

Summary
Inner Mongolia Cashmere goat is a well‐known local cashmere goat breed in China. It is famous for excellent fleece quality and a significant advantage in cashmere yield compared to other cashmere goat breeds. In this study, a genome‐wide association study was used to investigate fiber length, fiber diameter, and cashmere yield of 192 Inner Mongolia Cashmere goats using the Illumina GoatSNP52K Beadchip panel. We discovered that four single nucleotide polymorphisms (SNPs) reached genome‐wide significance levels. These SNPs were located in some genes, e.g. FGF12, SEMA3D, EVPL, and SOX5, possibly related to fleece traits in Inner Mongolia Cashmere goat. Gene ontology enrichment analysis revealed that these genes were enriched in several biological pathways that were involved in hair follicle development in cashmere goats. In summary, the identified significant SNPs and genes provide useful information to explore genetic mechanisms underlying the variation in fleece traits and genomic selection of Chinese cashmere goat.
Keywords: fleece traits, genome‐wide association study, GoatSNP52K Beadchip, Inner Mongolia Cashmere goats
Inner Mongolia Cashmere goats (IMCGs) are an important breed for cashmere and meat and a crucial genetic resource of Chinese goat population (Li et al. 2017; Islam et al. 2020). The cashmere produced by IMCGs is famous worldwide for its brightness, color, elasticity, thin diameter, and softness, notwithstanding the harsh environmental conditions where IMCGs usually lives (Zhang et al. 2014). There are few reports concerning fleece traits of IMCGs especially fiber length (FL) and fiber diameter (FD). Fleece traits determine the quality of cashmere products and are closely related to weaving products and economic profits. How to reduce the fiber fineness while maintaining the cashmere yield (CY) is the main aim of the breeding of cashmere goats.
Genome‐wide association studies (GWAS) has become an effective strategy to identify candidate genes for important economic traits in livestock (Wittenburg et al. 2020). In recent years, various genes or associated genetic markers affecting important traits have been identified in farm animals, e.g. pigs (Yao et al. 2019; Jiang et al. 2020), cattle (Sahana et al. 2010; Sarlo Davlia et al. 2020), sheep (Li et al. 2020; Hernandez‐montiel et al. 2020), and chickens (Liu et al. 2020; Zhang et al. 2020), etc.
Goats used in the present study (n = 192, 3 years old from 4 herds) were selected randomly from the Arbas Stock Farm (latitude 39°06′N and longitude 107°59′E) in southwestern Inner Mongolia, China. Before cashmere combing in May, patch samples of 10 cm2 on the side of the shoulder were obtained by shaving the area. The diameter of fiber samples were measured with an optical FD analyzer. The ear tissue samples were collected by ear deficiency forceps and quickly placed in a prepared cryopreservation tube containing 75% alcohol for storage at −80°C. Genomic DNA was extracted from the ear tissue samples with the AXYGEN Blood and Tissue Extraction Kit (Corning) according to the manufacturer’s instructions. The extracted DNA were subjected to electrophoresis in 2% agarose gel and stained with ethidium bromide to assess overall quality. All experimental procedures were approved by the Animal Care and Use Committee of the Inner Mongolia Agricultural University and conducted in strict accordance with the animal welfare and ethics guidelines.
The samples were genotyped using the Illumina GoatSNP52K Beadchip panel including 53 347 SNPs (Illumina Inc.). The samples with call rates <90% were removed from the analysis. The SNPs with GenCall (GC) scores <0.60, genotype call rates <90% (‐‐geno 0.10), minor allele frequencies <0.01 and significant Hardy–Weinberg disequilibrium at 10−6 were removed from the analysis (‐‐maf 0.01 –hwe 1e‐6). plink 1.90 beta (Chang et al., 2015) and R software were used for quality control. A total of 191 animals and 48 739 variants remained after quality control.
The associations among SNPs and traits were tested using mixed linear models in GEMMA software version 0.98 (Yu et al. 2006; Zhou & Stephens 2012). Herd and age were combined as a fixed effect, as they all significantly related to fleece traits (Wang et al. 2013). The statistical model used in this study was y = Xα + Zβ + Wµ + e, where y was the phenotypic of FL, FD, and CY, X was a matrix of fixed effects, α was the estimation parameter of the fixed effects, Z was a matrix of SNPs, β was the effect of the SNPs, W was a matrix of random effects, µ was the predicted random individuals, and e was the random error, with the distribution e ~ N (0, ). The significance threshold for the GWAS was defined using the Bonferroni correction method. The suggestive genome‐wide association significance threshold was P < 1.03 × 10−6 (0.05/48 739) and chromosome‐wide significance level threshold was P < 2.97 × 10−5 (0.05/48 739/29). Chromosome‐wide significance level SNPs were also defined to call chromosome‐wide significant associations, with a suggestive association cutoff P‐value <10−4 (Sahana et al. 2010). The Manhattan and quantile–quantile graphics were plotted with r v. 3.5.2 (Turner 2014). After the quality control was performed on the raw genotypes, a total of 48 739 SNPs were obtained, and were distributed over the 29 goat chromosomes (Fig. S1).
The descriptive statistics of the studied traits are set out in Table S1. All traits are normally distributed (Fig. S2). The principal component analysis result demonstrated that there was no genetic difference between the samples (Fig. S3). The results of GWAS can be seen in Fig. 1. A total of four SNPs reached the genome‐wide significance levels and 130 SNPs reached the chromosome‐wide significance levels for the 3 fleece traits. Among the 130 SNPs, 27 were associated with FL, 51 were associated with FD, and 52 were associated with CY. The quantile–quantile plot showed that genomic inflation factor (λ) of FL, FD, and CY were 1.057, 1.006, and 1.082 respectively, which are close to one, indicating there was no difference between observed vs. expected value.
Figure 1.
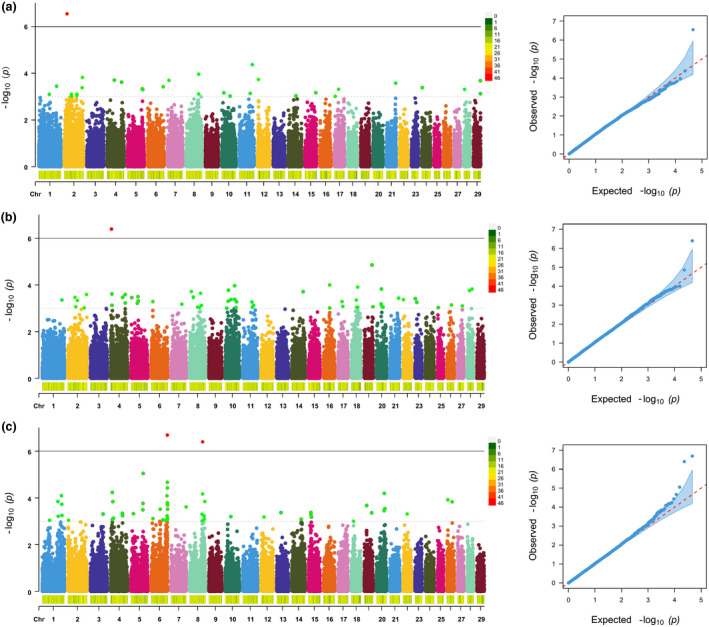
Manhattan plots and quantile–quantile (Q‐Q) plots of fleece traits for Inner Mongolia Cashmere goats. Black and grey lines indicate the thresholds for genome‐wide significance levels and chromosome‐wide significance levels respectively. The Q‐Q plots show the observed vs expected log P‐values. (a) Fiber length; (b) fiber diameter; (c) cashmere yield.
Relatively significant SNP loci were found within a distance of 100 kb region of the searching peak SNP. Some genes were also of great significance for skin or hair follicle growth and development, e.g. FGF12, SOX5, and EVPL, while some new genes were identified to be related to fleece traits in this study, e.g. GALNTL5, FBF1, SPHKAP, and RGS12.
Gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed on these candidate genes (Fig. 2). A total of 267 candidate genes were significantly enriched in 96 gene ontology terms, including 78 biological processes, 10 cellular components, and eight molecular functions. The KEGG pathway analysis revealed that the genes associated with the SNPs were significantly enriched in 25 pathways. These enriched pathways were associated with the growth and development of hair follicles. These results are consistent with the biological laws that with the increase of FL, the CY also increased. In this study, interestingly, we found that some significant genes (CCDC150, GTF3C3, DSCAM, CNTNAP2, and CAMK2D) are related to FL, FD, and CY. This further confirms the hypothesis that cashmere traits are regulated by multiple genes.
Figure 2.
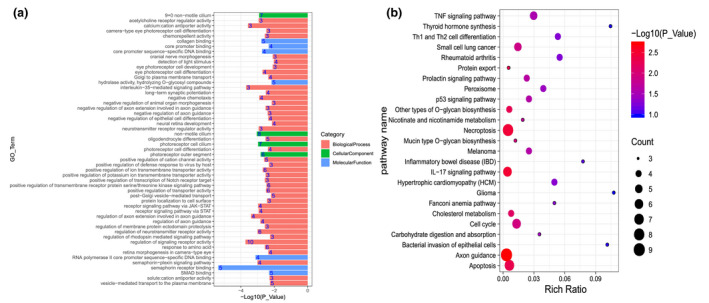
Gene ontology (GO: a) and Kyoto Encyclopedia of Genes and Genomes (b) enrichment analysis for the regional candidate genes with chromosome‐wide significant association.
In this study, we discovered a variety of functional genes and signaling pathways associated with the fleece traits in cashmere goats (Table S2). For instance, EVPL (Envoplakin) is a protein coding gene associated with follicle differentiation (Ahlawat et al. 2020). SOX5 is the developmental transcription factor, which is expressed in growing hair follicles in skin as well as in sebaceous and eccrine sweat glands (Kuhn et al. 1999). Semaphorin 3D (SEMA3D) is associated with Axon guidance, which depends upon mechanosensory transduction by hair cells to percept sensory information (Scott et al. 2019). FGF12 (fibroblast growth factor 12) is a member of the fibroblast growth factor family that regulates MAPK, PI3K‐Akt, and Ras signaling pathways involved in the sheep hair follicle development (Lv et al. 2020). The TNF signaling pathway is important hair follicle developmental pathway that affects the growth cycle of hair follicles (Pérez‐Garijo et al. 2013; Sulayman et al. 2019).
This is the first GWAS on cashmere traits of IMCGs based on GoatSNP52K Beadchip. Only four statistically significant loci were identified to be associated with cashmere traits in this population. A reason is that the earliest released SNP data of GoatSNP52K Beadchip were designed by six foreign goat breeds of 97 individuals that are not specific to Chinese goat population, especially cashmere goat. Another reason may be that the number of cashmere goat population in this study is small, and cashmere traits are complex quantitative traits controlled by genetic and environmental factors. The discovery of these genetic variation sites can be used as candidate SNP sites for development and validation of genetic markers for important economic traits of cashmere fineness, as well as accumulation of important genetic marker data and genetic materials for future cashmere goat breeding.
In summary, the first GWAS of IMCGs was performed to identify gene‐associated biological pathways related to three fleece traits. The candidate genes in this study were closely correlated to hair follicle development. These findings will make a significant contribution to the understanding of the mechanisms underlying fleece traits and to the application of genomic selection in IMCGs.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Figure S1 SNP markers distributed over the 29 goat chromosomes.
Figure S2 Normal distribution map of all traits. (a) Fiber length, (b) fiber diameter and (c) cashmere yield.
Figure S3 Principal component analysis (PCA) using all identified SNPs as markers.
Table S1 Significant SNPs associated with phenotypic value for fleece traits in Inner Mongolia cashmere goats.
Table S2 Descriptive statistics of fleece traits in Inner Mongolia cashmere goats.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31860637), China Agriculture Research System (No. CARS‐39‐06), The Central Government Guiding Special Funds for Development of Local Science and Technology (2020ZY0007), Youth Foundation in Inner Mongolia Agricultural University (No. QN202003), Science and Technology Project of Inner Mongolia Autonomous Region (2019GG243), and Research and Innovation Projects for Postgraduates (B20191136Z).
Contributor Information
R. Su, Email: siruiyu@126.com.
Y. H. Zhao, Email: 13947196432@163.com.
J. Q. Li, Email: lijinquan_nd@126.com.
Data availability statement
The genotype data are available in the Figshare Repository (https://figshare.com/s/d1e08c19cb642e538922).
References
- Ahlawat S., Arora R., Sharma R., Sharma U., Kaur A., Singh K.V., Singh M.K. & Vijh R.K. (2020) Skin transcriptome profiling of Changthangi goats highlights the relevance of genes involved in Pashmina production. Scientific Reports 10, 6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M. & Lee L.L. (2015) Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐montiel W., Martinez‐Nunez M.A., Ramon‐Ugalde J.P., Roman‐Ponce S.I., Calderon‐Chagoya R. & Zamora‐Bustillos R. (2020) Genome‐wide association study reveals candidate genes for litter size traits in Pelibuey sheep. Animals 10, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam R., Liu X.X., Gebreselassie G., Abied A., Ma Q. & Ma Y.H. (2020) Genome‐wide association analysis reveals the genetic locus for high reproduction trait in Chinese Arbas Cashmere goat. Genes Genomics 42, 893–9. [DOI] [PubMed] [Google Scholar]
- Jiang B., Wang M., Tang Z., Du X., Feng S., Ma G., Ye D., Cheng H., Wang H. & Liu X. (2020) Genome‐wide association study of bone mineral density trait among three pig breeds. Animal 14, 2443–51. [DOI] [PubMed] [Google Scholar]
- Kuhn F., Lassing C., Range A., Mueller M., Hunziker T., Ziemiecki A. & Andres A.C. (1999) Pmg‐1 and Pmg‐2 constitute a novel family of KAP genes differentially expressed during skin and mammary gland development. Mechanisms of Development 86, 193–6. [DOI] [PubMed] [Google Scholar]
- Li H., Wu X.L., Tait R.G. Jr, Bauck S., Thomas D.L., Murphy T.W. & Rosa G.J.M. (2020) Genome‐wide association study of milk production traits in a crossbred dairy sheep population using three statistical models. Animal Genetics 51, 624–8. [DOI] [PubMed] [Google Scholar]
- Li X.K., Su R., Wan W.T. et al. (2017) Identification of selection signals by large‐scale whole‐genome resequencing of cashmere goats. Scientific Report 7, 15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.J., Lu L., Wang J., Cui H.X., Chu H.H., Bi H.J., Zhao G.P. & Wen J. (2020) Genome‐wide association study of muscle glycogen in Jingxing yellow chicken. Genes 11, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X.L., Chen W.H., Sun W., Hussain Z., Wang S.H. & Wang J.Y. (2020) Analysis of lncRNAs expression profiles in hair follicle of hu sheep lambskin. Animals 10, E1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Garijo A., Fuchs Y. & Steller H. (2013) Apoptotic cells can induce non‐autonomous apoptosis through the TNF pathway. Elife 2, e01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahana G., Guldbrandtsen B., Bendixen C. & Lund M.S. (2010) Genome‐wide association mapping for female fertility traits in Danish and Swedish Holstein cattle. Animal Genetics 41, 579–88. [DOI] [PubMed] [Google Scholar]
- Sarlo Davila K.M., Howell A., Nuez A., Orelien A., Roe V., Rodriguez E., Dikmne S. & Mateescu R.G. (2020) Genome‐wide association study identifies variants associated with hair length in Brangus cattle. Animal Genetics 4, 12970. [DOI] [PubMed] [Google Scholar]
- Scott M.K., Yue J., Biesemier D.J., Lee J.W. & Fekete D.M. (2019) Expression of class III Semaphorins and their receptors in the developing chicken (Gallus gallus) inner ear. The Journal of Comparative Neurology 527, 1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulayman A., Tian K., Huang X.X. et al. (2019) Genome‐wide identification and characterization of long non‐coding RNAs expressed during sheep fetal and postnatal hair follicle development. Scientific Reports 9, 8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S.D. (2014) qqman: an R package for visualizing GWAS results using Q‐Q and manhattan plots. Biorxiv 005165.
- Wang Z.Y., Wang R.J., Zhang W.G., Wang Z.X., Wang P.J., Liu H., Gao L.X., Bai K., Meng R.Q. & Zhou J. (2013) Estimation of genetic parameters for fleece traits in yearling Inner Mongolia Cashmere goats. Small Ruminant Research 109, 15–21. [Google Scholar]
- Wittenburg D., Bonk S., Doschoris M. & Reyer H. (2020) Design of experiments for fine‐mapping quantitative trait loci in livestock populations. BMC Genetics 21, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Qing T.S., Wei X., Peng Y. & Dong D.X. (2019) A genome‐wide association study of reproduction traits in four pig populations with different genetic backgrounds. Asian‐Australasian Journal of Animal Sciences 33, 1400–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.M., Pressoir G., Briggs W.H. et al. (2006) A unified mixed‐model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics 38, 203–8. [DOI] [PubMed] [Google Scholar]
- Zhang H., Shen L.Y., Xu Z.X., Kramer L.M., Yu J.Q., Zhang X.Y., Na W., Yang L.L., Cao Z.P. & Ping H. (2020) Haplotype‐based genome‐wide association studies for carcass and growth traits in chicken. Poultry Science 99, 2349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.J., Wang Z.Y., Lei H. et al. (2014) Estimates of genetic parameters and genetic changes for fleece traits in Inner Mongolia cashmere goats. Small Ruminant Research 117, 41–6. [Google Scholar]
- Zhou X. & Stephens M. (2012) Genome‐wide efficient mixed‐model analysis for association studies. Nature Genetics 44, 821–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SNP markers distributed over the 29 goat chromosomes.
Figure S2 Normal distribution map of all traits. (a) Fiber length, (b) fiber diameter and (c) cashmere yield.
Figure S3 Principal component analysis (PCA) using all identified SNPs as markers.
Table S1 Significant SNPs associated with phenotypic value for fleece traits in Inner Mongolia cashmere goats.
Table S2 Descriptive statistics of fleece traits in Inner Mongolia cashmere goats.
Data Availability Statement
The genotype data are available in the Figshare Repository (https://figshare.com/s/d1e08c19cb642e538922).