

Abstract
Background
Genetic characteristics and genetic carrier diagnosis in Japanese hemophilia female carriers have not been evaluated.
Objectives
To provide genetic information on Japanese hemophilia female carriers and demonstrate the advantages of genetic testing in carrier diagnosis.
Methods
DNA sequencing combined with long polymerase chain reaction for inversion and multiplex ligation‐dependent probe amplification for large mutations.
Results
Genetic analysis was performed in 69 male hemophiliac patients (48 hemophilia A [HA] and 21 hemophilia B [HB]) and 112 female family members (FFM) (80 from 50 families with HA and 32 from 22 families with HB). In 72 hemophiliac families, the identified F8 mutations were inversion (42%), missense (26%), and other variations (32%), while 74% of F9 mutations were point mutations. Among the 112 FFM, 53/80 (66%) with HA and 21/32 (66%) with HB were diagnosed genetically as carriers based on detection of heterozygous mutations. Low factor VIII activity (FVIII:C) levels (<50 IU/dL) were detected in only 10% of gene‐confirmed carriers, suggesting that FVIII:C is not suitable for HA carrier prediction. Low FVIII/von Willebrand factor ratio (<0.9) was observed in 67% of gene‐confirmed carriers. Half of the gene‐confirmed HB carriers had low FIX:C (<60 IU/dL). Importantly, 32 mothers of 37 sporadic cases (86%) (24/27 [89%] HA and 8/10 [80%] HB) showed the relevant mutations, suggesting low incidence of de novo mutations in males.
Conclusions
This study is the first to provide genetic information on Japanese hemophilia female carriers. Gene analysis is the gold standard for carrier diagnosis as it well identifies undetected female carriers based on pedigree information and hemostatic measurements.
Keywords: carrier diagnosis, de novo mutation, female carriers, genetic analysis, hemophilia
Essentials.
Genetic characteristics and factor activities in Japanese hemophilia carriers have not been evaluated yet.
Genetic analysis was conducted in 112 female members of 72 hemophilia families to determine carrier status.
Factor activities are not reliable for carrier diagnosis while factor VIII/von Willebrand factor is somewhat useful but with limited applicability.
The estimated rate of de novo mutations in males in 37 hemophiliacs with a negative family history of hemophilia was 14%.
1. INTRODUCTION
Hemophilia A and B are X‐linked recessive bleeding disorders resulting from more than 3000 and 1200 different DNA variants 1 encoding clotting factors VIII (FVIII) and IX (FIX), respectively. Females carrying the hemophilia mutant gene in one of the X chromosomes and a normal allele on the other X chromosome are defined as carriers. Hemophilia carriers are classically categorized as obligate carriers and possible carriers based on the pedigree information. Obligate carriers are women who: (1) have hemophiliac fathers, (2) have more than one hemophiliac son, (3) have a hemophiliac son and another hemophiliac relative in the maternal family. On the other hand, possible carriers are women who have only one hemophiliac son or a relative with known hemophilia or known carrier for hemophilia in the maternal family. The carrier status is usually determined by carrier diagnosis testing.
Determination of the hemophilia carrier status, that is, “carrier diagnosis,” involves coagulation factor assay and genetic testing. 2 , 3 , 4 , 5 , 6 , 7 Measurement of coagulation factor activity levels was the only test performed traditionally; the carrier status was confirmed by the finding of coagulation factor activity of approximately half that of healthy normal women. However, subsequent advances in biotechnology demonstrated that FVIII or FIX activities in women are often affected by X‐inactivation, in which random suppression occurs in one of the two X chromosomes (i.e., Lyonization). 8 , 9 Extremely skewed X‐inactivation leads to profoundly low factor activity even in female hemophilia carriers. 10 , 11 In addition, clotting factor activity levels in females were also found to fluctuate under both physiological and pathological conditions, such as menstrual cycle, pregnancy, aerobic exercise, and chronic inflammation, and even show diurnal variation. 2 , 5 In 1971, Zimmerman et al. 12 used immunologic assay for antihemophilic factor like antigen (now, von Willebrand factor [VWF] antigen) to define hemophilia A (HA) carriers. They demonstrated that HA carriers had lower ratio of FVIII activity (FVIII:C) to VWF antigen (VWF:Ag) than non‐carrier females. The ratio has since been widely used to improve the sensitivity and specificity of the carrier diagnosis for HA. On the other hand, hemophilia B (HB) carrier is generally diagnosed based on factor IX activity (FIX:C) itself, because FIX is not bound to carrier protein. At present, genetic testing, including DNA sequencing for the detection of mutations in F8 and F9 genes, is considered the gold standard for the diagnosis of hemophiliac carriers. However, there is little or no information on screening for HA and HB female carriers by DNA sequencing.
Recent progress in genetic analysis has identified various mutations and/or gene variants of F8 and F9 in hemophiliacs. 1 , 13 , 14 To our knowledge, there are yet no genetic analysis‐based studies that have examined the carrier status of Japanese hemophilia female family members. Furthermore, the clinical severity, types of genetic variations, and clotting factor activity levels in Japanese hemophilia female carriers have not yet been evaluated. The present study was designed to provide genetic information on Japanese hemophilia female carriers based on available clinical data, as well as the relation between such genetic status and the levels of FVIII:C or FIX:C, using gene mutation analysis and DNA sequencing analysis.
2. MATERIALS AND METHODS
2.1. Subjects
Genetic analysis was performed in 69 (48 HA and 21 HB) hemophiliac males (patients) and 112 (80 HA and 32 HB) female family members (FFM) among 72 (50 HA and 22 HB) families in 28 hemophilia treatment clinical facilities between 2007 and end of 2019 (see the Acknowledgments section for a list of these facilities). Genetic tests were not applied for prenatal diagnosis or used in any decision process for abortion of hemophiliac children. The hemophiliac mutations in three obligate carriers who were genetically apparent carriers were defined as mutations in each family, because 3 patients died in each family (2 HA and 1 HB).
The study protocol was approved by the Ethics Committee on Medical Research of Tokyo Medical University (approval number #2017‐068). All study subjects provided written informed consent to participate in this study and were enrolled after receiving full explanation of the purpose of the study and the methods to be used. Of the 112 FFM, 106 were interviewed directly at Tokyo Medical University Hospital and details of the clinical features, phenotypes, and family pedigrees were recorded. Six FFM (2 HA and 4 HB) were interviewed at Hiroshima University Hospital and the Hospital of Hyogo College of Medicine.
2.2. Blood coagulation tests
FVIII:C and FIX:C levels were measured using one‐stage clotting assay (HemosIL™ APTT‐SP; Instrumentation Laboratory and Coagpia® APTT‐N; Sekisui Medical Co.) on an ACL 9000 Automated Coagulometer and CP3000™. VWF:Ag levels were measured using latex coagulating nephelometry (STA Listest vWF[FR]; Diagnostica Stago, Inc.) on a JCA‐BM8020 (JEOL).
2.3. Protocol for gene analysis and carrier diagnosis
2.3.1. F8 and F9 mutations for hemophilia in each family
Genetic analysis of F8 and F9 mutations was conducted in 50 subjects of 50 HA families and in 22 subjects of 22 HB families. The subjects of families with severe and moderate HA were screened for intron‐22 inversion analysis (Figure 1). This approach can identify inversion as the underlying mutation in approximately 40% of severe HA patients. 13 , 15 The inversion analysis was also applied in moderate HA patients because inversion is often detected in moderate HA patients whose FVIII:C is approximately 1 IU/dL. The remaining HA and HB cases underwent direct sequencing. Subjects negative for mutations by sequencing underwent F8 and F9 multiplex ligation‐dependent probe amplification (MLPA) that can detect gross rearrangements, such as large deletions, large insertions, and duplications.
FIGURE 1.
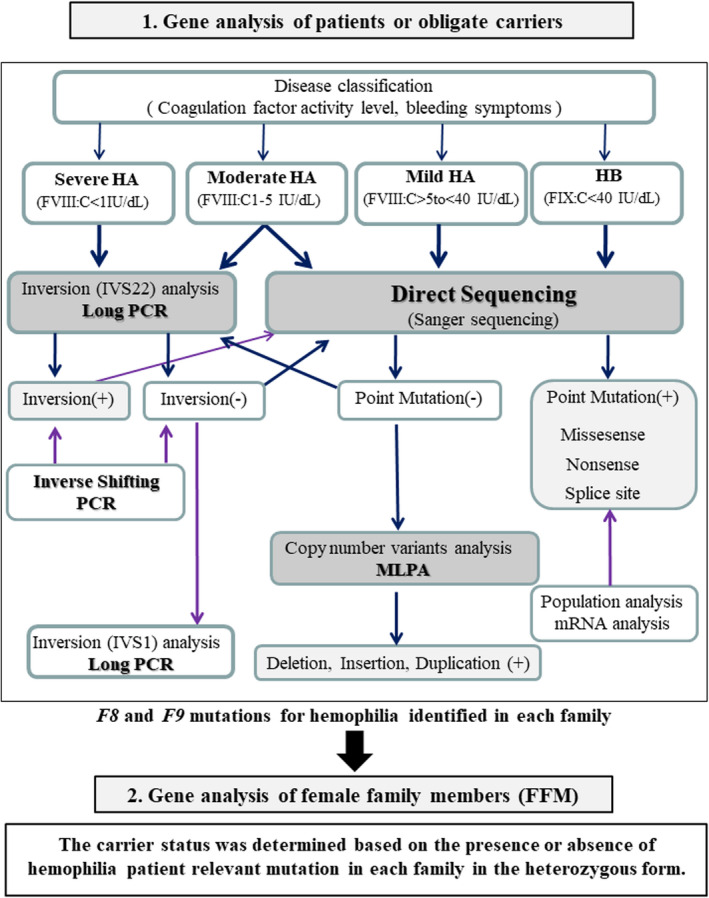
Proposed procedure of gene analysis for carrier diagnosis. (1) Patients or obligate carriers of hemophiliac family were approached for gene analysis. Subjects with severe and moderate hemophilia A (HA) were screened for intron 22 inversion analysis. The remaining HA and hemophilia B (HB) cases underwent direct sequencing. Patients who were found negative on direct sequencing were also analyzed by multiplex ligation‐dependent probe amplification (MLPA). The hemophilia‐related mutations were identified in each family. (2) Female family members (FFM) underwent gene analysis. The carrier status was determined based on the presence or absence of the hemophilia patient relevant mutation in each family in the heterozygous form. All gene analyses were conducted at least twice to validate the results. FIX:C, factor IX activity; FVIII:C, factor VIII activity; VWF:Ag, von Willebrand factor antigen; PCR, polymerase chain reaction
2.3.2. Diagnosis of the carrier status
The carrier status was determined based on the presence or absence of hemophilia patient relevant mutation in each family in the heterozygous form. All gene analyses were conducted at least twice.
2.4. Genetic analysis
Genomic DNA was extracted from leukocytes, and the whole coding regions and the exon‐‐intron boundaries in F8 and F9 were amplified using polymerase chain reaction (PCR). 16 , 17 The purified samples were sequenced using BigDye® Terminator v3.1 Sequencing kit and 3730 DNA Analyzer (Applied Biosystems‐ThermoFisher Scientific Inc.). All sequence variations were validated at least twice. The resultant sequences were compared to those of F8 and F9 (Ensemble number ENST00000360256.8 and ENST00000218099.6). FVIII and FIX Gene Variant Databases (https://f8‐db.eahad.org/; https://f9‐db.eahad.org/) were the two main databases used to determine the mutation. 1 For assessment of unreported mutations, the variations were examined in 50 normal Japanese individuals.
F8 inversions were detected using the Long‐PCR described previously with some modifications. 18 We used the SALSA MLPA P178 F8 and P207‐C1 F9 probe mix kits (MRC‐Holland), and the procedure provided by the manufacturer.
2.5. Statistical analysis
Differences between carrier and non‐carrier groups were analyzed by the Mann‐Whitney U test for continuous variables, and chi‐square test for categorical variables. A P value less than or equal to 0.05 was considered statistically significant. Statistical analyses were performed using IBM SPSS Statistics software, version 25 (IBM SPSS Statistics).
3. RESULTS
3.1. Subject characteristics
The study included 69 hemophiliac male patients (48 HA and 21 HB) and 112 FFM (80 HA and 32 HB) from 72 families (50 HA and 22 HB; Table 1). The disease status was classified as severe in 40 of 48 (83%) HA and 11 of 21 (52%) HB patients.
TABLE 1.
Subject characteristics
| Hemophilia A | Hemophilia B | |
|---|---|---|
| Families, n | 50 | 22 |
| Males (patients), n | 48 | 21 |
| Mean age, years | 18.8 (1–75) | 23.0 (1–79) |
| Clinical severity* | ||
| Severe | 40 (83%) | 11 (52%) |
| Moderate | 3 (6%) | 7 (33%) |
| Mild | 5 (10%) | 3 (14%) |
| Females, n | 80 | 32 |
| Possible carriers | 75 (94%) | 27 (84%) |
| Obligate carriers | 5 (6%) | 5 (16%) |
| Mean age, years | 37.7 (1–78) | 36.3 (22–63) |
| Relationship to patients | ||
| Mothers | 32 | 12 |
| Sisters | 23 | 8 |
| Grandmothers | 4 | 0 |
| Aunts | 9 | 3 |
| Cousins | 4 | 2 |
| Nieces | 5 | 0 |
| Daughters | 1 | 4 |
| Granddaughters | 1 | 1 |
| Oneself | 1 | 0 |
| Cousin‐daughters | 0 | 2 |
| Number of children a | ||
| Males with hemophilia b | 33 | 12 |
| (Sporadic hemophilia) | (27) | (10) |
| Female infants c | 30 | 11 |
| Pregnant women | 9 | 2 |
| Bleeding history in females, n | 80 | 32 |
| Unknown d | 37 (46%) | 12 (38%) |
| No bleeding | 29 (36%) | 14 (44%) |
| Bleeding | 14 (18%) | 6 (19%) |
| Nasal bleeding | 0 | 1 |
| Menorrhagia | 6 | 3 |
| Purpura | 4 | 0 |
| Postoperative bleeding | 0 | 0 |
| Tooth extraction bleeding | 3 | 1 |
| Postpartum bleeding | 5 | 1 |
| Joint bleeding | 1 | 0 |
| Injury bleeding | 1 | 1 |
| Anemia | 1 | 1 |
Data are number of subjects, range, or percentages. Data were recorded at the time of genetic testing for carrier diagnosis. “Males” represents patients. Seventy‐two family numbers do not match the 69 patients’ number because 3 patients died in each family (2 hemophilia A and 1 hemophilia B).
“Females” represents female family members (FFM) who participated in the study.
Mothers who gave birth to the indicated number of children.
Mothers who gave birth to males with hemophilia.
Mothers who gave birth to female infants or infants.
No documented report. Bleeding symptoms were obtained at clinical interviews conducted at the time of genetic testing for carrier diagnosis.
Clinical severity was classified based on coagulation factor activity level, into severe (<1 IU/dL), moderate (1–5 IU/dL), and mild (>5 IU/dL).
In the 80 HA FFM, 75 (94%) were possible carriers and 5 (6%) were obligate carriers. In the 32 HB FFM, 27 (84%) were possible carriers and 5 (16%) were obligate carriers (Table 1). The family relationships of the HA FFM to the patients were: mothers (40%), sisters (29%), and others (e.g., aunts, cousins, grandmothers; 31%), while for the HB FFM these were: mothers (38%), sisters (25%), and others (38%). Before maternal genetic testing, 32 mothers gave birth to 33 boys with HA; in 27 (82%) there was no family history of hemophilia and as such these cases were considered to be sporadic hemophilia. For HB, 12 mothers gave birth to 12 HB boys, and of those, 10 (83%) had been considered sporadic hemophilia. At the time of consultation, 9 HA and 2 HB FFM were pregnant and preparing for deliveries.
The results of clinical interviews with 112 FFM on bleeding histories at the time of genetic testing included no answer (no appropriate answer or unsuccessful interviews) from 37 (46%) HA and 12 (38%) HB FFM, no bleeding from 29 (36%) HA and 14 (44%) HB FFM, and symptomatic bleeding from 14 (18%) HA and 6 (19%) HB FFM (Table 1).
3.2. F8 and F9 mutations with hemophilia in 72 families
The F8 mutations in 50 subjects (48 patients and 2 obligate carriers) were as follows: intron‐22 inversions (n = 21), missense mutations (n = 13), nonsense mutations (n = 4), small deletions (n = 3), small insertions (n = 2), large deletions (n = 2), large insertions (n = 2), and splice site mutations (n = 2; Figure 2A). 19 It is noteworthy that 70% of the F8 genotype in 50 families showed null mutations, as defined by Gouw et al. 20 We detected 30 different unique F8 mutations in HA (Table 2), including the following 11 novel mutations: 3 missense mutations (p. Leu216Pro, p. Tyr414His, p. Trp222Leu), 1 acceptor splice site mutation (c.1010‐1G>A), 2 large deletions (exons 8–9 and exons 12–22), 2 small deletions (c.1203‐1206delCT and c.6102‐6103delG), 1 large insertion (LINE insertion in exon 19), and 2 small insertions (c.5452‐5453insGA, c.5986‐5987insA), which were not listed in the FVIII Gene Variant Databases. The phenotype of three HA patients with novel missense mutations in exons 5, 8, and 24, which correspond to A1, A2, and C2 domains in the FVIII proteins, were two mild and one severe type, respectively. No such mutations were detected in 50 normal Japanese individuals. Furthermore, in silico analyses of three newly found missense mutations, the predicting systems estimated the function as deleterious. Another eight previously unreported mutations were all null mutations, with the phenotype classified as severe type of hemophilia. These results add support to the above conclusion that the novel mutations were clinically relevant mutations in HA families.
FIGURE 2.
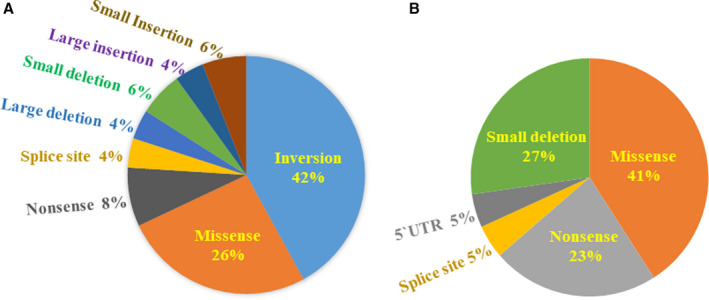
Distribution of F8 and F9 mutation types identified in hemophilia families. A, F8 mutations in 50 hemophilia A (HA) families. B, F9 mutations in 22 hemophilia B (HB) families. Mutations and variants were described according to the guideline of nomenclature by the Human Genome Variation Society. 19 Null and non‐null classification was based on the definition by Gouw et al. 20
TABLE 2.
F8 and F9 mutations in hemophilia in 72 families
| Mutation type in 50 HA families | FVIII protein domain | F8 Gene location | Nucleotide and amino acid change (HGVS) | Legacy no. | Novel mutation | de novo mutation |
|---|---|---|---|---|---|---|
| Inversion (21 families) | Intron 22 | Inversion | de novo mutation | |||
| Missense mutation (13 families) | ||||||
| A1 | Exon 5 | c.605G>A; p. Ser202Asn | 183 Ser(agt) > Asn(aat) | |||
| A1 | Exon 5 | c.643 T > C; p. Leu216Pro | 197 Leu(cta) > Pro(cca) | Novel mutation | ||
| a1 | Exon 8 | c.1172G>A; p. Arg391His | 372 Arg(cgc) > His(cac) | |||
| A2 | Exon 8 | c.1240 T > C; p. Tyr414His | 395 Tyr(tat) > His(cat) | Novel mutation | ||
| A2 | Exon 10 | c.1475A>G; p. Tyr492Cys | 473 Tyr(tat) > Cys(tgt) | |||
| A2 | Exon 12 | c.1798G>A; p. Glu600Lys | 581 Glu(gag) > Lys(aag) | |||
| C1 | Exon 16 | c.5399G>A; p. Arg1800His | 1781 Arg(cgt) > His(cat) | |||
| C1 | Exon 16 | c.6046C>T; p. Arg2016Trp | 1997 Arg(cgg) > Trp(tgg) | |||
| C1 | Exon 19 | c.6065G>A; p. Gly2022Asp | 2003 Gly(ggc) > Asp(gac) | |||
| C1 | Exon 23 | c.6505C>T; p. Arg2169Cys | 2150 Arg(cgt) > Cys(tgt) | |||
| C1 | Exon 23 | c.6544C>T; p. Arg2182Cys | 2163 Arg(cgc) > Cys(tgc) | de novo mutation | ||
| C2 | Exon 24 | c.6665G>T; p. Trp2222Leu | 2203 Trp(tgg) > Leu(ttg) | Novel mutation | ||
| C2 | Exon 26 | c.6977G>A; p. Arg2326Gln | 2307 Arg(cga) > Gln(caa) | |||
| Nonsense mutation (4 families) | ||||||
| A1 | Exon 8 | c.1063C>T; p. Arg355* | 336 Arg(cga) > Stop(tga) | |||
| A2 | Exon 13 | c.2099C>A; p. Ser700* | 681 Ser(tcg) > Stop(tag) | |||
| B | Exon 14 | c.2373G>A; p. Trp791* | 772 Trp(tgg) > Stop(tga) | de novo mutation | ||
| C2 | Exon 24 | c.6682C>T; p. Arg2228* | 2209 Arg(cga) > Stop(tga) | |||
| Splice site mutation (2 families) | ||||||
| Intron 7 | c.1010‐1G>A | Acceptor splice site mutation | Novel mutation | |||
| Intron 24 | c.6724‐1G>A | Acceptor splice site mutation | ||||
| Large deletion (2 families) | ||||||
| Exon 8–9 | Exon 8–9 deletion | Novel mutation | ||||
| Exon 12–22 | Exon 12–22 deletion | Novel mutation | ||||
| Small deletion (3 families) | ||||||
| A1 | Exon 2 | c.1203‐1206del CT | 2‐bp deletion (CT) | Novel mutation | ||
| B | Exon 14 | c.3629‐3637del A | 1‐bp deletion (A) | |||
| A3 | Exon 19 | c.6102‐6103del G | 1‐bp deletion (G) | Novel mutation | ||
| Large insertion (2 families) | ||||||
| Intron 18 | Alu insertion | |||||
| Exon 19 | LINE insertion | Novel mutation | ||||
| Small insertion (3 families) | ||||||
| B | Exon 14 | c.3629‐3637ins A | 1‐bp insertion (A) | |||
| A3 | Exon 16 | c.5452‐5453ins GA | 2‐bp insertion (GA) | Novel mutation | ||
| A3 | Exon 18 | c.5986‐5987ins A | 1‐bp insertion (A) | Novel mutation |
| Mutation type in 22 HB families | FIX protein domain | F9 Gene location | Nucleotide and amino acid change (HGVS) | Legacy No. | Novel mutation | de novo mutation |
|---|---|---|---|---|---|---|
| Missense mutation (9 families) | ||||||
| Pro‐peptide | Exon 2 | c.127C>T; p. Arg43Trp | −4 Arg(cgg) > Trp(tgg) | |||
| EGF1 | Exon 4 | c.284A>G; p. Asp95Gly | 49 Asp(gat) > Gly(ggt) | |||
| EGF2 | Exon 5 | c.412A>C; p. Asn138His | 92 Asn(aaa) > His(caa) | |||
| Linker | Exon 6 | c.571C>T; p. Arg191Cys | 145 Arg(cgt) > Cys(tgt) | |||
| Act‐peptide | Exon 6 | c.677G>A; p. Arg226Gln | 180 Arg(cga) > Gln(caa) | |||
| Serine protease | Exon 8 | c.881G>A; p. Arg294Gln | 248 Arg(cga) > Gln(caa) | |||
| Serine protease | Exon 8 | c.987C>G; p. Ser329Arg | 283 Ser(agc) > Arg(agg) | |||
| Serine protease | Exon 8 | c.1010C>T; p. Ala337Val | 291 Ala(gct) > Val(gcc) | |||
| Serine protease | Exon 8 | c.1135C>T; p. Arg379Gln | 333 Arg(cga) > Gln(caa) | |||
| Nonsense mutation (5 families) | ||||||
| Gla | Exon 2 | c.181G>T; p. Glu61* | 15 Glu(gag) > Stop(tag) | Novel mutation | ||
| Gla | Exon 2 | c.223C>T; p. Arg75* | 29 Arg(cga) > Stop(tga) | |||
| Serine protease | Exon 8 | c.880C>T; p. Arg294* | 248 Arg(cga) > Stop(tga) | |||
| Serine protease | Exon 8 | c.1150C>T; p. Arg384* | 338 Arg(cga) > Stop(tga) | |||
| Serine protease | Exon 8 | c.1150C>T; p. Arg384* | 338 Arg(cga) > Stop(tga) | |||
| Splice site mutation (1 family) | ||||||
| Intron 2 | c.252+5G>A | Donor splice site mutation | de novo mutation | |||
| 5`UTR point mutation (1 family) | ||||||
| F9 5`UTR | c.−48G>C | −19 G > C | ||||
| Small deletion (6 families) | ||||||
| Pro‐peptide | Exon 2 | c.103del G | 1‐bp deletion (G) | Novel mutation | ||
| Gla | Exon 2 | c.159‐160del AG | 2‐bp deletion (AG) | |||
| Gla | Exon 2 | c.159‐160del AG | 2‐bp deletion (AG) | |||
| Serine protease | Exon 8 | c.1129‐1132del GTT or TTG | 3‐bp deletion (GTT or TTG) | Novel mutation | ||
| Serine protease | Exon 8 | c.1258‐1259del AG | 2‐bp deletion (AG) | Novel mutation | de novo mutation | |
| Serine protease | Exon 8 | c.1318‐1320del A | 1‐bp deletion (A) | Novel mutation |
Abbreviations: FVIII, factor VIII; FIX, factor IX; HA, hemophilia A; HB, hemophilia B.
The F9 mutations in 22 subjects (21 patients and 1 obligate carrier) were as follows: 9 missense mutations, 6 small deletions, 5 nonsense mutations, 1 point mutation in 5’UTR, and 1 splice site mutations (Figure 2B). Thus, the type of F9 mutation equivalent to 74% was point mutation. Fifteen unique types of point mutations and 5 small deletions in the HB were detected, including 5 novel mutations (p. Glu61*, c.103delG, c.1129‐1132delGTTorTTG, c.1258‐1259delAG, c.1318‐1320delA; Table 2). All of the above previously unreported mutations were classified as null mutations.
3.3. Determination of carrier status in 112 FFM based on mutation
Genetic analysis was performed in all 112 FFM (80 HA and 32 HB). The analysis successfully determined the carrier status of all 112 FFM based on whether they showed the relevant mutation in the family. Based on the results of genetic analysis, 74 (66%), including 7 obligate carriers, were carriers while 38 (34%) were non‐carriers. For HA FFM, 53 (66%) were genetically confirmed as carriers while 27 (34%) were non‐carriers. For HB FFM, 21 (66%) were carriers and 11 non‐carriers (34%).
3.4. Assessment of carrier status by blood coagulation tests in gene‐confirmed carriers and non‐carriers
Coagulation factor activity levels were measured in 83 of the 112 FFM (61 HA and 22 HB, Table 3). Eleven (9 HA and 2 HB) pregnant women were excluded from this validation beforehand.
TABLE 3.
Assessment of carrier status using blood coagulation tests in gene‐confirmed carriers and non‐carriers
| Hemophilia A | Carriers | Non‐carriers | P value |
|---|---|---|---|
| Subjects of age at testing | n = 40 | n = 21 | |
| Mean age, years (IQR, range) | 38 (17.8,1–69) | 36 (20.0,13–66) | 0.382 |
| Subjects of ABO blood group | n = 39 | n = 21 |
0.156 χ2 = 2.017 |
| Blood group non‐O | 19 (49%) | 15 (71%) | |
| Blood group O | 20 (51%) | 6 (29%) | |
| Subjects who underwent FVIII:C analysis | n = 40 | n = 21 | |
| Mean FVIII:C, IU/dL (range) | 76 (9–144) | 122 (72–205) | <0.001 |
| <50 IU/dL | 4/40 (10%) | 0/21 (0%) | NA |
| ≥50 IU/dL | 36/40 (90%) | 21/21 (100%) | <0.001 |
| Subjects who underwent VWF:Ag analysis | n = 33 | n = 19 | |
| Mean VWF:Ag, IU/dL (range) | 104 (60–204) | 95 (57–172) | 0.332 |
| Subjects who underwent FVIII/VWF ratio analysis | n = 33 | n = 19 | |
| Mean FVIII/VWF ratio (range) | 0.79 (0.09–1.48) | 1.35 (0.92–1.78) | <0.001 |
| <0.9 | 22/33 (67%) | 0/19 (0%) | NA |
| 0.9–1.5 | 11/33 (33%) | 13/19 (68%) | 0.063 |
| ≥1.5 | 0/33 (0%) | 6/19 (32%) | NA |
| Hemophilia B | |||
| Subjects who underwent FIX:C analysis | n = 12 | n = 10 | |
| Mean FIX:C, IU/dL (range) | 51 (11–67) | 95 (67–173) | <0.001 |
| <60 IU/dL | 6/12 (50%) | 0/10 (0%) | NA |
| ≥60 IU/dL | 6/12 (50%) | 10/10 (100%) | <0.001 |
| <65 IU/dL | 11/12 (92%) | 0/10 (0%) | NA |
| ≥65 IU/dL | 1/12 (8%) | 10/10 (100%) | 0.364 |
Low FVIII:C levels were defined as 50 IU/dL according to Labarque et al. 3 The cut‐off levels of FVIII:C/VWF:Ag ratio were determined as 0.9 and 1.5 according to ROC analysis (AUC = 0.992; see Figure S4). The FIX:C cut‐off levels were defined as 60 IU/dL by Plug et al. 5 and as 65 IU/dL by ROC analysis (AUC = 0.992, Figure S5).
Abbreviations: AUC, area under the curve; FVIII:C, factor VIII activity; FVIII/VWF ratio; FVIII:C to VWF:Ag ratio, FIX:C, factor IX activity, NA; not applicable; ROC, receiver operating characteristic; VWF:Ag, von Willebrand factor antigen.
Carriers and non‐carriers were diagnosed by gene analysis.
3.4.1. Differences in FVIII:C levels between HA carriers and non‐carriers
The results of FVIII:C analysis were compared between 40 HA gene‐confirmed carriers versus 21 non‐carriers. Low FVIII:C levels were defined as 50 IU/dL or less, according to Labarque et al., 3 because Receiver operating characteristic (ROC) analysis could not determine the cut‐off value of FVIII:C (Figure S1 in supporting information). Although the mean value of FVIII:C in 40 HA carriers (76 IU/dL: range 9–144) was lower than in non‐carriers (122 IU/dL: range 72–205; P < 0.001), FVIII:C in the majority of gene‐confirmed carriers (36/40, 90%) was higher than 50 IU/dL. Because FVIII levels are known to increase with age, we evaluated the impact of age at testing. The HA gene‐confirmed carriers (mean age 38 years) were not significantly older than those of non‐carriers (mean age 36 years; P = 0.382; Table 3). In addition, the mean FVIII:C value was 65.9 IU/dL (higher than 50 IU/dL) in 14 symptomatic HA women (Table 1).
3.4.2. Differences in VWF:Ag levels between HA carriers and non‐carriers
The mean VWF:Ag in 33 HA carriers and 19 non‐carriers were 104 IU/dL (range 60–204) and 95 IU/dL (range 57–172), respectively (Table 3). Healthy blood group O individuals are known to have lower VWF:Ag and FVIII levels compared to blood group non‐O individuals. 21 , 22 In our study, the proportions of blood group O in HA carriers (n = 39) and non‐carriers (n = 21) were 51% and 29%, respectively (Table 3).
Furthermore, blood group O carriers had significantly lower VWF:Ag (mean 88 IU/dL, n = 17) than blood group non‐O carriers (mean 120 IU/dL, n = 15, p < 0.01, Figure S2 in supporting information). However, the mean FVIII:C and FVIII/VWF ratio were not significantly different between blood group O (mean 68 IU/dL, 0.87) and blood group non‐O carriers (mean 83 IU/dL, 0.70). A similar trend was seen in HA non‐carriers; the mean FVIII:C and FVIII/VWF ratio were not significantly different between blood groups O and non‐O (Figure S3 in supporting information). At this stage, we have no explanation for the higher mean VWF:Ag in HA carriers than non‐carriers.
3.4.3. Differences in FVIII:C/VWF:Ag ratio between HA carriers and non‐carriers
The mean of FVIII:C/VWF:Ag (FVIII/VWF) ratio was lower in the carriers than non‐carriers (0.79 versus 1.35; P < 0.001; Table 3). In addition, the mean ratio in 14 symptomatic women was 0.81 (Table 1). ROC analysis successfully confirmed the cutoff value of FVIII/VWF ratio (area under the curve (AUC)= 0.992, Figure S4 in supporting information). The lower cut‐off value of FVIII/VWF ratio was 0.9 as determined by the ROC analysis, with sensitivity and specificity for HA carrier of 67% and 100%, respectively. On the other hand, the higher cut‐off value was 1.5, with sensitivity and specificity for non‐carrier of 100% and 32%, respectively. The proportion of subjects with the FVIII/VWF ratio between 0.9 and 1.5 was not significantly different between carriers (11/33, 33%) and non‐carriers (13/19, 68%, P = 0.063). These results suggest that determining the carrier status is difficult in subjects with FVIII/VWF ratio ranging from 0.9 to 1.5, while HA non‐carriers can be accurately predicted in subjects with FVIII/VWF ratio of >1.5. Considered together, the above results suggest that FVIII:C alone is not reliable for the diagnosis of carrier status while the FVIII/VWF ratio is somewhat useful though with limited applicability.
3.4.4. Differences in FIX:C levels between HB carriers and non‐carriers
The mean of FIX:C was significantly lower in carriers (51 U/dl, range, 11–67, n = 12 HB) than non‐carriers (95 IU/dL; range, 67–173; n = 10; P < 0.001; Table 3). The FIX:C cut‐off levels were defined as 60 IU/dL by Plug et al. 5 , 14 and 65 IU/dL by the ROC analysis conducted in the present study (Figure S5 in supporting information). The mean FIX:C of six symptomatic gene‐confirmed carrier women was 44.6 IU/dL (Table 1). Using the above cut‐off value established by Plug et al., 5 6/12 (50%) of our subjects were considered carriers (with FIX:C > 60 IU/dL). In comparison, using the cut‐off value determined by our ROC analysis (65 IU/dL), 11/12 (92%) were labelled HB carriers (with FIX:C < 65 IU/dL), whereas 10/10 (100%) were non‐carriers (with FIX:C ≥ 65 IU/dL). Thus, the cut‐off level of 65 IU/dL based on ROC analysis is useful in predicting the carrier status. Interestingly, the sensitivity for the diagnosis of carrier state varied significantly between the cut‐off value at 60 IU/dL reported by Plug et al. 5 and that of 65 IU/dL determined in our study, suggesting that the cut‐off value for the diagnosis varies according to the sample population.
3.5. Verification of sporadic hemophilia and de novo mutation
Based on the pedigree information, 37 hemophiliac sons (27 HA and 10 HB) from 72 families were diagnosed as sporadic hemophilia (Table 4). In genetic analysis, 24 HA mothers (89%) had the patient relevant mutation in the heterozygote form while 3 HA mothers (11%) did not have the relevant mutation. Therefore, all 27 represent sporadic HA but in only 11% was the sporadic mutation in the affected male while in the other 24 cases of sporadic HA, the sporadic mutation was in the child's mother and potentially in the child's grandmother. In genetic analysis of 10 HB cases, 8 HB mothers (80%) had the patient relevant mutation while 2 HB mothers (20%) did not have the relevant mutation. Therefore, all 10 represent sporadic HB but in only 20% was the sporadic mutation in the affected male while in the other 8 cases of sporadic HB, the sporadic mutation was in the child's mother and potentially in the child's grandmother.
TABLE 4.
Verification of sporadic hemophilia and de novo mutations
| Status of mothers | de novo mutation in children | ||
|---|---|---|---|
| Carriers | Non‐carriers | ||
| Hemophilia A |
24/27 (89) |
3/27 (11) |
Inversion (intron 22) p. Arg2182Cys (exon 23) p. Trp791* (exon 14) |
| Hemophilia B |
8/10 (80) |
2/10 (20) |
c.252+5G>A (intron 2) c.1258‐1259delAG (exon8) |
| Total |
32/37 (86) |
5/37 (14) |
|
Data are number of affected subjects/total (%).
These results suggest that 5 children (3 HA and 2 HB) had de novo mutations (F8 inversion, F8 p. Arg2182Cys, F8 p. Trp791*, F9 c.252+5G>A, F9 c.1258‐1259delAG). In summary, 32 mothers of 37 sporadic cases showed relevant mutations, and consequently, the incidence of de novo mutations in male infants in the present study was 14%, suggesting a low frequency for de novo mutations in male hemophiliacs.
4. DISCUSSION
The present study provided comprehensive genetic information on Japanese hemophilia female carriers. First, we successfully identified the F8 and F9 mutations in all 72 hemophilic families. All the identified mutations in HA patients, except for missense mutations, were null mutations (74%). In addition, two missense mutations (p. Tyr414His and p. Arg2182Cys) resulted in severe HA phenotype. Thus, the genetic tests showed a reasonable proportion of severe HA (83%) in our study. In HB patients, although F9 large mutations were not detected, 73% of the F9 mutations were point mutations. The distribution of point mutations was almost similar to those reported in other countries. 14 , 23 Since the early 1980s, when Sanger capillary sequencing made it possible to establish carrier status, DNA direct sequencing has evolved from haplotyping to mutation analysis, 24 , 25 offering certainty about the carrier status. 26 , 27 , 28 Despite the clear advantage of the next generation sequencing in various settings, including exploration of deep intron mutation, Sanger sequencing remains a better option for genetic testing and diagnosis of hemophiliac carriers, because there still can be a variety of unknown mutations of F8 and F9 target genes. In this study, carrier status was determined in 112 FFM based on the presence of patient relevant mutation in each family in the heterozygous form; 53/80 HA FFM (66%) and 21/32 HB FFM (66%) were diagnosed as carriers. All FFM with hemophilic relatives suffering bleeding tendencies were found to be carriers (Table 1). The numbers and proportions of subjects were limited and biased in this study with male patients from 44 mothers among the 112 subjects. In addition, there was no significant association between mutation patterns and genetic transmission.
FVIII levels show considerable variability, and normal levels do not always represent non‐carrier status. Labarque et al. 3 measured the FVIII:C and FVIII/VWF ratio in 277 HA carriers and found normal FVIII:C (>0.5 U/ml) in 77% of the carriers. In our study, the FVIII:C level was ≥50 IU/dL in 36 of 40 (90%) HA carriers. Furthermore, the mean FVIII:C of 14 symptomatic women (65.9 IU/dL) was higher than 50 IU/dL. Although we examined the impact of age and blood group on FVIII:C level, we can offer no explanation for the relatively high levels of FVIII:C encountered in our subjects.
Four HA gene‐confirmed carriers with low FVIII:C had large deletions (exons 12–22), 1‐bp deletion (c.6102‐6103delG), acceptor splice site mutation (c.1010‐1G>A), and inversion; namely, four strong mutations were so‐called null mutation. On the other hand, six HB gene‐confirmed carriers with FIX:C below 60 IU/dl were p. Arg43Trp, p. Arg75*, p. Arg191Cys, p. Arg226Gln, p. Arg294*, and p. Arg294Gln. The two missense mutations, p. Arg191Cys and p. Arg226Gln, generated the site of activation peptide known to play a critical role in cleavage by factor XIa or factor VIIa/tissue factor. These findings support the notion that carrier women with null or missense mutation that correspond to special functions often have significantly lower coagulation factor activity.
Our study showed that 22 of 33 (67%) carriers had low FVIII/VWF ratio (<0.9), which is the lower cut‐off value for the diagnosis of carriers selected by both our ROC analysis and Labarque et al. 3 In comparison, none of the 33 (0%) carriers in our study and only 3 of the 33 (8.3%) carriers in the study of Labarque et al. 3 had high FVIII/VWF ratio (defined as >1.5 by ROC analysis and >1.2, respectively). These results suggest that a high FVIII/VWF ratio (>1.5) can exclude HA carriers while a low ratio (<0.9) can exclude non‐carriers. However, mid‐range ratio (0.9–1.5), which was observed in 24/52 (46%) of HA FFM, cannot predict carrier status well. Moreover, the cut‐off value varies widely according to the study population and test reagents. Taken together, the FVIII/VWF ratio is somewhat useful but with limited applicability. With regard to the FIX:C value, the sensitivity using the cut‐off value of 65 IU/dL, as identified by ROC curve, was much better than that of the commonly used level of 60 IU/dL. We conclude that FIX level is satisfactory for the prediction of the carrier state, but is not a perfect tool due to the difficulty in determining the cut‐off value.
Although carrier women are generally considered asymptomatic, some reported abnormal bleeding, such as epistaxis, prolonged bleeding after dental procedures, purpura/ecchymoses, or post‐/perioperative bleeding. The reported rate of these abnormal bleeding patterns is about 30% among carrier women. 5 , 6 , 7 Postpartum hemorrhage is a serious problem, even in non‐hemophilic women. If the pregnant woman and the health‐care provider are aware of the hemophilia carrier status, safe childbirth and proper management of postpartum bleeding, including management of hemostasis in Cesarean section, are possible. 29 Thus, the significance of gene analysis for hemophilia carrier diagnosis includes accurate assessment of the carrier status.
Miesbach et al. 6 compared bleeding symptoms and phenotypes in 46 HA carriers by F8 mutations, and reported strong bleeding tendency in many carriers with inversion and/or deletion. They concluded that the severity of bleeding in HA carriers correlated with the phenotype of the male hemophilic relative and the underlying F8 mutation. In the present study, the carrier status was confirmed in all FFM with history of bleeding symptoms by genetic analysis. Our results also suggest that null mutation correlates with lower coagulation factor activity. However, we could not analyze the relationship between severity of bleeding in the carriers and gene mutation.
Sporadic hemophiliacs are defined as patients with negative family history, and they form about one third of all cases of hemophilia. 7 Kasper and Lin 30 reported that 88% of the mothers of the sporadic cases had the relevant mutation in their leukocytes, but only 19% of the maternal grandmothers carried the same mutation. Thus, the rate of de novo mutation was 12% among male sporadic hemophiliacs in the above study. In other studies, the reported rate of de novo mutation was 18% 31 and 28% 32 in children with sporadic hemophilia. In the present study, no hemophilia‐relevant mutation was found in 5 of 37 mothers of children with sporadic hemophilia. This finding indicates that sporadic hemophilia was due to de novo mutation in 14% (HA: 11%, HB: 20%) of male infants. Actually, in all families with hemophilia, the latter was associated with sporadic mutation at some point, but that could have been many generations ago. The above 14% represents the proportion of male hemophiliacs with de novo mutation and no family history of hemophilia. Therefore, the chance of de novo mutation in a boy with sporadic hemophilia is expected to be lower than that of his mother. Even in the absence of family history of hemophilia, the mother of a child with sporadic hemophilia might be found to have relevant high‐frequency mutation by genetic analysis. None of the maternal grandmothers involved in this study had inversion, although inversion was detected in two mothers of sporadic HA males in two unrelated families. It is possible that the inversion was generated during the process of gametogenesis in either of the maternal grandparents, based on the study of Becker et al., 32 who showed a higher frequency of inversion in males than females.
Several limitations of this study should be noted. First, non‐carriers were diagnosed based on the absence of F8 and F9 mutations in the heterozygous form. However, the affected allele might not have been detected due to technical limitations, although all analyses were validated at least twice. Second, this study mainly included patients with severe HA (83%); as such, the results cannot be extrapolated to mild and moderate HA. Third, because pedigree information plays a critical role in assessing the carrier status for hemophilia, an accurate family history is essential for carrier diagnosis. Although the present study carefully assessed pedigree information from medical interviews, a full access to the official records was not available, resulting in a higher incidence of pedigree‐based sporadic hemophilia than in previous studies. Also, details of bleeding symptoms were obtained from 63 of the 112 (56%) by self‐reports rather than by direct medical interview. Furthermore, we did not employ a bleeding evaluation tool, such as pictorial blood assessment chart for menorrhagia. 33 Fourth, the number of FFM who underwent measurement of coagulation factor activity was limited and the measurement reagents used in the assays were sometimes different.
In conclusion, the present study is the first to present comprehensive genetic information on Japanese hemophilia female carriers and demonstrated the usefulness of genetic analysis in carrier diagnosis by comparing gene testing with pedigree information and measurement of coagulation factor activity levels. Interestingly, our results showed a very low rate of de novo mutations in patients with sporadic hemophilia. Taken together, the significance of gene analysis for carrier diagnosis in hemophilia includes accurate assessment of the carrier status.
CONFLICTS OF INTEREST
K. Shinozawa was an endowed assistant professor funded by Baxter/Baxalta/Shire and CSL Behring (until March 2020). K. Amano has received honoraria from Chugai Pharmaceutical, Sanofi Genzyme, Bayer, Takeda Pharmaceutical, Novo Nordisk Pharma, CSL Behring, KM Biologics, and Pfizer. T. Hagiwara has received research funding from Sanofi Genzyme, and honoraria from Chugai Pharmaceutical, Bayer, Takeda Pharmaceutical, Novo Nordisk Pharma, CSL Behring, and Janssen Pharma. M. Bingo has received honoraria from Chugai Pharmaceutical, Sanofi Genzyme, Bayer, Takeda Pharmaceutical, Novo Nordisk Pharma, and CSL Behring. Y. Chikasawa has received honoraria from Chugai Pharmaceutical, Sanofi Genzyme, Bayer, Takeda Pharmaceutical, Novo Nordisk Pharma, and CSL Behring. H. Inaba declares no conflict of interest. E. Kinai has received honoraria and research grants from Chugai Pharmaceutical and also honoraria from Sanofi Genzyme, Bayer, Takeda Pharmaceutical, Novo Nordisk Pharma, and CSL Behring. K. Fukutake has received grants and personal fees from Takeda Co. Ltd. (Baxalta/Shire), Bayer, Pfizer, CSL Behring, Novo Nordisk, Sanofi S.A. (Biogen/Bioverativ), KM Biologics (Kaketsuken), and Chugai Pharmaceutical Co. Ltd, and grants from Japan Blood Products Organization, and CMIC Holdings Co., Ltd, and personal fees from SRL Inc., LSI Medience, Roche Diagnostics, Siemens, Sekisui Medical, Fujirebio Inc., Torii Pharmaceuticals, Octapharma, Sysmex, MSD outside the submitted work.
AUTHOR CONTRIBUTIONS
K. Shinozawa designed the research, performed gene analysis, analyzed the results, and wrote the manuscript. K. Amano designed the research, performed clinical work, and provided advice. T. Hagiwara, M. Bingo, and Y. Chikasawa performed clinical work. H. Inaba analyzed data and provided advice. E. Kinai performed clinical work, interpreted the data, and edited the manuscript. K. Fukutake designed the research, performed clinical work, and supervised the study.
Supporting information
Fig S1‐S5
ACKNOWLEDGMENTS
The authors thank the staff of all the facilities and the collaborators: Takashi Kaneko (Tokyo Metropolitan Children’s Medical Center, Tokyo), Michio Sakai (Hospital of the University of Occupational and Environmental Health, Fukuoka), Kyoko Suzuki (Juntendo University Urayasu Hospital, Chiba), Yoko Mizoguchi and Masao Kobayashi (Hiroshima University Hospital, Hiroshima), Akira Ishiguro (National Center for Child Health and Development, Tokyo), Setsuko Kitaoka (National Hospital Organization Sendai Medical Center, Fukushima), Yuji Kikuchi (Mitsui Memorial Hospital, Tokyo), Isao Komori (Matsudo City General Hospital, Chiba), Taemi Ogura (Shizuoka Children’s Hospital, Shizuoka), Yasushi Nagai (Nagai Mothers Hospital, Saitama), Tazuko Tokugawa and Akihiro Sawada (The Hospital of Hyogo College of Medicine, Hyogo), Yukitosi Simizu (Yamagata City Hospital Saiseikan, Yamagata), Daisuke Hasegawa (St. Luke’s International Hospital, Tokyo), Yuri Okimoto and Harumi Kakuda (Chiba Children’s Hospital, Chiba), Yuni Yamaki (University of Tsukuba Hospital, Ibaraki), Tomoki Kosho (Shinshu University Hospital, Nagano), Takashi Suzuki (Ogikubo Hospital, Tokyo), Asashi Tanaka (Tokyo Medical University Hachioji Medical Center, Tokyo), Kenji Shirai (Seirei Mikatahara General Hospital, Shizuoka), Katsutsugu Umeda (Kyoto University Hospital, Kyoto), Mayuko Iijima (Gunma University Hospital, Gunma), Atsushi Shibuya (Shibuya Children’s Clinic, Saitama), Yasuyuki Hirano (Hirano Clinic, Chiba), Hiroyuki Kawaguchi (National Defense Medical College Hospital, Saitama), Takahito Inoue (Fukuoka University Chikushi Hospital, Fukuoka), Yoshinobu Seki (Uonuma‐Kikan Hospital, Niigata), and Yoshiyuki Kosaka (Hyogo Prefectural Kobe Children's Hospital, Hyogo).
Funding information
This study was supported by funds from the endowed course (Department of Molecular Genetics of Coagulation Disorders, Tokyo Medical University) for Baxter/Baxalta/Shire and CSL Behring, the Japanese Society of Laboratory Medicine Fund for the Promotion of Scientific Research, and the Japan Agency for Medical Research and Development (AMED) (grant #JP20fk0410017).
Manuscript handled by Katsue Suzuki‐Inoue
Final decision:Katsue Suzuki‐Inoue, 8 March 2021
REFERENCES
- 1. McVey JH, Rallapalli PM, Kemball‐Cook G, et al. The European Association for Haemophilia and Allied Disorders (EAHAD) coagulation factor variant databases: important resources for haemostasis clinicians and researchers. Haemophilia. 2020;26:306‐313. [DOI] [PubMed] [Google Scholar]
- 2. Graham JB, Rizza CR, Chediak J, et al. Carrier detection in hemophilia A: a cooperative international study. I. The carrier phenotype. Blood. 1986;67:1554‐1559. [PubMed] [Google Scholar]
- 3. Labarque V, Perinparajah V, Bouskill V, et al. Utility of factor VIII and factor VIII to von Willebrand factor ratio in identifying 277 unselected carriers of hemophilia A. Am J Hematol. 2017;92:E94‐E96. [DOI] [PubMed] [Google Scholar]
- 4. Rizza CR, Rhymes IL, Austen DE, Kernoff PB, Aroni SA. Detection of carriers of haemophilia: a ‘blind’ study. Br J Haematol. 1975;30:447‐456. [DOI] [PubMed] [Google Scholar]
- 5. Plug I, Mauser‐Bunschoten EP, Bröcker‐Vriends AHJT, et al. Bleeding in carriers of hemophilia. Blood. 2006;108:52‐56. [DOI] [PubMed] [Google Scholar]
- 6. Miesbach W, Alesci S, Geisen C, Oldenburg J. Association between phenotype and genotype in carriers of haemophilia A. Haemophilia. 2011;17:246‐251. [DOI] [PubMed] [Google Scholar]
- 7. Hermans C, Kulkarni R. Women with bleeding disorders. Haemophilia. 2018;24(Suppl 6):29‐36. [DOI] [PubMed] [Google Scholar]
- 8. Lyon MF. Sex chromatin and gene action in the mammalian X‐chromosome. Am J Hum Genet. 1962;14:135‐148. [PMC free article] [PubMed] [Google Scholar]
- 9. Lyon MF. Lyonisation of the X‐chromosome. Lancet. 1963;23(2):1120‐1121. [DOI] [PubMed] [Google Scholar]
- 10. Valleix S, Vinciguerra C, Lavergne JM, Leuer M, Delpech M, Negrier C. Skewed X‐chromosome inactivation in monochorionic diamniotic twin sisters results in severe and mild hemophilia A. Blood. 2002;100:3034‐3036. [DOI] [PubMed] [Google Scholar]
- 11. Okumura K, Fujimori Y, Takagi A, et al. Skewed X chromosome inactivation in fraternal female twins results in moderately severe and mild haemophilia B. Haemophilia. 2008;14:1088‐1093. [DOI] [PubMed] [Google Scholar]
- 12. Zimmerman TS, Ratnoff OD, Littell AS. Detection of carriers of classic hemophilia using an immunologic assay for antihemophilic factor. Clin Invest. 1971;50:255‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gouw SC, van den Berg HM, Oldenburg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta‐analysis. Blood. 2012;119:2922‐2934. [DOI] [PubMed] [Google Scholar]
- 14. Goodeve AC. Hemophilia B: FIX: molecular pathogenesis and mutation analysis. J Thromb Hemost. 2015;13:1184‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spena S, Garagiola I, Cannavò A, et al. Prediction of factor VIII inhibitor development in the SIPPET cohort by mutational analysis and factor VIII antigen measurement. J Thromb Hemost. 2018;16:778‐790. [DOI] [PubMed] [Google Scholar]
- 16. Williams IJ, Abuzenadah A, Winship PR, et al. Precise carrier diagnosis in families with haemophilia A: use of conformation sensitive gel electrophoresis for mutation screening and polymorphism analysis. Thromb Hemost. 1998;79:723‐726. [PubMed] [Google Scholar]
- 17. Sasaki A, Nagaizumi K, Inaba H, Suzuki T, Arai M, Fukutake K. Eighteen point mutations in Japanese patients with hemophilia B. Jpn J Thromb Hemost. 2004;15:107‐113. [Google Scholar]
- 18. Liu Q, Sommer SS. Subcycling‐PCR for multiplex long‐distance amplification of regions with high and low GC content: application to the inversion hotspot in the factor VIII gene. Biotechniques. 1998;25:1022‐1028. [DOI] [PubMed] [Google Scholar]
- 19. den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat. 2016;37:564‐569. [DOI] [PubMed] [Google Scholar]
- 20. Gouw SC, van der Bom JG, van den Berg HM, Zewald RA, Ploos van Amstel JK, Mauser‐Bunschoten EP. Influence of the type of F8 gene mutation on inhibitor development in a single centre cohort of severe haemophilia A patients. Haemophilia. 2011;17:275‐281. [DOI] [PubMed] [Google Scholar]
- 21. Gill JC, Endres‐Brooks J, Bauer PJ, Marks WJ Jr, Montgomery RR. The effect of ABO blood group on diagnosis of won Willebrand disease. Blood. 1987;69:1691‐1695. [PubMed] [Google Scholar]
- 22. Sousa NC, Anicchino‐Bizzacchi JM, Locatelli MF, Castro V, Barjas‐Castro ML. The relationship between ABO groups and subgroups, facto VIII and von Willebrand factor. Haematologica. 2007;92:236‐239. [DOI] [PubMed] [Google Scholar]
- 23. Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021;106:123‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keeney S, Mitchell M, Goodeve A. UK haemophilia center doctors’ organization haemophilia genetics laboratory network. The molecular analysis of haemophilia A: a guideline from the UK haemophilia centre doctors’ organization haemophilia genetics laboratory network. Haemophilia. 2005;11:387‐397. [DOI] [PubMed] [Google Scholar]
- 25. Mitchell M, Keeney S, Goodeve A. UK haemophilia centre doctors’ organization haemophilia genetics laboratory network. The molecular analysis of haemophilia B: a guideline from the UK haemophilia centre doctors’ organization haemophilia genetics laboratory network. Haemophilia. 2005;11:398‐404. [DOI] [PubMed] [Google Scholar]
- 26. Camerino G, Grzeschik KH, Jaye M, et al. Regional localization on the human X chromosome and polymorphism of the coagulation factor IX gene (hemophilia B locus). Proc Natl Acad Sci USA. 1984;81:498‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peake IR, Lillicrap DP, Boulyjenkov V, et al. Haemophilia: strategies for carrier detection and prenatal diagnosis. Bull World Health Organ. 1993;71:429‐458. [PMC free article] [PubMed] [Google Scholar]
- 28. Peake IR, Lillicrap DP, Boulyjenkov V, et al. Report of a joint WHO/WFH meeting on the control of haemophilia: carrier detection and prenatal diagnosis. Blood Coagul Fibrinolysis. 1993;4:313‐344. [DOI] [PubMed] [Google Scholar]
- 29. Kulkarni R, Presley RJ, Lusher JM, et al. Complications of haemophilia in babies (first two years of life): a report from the centers for disease control and prevention universal data collection system. Haemophilia. 2017;23:207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kasper CK, Lin JC. Prevalence of sporadic and familial haemophilia. Haemophilia. 2007;13:90‐92. [DOI] [PubMed] [Google Scholar]
- 31. Ljung RC, Sjörin E. Origin of mutation in sporadic cases of haemophilia A. Br J Hematol. 1999;106:870‐874. [DOI] [PubMed] [Google Scholar]
- 32. Becker J, Schwaab R, Möller‐Taube A, et al. Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type‐dependent sex ratio of mutation frequencies. Am J Hum Genet. 1996;58:657‐670. [PMC free article] [PubMed] [Google Scholar]
- 33. Higham JM, O'Brien PM, Shaw RW. Assessment of menstrual blood loss using a pictorial chart. Br J Obstet Gynaecol. 1990;97:734‐739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5