

Abstract
The identification of a green, versatile, user‐friendly, and efficient methodology is necessary to facilitate the use of Heck‐Cassar‐Sonogashira (HCS) cross‐coupling reaction in drug discovery and industrial production in the pharmaceutical segment. The Heck‐Cassar and Sonogashira protocols, using N‐hydroxyethylpyrrolidone (HEP)/water/N,N,N′,N′‐tetramethyl guanidine (TMG) as green solvent/base mixture and sulfonated phosphine ligands, allowed to recycle the catalyst, always guaranteeing high yields and fast conversion under mild conditions, with aryl iodides, bromides, and triflates. No catalyst leakage or metal contamination of the final product were observed during the HCS recycling. To our knowledge, a turnover number (TON) up to 2375, a turnover frequency (TOF) of 158 h−1, and a process mass intensity (PMI) around 7 that decreased around 3 after solvent, base, and palladium recovery, represent one of the best results to date using a sustainable protocol. The Heck‐Cassar protocol using sSPhos was successfully applied to the telescoped synthesis of Erlotinib (TON: 1380; TOF: 46 h−1).
Keywords: cross-coupling, green chemistry, Heck-Cassar-Sonogashira, homogeneous catalysis, TPPTS
Recycle and reuse: Heck‐Cassar‐Sonogashira cross‐coupling using the green N‐hydroxyethylpyrrolidone (HEP)/H2O/N,N,N′,N′‐tetramethyl guanidine (TMG) blend, sulfonate phosphines, and aryl iodides, bromides, and triflates affords the corresponding products in high yield free from metal contamination. The catalyst/HEP/H2O solution can be recycled, increasing turnover number and frequency up to 2375 and 158 h−1, respectively, with a low process mass intensity close to 3 after solvent, base, and palladium recovery. Additionally, Erlotinib synthesis is described.

Introduction
Catalysis has been listed by the fathers of green chemistry as a fundamental tool to shift the paradigm of chemical processes from classical to sustainable methodologies. [1] Green metrics parameters can rapidly reveal that the use of stoichiometric technologies and large volume of solvents are the primary source of concern regarding waste output. Switching to catalysis for carbon‐carbon bond formation often involves, as the only solution, the use of transition metals. [2] In this context, palladium‐catalyzed reactions have a prominent role. [3] However, due to the high price and low availability of palladium catalysts, efforts to reduce their loading and to perform their recycling are mandatory, in particular aiming to transfer bench reaction protocols to technologically advanced industrial processes. The development path has clearly to respect also other principles of greenness and sustainability such as the selection of green solvents and reagents. Since a great challenge is the transition from a fossil‐resources‐based economy to a renewable‐biomass‐based one, biogenic solvents represent an attractive alternative. Among the palladium‐catalyzed cross‐coupling methodologies, the Heck‐Cassar‐Sonogashira coupling [4] (HCS; Scheme 1) is one of the most useful reactions in the pharmaceutical segment, [5] and making it greener would offer great productive advantages.
Scheme 1.
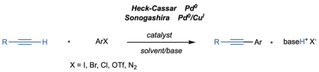
General scheme of the HCS coupling.
The reaction was independently reported in 1975 by Sonogashira et al. [6] as Pd0/CuI‐catalyzed cross‐coupling and a few months before by Dieck and Heck [7] and Cassar [8] as an extension of the classical Heck reaction to alkynes catalyzed by Pd0 complexes. [9] Several studies have investigated the influence of leaving groups, palladium ligands, co‐catalysts, solvents, and bases. [4] The wide applicability of palladium cross‐coupling to complex polyfunctionalized structures has recommended its use in many industrial synthetic protocols, in particular in the multistep synthesis of active pharmaceutical ingredients (API).
Since solvents and water represent 80–90 % of waste in the pharmaceutical industry,[ 1b , 10 ] the identification of green and biogenic alternatives is the prerequisite for the development of efficient, industrially sustainable methodologies in organometallic catalysis. [11] However, the solvent effect must be carefully studied since it can affect catalyst stability and any step of the reaction mechanism. [12] The reaction protocol must be user‐friendly to favor the introduction of versatile organometallic catalysis starting from the molecular design. In particular, stable PdII precatalysts and ligands should always guarantee high yields and high chemoselectivity, necessary to design efficient synthetic strategies avoiding the use of protective groups. The reaction must also be efficient under mild conditions in order to decrease energy consumption, to guarantee chemoselectivity and to be compatible with complex and sensitive chemical architectures. In addition, the overall process should be cost competitive. Concerning the palladium fate, there must be no leakage of the catalyst during the process to avoid product contamination, and the metal must be fully recovered also for environmental reasons. Finally, the reaction protocol must guarantee a rapid, easy, and efficient scale up.
A statistical analysis of the literature data showed that N,N‐dimethylformamide (DMF) is the solvent of choice for HCS coupling. [13] However, this versatile solvent that is widely used in the pharmaceutical industry is toxic [14] and a source of concern since it generates dimethylamine, favoring the formation of highly genotoxic nitrosamines. [15] Other very popular solvents for HCS coupling are N‐methyl pyrrolidone (NMP), dioxane, tetrahydrofuran (THF), dimethylsulfoxide (DMSO), dimethoxyethane (DME), and neat bases. [4] Unfortunately, these solvents share with DMF several drawbacks in terms of environmental health and safety (EHS). [16] In a recent paper, organic chemists coming from the major pharmaceutical industries voted “the substitution of dipolar organic solvents with green alternatives” as a top priority. [17] Several reported HCS protocols required a small amount of catalyst, and Plenio and co‐workers achieved the outstanding turnover number (TON) and turnover frequency (TOF) of >19600 and up to 9900 h−1, respectively, using the Sonogashira protocol in diisopropyl amine.[ 4 , 18 ] Concerning HCS cross‐coupling reactions, the use of greener alternatives like water, ionic liquids, dimethylisosorbide, γ‐valerolactone, deep eutectic solvents (DES), and CyreneTM has been reported. [19] We recently contributed with our preliminary results on the use of several green solvents for the HCS coupling, namely, N‐octylpyrrolidone (NOP), N‐benzylpyrrolidone (NBnP), N‐cyclohexylpyrrolidone (NCP), N‐hydroxyethylpyrrolidone (HEP), anisole, and tert‐butyl acetate (tBuOAc). [20] However, with the exception of Lipshutz's micellar approach in water [TON 1740; TOF 217 h−1; process mass intensity (PMI)≈15], [21] the amount of catalyst used in green alternative solvents, reaction conditions, yields, and PMI are not in line with the design of versatile, efficient, green, sustainable, and low‐cost reactions.[ 4 , 18 , 22 ] Last but not least, the metal must be readily separated/removed from the product, especially for pharmaceutical applications, since the International Conference of Harmonization Guidelines Q3D (ICH Q3D) set very low limits for elemental impurities in medicines. [23] Consequently, there is a need for a user‐friendly, sustainable, efficient, and flexible protocol for the HCS cross‐coupling that could be easily used by chemists and easily optimized to design an industrial process. In addition, the identification of recycling conditions for the palladium catalysts remains a key goal to decrease costs while increasing reaction greenness and industrial potential. [24]
The target of this study was the identification of a flexible and sustainable procedure that can be applied in parallel synthesis for drug discovery and that allows to easily optimize the reaction conditions in order to achieve high yields, high TON/TOF values and competitive PMI recalculated considering solvent, base, and palladium recovery.
Results and Discussion
We have recently described an efficient Sonogashira protocol based on the use of 2 mol % of Pd(PPh3)2Cl2 as pre‐catalyst, 1 mol % of CuI as co‐catalyst, and N,N,N′,N′‐tetramethyl guanidine (TMG) [25] as base in several green and biogenic solvents. [20] Among them, HEP in combination with TMG showed the best performance in terms of reaction time and yield. HEP is a biogenic solvent [26] that is already available in large quantities, being an intermediate for the synthesis of N‐vinyl‐pyrrolidone. In addition to its excellent dissolution capacity, HEP has a very high affinity for water that allows a smooth product extraction during work up, using standard organic solvents. From an EHS point of view, this solvent has interesting properties, displaying a flash point close to 212 °C and a rat oral LD50 >14400 mg kg−1. [27] Moreover, HEP is not genotoxic, not reprotoxic, and does not raise any environmental concern. The main concern on the use of DMF is related to workers exposure since it is reprotoxic, has a rat oral LD50 of 3010 mg kg−1, and can lead to damages to various organs in humans, among which the liver is the primary target.[ 14 , 28 ] Since solvent metabolic fate is a critical parameter to define their EHS impact and greenness, we decided to explore this aspect that was not reported in the literature for HEP.
In particular, our hypothesis was that the low LD50 could be related to a different metabolic fate with respect to the parent NMP. Therefore, we explored the metabolism of HEP in comparison to NMP (LD50 4100 mg kg−1). [29] Both in rat and human liver microsomes HEP turned out to be more stable than NMP (Figure 1a). Under these in‐vitro experimental conditions, NMP was converted by hydroxylation to 5‐hydroxy‐N‐methyl‐2‐pyrrolidone (5‐HNMP) and, to a minor extent, to 2‐pyrrolidone (2‐P) via N‐demethylation (Figure 1b). In fact, 5‐HNMP is a well‐known biomarker of environmental exposure to NMP [30] and it is a major in‐vivo urinary metabolite in rats [31] and humans. [32] Moreover, 2‐P has been reported as a minor in‐vivo metabolite both in rats and humans.[ 30 , 33 ]
Figure 1.
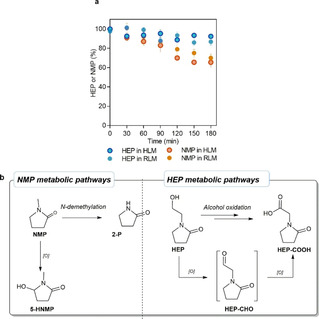
In‐vitro metabolism of NMP and HEP in rat (RLM) and human (HLM) liver microsomes. (a) Comparison of metabolic stability under the same experimental conditions. Error bars correspond to one standard deviation. (b) Proposed metabolic pathways.
HEP revealed a metabolic pathway different from NMP, since it was converted into the corresponding acid (HEP‐COOH) by oxidation of the hydroxy group on the N‐hydroxyethyl side chain, likely via an aldehyde intermediate. The metabolites 5‐HNMP, 2‐P and HEP‐COOH were identified by comparison of their liquid chromatography and high‐resolution mass spectrometry features with those of synthetic reference standards, and all the experimental data supporting these findings are available in the Supporting Information files. Thus, not only is HEP more stable toward microsomal oxidation than NMP, but it also gives a polar acidic metabolite that can be easily eliminated by the kidneys. Therefore, HEP could represent a valid alternative to dipolar solvents in organometallic catalysis. Our data clearly indicate that the primary metabolic oxidation site of HEP is the hydroxyethyl side chain. The glycine derivative can then undergo a further pyrrolidone ring degradation, generating a succinyl‐glycine derivative in analogy with NMP ring oxidation/hydrolysis. Since it is considered only as an intermediate for the synthesis of N‐vinyl pyrrolidone, HEP is not included in any solvent database. However, we have used for its classification the CHEM21 selection guide for classical and less classical solvents (Table 1). [10b] Although HEP is a biogenic solvent with a good toxicological and ecotoxicological profile, it is penalized in the EHS criteria classification for green solvents by the fact that it has boiling point >200 °C, which by definition increases also the cumulative energy demand (CED). Most of the recently reported biogenic solvents such as cyrene, DES (that contain glycerol), dimethylisosorbide, and γ‐valerolactone have boiling points >200 °C. However, in our opinion, with the actual distillation technologies especially when the product does not require any rectification, the impact of the boiling point is mitigated at industrial level. [10d]
Table 1.
Solvents ranking according to CHEM21 selection guide.
|
Solvent |
E |
H |
S |
|---|---|---|---|
|
DMF |
5 |
9 |
3 |
|
NMP |
7 |
9 |
1 |
|
HEP |
7 |
2 |
1 |
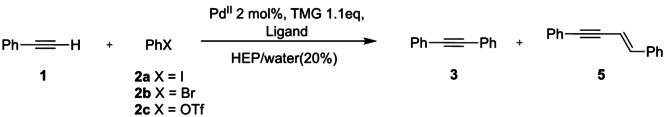
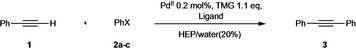
In order to identify the best reaction conditions aimed at recycling the catalyst with different substrates, we have explored the effect of water using simple and commercially available sulfonated phosphines as ligands. [34] The model reaction between phenylacetylene 1 and aryl derivatives 2 a–c was performed in HEP using TMG as organic base, following both the Heck‐Cassar and the Sonogashira protocols with PdII pre‐catalysts. The beneficial effect of TMG to the HCS coupling can be ascribed to its very high pK a (15.2 in water and 23.3 in acetonitrile) [35] that favors a rapid rearrangement from a π to a σ complex between the alkyne and the metal, generating the suitable species for the transmetalation step (TM, Scheme 2).
Scheme 2.

HCS cross coupling general mechanism. OA=oxidative addition; TM=transmetalation; RE=reductive elimination.
The coupling between phenylacetylene 1 and 2 a–c was studied as a model reaction in order to identify the optimal conditions (concentration, cocatalyst, temperature, and stoichiometry) to be used in the recycling protocols. The results reported in Table 2 showed that using the Sonogashira conditions, the conversion was always complete within 1 h, independently from the presence of water and from the nature of the sulfonated phosphine (entries 1–5). The study was focused on readily available sulfonated ligands, namely sodium 3‐(diphenylphosphino)benzenesulfonic acid sodium salt (TPPMS), 4,4′‐(phenylphosphinidene)bis(benzenesulfonic acid) dipotassium salt hydrate (TPPDS), and 3,3′,3′′‐phosphanetriyltris‐(benzenesulfonic acid) trisodium salt (TPPTS). Since TPPTS is the cheapest and largely available water‐soluble phosphine, being employed in the biphasic hydroformylation with rhodium catalysts (Ruhrchemie/Rhône‐Poulenc process), [36] and it induces a faster conversion using only 5 % excess of 1 (entry 3), we did not consider TPPMS and TPPDS for further studies. Interestingly, the Sonogashira coupling still provided excellent results decreasing the CuI cocatalyst amount down to 0.1 mol % (entries 6 and 7). In addition, the reactions performed with CuI, CuBr, and CuCl gave comparable results (entries 6–9). The Pd(0)/TPPTS catalyst proved to be efficient even in the absence of CuI, being able to smoothly generate diphenylacetylene 3 at 60 °C in 1 h (entry 10) and at 30 °C in 14 h (entry 11). However, the process can be accelerated by increasing the amount of acetylene (entry 12).
Table 2.
Screening for the HCS in HEP/water/TMG system conditions.
|
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Entry |
2 |
1 [equiv.] |
PdII |
Ligand [mol %] |
CuX [mol %] |
T [°C] |
t [h] |
Conv.[a] [%] |
3/5[a] |
3 yield[b] [%] |
|
1 |
2 a |
1.05 |
Pd(PPh3)2Cl2 |
– |
CuI (1) |
30 |
0.5 |
>99 |
>99 : 1 |
97[c] |
|
2 |
2 a |
1.05 |
Pd(PPh3)2Cl2 |
– |
CuI (1) |
30 |
0.5 |
>99 |
>99 : 1 |
95 |
|
3 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
CuI (1) |
30 |
0.5 |
>99 |
>99 : 1 |
95[d] |
|
4 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPMS (4) |
CuI (1) |
30 |
1 |
>99 |
>99 : 1 |
92 |
|
5 |
2 a |
1.5 |
Pd(CH3CN)2Cl2 |
TPPDS (4) |
CuI (1) |
30 |
1 |
>99 |
>99 : 1 |
95 |
|
6 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
CuI (0.25) |
30 |
0.5 |
>99 |
>99 : 1 |
93 |
|
7 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
CuI (0.1) |
30 |
1 |
>99 |
>99 : 1 |
95 |
|
8 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
CuBr (0.25) |
30 |
0.5 |
>99 |
>99 : 1 |
95 |
|
9 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
CuCl (0.25) |
30 |
0.5 |
>99 |
>99 : 1 |
92 |
|
10 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
– |
60 |
1 |
>99 |
>99 : 1 |
95 |
|
11 |
2 a |
1.05 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
– |
30 |
14 |
>99 |
>99 : 1 |
97 |
|
12 |
2 a |
1.5 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
– |
30 |
3 |
>99 |
>99 : 1 |
94 |
|
13 |
2 b |
1.5 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
– |
60 |
4 |
>99 |
88 : 12 |
91 |
|
14 |
2 b |
1.2 |
Pd(CH3CN)2Cl2 |
TPPTS (4) |
– |
60 |
4 |
>99 |
>99 : 1 |
96[e] |
|
15 |
2 b |
1.5 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
– |
60 |
2 |
>99 |
90 : 10 |
95 |
|
16 |
2 b |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
– |
60 |
2 |
>99 |
>99 : 1 |
93[e] |
|
17 |
2 b |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
– |
30 |
24 |
80 |
>99 : 1 |
– |
|
18 |
2 b |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
CuI (1) |
30 |
2 |
>99 |
>99 : 1 |
94 |
|
19 |
2 b |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
CuI (0.1) |
30 |
3 |
>99 |
>99 : 1 |
97 |
|
20 |
2 c |
1.2 |
Pd(CH3CN)2Cl2 |
sSPhos (4) |
– |
60 |
1 |
>99 |
90 : 10 |
92 |
|
21 |
2 c |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (6) |
– |
60 |
3 |
>99 |
>99 : 1 |
92[e] |
|
22 |
2 c |
1.05 |
Pd(CH3CN)2Cl2 |
sSPhos (6) |
CuI (0.25) |
30 |
4 |
>99 |
>99 : 1 |
95 |
[a] Determined by HPLC. [b] The products were isolated after cyclohexane extraction and purification by flash chromatography only if the conversion exceeded 90 %. [c] The reaction was performed under anhydrous conditions. [d] The reaction was performed also using the recovered catalyst kept for one week under nitrogen, obtaining the same reaction yield. [e] Phenylacetylene 1 was added using a syringe pump over the reaction time.
With aryl iodides the rate‐determining step of the reaction is the transmetalation process (Scheme 2). [37] Interestingly, this key step proved to be more efficient in the Sonogashira protocol than in the Heck‐Cassar one, where a second PdII complex performs the transmetalation. Košmrlj and co‐workers [38] were able to detect the PdIIalkyne2 intermediate H supporting the mechanism reported below.
Moving to bromide 2 b, the Pd0/TPPTS catalyst was able to induce complete conversion at 60 °C in 4 h (entry 13). The homocoupling product 4 (Scheme 3) was not detected, but, on the contrary, 12 % of [(E)‐4‐phenylbut‐1‐en‐3‐ynyl]benzene] 5 was found in the final reaction mixture. The side product was suppressed by slow addition of phenylacetylene 1 in 4 h (entry 14), which also allowed to decrease the need for alkyne excess.
Scheme 3.
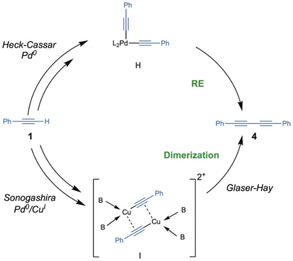
Mechanisms for the formation of the homocoupling product 4
The catalyst generated with Buchwald's ligand, sodium 2’‐dicyclohexylphosphino‐2,6‐dimethoxy‐1,1′‐biphenyl‐3‐sulfonate (sSPhos), [39] afforded product 3 in high yield without copper at 60 °C (entry 15). The formation of enyne 5 was again controlled decreasing the excess of 1 and using the slow addition mode (entry 16).
Only the Sonogashira protocol could be successfully performed at 30 °C and the reaction afforded selectively the coupling product 3 in a few hours even with 0.1 mol % of CuI (entries 17–19). Phenyl triflate 2 c behaved similarly to the corresponding bromide 2 b. In fact, the copper free reaction at 60 °C selectively afforded 3 in 1 h only with the slow addition of acetylene 1 (entries 20 and 21).
At 30 °C with the Pd0/CuI catalytic system the reaction was completed in 4 h (entry 22). In order to have a clear understanding of the reaction performed in HEP/water/TMG, 31P NMR spectroscopy was used to monitor the fate of the phosphine. The experiment described by Košmrlj and co‐workers on the copper‐free protocol [Pd(PPh3)2Cl2, pyrrolidine, CH2Cl2, aryl iodide 2 a, and phenylacetylene 1] [38] was performed replacing pyrrolidine with TMG as base, and formation of complex H was similarly detected. On the contrary, repeating the reaction in the presence of CuI, complex H was never observed (Scheme 2). Traces of complex H were instead detected when the HEP/water/TMG protocol was used in Heck‐Cassar conditions. The formation of the homocoupling product 4 is considered the main side reaction of the HCS, [40] and it can be generated by both protocols as described in Scheme 3.
Specifically, 4 could arise from the dimerization of complex I catalyzed by CuI salts (Glaser‐Hay coupling) [41] or from a reductive elimination originated by PdII complex H. However, in our case all the reactions were performed under nitrogen atmosphere with degassed solvents and the Glaser‐Hay coupling, which would require oxidative conditions, was completely suppressed. On the other hand, when copper‐free HCS with HEP/water/TMG protocol was applied, the 31P signal of complex H was very low and the homocoupling product 4 was never observed.
This evidence could be attributed to the matched effect of protic solvents and based on the reaction mechanism, even if the direct coordination of the acetylene on complex B cannot be totally ruled out and requires further investigations (Scheme 4). [42]
Scheme 4.
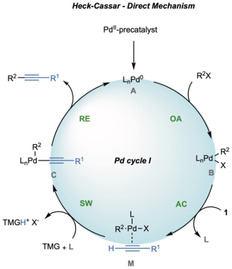
The direct mechanism for Heck‐Cassar protocol. OA=oxidative addition; AC=acetylene coordination; SW=switch from π to σ complex.
In a few reactions (entries 13, 15, 20), the presence of compound 5 was observed, coming from self‐hydroalkynylation of phenylacetylene 1 (Scheme 5). [43] When the HCS coupling is inefficient and the reaction requires high temperature or a large excess of acetylene, the formation of enyne 5 can become a competitive process. [43]
Scheme 5.
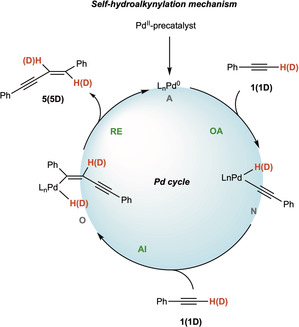
Mechanism of the Pd0‐catalyzed self‐hydroalkynylation of phenylacetylene 1. OA=oxidative addition; AI=alkyne insertion; RE=reductive elimination.
This is observed only for couplings performed using the Heck‐Cassar protocol with bromides and triflates as leaving groups. The reaction outcome is related to the efficiency of the oxidative addition of the Pd0 complex on the aryl derivative 2 versus the one on the alkyne 1. With iodide 2 a the HCS is fast and selective, while with less efficient 2 b and 2 c the competition with alkyne 1 was emerging in some cases, generating variable amount of enyne 5. However, the simple slow addition of alkyne allowed to suppress this side reaction and selectively afforded diphenylacetylene 3 in high yield. When the reaction was performed using deuterated phenylacetylene 1D in the absence of aryl halide and copper cocatalyst, the corresponding deuterated enyne 5D was isolated, thus confirming that the vinylic hydrogens are coming exclusively from the acetylene moiety. In fact, enyne 5 is the first step toward acetylene oligomerization. [43]
The catalyst generated using PPh3 was stable in the presence of TMG and HEP/water as solvent. In fact, the 31P NMR spectrum recorded after addition of the base to the pre‐catalyst/ligand‐containing solution was identical to the one recorded at the end of the reaction after product extraction with cyclohexane. It is also important to stress that the reactions of Table 2 (entries 10 and 16) carried out using 0.2 mol % of catalyst instead of 2 mol % under Heck‐Cassar conditions did not reach complete conversion after 14 h (see Table S2, entries 41 and 42). These observations supported the idea to recycle the catalyst using sulfonated phosphines with short reaction time, thus opening the possibility to increase the TON of the HCS reaction in HEP/water.
Since the HCS cross‐coupling and, in general, Pd0‐catalyzed reactions are compatible with several functional groups on the aromatic ring, we decided to explore substrate scope by performing the reaction on bromides and triflates bearing electron‐donating substituents that could potentially decrease the efficiency of the oxidative addition step.
In addition, as highlighted by our recent results, [19] the HCS outcome is mainly affected by the nature of the alkyne, and the attention was therefore focused on differently substituted acetylenes. The results reported in Table 3 show that the catalyst, generated in the HEP/water/TMG system, could be easily recycled independently from the leaving group, the presence of electron‐donating moiety on the aryl derivative, the sulfonated phosphine ligand, or the protocol used. The simple extraction with an immiscible solvent allows to easily recover the final product, leaving the catalyst intact in the HEP/water phase.
Table 3.
HCS catalyst recycling: effect of ligand, leaving group, and alkyne substitution.[a]
|
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Entry |
Alkyne |
R2X |
Alkyne [equiv.] |
Ligand [mol %] |
CuI [mol %] |
T [°C] |
Cycle time [h] |
Alkyne addition |
Cycles |
TON final |
Product |
Overall yield[b] [%] |
|
1 |
1 |
2 a |
1.05 |
TPPTS (4) |
0.5 |
30 |
0.5 |
rapid |
10 |
470 |
3 |
94[c] |
|
2 |
1 |
2 a |
1.5 |
TPPDS (4) |
0.5 |
30 |
1 |
rapid |
10 |
465 |
3 |
93 |
|
3 |
1 |
2 a |
1.05 |
TPPMS (4) |
0.5 |
30 |
1 |
rapid |
10 |
475 |
3 |
95 |
|
4 |
6 |
2 a |
1.5 |
TPPTS (4) |
0.5 |
30 |
1 |
rapid |
10 |
460 |
14 |
92 |
|
5 |
7 |
2 a |
1.5 |
TPPTS (4) |
0.5 |
30 |
1 |
rapid |
10 |
470 |
15 |
94 |
|
6 |
8 |
2 a |
1.5 |
TPPTS (4) |
0.5 |
30 |
1 |
rapid |
10 |
475 |
16 |
95 |
|
7 |
9 |
2 a |
1.5 |
TPPTS (4) |
0.5 |
30 |
1 |
rapid |
10 |
480 |
17 |
96 |
|
8 |
10 |
2 a |
1.5 |
TPPTS (4) |
0.5 |
50 |
1 |
rapid |
10 |
465 |
18 |
93[d] |
|
9 |
1 |
2 a |
1.05 |
TPPTS (4) |
– |
60 |
1 |
rapid |
15 |
705 |
3 |
94[c] |
|
10 |
1 |
2 b |
1.05 |
sSphos (4) |
– |
60 |
2 |
2 h |
10 |
470 |
3 |
94 |
|
11 |
1 |
2 b |
1.05 |
sSphos (4) |
0.5 |
30 |
2 |
rapid |
7 |
330 |
3 |
94 |
|
12 |
1 |
11 b |
1.05 |
sSphos (4) |
– |
60 |
2 |
2 h |
10 |
460 |
19 |
92 |
|
13 |
1 |
12 b |
1.05 |
sSphos (4) |
– |
60 |
2 |
2 h |
10 |
475 |
20 |
95 |
|
14 |
9 |
2 b |
1.5 |
sSphos (4) |
– |
60 |
2 |
rapid |
10 |
460 |
17 |
92 |
|
15 |
10 |
2 b |
2 |
sSphos (6) |
– |
60 |
2 |
rapid |
5 |
238 |
18 |
95[d] |
|
16 |
6 |
13 b |
2 |
sSphos (6) |
– |
60 |
3 |
rapid |
8 |
376 |
22 [e] |
94 |
|
17 |
1 |
2 c |
1.05 |
sSphos (6) |
– |
60 |
3 |
3 h |
10 |
480 |
3 |
96 |
|
18 |
1 |
11 c |
1.05 |
sSphos (6) |
– |
60 |
3 |
3 h |
10 |
475 |
19 |
95 |
|
19 |
1 |
12 c |
1.05 |
sSphos (6) |
– |
60 |
3 |
3 h |
10 |
480 |
20 |
96 |
|
20 |
7 |
2 c |
2 |
sSphos (6) |
– |
60 |
2 |
rapid |
10 |
460 |
15 |
92 |
|
21 |
10 |
2 c |
2 |
sSphos (6) |
– |
60 |
2 |
rapid |
7 |
330 |
18 |
94[d] |

[a] All HCS couplings were carried out under nitrogen atmosphere. At the given time the reactions were cooled at RT, extracted with cyclohexane, and the HEP/water phase containing the catalyst was recycled. [b] The combined cyclohexane extracts were distilled and the crude was subsequently purified by flash chromatography generating an average yield. [c] Entries 1 and 9 were carried out also using toluene, 2‐MeTHF, methyl tert‐butyl ether, and isobutyl acetate as extraction solvents giving the same result. [d] Performed with 30 % of water as cosolvent. [e] The work‐up was carried out with toluene and deprotection of the intermediate 21 was directly performed by treatment with NaOH under reflux affording 22 (see the Supporting Information). With quantitative deprotection, yields of 21 and 22 are identical. After deprotection toluene was recovered.
The catalyst solution was then used in the following reaction cycle by simply adding the two reagents and the TMG base (Figure 2). The recycling time was fixed (0.5, 1, 2, or 3 h) according to the reaction rate, and the conversion, measured after each cycle workup, was >95 % in all cases.
Figure 2.
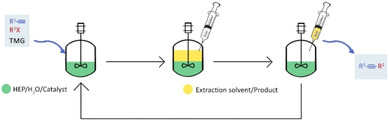
General recycling process.
It is worth noting that the Sonogashira iterative protocol was stopped after 10 recycles with 2 a even if in many cases the catalyst was still active (Table 3, entries 1–8), whereas the copper‐free protocol of the reaction between 1 and aryl iodide 2 a at 60 °C was recycled 15 times (entry 9).
Moving to aryl bromides and triflates, the catalyst was easily recycled in all cases excluding the reactions containing 1‐hexyne 10 as starting alkyne (Table 3, entries 15 and 21). Moreover, in order to suppress the formation of the enynes, some reactions required the slow addition of the acetylene (entries 10, 12, 13, 17, 18, 19). To further confirm the robustness of the procedure, the key intermediate for the synthesis of Erlotinib, 22, was successfully isolated with an average yield of 94 % after 8 cycles and quantitative deprotection from 21 (entry 16). The HEP/water system allowed to completely recycle the catalyst avoiding any leakage. In fact, the combined crude materials coming from the reactions, obtained by extraction/solvent distillation, were analyzed by inductively coupled plasma optical emission spectrometry (ICP‐OES) and contained <0.25 ppm of palladium metal and <0.02 ppm of copper when Sonogashira protocol was applied, hence satisfying an essential requirement for pharmaceutically industrial applications.
The recycling sequence (10 cycles) normally took 2–3 days overall in the lab, and the catalyst solution in HEP/water was stored overnight at room temperature under nitrogen without any loss in the catalytic activity. Furthermore, in order to increase the flexibility of the protocol, alternative solvents for final product extraction were also tested, repeating the reaction reported in entries 1 and 9 of Table 3. Toluene, CH2Cl2, 2‐methyl tetrahydrofuran (2‐MeTHF), methyl tert‐butyl ether (MTBE), ethyl acetate (EA), and the greener isobutyl acetate (IBA) were explored to test work‐up efficiency. With the only exception of CH2Cl2 and EA, all the solvents were efficient in extracting the product from the reaction mixture and the possible organic solvent residue in the HEP/water phase did not affect the catalyst recycling. The extraction, phase separation and catalyst recycling can be easily scaled up in a continuous process at industrial level with currently available technologies. Finally, with the optimized conditions in hand, we scaled up 10 times the substrate amount reaching a 5 mmol scale and increased the reaction concentration from 1 to 2.5 m. The catalyst amount was decreased to 0.2 mol % still giving complete conversions in a few hours (3–4 h) independently from the leaving group and the protocol used, allowing up to 5 recycles (Table 4). TON and TOF ranged between 1380–2375 and 110–158 h−1, respectively, while the PMI ranged from 7 to 8. In order to increase sustainability, the spent catalyst solution was treated with sodium formate to generate palladium black that was filtered out with the aid of charcoal (90 % palladium recovery). The remaining mixture was than treated with solid NaOH and distilled to recover TMG and HEP with a 95 % yield. The PMI after recovery was close to 3 in all entries of Table 4.
Table 4.
TON, TOF, and PMI values of optimized HCS.[a]
|
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Entry |
2 |
1 [equiv.] |
Ligand [mol %] |
CuI [mol %] |
T [°C] |
t [h] |
Cycles |
Overall yield[b] [%] |
TON[c] |
TOF[c] [h−1] |
PMI |
PMI[d] with recovery |
|
1 |
2 a |
1.05 |
TPPTS (0.6) |
0.05 |
30 |
3 |
5 |
95 |
2375 |
158 |
7.3 |
2.9 |
|
2 |
2 a |
1.05 |
TPPTS (0.4) |
– |
60 |
3 |
4 |
93 |
1860 |
155 |
7.6 |
3.0 |
|
3 |
2 b |
1.2 |
sSPhos (0.6) |
0.05 |
60 |
4 |
3 |
90 |
1380 |
112 |
7.9 |
3.0 |
|
4 |
2 c |
1.2 |
sSPhos (0.6) |
– |
60 |
4 |
4 |
88 |
1760 |
110 |
8.2 |
3.4 |
[a] All reactions were carried out under a nitrogen atmosphere with a 2.5 m concentration; the conversions were measured by HPLC at the end of each cycle, being always higher than 90 %. [b] The yield was obtained combining all the crude extracts. The reaction could be also performed by recycling the distilled cyclohexane in the following extraction process; however, this option was not used for PMI calculation. [c] TON and TOF were calculated considering the average yield and the overall reaction time. [d] PMI was recalculated after recovery of cyclohexane (95 %), TMG (95 %), HEP (95 %), and palladium (90 %), see the Supporting Information for the detailed calculation.
The protocol was successfully applied to the telescoped synthesis of Erlotinib 24 (Scheme 6). The HCS was carried out following a procedure similar to the ones in Table 3 (3 recycles; TON: 1380; TOF: 46 h−1) and using toluene for the extraction of 21.
Scheme 6.
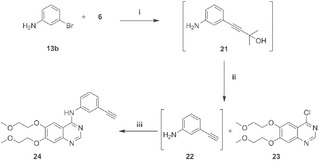
Erlotinib synthesis. (i) Pd(CH3CN)2Cl2 (0.2 mol %), sSPhos (0.6 mol %), 6 (2 equiv.), HEP/H2O/TMG, 80 °C 10 h; 3 recycles. (ii) Toluene/NaOH (1.5 equiv.), 1 h reflux. (iii) 23 (0.9 equiv.), toluene/isopropanol 1 : 1, 4 h, 55 °C, 75 % yield.
The limited amount of recycles is related to the presence of the aniline moiety that interferes with the palladium catalyst. The toluene solution of intermediate 21 was treated with NaOH at reflux for 1 h to get 22, washed with water, concentrated to 0.5 mol solution, and immediately used in the coupling with 23 (toluene/isopropanol) to get Erlotinib 24 with a 75 % overall yield from 13 b.
Conclusion
The palladium‐catalyzed Heck‐Cassar‐Sonogashira (HCS) cross‐coupling using sulfonated phosphines in N‐hydroxyethylpyrrolidone (HEP)/water as solvent mixture and N,N,N′,N′‐tetramethyl guanidine (TMG) as base proved to be applicable on aryl iodides, bromides, and triflates. The catalyst, being stable and perfectly soluble in HEP/water, could be recycled, and the product, after a simple extraction and solvent evaporation, was recovered free from metal contamination.
The process was further optimized to drastically decrease the required amount of Pd catalyst and increase the turnover number and frequency up to 2375 and 158 h−1, respectively. [45] Palladium metal, TMG, and HEP were recovered from the exhausted catalyst solution, and the recalculated process mass intensity was close to 3 based on the crude.
The identification of the side reaction that generates the enyne derivatives with the copper‐free Heck‐Cassar protocol was limited by the simple control of the alkyne addition rate to optimize the stoichiometry. Concerning the transmetalation process, HEP/water/TMG system plays an important role on the copper‐free reaction mechanism; nevertheless, the occurrence of the alkyne direct coordination on the oxidative addition complex or the transmetalation process between two PdII complexes cannot be ascertained by our study and deserves further investigations. Tyrosine kinase inhibitor Erlotinib 24 was obtained in high yield from 13 b via a three‐step telescoping process starting from a sustainable HCS coupling procedure.
Experimental Section
The reaction scale and concentrations for all the experiments and the protocol for in‐vivo metabolism in rat and human liver microsomes are described in the Supporting Information.
General procedure for HCS cross‐coupling recycling protocol (Table 4)
To a 10 mL Schlenk tube purged under N2 atmosphere, palladium pre‐catalyst (0.2 mol %), phosphine ligand (TPPTS or sSPhos, 0.4–0.6 %), and CuX co‐catalyst (0.05 mol % only with Sonogashira protocol) were dissolved in 2 mL of HEP and water as co‐solvent. The other reagents were then added in the following order: TMG (635 mg, 690 μL, 5.5 mmol, 1.1 equiv.), aryl halide 2 a–c (5 mmol, 1 equiv.), and phenyl acetylene 1 (1.05–1.2 equiv.). The reaction mixture was heated to 30 or 60 °C with an oil bath and maintained at this temperature under stirring. After complete conversion (monitored with HPLC‐UV at 210 nm), the mixture was extracted three times under N2 with cyclohexane. The organic layer was removed with a syringe, and another portion of TMG, aryl halide, and alkyne were added to the HEP/water phase and another catalytic cycle was performed at 30 or 60 °C. The conversion of the new cycle of reaction was monitored by the previously mentioned analysis. The extraction solvent could be distilled and directly recovered into the next extraction cycle, or the organic phases obtained from the different cycles could be combined, distilled to recover cyclohexane, and the residue purified, if necessary, by flash chromatography. The HEP/water solution containing the spent catalyst was treated with sodium formate (0.04 mmol, 2.8 mg) for 1 h at 60 °C in order to precipitate the palladium catalyst. The mixture was then filtered on charcoal (60 mg), and the palladium metal was recovered in quantitative yields. The filtrate was basified with 3 g of NaOH to achieve a pH around 13–14 and then distilled under reduced pressure to recover 95 % of HEP and TMG.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank for the support Faggi Enrico S.p.A. for catalysts supply and elemental impurity analysis. We kindly acknowledge Alma Mater Studiorum University of Bologna and Fresenius kabi for financial support. We thank Govind Singh from FKOL, Gurgaon India for the collaboration on Erlotinib.
T. Fantoni, S. Bernardoni, A. Mattellone, G. Martelli, L. Ferrazzano, P. Cantelmi, D. Corbisiero, A. Tolomelli, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor, A. Ricci, ChemSusChem 2021, 14, 2591.
References
- 1.
- 1a. Anastas P. T., Warner J. C., Green Chemistry: Theory and Practice, Oxford University Press, New York: 1998; [Google Scholar]
- 1b. Roschangar F., Sheldon R. A., Senanayake C. H., Green Chem. 2015, 17, 752. [Google Scholar]
- 2.
- 2a. Sheldon R. A., Chem. Commun. 2008, 3352–3365; [DOI] [PubMed] [Google Scholar]
- 2b. Watson W. J. W., Green Chem. 2012, 14, 251; [Google Scholar]
- 2c. McElroy C. R., Constantinou A., Jones L. C., Summerton L., Clark J. H., Green Chem. 2015, 17, 3111–3121; [Google Scholar]
- 2d. Biffis A., Centomo P., Del Zotto A., Zecca M., Chem. Rev. 2018, 118, 2249–2295; [DOI] [PubMed] [Google Scholar]
- 2e. Colacot T. J., Johansson Seechurn C. C. C., Organometallic in Industry, Wiley-VCH, Weinheim: 2020. [Google Scholar]
- 3.
- 3a. Cabri W., Catal. Today 2009, 140, 2–10; [Google Scholar]
- 3b. Dumrath A., Lübbe C., Beller M., Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments (Ed: Molnar Á.), Wiley-VCH, Weinheim: 2013, pp. 445–489. [Google Scholar]
- 4.
- 4a. Schilz M., Plenio H., J. Org. Chem. 2012, 77, 2798–2807; [DOI] [PubMed] [Google Scholar]
- 4b. Chinchilla R., Nájera C., Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations (Eds.: Trost B. M., Li C.-J.), Wiley-VCH; 2015, pp. 269–297; [Google Scholar]
- 4c. Alonso D. A., Baeza A., Chichilla R., Gomez C., Guillena G., Pastor I. M., Ramon D. J., Catalysts 2018, 8, 202. [Google Scholar]
- 5.Ponatinib:
- 5a. Huang W. S., Metcalf C. A., Sundaramoorthi R., Wang Y., Zou D., Thomas R. M., Zhu X., Cai L., Wen D., Liu S., Romero J., Qi J., Chen I., Banda G., Lentini S. P., Das S., Xu Q., Keats J., Wang F., Wardwell S., Ning Y., Snodgrass J. T., Broudy M. I., Russian K., Zhou T., Commodore L., Narasimhan N. I., Mohemmad Q. K., Iuliucci J., Rivera V. M., Dalgarno D. C., Sawyer T. K., Clackson T., Shakespeare W. C., J. Med. Chem. 2010, 53, 4701–4719; [DOI] [PubMed] [Google Scholar]
- 5b. Dong Z., Huang W.-S., Thomas R. M., Romero J. A. C., Qi J., Wang Y., Zhu X., Shakespeare W. C., Sundaramoorthi R., Metcalf C. A., Dalgarno D. A., Sawyer T. K., (Ariad Pharmaceutical Inc.), US9278971, 2016;
- 5c. Handa S., Jin B., Bora P. P., Wang Y., Zhang X., Gallou F., Reilly J., Lipshutz B. H., ACS Catal. 2019, 9, 2423–2431; Erlotinib:; [Google Scholar]
- 5d. Cabri W., Oldani E., (Secifarma), US5902902, 1999;
- 5e. Caporale A., Tartaggia S., Castellin A., De Lucchi O., Beilstein J. Org. Chem. 2014, 10, 384–393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Chandraratna R., (Allergan Inc.), US5602130, 1997; Tazarotene:
- 5g. Frigoli S., Fuganti C., Malpezzi L., Serra S., Org. Process Res. Dev. 2005, 9, 646–650; Alectinib:; [Google Scholar]
- 5h. Hughes D. L., Org. Process Res. Dev. 2016, 20,1855–1869 and references therein; [Google Scholar]
- 5i.A. Tolomelli, L. Ferrazzano, A. De Nisi, W. Cabri, (Fresenius Kabi & University of Bologna) EP18211036 2016; Eniluracil:
- 5j. Cooke J. W. B., Bright R., Coleman M. J., Jenkins K. P., Org. Process Res. Dev. 2001, 5, 383–386. [Google Scholar]
- 6. Sonogashira K., Tohda Y., Hagihara N., Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]
- 7. Dieck H. A., Heck F. R., J. Organomet. Chem. 1975, 93, 259–263. [Google Scholar]
- 8. Cassar L., J. Organomet. Chem. 1975, 93, 253–257. [Google Scholar]
- 9.
- 9a. Heck R. F., Nolley J. P., J. Org. Chem. 1972, 37, 2320; [Google Scholar]
- 9b. Cabri W., Candiani I., Acc. Chem. Res. 1995, 28, 2–7; [Google Scholar]
- 9c. Cabri W., Candiani I., Bedeschi A., Santi R., Synlett 1992, 871–872. [Google Scholar]
- 10.
- 10a. Clarke C. J., Tu W., Levers O., Bröhl A., Hallett H. J. P., Chem. Rev. 2018, 118, 747–800; [DOI] [PubMed] [Google Scholar]
- 10b. Prat D., Wells A., Hayler J., Sneddon H., McElroy C. R., Abou-Shehada S., Dunn P. J., Green Chem. 2016, 18, 288–296; [Google Scholar]
- 10c. Cicco L., Dilauro G., Perna F. M., Vitale P., Capriati V., Org. Biomol. Chem. 2021, 19, 2558–2577; [DOI] [PubMed] [Google Scholar]
- 10d. Byrne F. P., Saimeng J., Paggiola G., Petchey T. H. M., Clark J. H., Farmer T. J., Hunt A. J., McElroy C. R., Sherwood J., Sustainable Chem. Process 2016, 4, 7; [Google Scholar]
- 10e. Hooshmand S. E., Afshari R., Ramón D. J., Varma R. S., Green Chem. 2020, 22, 3668–3692. [Google Scholar]
- 11. Dyson P. J., Jessop P. G., Catal. Sci. Technol. 2016, 6, 3302–3316. [Google Scholar]
- 12. Sherwood J., Clark J. H., Fairlamb I. J. S., Slatter J. M., Green Chem. 2019, 21, 2164–2213. [Google Scholar]
- 13. Wilson K. L., Kennedy A. R., Murray J., Greatrex B., Jamieson C., Watson A. J. B., Beilstein J. Org. Chem. 2016, 12, 2005–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim T. H., Kim S. G., Saf. Health Work 2011, 2, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lessons learnt from presence of N-nitrosamine impurities in sartan medicines EMA/526934/2019 2020.
- 16.
- 16a. Amelio A., Genduso G., Vreysen S., Luis P., Van der Bruggen B., Green Chem. 2014, 16, 3045–3063; [Google Scholar]
- 16b. Prat D., Hayler J., Wells A., Green Chem. 2014, 16, 4546–4551; [Google Scholar]
- 16c. Benazzouz A., Moity L., Pierlot C., Sergent M., Molinier V., Aubry J. M., Ind. Eng. Chem. Res. 2013, 52, 16585–16597; [Google Scholar]
- 16d. Prat D., Pardigon O., Flemming H. W., Letestu S., Ducandas V., Isnard P., Guntrum E., Senac T., Ruisseau S., Cruciani P., Hosek P., Org. Process Res. Dev. 2013, 17, 1517–1525; Specifically, on the future fate of NMP: [Google Scholar]
- 16e. Sherwood J., Farmer T. J., Clark J. H., Chem. 2010, 4, 2004–2012. [Google Scholar]
- 17. Bryan M. C., Dunn P. J., Entwistle D., Gallou F., Koenig S. G., Hayler J. D., Hickey M. R., Hughes S., Kopach M. E., Moine G., Richardson P., Roschangar F., Steven A., Weiberth F. J., Green Chem. 2018, 20, 5082–5103. [Google Scholar]
- 18. Köllhofer A., Plenio H., Adv. Synth. Catal. 2005, 347, 1295–1300. [Google Scholar]
- 19.Ionic Liquid:
- 19a. Reddy A. S., Laali K. K., Tetrahedron Lett. 2015, 56, 4807–4810; Water: [Google Scholar]
- 19b. Herve G., Len C., Sustainable Chem. Processes 2015, 3, 3; Dimethylisosorbide: [Google Scholar]
- 19c. Wilson K. L., Murray J., Sneddon H. F., Jamieson C., Synlett 2018, 29, 2293–2297; γ-Valerolactone: [Google Scholar]
- 19d. Strappaveccia G., Luciani L., Bartollini E., Marrocchi A., Pizzo F., Vaccaro L., Green Chem. 2015, 17, 1071–1076; Deep eutectic solvents: [Google Scholar]
- 19e. Messa F., Dilauro G., Perna F. M., Vitale P., Capriati V., Salomone A., ChemCatChem 2020, 12, 1979–1984. [Google Scholar]
- 20. Ferrazzano L., Martelli G., Fantoni T., Daka A., Corbisiero D., Viola A., Ricci A., Cabri W., Tolomelli A., Org. Lett. 2020, 22, 3969–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin B., Gallou F., Reilly J., Lipshutz B. H., Chem. Sci. 2019, 10, 3481–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Doucet H., Hierso J.-C., Angew. Chem. Int. Ed. 2007, 46, 834–871; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 850–888; [Google Scholar]
- 22b. Martek B. A., Gazvoda M., Urankar D., Košmrlj J., Org. Lett. 2020, 22, 4938–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guideline for Elemental Impurities ICH Q3D (R1) 2019.
- 24. Dumrath A., Wu X., Neumann H., Spannenberg A., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2010, 49, 8988–8992; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9172–9176. [Google Scholar]
- 25.GSK's acids and bases selection guide reported a high green score for TMG. See: Henderson R. K., Hill A. P., Redman A. M., Sneddon H. F., Green Chem. 2015, 17, 945–949. [Google Scholar]
- 26. Haus M. O., Louven Y., Palkovits R., Green Chem. 2019, 21, 6268–6276. [Google Scholar]
- 27.
- 27a.ECHA reference for HEP: https://echa.europa.eu/it/registration-dossier/-/registered-dossier/13179. The registration that is only referred to European countries is supporting use, storage and transportation of≥1000 to <10000 tonnes per annum; For the most recent data for LD50 >14400 see:
- 27b. Ansell J. M., Fowler J. A., Food Chem. Toxicol. 1988, 26, 475–479. [DOI] [PubMed] [Google Scholar]
- 28.ECHA reference for DMF: https://echa.europa.eu/it/registration-dossier/-/registered-dossier/13179. The registration that is only referred to European countries is supporting use, storage and transportation of≥10 000 to <100 000 tonnes per annum.
- 29.NMP was used in several reactions as an alternative, however, also NMP has several issues from a safety point of view:
- 29a.ECHA reference for NMP see: https://echa.europa.eu/it/registration-dossier/-/registered-dossier/15493/1. The registration that is only referred to European countries is supporting use, storage and transportation of≥10 000 to <100 000 tonnes per annum;
- 29b. Sherwood J., Farmer T. J., Clark J. H., Chem 2018, 4, 2004–2010. [Google Scholar]
- 30. Carnerup M. A., Spanne M., Jonsson B. A., Toxicol. Lett. 2006, 162, 139–145. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Wells D. A., Digenis G. A., Drug Metab. Dispos. 1988, 16, 243–249; [PubMed] [Google Scholar]
- 31b. Wells D. A., Hawi A. A., Digenis G. A., Drug Metab. Dispos. 1992, 20, 124–126. [PubMed] [Google Scholar]
- 32. Åkesson B., Jonsson B. A., Drug Metab. Dispos. 1997, 25, 267–269. [PubMed] [Google Scholar]
- 33. Carnerup M. A., Saillenfait A. M., Jonsson B. A., Food Chem. Toxicol. 2005, 43, 1441–1447. [DOI] [PubMed] [Google Scholar]
- 34. Amatore C., Blart E., Genet J. P., Jutand A., Lemaire-Audoire S., Savignac M., J. Org. Chem. 1995, 60, 6829–6839. [Google Scholar]
- 35. Ishikawa T., Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts (Eds.: Ishikawa T.), Wiley; 2009, pp. 93–143. [Google Scholar]
- 36.
- 36a. Kunz E. G. (Rhône-Poulenc, Paris), FR2349562, 1976;
- 36b. Kohlpaintner C. W., Fischer R. W., Cornils B., Appl. Catal. A 2001, 221, 219–225. [Google Scholar]
- 37. He C., Ke J., Xu H., Lei A., Angew. Chem. Int. Ed. 2013, 52, 1527–1530; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1567–1570. [Google Scholar]
- 38. Gazvoda M., Virant M., Pinter B., Košmrlj J., Nat. Commun. 2018, 9, 4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anderson K. W., Buchwald S. L., Angew. Chem. Int. Ed. 2005, 44, 6173–6177; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6329–6333. [Google Scholar]
- 40. Elangovan A., Wang Y.-H., Ho T.-I., Org. Lett. 2003, 5, 1841–1844. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Glaser C., Ber. Dtsch. Chem. Ges. 1869, 2, 422–424; [Google Scholar]
- 41b. Hay A. S., J. Org. Chem. 1962, 27, 3320–3321; [Google Scholar]
- 41c. Siemsen P., Livingston R. C., Diederich F., Angew. Chem. Int. Ed. 2000, 39, 2632–2657; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2740–2767; [Google Scholar]
- 41d. Jover J., Spuhler P., Zhao L., McArdleb C., Maseras F., Catal. Sci. Technol. 2014, 4, 4200–3209. [Google Scholar]
- 42. Tougerti A., Negri S., Jutand A., Chem. Eur. J. 2007, 13, 666–676. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Trost B. M., Sorum M. T., Chan C., Harms A. E., Ruhter G., J. Am. Chem. Soc. 1977, 119, 698–708; [Google Scholar]
- 43b. Grunwald A., Heinemann F. W., Munz D., Angew. Chem. Int. Ed. 2020, 59, 21088–21095; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21274–21281. [Google Scholar]
- 44. Wu Y.-T., Lin W.-C., Liu C.-J., Wu C.-Y., Adv. Synth. Catal. 2008, 350, 1841–1849. [Google Scholar]
- 45.Catalyst recycling to increase the TON is still a challenge, see: Huang J., Wang W., Li H., ACS Catal. 2013, 3, 1526–1536. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary