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J. R., J. Am. Chem. Soc. 1996, 118, 6317–6318. [DOI] [PubMed] [Google Scholar]
- 20. Herges R., Geuenich D., J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar]
- 21.
- 21a. Lombardi F., Lodi A., Ma J., Liu J., Slota M., Narita A., Myers W. K., Müllen K., Feng X., Bogani L., Science 2019, 366, 1107–1110; [DOI] [PubMed] [Google Scholar]
- 21b. Slota M., Keerthi A., Myers W. K., Tretyakov E., Baumgarten M., Ardavan A., Sadeghi H., Lambert C. J., Narita A., Müllen K., Bogani L., Nature 2018, 557, 691–695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary