

Abstract
Purpose
Hedgehog (Hh) signaling promotes castration‐resistant prostate cancer by supporting androgen‐independent prostate cancer cell development and growth; however, its role in neuroendocrine prostate cancer (NEPC) has not yet been explored. In this study, we assessed the expression of key genes involved in Hh signaling in prostate cancer and investigated the potential role of smoothened (SMO) in the pathogenesis of NEPC.
Methods
Six public datasets, each containing cases of prostate adenocarcinoma (AdPC) and NEPC, were analyzed to compare the differential messenger RNA (mRNA) expression of six classic Hh signaling genes. The SMO, synaptophysin, chromogranin A (CHGA) and androgen receptor (AR) proteins were evaluated in human tissues from 5 cases of NEPC, 2 cases of AdPC mixed with NEPC, 2 cases of AdPC with neuroendocrine differentiation and 22 cases of high‐grade AdPC as determined by an immunohistochemistry assay. Gene set enrichment analysis (GSEA) was performed to identify relevant genetic signatures associated with SMO expression based on the public datasets. Stable SMO‐knockdown LNCaP and C4‐2B cells were established with a lentiviral system, and the expression of SMO, Gli1, AR, prostate‐specific antigen (PSA), and REST was assessed by real‐time polymerase chain reaction and western blot. Secreted PSA in the conditioned medium was assessed by ELISA. Gli1 was ectopically expressed performed by the transfection of Gli1 complementary DNA into SMO‐knockdown LNCaP cells, and western blot was used to assess of AR and PSA expression.
Results
The mRNA level of SMO was dramatically downregulated in NEPC samples compared with AdPC samples in all 6 public datasets. SMO protein loss was observed in 100% of NEPC samples but in only 9% (2 of 22) of high‐grade AdPC samples. GSEA results showed that SMO loss was closely correlated with AR signaling activity. Stable SMO knockdown significantly attenuated AR signaling activity and suppressed AR expression, while Gli1 overexpression partially reversed the inhibitory effects of SMO knockdown on AR signaling activity and AR expression in LNCaP and C4‐2B cells.
Conclusion
These results demonstrate that SMO loss is a characteristic of NEPC and that detecting SMO by IHC could aid pathologists in NEPC diagnosis. SMO loss may promote NEPC pathogenesis by modulating AR signaling.
Keywords: androgen receptor signaling, Hedgehog signaling, neuroendocrine prostate cancer, prostate adenocarcinoma, smoothened
1. INTRODUCTION
Prostate cancer is one of the most common and deadly cancers in males worldwide, and more than 34 thousand men are estimated to die from prostate cancer in 2021. 1 Antiandrogen therapies are standardized approaches to the treatment of metastatic prostate cancers. However, after months or years of remission, nearly all patients relapse, and these cancers are termed castration‐resistant prostate cancer (CRPC), 2 the majority of which still depend on androgen receptor signaling (AR) for survival. Currently, with the widespread clinical usage of potent AR pathway inhibitors (APIs), the incidence of treatment‐emergent neuroendocrine prostate cancer (NEPC) has paradoxically increased, 3 with up to 20% of advanced CRPCs ultimately developing small‐cell neuroendocrine pathological features. 4 NEPC tumors are AR signaling independent and exhibit certain neuroendocrine signatures, making them resistant to APIs, and the platinum‐based chemotherapeutic regimen is effective for only a short period 5 ; thus, NEPC is the most lethal subtype of CRPC.
Tremendous progress has been made on understanding NEPC in the last decade. Genomic loss of TP53 and RB1 is more prevalent in NEPC (50%–75%) than in adenocarcinoma (5%–15%) and facilitates the activation of pluripotent networks mediated by derepression of SOX2 and EZH2. 6 , 7 Genomic aberrations cooperate with epigenetic modifiers, such as REST, 8 and neurolineage pioneering transcription factors, such as BRN2, 9 ONECUT2, 10 SRRM4, 11 and MYCN 12 drive the emergence of NEPC. Loss of luminal lineage transcription factors, such as AR and FOXA1 breaches the barriers of prostate adenocarcinoma (AdPC) reprogramming. 13 However, the evolution of NEPC is a complex process and a spectrum of intermediate differentiation states constitute a continuum between the AdPC and NEPC phenotypes; therefore, the underlying molecular mechanisms remain largely elusive, and druggable targets for clinical application need to be identified.
Smoothened (SMO) is a class Frizzled (Class F) G protein‐coupled receptor that is a key signal transducer of the Hedgehog (Hh) pathway. 14 In the presence of secreted Hh ligands (e.g., SHH, DHH, and IHH), SMO is released by an inhibitory receptor, PTCH1, which leads to the activation of glioma‐associated oncogene (Gli) transcription factors (TFs), namely, Gli1, Gli2, and Gli3. 15 The Hh signaling pathway plays fundamental morphogenic and mitogenic roles in the process of embryonic development 16 and a vital role in the regulation of cell fate and ductal morphogenesis in the prostate; however, at the completion of embryogenesis, Hh signaling becomes quiescent. 17 , 18 Hyperactive Hh caused by recurrent gain‐of‐function mutations in SMO is recognized as an oncogenic driver of cancers, including basal cell carcinoma (BCC) 19 and medulloblastoma (MB). 20 Most Hh signaling inhibitors (HHIs) suppress Hh signaling by directly targeting SMO, vismodegib and sonidegib and have been approved by the US Food and Drug Administration for use in advanced BCC. 21 , 22 Investigations of other HHIs in other cancers, including pancreatic, hematological, and prostate cancer, have also yielded attractive therapeutic results. 22
In prostate cancer, Hh pathway activity is required for the propagation and metastasis of tumors. 23 Hh and AR signaling are intimately intertwined, and Hh ligand expression is directly suppressed by AR signaling but is released in androgen‐depleted conditions, 24 suggesting that Hh signaling plays roles in prostate cancer cell survival after androgen ablation; in return, activated Hh signaling supports the progression of prostate cancer by sustaining the activity of AR signaling, which demonstrates the potential clinical feasibility of HHIs in treating CRPC. 25 , 26 However, the significance of Hh signaling in NEPC has not yet been investigated. In this study, we determined that the expression of SMO, the key transducer of Hh, was reduced/lost in NEPC and that SMO loss was associated with attenuated AR signaling, suggesting a novel role for SMO in the pathogenesis of NEPC.
2. MATERIALS AND METHODS
2.1. Data acquisition and processing
Transcriptome and clinical data were obtained from the publicly available cBioPortal web server, 27 the Gene Expression Omnibus database, and the Living Tumor Laboratory (LTL) database (www.living tumor laboratory. com). A total of six datasets (from five independent studies) containing clinical AdPC and NEPC samples and two independent datasets containing patient‐derived xenograft (PDX) AdPC and NEPC samples were analyzed. The expression of selected genes was compared between AdPC and NEPC. Data on copy number variations (CNVs) were obtained from cBioPortal. An enrichment analysis was performed with GSEAv4.0.3 software, 28 and hallmark gene sets from the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb) were used for pathway analysis.
2.2. Clinical sample collection
Tissue collection protocols were approved by the Ethics Committee of Shandong Provincial Hospital affiliated to Shandong University. Due to the retrospective nature of the study, patient consent was not required. All data were analyzed anonymously. A total of seven samples, including five from patients with morphologically identified NEPC and two from patients with AdPC mixed with NEPC who visited our hospital between January 2015 and April 2020, were subjected to immunohistochemistry (IHC) analysis. Among these seven patients, two with NEPC were previously treated with hormonal therapy, three with NEPC were diagnosed with primary treatment‐naïve prostate cancer, and two with AdPC mixed with NEPC were previously treated with hormonal therapy. Samples from 22 patients with high‐grade (Gleason score ≥7) AdPC and 5 patients with AdPC with neuroendocrine differentiation were also collected as part of this study. All tissues were obtained from surgical resection or needle biopsy.
2.3. Immunohistochemistry
IHC was performed using routine protocols at the Department of Pathology of our hospital. Paraffin‐embedded blocks were cut into 4‐μm serial sections, which were deparaffinized and rehydrated routinely. Antigen repair was conducted in Tris‐EDTA (pH 9.0) with high‐pressure steam for 3 min. Endogenous peroxidase activity was blocked with 3.0% hydrogen peroxide for 20 min. Then, the sections were incubated with antibodies against SMO, AR, CHGA, and synaptophysin (SYP). The list of primary antibodies and dilution ratios is given in Table S1. Hematoxylin and eosin staining was also conducted to obtain a clear view of the tissues. Images of these sections were acquired under a microscope and scored independently by two pathologists with more than 10 years of experience as reported previously. 29 Briefly, the proportion score represented the proportion of positive‐staining tumor cells, and was assigned as 0, none; 1, 1⁄100; 2, 1⁄100 to 1⁄10; 3, 1⁄10 to 1⁄3; 4, 1⁄3 to 2⁄3; and 5, 2⁄3. Next, the average intensity of positive tumor cells was assigned as an intensity score as below: none (−), weak (1+), moderate (2+), and strong (3+). The proportion and intensity scores were then added to obtain a total score.
2.4. Cell culture and reagents
LNCaP cells were purchased from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences, and C4‐2B cells were kindly provided by Xing Yutong from Nankai University. These cells were cultured in Roswell Park Memorial Institute 1640 (RPMI‐1640) (Gibco) supplemented with 10% fetal bovine serum (Gibco). RPMI‐1640 without phenol red and charcoal‐stripped fetal bovine serum was used to mimic androgen deprivation conditions in vitro. SAG, JQ1, ENZ, and DHT were purchased from Selleckchem, and were dissolved in dimethyl sulfoxide at stock concentrations of 200, 0.5, 10, and 10 µM, respectively. GANT61 (Selleckchem) was diluted in ethanol at a stock concentration of 10 mM.
2.5. Gene silencing
LNCaP cells were transfected with an short hairpin RNA (shRNA) to knockdown the expression of SMO. sh‐SMO and sh‐NC (GV115 vector) recombinant lentiviruses were purchased from GeneChem. The target sequence of sh‐SMO was 5′‐ATCGCTACCCTGCTGTTAT‐3′, with the scramble RNA serving as a control (sh‐NC, 5′‐TTCTCCGAACGTGTCACGT‐3′). Transfection efficiency was monitored by determining the percentage of green fluorescent protein‐positive cells and performing real‐time polymerase chain reaction (PCR) and western blot analyses.
2.6. Gene overexpression
LNCaP cells were transfected with Gli1 complementary DNA (cDNA) to overexpress Gli1. The Gli1 cDNA (GV492 vector) was purchased from GeneChem, and the transfection experiment was performed according to the manufacturer's instructions. Transfection efficiency was monitored by real‐time PCR and western blot analyses.
2.7. Western blot analysis
Cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes, which were then incubated with primary antibodies against AR, SMO, prostate‐specific antigen (PSA), Gli1, SYP, RE1 silencing transcription factor (REST), and β‐actin. All membranes were incubated with an horseradish peroxidase (HRP)‐conjugated secondary antibody. Proteins were revealed by using an Immobilon Western Chemiluminescent HRP Substrate Kit (Merck Millipore) and visualized by autoradiography. The list of primary antibodies and dilution ratios is given in Table S1. All western blot experiments were performed at least three times.
2.8. RNA isolation and real‐time polymerase chain reaction
Total RNA was extracted from LNCaP and C4‐2B cells using TRIzol reagent (Takara) according to the manufacturer's guidelines. One microgram of total RNA was used to generate first‐strand cDNA, and the relative expression of target genes was determined after normalization against β‐actin. A list of real‐time PCR primers used in this study is shown in Table S2. All experiments were performed in triplicate and repeated three times.
2.9. ELISA
LNCaP and C4‐2B cells were cultured in 6‐well plates until they were 60% confluent. Then the monolayers were washed three times with phosphate‐buffered saline and maintained in fresh medium for 2 days. The culture supernatants were collected and measured using an ELISA kit (JingMei Biological Engineering Co., Ltd) according to the manufacturer's instructions. The number of cells in each well was counted, and the PSA expression level in each well was normalized according to the cell number. The experiment was performed three times.
2.10. Statistical methods
Comparisons between two independent groups were performed with Student's t test unless stated otherwise. The data were statistically analyzed by using SPSS software (SPSS standard, version 18.0; SPSS, Inc.). Values of p < .05 were considered statistically significant unless stated otherwise.
3. RESULTS
3.1. SMO mRNA expression is downregulated in NEPC
Recent studies have shown that NEPC has a unique transcriptional profile compared to that of AdPC. To identify differences in Hh pathway genes between AdPC and NEPC, we examined the expression of six key gene components of the Hh axis (SHH, SMO, PTCH1, Gli1, Gli2, and Gli3) (Figure S1A) in three public RNA‐seq datasets of prostate cancer 30 , 31 , 32 and identified SMO as the gene with the most significant differential (lower) expression in NEPC compared to AdPC (Figure 1A–C). Three other datasets of prostate cancer from two studies 33 , 34 were also analyzed, the results of which confirmed the loss of SMO in NEPC (Figure 1D,E). Similar results were also obtained in the prostate cancer PDXs from the LTL dataset 35 (Figure S1D) and prostate cancer cell lines (Figure S1E).
Figure 1.
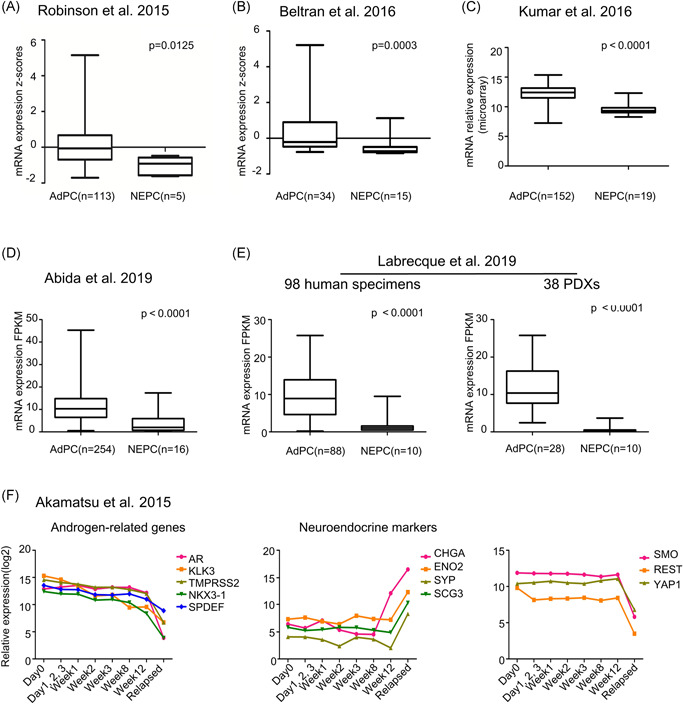
The messenger RNA (mRNA) of smoothened (SMO) is downregulated in neuroendocrine prostate cancer (NEPC). Differential SMO mRNA expression between prostate adenocarcinoma (AdPC) and NEPC in datasets provided by (A) Robinson et al., 32 (B) Beltran et al., 30 (C) Kumar et al., 31 (D) Abida et al., 34 and (E) Labrecque et al. 52 The Mann–Whitney test was used to compare the differences and p < .05 was considered significantly different. (F) Relative expression of androgen‐related genes (AR, KLK3, TMPRSS2, NKX3‐1, and SPDEF), neuroendocrine‐related genes (CHGA, ENO2, SYP, SCG3), REST, YAP1, and SMO at different time points in the LTL331R model after xenografting into castrated mice. REST, RE1 silencing transcription factor; SYP, synaptophysin [Color figure can be viewed at wileyonlinelibrary.com]
LTL331/331R 35 is a unique NEPC PDX used to study the transdifferentiation process of NEPC. The changes in tumor volume are shown in Figure S1F, which indicates disease progression. We next analyzed the dynamic changes in SMO gene expression in this model. Figure 1E shows that the mRNA levels of AR‐related genes (AR, KLK3, TMPRSS2, NKX3‐1, and SPDEF) were decreased while the levels of neuroendocrine markers (CHGA, ENO2, SYP, and SCG3) were increased during the transdifferentiation of NEPC. Previously reported NE‐associated TFs (ASCL1, SOX2, PEG10, BRN2, and SRRM4) 9 , 11 , 35 , 36 , 37 were also analyzed in this model and exhibited increased expression to varying degrees at different time points (Figure S1G). Interestingly, the expression of SMO remained stable for 12 weeks postcastration and was markedly downregulated at the terminal time points of NEPC relapse. The profile of SMO expression dynamics was highly similar to those of REST and YAP1, which were recently reported to be selectively lost in NEPC, 8 , 38 thus suggesting a role of SMO loss in the emergence of NEPC.
Analysis of CNVs in these datasets revealed no evidence of copy number loss for SMO in NEPC (Figure 2A–C), indicating a possible epigenomic mechanism underlying the downregulation of SMO. BRD4, a member of the bromodomain and extraterminal (BET) family, is an epigenetic reader that recognizes acetylated lysine residues of histone proteins and acts as a transcriptional regulator. 39 Reportedly, BRD4 plays an important role in prostate cancer progression, 40 and Hh signaling was shown to be an epigenetic target of BET in a mouse cell line. 41 To uncover the upstream signaling governing SMO expression in prostate cancer, we treated LNCaP and C4‐2B cells with a BET inhibitor (JQ1). Intriguingly, as expected, JQ1 treatment downregulated the SMO mRNA and protein levels in both cell lines (Figure 2D), suggesting that SMO expression is positively regulated by BET proteins.
Figure 2.
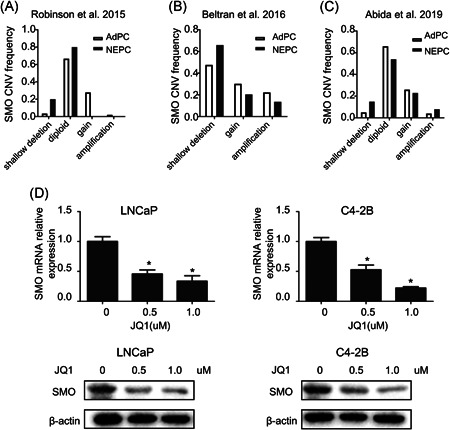
SMO may be regulated by BET inhibitor. Available CNV data for the SMO gene in prostate cancer from the datasets of (A) Robinson et al. 32 , (B) Beltran et al. 30 , and (C) Abida et al., 34 (D) LNCaP and C4‐2B cells were treated with JQ1 for 48 h, and the expression of SMO was assessed by real‐time PCR and western blot. *p < .05, versus the control group. AdPC, prostate adenocarcinoma; BET, bromodomain and extraterminal; CNV, copy number variation; NEPC, neuroendocrine prostate cancer; PCR, polymerase chain reaction; SMO, smoothened
3.2. Loss of SMO protein expression is prevalent in NEPC but rare in high‐grade AdPC
Given the specific downregulation of SMO mRNA in NEPC, we next assessed SMO protein levels in human prostate cancer tissues by IHC. The percentages and intensities of SMO, AR, SYP, and CHGA staining are summarized in Table 1 and Figure 3F. Among the morphologically identified cases of NEPC, 100% (5 of 5) showed complete loss of SMO, and heterogeneous positive staining for the classic NE markers SYP, and CHGA (Table 1; Figure 3B,C). In contrast, only 9% (2 of 22) of the high‐grade AdPC showed loss of SMO, and most AdPC tissues showed weak to strong staining of SMO (Table 1; Figure 3A). Interestingly, in two tissue samples of AdPC mixed with NEPCs, the NEPC components showed SMO loss, while the adenocarcinoma components showed strong positivity (Figure 3D,E), AR/SMO and SYP/CHGA staining were mutually exclusive. Moreover, the tissue samples of AdPC with neuroendocrine differentiation tissues were positively stained for SMO, AR, SYP, and CHGA in luminal cancer cells (Figure S2). Collectively, these results demonstrate that SMO expression is lost specifically in NEPC tissues.
Table 1.
Expression of SMO, AR, SYP, and CHGA in human prostate cancer
| Patient ID | Pathological classification | SMO | AR | SYP | CHGA |
|---|---|---|---|---|---|
| Patient #1 | AdPC | 90%, 3+ | 85%, 3+ | ‐ | ‐ |
| Patient #2 | AdPC | 65%, 1+ | 60%, 1+ | ‐ | ‐ |
| Patient #3 | AdPC | 85%, 3+ | 85%, 3+ | ‐ | ‐ |
| Patient #4 | AdPC | 90%, 3+ | 90%, 3+ | ‐ | ‐ |
| Patient #5 | AdPC | 75%, 1+ | 70%, 2+ | ‐ | ‐ |
| Patient #6 | AdPC | 90%, 3+ | 90%, 3+ | ‐ | ‐ |
| Patient #7 | AdPC | 55%, 1+ | ‐ | ‐ | ‐ |
| Patient #8 | AdPC | 65%, 2+ | 75%, 2+ | ‐ | ‐ |
| Patient #9 | AdPC | 45%, 3+ | 40%, 2+ | ‐ | ‐ |
| Patient #10 | AdPC | 90%, 2+ | 90%, 1+ | ‐ | ‐ |
| Patient #11 | AdPC | 90%, 1+ | 80%, 2+ | ‐ | ‐ |
| Patient #12 | AdPC | 65%, 3+ | 70%, 2+ | ‐ | ‐ |
| Patient #13 | AdPC | 60%, 1+ | ‐ | ‐ | ‐ |
| Patient #14 | AdPC | 90%, 2+ | 80%, 3+ | ‐ | ‐ |
| Patient #15 | AdPC | 80%, 1+ | 80%, 1+ | ‐ | ‐ |
| Patient #16 | AdPC | 85%, 3+ | 85%, 2+ | ‐ | ‐ |
| Patient #17 | AdPC | 50%, 2+ | 50%, 2+ | ‐ | ‐ |
| Patient #18 | AdPC | 70%, 1+ | 80%, 1+ | ‐ | ‐ |
| Patient #19 | AdPC | 80%, 2+ | 60%, 1+ | ‐ | ‐ |
| Patient #20 | AdPC | 75%, 2+ | 75%, 2+ | ‐ | ‐ |
| Patient #21 | AdPC | 85%, 3+ | 85%, 3+ | ‐ | ‐ |
| Patient #22 | AdPC | 75%, 2+ | 75%, 2+ | ‐ | ‐ |
| Patient #23 | AdPC with NE differentiation | 60%, 2+ | 70%, 2+ | 35%, 1+ | 40%, 1+ |
| Patient #24 | AdPC with NE differentiation | 90%, 2+ | 85%, 3+ | 40%, 2+ | 35%,1+ |
| Patient #25 | AdPC with NE differentiation | 65%, 1+ | 65%, 2+ | 45%, 2+ | 40%, 3+ |
| Patient #26 | AdPC with NE differentiation | 90%, 2+ | 90%, 3+ | 80%, 2+ | 80%, 2+ |
| Patient #27 | AdPC with NE differentiation | 70%, 1+ | 60%, 3+ | 10%, 1+ | ‐ |
| Patient #28 | NEPC | ‐ | ‐ | 80%, 3+ | 80%, 3+ |
| Patient #29 | NEPC | ‐ | ‐ | 80%, 2+ | 80%, 2+ |
| Patient #30 | NEPC | ‐ | ‐ | 5%, 3+ | 5%, 2+ |
| Patient #31 | NEPC | ‐ | ‐ | 5%, 2+ | 5%, 2+ |
| Patient #32 | NEPC | ‐ | ‐ | 80%, 2+ | 85%,2+ |
| Patient #33 | Mixture of AdPC and NEPC | 70%, 3+ | 60%, 3+ | 10%, 3+ | 10%, 3+ |
| Patient #34 | Mixture of AdPC and NEPC | 70%, 1+ | 60%, 2+ | 20%, 3+ | 20%, 3+ |
Abbreviations: AdPC, prostate adenocarcinoma; AR, androgen receptor; CHGA, chromogranin A; NE, neuroendocrine; NEPC, neuroendocrine prostate cancer; SMO, smoothened; SYP, synaptophysin.
Figure 3.
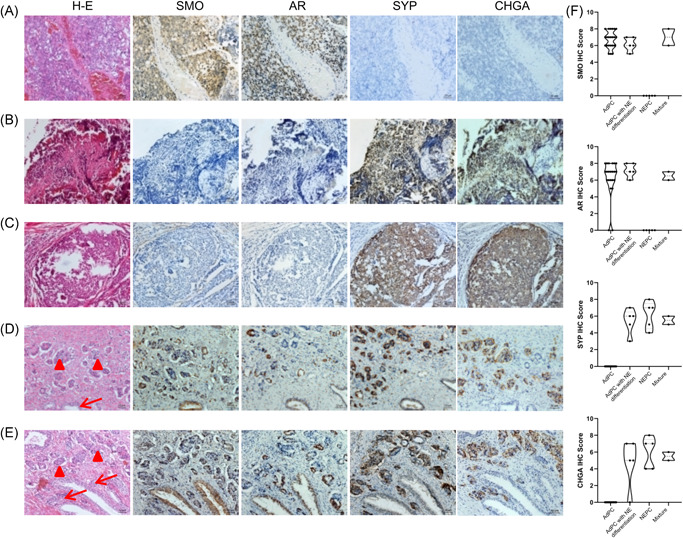
SMO is selectively underexpressed in human NEPC tissues. Representative IHC staining images of SMO, AR, SYP, and CHGA in tissue samples of (A) AdPC, (B, C) NEPC, and (D, E) AdPC mixed with NEPC. The arrows mark the AdPC region, and triangles mark the NEPC region. Scale bar 50 μm. (F) The IHC scores are summarized. AdPC, prostate adenocarcinoma; AR, androgen receptor; CHGA, chromogranin A; IHC, immunohistochemistry; NEPC, neuroendocrine prostate cancer; SMO, smoothened; SYP, synaptophysin [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Gene set enrichment analysis of global transcriptomic variations associated with SMO expression
The downregulated SMO expression in NEPC suggests that SMO may play roles in the transdifferentiation of NEPC. To confirm this, we performed GSEA to evaluate the global variations in SMO gene expression based on the data from the six published datasets listed in Figure 1. An SMO mRNA expression level below the lower quartile was defined as “SMO‐lo”; otherwise, it was defined as “SMO‐hi.” Gene sets with a normalized enrichment score >1.50 or <−1.50 and a q value ≤.002 were considered significant, as shown in Figures 4 and 5. In the six datasets, the most frequent molecular signatures that were positively associated with SMO expression were “ANDROGEN_RESPONSE” (6/6) and “CHOLESTER_HOMEOSTASIS” (5/6), while the most frequent signatures that were negatively associated with SMO expression were “E2F_TARGETS” (6/6), “PANCREAS_BETA_CELLS” (5/6), and “G2M_CHECKPOINT” (4/6) (Figure 4A–E). These results indicate that SMO downregulation is associated with several key signatures of NEPC, suggesting that SMO is a barrier in the transdifferentiation of AdPC and that SMO loss may drive the emergence of the NEPC phenotype by involving these pathways.
Figure 4.
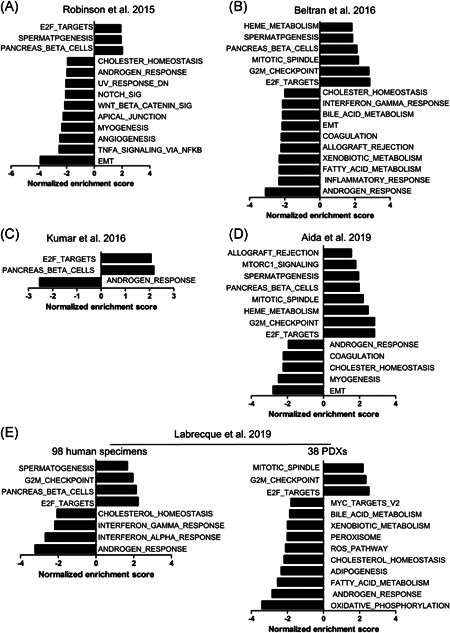
GSEA reveals that SMO downregulation is linked to key signatures of NEPC. GSEA was used in the comparison of “SMO‐lo” versus “SMO‐hi” in the datasets of (A) Robinson et al., 32 (B) Beltran et al., 30 (C) Kumar et al., 31 (D) Aida et al., 34 and (E) Labrecque et al., 52 . Representative pathways that were most significantly up‐or downregulated are shown. SMO mRNA expression levels below the lower quartile level were defined as “SMO‐lo,” and all other levels were defined as “SMO‐hi.” GSEA, gene set enrichment analysis; mRNA, messenger RNA; NEPC, neuroendocrine prostate cancer; SMO, smoothened
Figure 5.
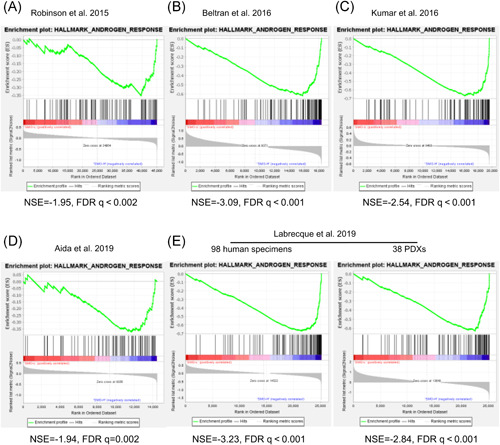
SMO expression is linked to the activation of AR signaling. GSEA (hallmark pathways) comparing “SMO‐lo” versus “SMO‐hi” prostate cancer transcriptomic samples revealed the most negatively enriched gene set, “ANDROGEN_RESPONSE.” AR, androgen receptor; FDR, false discovery rate; GSEA, gene set enrichment analysis; NES, normalized enrichment score [Color figure can be viewed at wileyonlinelibrary.com]
3.4. SMO knockdown inhibits AR signaling activity and reduces AR expression
The above results show that the “ANDROGEN_RESPONSE” is the most negatively enriched signature of SMO expression (Figure 5), suggesting that an association exists between SMO and AR signaling. A previous study by Chen 24 reported that AR suppressed the expression of Hh ligands in prostate cancer cells, which was confirmed by our herein, as exogenous androgen deprivation relieved the repression of SHH expression (Figure S3A). In contrast, SMO expression was not significantly different between the androgen (DHT) and androgen antagonist (ENZ) treatment groups in our study (Figure S3B), and similar results were obtained in the prostate cancer PDXs from the LTL dataset (Figure S3C). Together, these results suggest that SMO, unlike its ligand SHH, is not a regulatory target of AR.
Next, we assessed the effect of SMO loss on AR signaling by constructing stable SMO‐knockdown LNCaP and C4‐2B cell lines with a lentiviral‐delivered shRNA (termed sh‐SMO). As shown in Figure 6A,B, AR, and PSA expression was decreased in sh‐SMO cells compared with control cells (sh‐NC), and the expression levels of NE markers were not significantly different, which was consistent with our GSEA results (Figure 4). Because SMO reportedly exerts its effects mainly by activating Gli TFs, 15 we next assessed the effects of the Gli antagonist GANT61 and the Hh signaling agonist SAG on AR expression in sh‐SMO cells. Figure 7A shows that GANT61 further reduced AR and PSA protein expression, while SAG treatment partially restored AR and PSA expression, suggesting that Gli TFs are important for the regulation of AR signaling by SMO.
Figure 6.
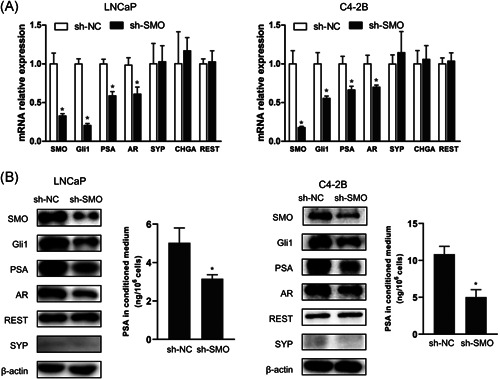
Knockdown of SMO inhibits AR pathway activity and reduces AR expression. LNCaP and C4‐2B cells were used to generate stable SMO‐knockdown cells using a lentiviral shRNA targeting SMO. (A) Real‐time PCR was used to assess the relative mRNA expression of markers related to Hh (SMO, Gli1), AR (AR, PSA), and NE (REST, SYP, CHGA); β‐actin was used as the loading control. *p < .05, versus sh‐NC. (B)Validation of the protein levels of candidate genes by western blot, and the secreted levels of PSA in the conditioned medium were assessed by enzyme‐linked immunosorbent assay (ELISA). *p < .05, versus sh‐NC (con); # p < .05, versus sh‐SMO (con). AR, androgen receptor; CHGA, chromogranin A; mRNA, messenger RNA; NC, negative control; NE, neuroendocrine; PCR, polymerase chain reaction; PSA, prostate‐specific antigen; REST, RE1 silencing transcription factor; shRNA, short hairpin RNA; SMO, smoothened; SYP, synaptophysin
Figure 7.
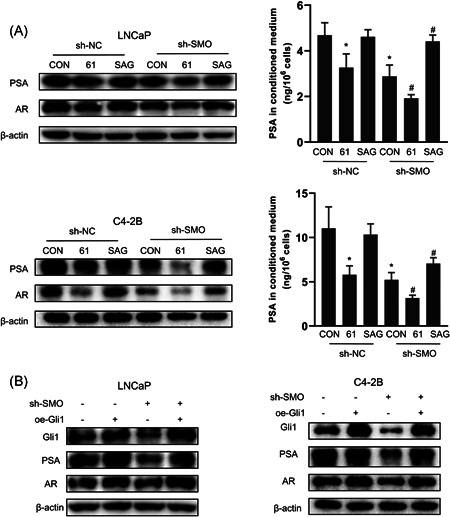
Knockdown of SMO suppresses AR signaling via Gli1. (A) LNCaP and C4‐2B cells were treated with an Hh antagonist (GANT61) or Hh activator (SAG) for 48 h, the expression of AR and PSA was assessed by western blot and ELISA. (B) sh‐SMO or sh‐NC LNCaP and C4‐2B cells were transfected with ectopic recombinant Gli1 cDNA (oe‐GLI1), and the expression of Gli1, AR, and PSA was assessed by western blot. AR, androgen receptor; cDNA, complementary DNA; NC, negative control; PSA, prostate‐specific antigen; SMO, smoothened
Gli1 is a well‐recognized downstream target as well as an effector TF of the Hh pathway, 42 and we consistently found that Gli1 expression was downregulated in sh‐SMO cells (Figure 6A,B). Notably, as shown in Figure 7B, overexpression of Gli1 in SMO‐knockdown cells rescued the expression of AR and PSA, indicating that Gli1 is the downstream effector of SMO that is involved in the regulation of AR signaling.
4. DISCUSSION
The development of highly aggressive NEPC in prostate cancer patients undergoing treatment with stringent AR blockade is a critical clinical issue. It is imperative to investigate the molecular mechanisms of NEPC pathogenesis and develop effective therapies for this lethal disease. In this study, we found that the expression of SMO, a key factor in Hh signaling, is dramatically downregulated or lost in NEPC by analyzing multiple public datasets of advanced prostate cancer. We confirmed this finding in clinical prostate cancer specimens obtained from our hospital by IHC analysis. In vitro experiments showed that knocking down SMO in LNCaP and C4‐2B cells downregulated the activity of AR signaling and AR gene expression, implying a supporting role of SMO loss in the transition from the AR‐positive luminal phenotype to the AR‐negative neural phenotype in prostate cancer.
SMO is a key transmembrane protein receptor of Hh signaling, and it was identified as an oncogene and a therapeutic target in BCC and MB. 19 , 20 In prostate cancer, Hh signaling was reported to play a driving role in the development of androgen blockade resistance, and HHIs targeting SMO were investigated as treatments for CRPC. 26 , 43 However, our results demonstrated that SMO was downregulated in NEPC compared with AdPC, suggesting a novel role of SMO in inhibiting the development of NEPC. As the widespread clinical application of potent APIs in CRPC led to the rising incidence of NEPC, the risk of HHIs causing NEPC should be reconsidered.
Currently, accumulating evidence indicates that the transition from AdPC to NEPC is a continuum encompassing a spectrum of intermediate cellular differentiation states. 44 Distinct molecular drivers of NEPC may exert their roles at different time points; for example, in the LTL331/331R PDX model, PEG10 was found to be activated in the very early stage of NEPC, 35 while we found that SMO expression was decreased in the late stage (Figure 1F), similar to the dynamic changes in REST and YAP1, two other repressors of NEPC development. 8 , 38 Our IHC data revealed that SMO protein expression was detectable in most AdPC tissues, two AdPC tissues with NE differentiation, and two AdPC loci with a mixture of AdPC and NEPC but was lost in NEPC tissues. These distinct temporal and spatial expression patterns of SMO suggest that it may be a useful biomarker of terminally differentiated NEPC cells, and additional studies with a large panel of clinical samples are warranted.
AR signaling plays a principal role in prostate cancer development and progression to CRPC and is notably attenuated in NEPC. 45 Our GSEA results indicated that “ANDROGEN_RESPONSE” had an extremely significant positive correlation with SMO expression. The relationship between AR and Hh signaling is complicated, as Hh signaling is suppressed by AR signaling under normal conditions, while Hh signaling is de‐repressed under androgen deprivation conditions, which plays a compensatory role in AR signaling to promote the survival of prostate cancer cells. 25 Our in vitro experiments using the sh‐SMO LNCaP and C4‐2B cell lines showed that SMO was closely related to AR signaling activity and AR gene expression, which is consistent with previous reports that Hh signaling supports the activity of AR signaling in androgen‐deprived and androgen‐independent prostate cancers. 25 We further found that the regulation of AR signaling by SMO was mediated by Gli1. These results provide new insights into the regulation of AR signaling and suggest that SMO acts as a barrier to the lineage transition of AdPC and that SMO loss may drive the transdifferentiation of NEPC characterized by the suppressing of AR signaling.
Our GSEA result is very interesting and implicative as it also indicated that SMO loss is associated with several other crucial molecular signatures of NEPC. “CHOLESTER_HOMEOSTASIS” was the second most frequent molecular signature positively correlated with SMO expression, and a close link exists among cholesterol, Hh signaling, and AR signaling. 46 , 47 , 48 , 49 Recently, a mass spectrometry‐based proteomic analysis revealed reduced expression level of proteins involved in lipid biosynthesis in NEPC PDXs, 50 suggesting a repressive role of lipid biosynthesis in NEPC pathogenesis. Given that NEPC is AR independent and resistant to APIs, how SMO loss affects steroidogenesis and androgen synthesis in prostate cancer cells is worthy of investigation. Our GSEA results also indicated that SMO loss is associated with the following enhanced molecular signatures: “E2F_TARGETS” and “G2M_CHECKPOINT.” Previous studies have reported aberrant features of cell cycle regulation, particularly increases in E2F activity and G2M checkpoint dysregulation 51 that distinguish NEPC from CRPC. Thus, we hypothesized that SMO loss is involved in the transdifferentiation of NEPC by remodeling prostate cancer cell proliferation. However, we did not observe a significant difference in cell proliferation rates between sh‐NC and sh‐SMO LNCaP cells (data not shown). Kaplan–Meier survival analysis of the Abida dataset 34 suggested that SMO loss was correlated with poor overall survival, although the difference was not statistically significant (Figure S4). We hypothesized that cell proliferation is not a direct downstream target of SMO, and that other factors are involved; however, further research is required.
In summary, our results indicate that SMO loss is characteristic of NEPC. SMO acts as a barrier to AdPC transdifferentiation and SMO loss may be drive NEPC by dysregulating AR signaling in a Gli1‐dependent manner. Thus, these results serve as a reminder to use caution when suing HHIs to treat CRPC.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Lili Wang, Chunxiao Wu, and Zhiming Lu conceived the experiments. Lili Wang, Haiying Li, Zhang Li, Ming Li, and Qi Tang carried out the experiments and analyzed data. Lili Wang wrote the manuscript under the guidance of Chunxiao Wu and Zhiming Lu.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank Hui Fan at Provincial Hospital Affiliated to Shandong University and Dr. Bo Han at the Qilu Hospital of Shandong University for their technical assistance. This study was supported by the National Natural Science Foundation of China (Grant number: 81372775).
Wang L, Li H, Li Z, et al. Smoothened loss is a characteristic of neuroendocrine prostate cancer. The Prostate. 2021;81:508–520. 10.1002/pros.24122
Contributor Information
Chunxiao Wu, Email: wuchx@126.com.
Zhiming Lu, Email: luzhiming@sdu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7‐33. [DOI] [PubMed] [Google Scholar]
- 2. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beltran H, Tomlins S, Aparicio A, et al. Aggressive variants of castration‐resistant prostate cancer. Clin Cancer Res. 2014;20(11):2846‐2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aggarwal R, Huang J, Alumkal JJ, et al. Clinical and genomic characterization of treatment‐emergent small‐cell neuroendocrine prostate cancer: a multi‐institutional prospective study. J Clin Oncol. 2018;36(24):2492‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conteduca V, Oromendia C, Eng KW, et al. Clinical features of neuroendocrine prostate cancer. Eur J Cancer. 2019;121:7‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kareta MS, Gorges LL, Hafeez S, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. 2015;16(1):39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X, Coleman IM, Brown LG, et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration‐resistant prostate cancer. Clin Cancer Res. 2015;21(20):4698‐4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bishop JL, Thaper D, Vahid S, et al. The master neural transcription factor BRN2 is an androgen receptor‐suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017;7(1):54‐71. [DOI] [PubMed] [Google Scholar]
- 10. Guo H, Ci X, Ahmed M, et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat Commun. 2019;10(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee AR, Gan Y, Tang Y, Dong X. A novel mechanism of SRRM4 in promoting neuroendocrine prostate cancer development via a pluripotency gene network. EBioMedicine. 2018;35:167‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dardenne E, Beltran H, Benelli M, et al. N‐Myc induces an EZH2‐mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30(4):563‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teng M, Zhou S, Cai C, Lupien M, He HH. Pioneer of prostate cancer: past, present and the future of FOXA1. Protein Cell. 2020;12:29‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang C, Wu H, Evron T, et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat Commun. 2014;5:4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arensdorf AM, Marada S, Ogden SK. Smoothened regulation: a tale of two signals. Trends Pharmacol Sci. 2016;37(1):62‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059‐3087. [DOI] [PubMed] [Google Scholar]
- 17. Berman DM, Desai N, Wang X, et al. Roles for Hedgehog signaling in androgen production and prostate ductal morphogenesis. Dev Biol. 2004;267(2):387‐398. [DOI] [PubMed] [Google Scholar]
- 18. Le V, He Y, Aldahl J, et al. Loss of androgen signaling in mesenchymal sonic hedgehog responsive cells diminishes prostate development, growth, and regeneration. PLOS Genet. 2020;16(1):e1008588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):342‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31(3):306‐310. [DOI] [PubMed] [Google Scholar]
- 21. Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cortes JE, Gutzmer R, Kieran MW, Solomon JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev. 2019;76:41‐50. [DOI] [PubMed] [Google Scholar]
- 23. Karhadkar SS, Steven Bova G, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707‐712. [DOI] [PubMed] [Google Scholar]
- 24. Chen M, Tanner M, Levine AC, Levina E, Ohouo P, Buttyan R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle. 2009;8(1):149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen M, Feuerstein MA, Levina E, et al. Hedgehog/Gli supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol Cancer. 2010;9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ibuki N, Ghaffari M, Pandey M, et al. TAK‐441, a novel investigational smoothened antagonist, delays castration‐resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int J Cancer. 2013;133(8):1955‐1966. [DOI] [PubMed] [Google Scholar]
- 27. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Nat Acad Sci. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand‐binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17(5):1474‐1481. [DOI] [PubMed] [Google Scholar]
- 30. Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration‐resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162(2):454. [DOI] [PubMed] [Google Scholar]
- 33. Labrecque MP, Coleman IM, Brown LG, et al. Molecular profiling stratifies diverse phenotypes of treatment‐refractory metastatic castration‐resistant prostate cancer. J Clin Invest. 2019;130:4492‐4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428‐11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akamatsu S, Wyatt AW, Lin D, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12(6):922‐936. [DOI] [PubMed] [Google Scholar]
- 36. Rapa I, Ceppi P, Bollito E, et al. Human ASH1 expression in prostate cancer with neuroendocrine differentiation. Mod Pathol. 2008;21(6):700‐707. [DOI] [PubMed] [Google Scholar]
- 37. Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53‐ and RB1‐deficient prostate cancer. Science. 2017;355(6320):84‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng S, Prieto‐Dominguez N, Yang S, et al. The expression of YAP1 is increased in high‐grade prostatic adenocarcinoma but is reduced in neuroendocrine prostate cancer. Prostate Cancer Prostatic Dis. 2020;23:661‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donati B, Lorenzini E, Ciarrocchi ABRD4. and Cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urbanucci A, Barfeld SJ, Kytölä V, et al. Androgen receptor deregulation drives bromodomain‐mediated chromatin alterations in prostate cancer. Cell Rep. 2017;19(10):2045‐2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846‐851. [DOI] [PubMed] [Google Scholar]
- 43. Gowda PS, Deng JD, Mishra S, et al. Inhibition of hedgehog and androgen receptor signaling pathways produced synergistic suppression of castration‐resistant prostate cancer progression. Mol Cancer Res. 2013;11(11):1448‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beltran H, Hruszkewycz A, Scher HI, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res. 2019;25(23):6916‐6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vashchenko N, Abrahamsson PA. Neuroendocrine differentiation in prostate cancer: implications for new treatment modalities. Eur Urol. 2005;47(2):147‐155. [DOI] [PubMed] [Google Scholar]
- 46. Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab. 2010;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang P, Nedelcu D, Watanabe M, et al. Cellular cholesterol directly activates smoothened in Hedgehog signaling. Cell. 2016;166(5):1176‐1187 e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luchetti G, Sircar R, Kong JH, et al. Cholesterol activates the G‐protein coupled receptor smoothened to promote Hedgehog signaling. eLife. 2016;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lubik AA, Nouri M, Truong S, et al. Paracrine sonic hedgehog signaling contributes significantly to acquired steroidogenesis in the prostate tumor microenvironment. Int J Cancer. 2017;140(2):358‐369. [DOI] [PubMed] [Google Scholar]
- 50. Flores‐Morales A, Bergmann TB, Lavallee C, et al. Proteogenomic characterization of patient‐derived Xxnografts highlights the role of REST in neuroendocrine differentiation of castration‐resistant prostate cancer. Clin Cancer Res. 2019;25(2):595‐608. [DOI] [PubMed] [Google Scholar]
- 51. McNair C, Xu K, Mandigo AC, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest. 2018;128(1):341‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Labrecque MP, Coleman IM, Brown LG, et al. Molecular profiling stratifies diverse phenotypes of treatment‐refractory metastatic castration‐resistant prostate cancer. J Clin Invest. 2019;129(10):4492‐4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.