

Abstract
Stilbenoids are natural compounds endowed with several biological activities, including cardioprotection and cancer prevention. Among them, (±)‐trans‐δ‐viniferin, deriving from trans‐resveratrol dimerization, was investigated in its ability to target DNA duplex and G‐quadruplex structures by exploiting NMR spectroscopy, circular dichroism, fluorescence spectroscopy and molecular docking. (±)‐trans‐δ‐Viniferin proved to bind both the minor and major grooves of duplexes, whereas it bound the 3’‐ and 5’‐ends of a G‐quadruplex by stacking on the outer quartets, accompanied by rearrangement of flanking residues. Specifically, (±)‐trans‐δ‐viniferin demonstrated higher affinity for the investigated DNA targets than its monomeric counterpart. Additionally, the methoxylated derivatives of (±)‐trans‐δ‐viniferin and trans‐resveratrol, i. e. (±)‐pterostilbene‐trans‐dihydrodimer and trans‐pterostilbene, respectively, were evaluated, revealing similar binding modes, affinities and stoichiometries with the DNA targets as their parent analogues. All tested compounds were cytotoxic at μM concentration on several cancer cell lines, showing DNA damaging activity consistent with their ability to tightly interact with duplex and G‐quadruplex structures.
Keywords: DNA, G-quadruplexes, resveratrol, stilbenoids, viniferin
The stilbenoid (±)‐trans‐δ‐viniferin, deriving from trans‐resveratrol dimerization, binds to minor and major grooves of B‐DNA, and to 3’‐ and 5’‐ends of G‐quadruplex DNA by stacking on the outer quartets accompanied by rearrangement of flanking residues. Consistent with the ability to tightly interact with different DNA secondary structures, (±)‐trans‐δ‐viniferin shows DNA damaging activity.
Introduction
Phenolic compounds are a class of plant secondary metabolites widely present in fruits, vegetables and legumes.[ 1 , 2 ] Over 8000 natural phenolic products have been described in edible plants, representing an important part of our diet as they are the most abundant antioxidants consumed by humans. [3]
Among natural phenolic compounds, stilbenoids are largely found in several fruits and crops, including blueberries, cranberries, peanuts and grapes. [4] Stilbenoids are both woody constitutive metabolites and phytoalexins, substances produced by plants in response to biotic and abiotic stress factors, such as microbes and fungi infections, UV radiations, physical trauma. [5] They share a common structure generally constituted by a C6‐C2‐C6 unit, i. e. a 1,2‐diphenylethylene moiety, with aromatic rings carrying one or more hydroxy groups. In the last decades, stilbenoids have been largely studied because of their diverse bioactivities, comprising cardioprotection, neuroprotection, anti‐diabetic and anti‐inflammatory properties, cancer prevention and treatment, [6] with trans‐resveratrol and its methoxylated analogue trans‐pterostilbene (Figure 1A, B) being the most deeply investigated compounds of this family.[ 7 , 8 , 9 , 10 , 11 ]
Figure 1.
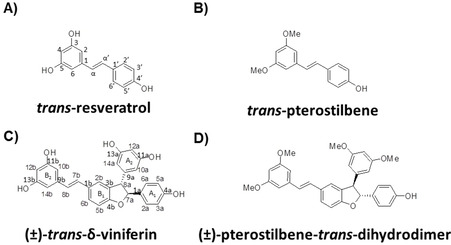
Chemical structures of A) trans‐resveratrol, B) trans‐pterostilbene, C) (±)‐trans‐δ‐viniferin and D) (±)‐pterostilbene‐trans‐dihydrodimer. Atom and ring numbering used in this study are reported in A) and C) according to Wilkens et al. [33]
A number of recent studies, covering a myriad of models, from cell cultures to animal studies, as well as human clinical trials, have brought to the fore their potential as anticancer compounds, mainly acting through epigenetic mechanisms.[ 12 , 13 , 14 , 15 , 16 , 17 , 18 ] Epigenetics regulates the genetic code at several levels. Among these are DNA changes, which include modifications to DNA methylation state, histones methylation, acetylation, ubiquitination and phosphorylation, and non‐coding RNA changes.
In this context, implications of G‐quadruplex formation in genomic DNA for epigenetic (re)programming and chromatin remodelling have been recently considered. [19] G‐quadruplexes are nucleic acid secondary structures originating from guanine‐rich oligonucleotide sequences which produce stacked arrangements of G‐quartets stabilized by Hoogsteen hydrogen bonding. Compared to duplex DNA, G‐quadruplexes exhibit higher structural polymorphism and can adopt various topologies, i. e. parallel, antiparallel and hybrid. These structures have received significant attention as potential therapeutic targets.[ 20 , 21 ] Indeed, G‐quadruplexes are found in the promoter regions of several genes associated with the development of cancer (e. g. c‐myc, bcl‐2 and c‐kit oncogenes), where stabilization of the folded G‐quadruplexes due to ligand interactions is proposed to inhibit the binding of transcription factors, leading to downstream silencing of oncogene expression.[ 22 , 23 , 24 ] Notably, also human telomeric sequences are able to form G‐quadruplex structures that are not recognized by telomerase, enzyme involved in telomere elongation. Since telomerase expression is upregulated in cancer cells, the discovery of ligands able to stabilize telomeric G‐quadruplex structures represents an appealing strategy for targeted anticancer therapies. [25]
Several groups have reported that trans‐resveratrol and its analogues, in addition to producing DNA damage, activate DNA repair mechanisms in various cancer cell lines, including prostate, colon and breast cancer cells.[ 26 , 27 , 28 ]
In this context, some of us have recently investigated the binding of trans‐resveratrol and its natural precursor trans‐polydatin to various DNA model systems forming duplex or G‐quadruplex structures. The biological effects of trans‐resveratrol and trans‐polydatin on melanoma cells well correlated with the biophysical data, proving that the interactions with DNA, observed for both compounds, can be somehow involved in the mechanisms explaining their anticancer activity.[ 29 , 30 ] In particular, trans‐resveratrol was found to induce higher antiproliferative effects, higher inhibition of telomerase activity and more efficient reduction of the expression of c‐myc oncogene compared to trans‐polydatin.
Encouraged by these results on the monomeric stilbenoid trans‐resveratrol, the DNA interactions and related activity on cancer and normal cells have been here in‐depth investigated for the dimeric derivative thereof, i. e. (±)‐trans‐δ‐viniferin (Figure 1C). This product, obtained through trans‐resveratrol self‐condensation, has a three‐dimensional skeleton which is intrinsically endowed with a higher number of functional groups and higher structural complexity than trans‐resveratrol itself, thus showing in principle higher potential for selective binding to a specific target. Additionally, the methoxylated analogues of both trans‐resveratrol and (±)‐trans‐δ‐viniferin, i. e. trans‐pterostilbene and (±)‐pterostilbene‐trans‐dihydrodimer (Figure 1D) respectively, have been also synthesized according to reported procedures [31] and here examined for comparison.
In detail, (±)‐trans‐δ‐viniferin as well as the pure enantiomers (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin [32] have been studied in their interaction with B‐DNA duplex and G‐quadruplex models by NMR, circular dichroism (CD), fluorescence spectroscopy and molecular docking, and compared in their binding behaviour to trans‐resveratrol, trans‐pterostilbene and (±)‐pterostilbene‐trans‐dihydrodimer. Two B‐DNA duplex models have been selected aiming at evaluating the ability of all the natural compounds of interest to interact with different kinds of duplex‐forming DNA sequences which can be typically found in vivo, i. e. CG‐ and AT‐rich sequences. Moreover, a parallel G‐quadruplex model has been also examined in order to assess the binding ability of the studied natural compounds towards the G‐quadruplex topology most commonly found in cells.[ 22 , 23 , 24 ] Then, biological assays have been performed to evaluate the antiproliferative effects on cancer cells and the DNA damage ability of the dimeric and monomeric stilbenoids. Altogether these data allowed a deep insight into the capacity of these compounds to bind different DNA models, in duplex as well as in G‐quadruplex form, which will be helpful for the design of novel selective DNA targeting drugs based on stilbenoid derivatives.
Results
NMR studies
To gain structural insights into the interactions of (±)‐trans‐δ‐viniferin with both DNA duplex and G‐quadruplex models, 31P and 1H NMR experiments were performed.
31P and 1H NMR experiments on (±)‐trans‐δ‐viniferin with B‐DNA duplex models
The self‐complementary oligonucleotides d(CGTACG)2 (forming the duplex structure here named ds6) and d(AAGAATTCTT)2 (forming the duplex structure here named ds10) were used as models for CG‐ and AT‐rich sequences, respectively. These duplex‐forming oligonucleotides have been extensively investigated by both 1H and 31P NMR proving to be good models, though short, of B‐DNA.[ 34 , 35 , 36 , 37 ] Folding of these oligomers into a duplex conformation was obtained at 15 °C in 110 mM Na+‐containing buffer. The 1H NMR spectra displayed signals in a region ranging from 12.4 to 13.9 ppm that were attributed to the imino protons involved in the CG and AT Watson‐Crick base pairs. In particular, only two and three signals were detected for ds6 and ds10 duplexes, respectively, because of the fraying process of the terminal base pairs (Figure S1A, B, bottom).
It is known that monitoring 31P resonance is a sensitive method to detect changes in the alfa=O(3’)‐P‐O(5’)‐C(5’) and zeta=C(3’)‐O(3’)‐P−O(5’) angles of the oligonucleotides from a gauche‐gauche conformation (−60°, −90°) to a gauche‐trans conformation (−60°, +180°) when an intercalation process occurs. Even small conformational changes at these angles are usually associated with a downfield shift up to 1.0–2.5 ppm for 31P resonances.[ 38 , 39 ] The 31P spectra acquired upon addition of (±)‐trans‐δ‐viniferin to a solution of ds6 duplex, at different ratios R=[(±)‐trans‐δ‐viniferin]/[DNA], did not show shift variation of any signal (Figure 2A and Table 1). Moreover, in the titration experiment, the 1H NMR spectra showed only generalized line broadening without displaying chemical shift changes (Figures S1A and S2 and Table S1). This behaviour suggested that complex equilibria, intermediate‐to‐fast on the NMR time scale, occurred among multiple, different species in solution. Consistently with the generalized line broadening observed in 1D‐NMR spectra, no intermolecular Nuclear Overhauser Effect (NOE) contacts between (±)‐trans‐δ‐viniferin and ds6 duplex were observed in 2D‐NOESY experiments.
Figure 2.
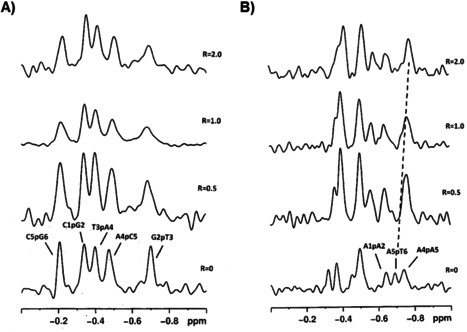
1H‐decoupled 31P NMR spectra of A) ds6 and B) ds10 duplexes treated with (±)‐trans‐δ‐viniferin at 15 °C in H2O/D2O (9 : 1), 100 mM NaCl, 10 mM sodium phosphate buffer (pH 7.0), at different ratios R=[(±)‐trans‐δ‐viniferin]/[DNA]. Dashed line in B) indicates the upfield shift of the A5pT6 signal.
Table 1.
31P NMR chemical shift assignments of the signals in the free ds6 or ds10 duplex and in their complexes with (±)‐trans‐δ‐viniferin.[a,b] |Δδ|≥0.10 are highlighted in bold.
|
ds6 |
|
Δδ[c] |
ds10 |
|
Δδ[c] |
|---|---|---|---|---|---|
|
C1pG2 |
−0.35 |
0.00 |
A1pA2 |
−0.65 |
+0.04 |
|
G2pT3 |
−0.70 |
0.00 |
A2pG3 |
−0.38 |
−0.07 |
|
T3pA4 |
−0.40 |
0.00 |
G3pA4 |
−0.50 |
0.00 |
|
A4pC5 |
−0.50 |
−0.01 |
A4pA5 |
−0.80 |
−0.05 |
|
C5pG6 |
−0.22 |
−0.02 |
A5pT6 |
−0.80 |
−0.10 |
|
|
|
|
T6pT7 |
−0.57 |
−0.07 |
|
|
|
|
T7pC8 |
−0.40 |
+0.03 |
|
|
|
|
C8pT9 |
−0.50 |
0.00 |
|
|
|
|
T9pT10 |
−0.40 |
−0.05 |
[a] Measured at 15 °C in H2O/D2O (9 : 1), 100 mM NaCl, 10 mM sodium phosphate buffer (pH 7.0) and R=2.0. [b] Measured in ppm from external DSS. [c] Δδ=δbound‐δfree.
1H and 31P NMR titration experiments performed with ds10 duplex gave different results: although also in this case no relevant chemical shift variation was detected in 1H NMR spectra, the signals belonging to its 5’AATT3’ tract were slightly perturbed (Figures S1B and S3 and Table S1). The 31P NMR spectra did not show downfield chemical shift variations of the signals indicating that no major, significant change in the backbone of the oligonucleotide occurred. Nevertheless, a small upfield chemical shift variation (Δδ=−0.10) was observed for the signal attributed to A5pT6 (Figure 2B and Table 1).
To obtain a deeper insight into the interactions between (±)‐trans‐δ‐viniferin and ds10 duplex, 2D‐NOESY experiments were performed. The resonances of (±)‐trans‐δ‐viniferin in the complex were attributed by TOCSY and ROESY experiments on the basis of the resonances of the free ligand (Figure S4 and Table S2). (±)‐trans‐δ‐Viniferin signals were mostly overlapped to anomeric protons of the oligomer. However, ligand protons at δ 6.79 (H3,5a), 6.42 (H10,14b) and 5.42 (H7a) ppm showed NOEs contacts with H2 and H1’ protons of A and T units in the 5’AATT3’ tract, located in the minor groove of the duplex (Figure 3 and Table 2). In addition, protons H3,5a and H10,14b of (±)‐trans‐δ‐viniferin displayed NOE interactions with the aromatic H6 of T6 located in the major groove (Figure 3 and Table 2). No NOE cross‐peaks were observed between the imino protons of ds10 duplex and the ligand protons.
Figure 3.
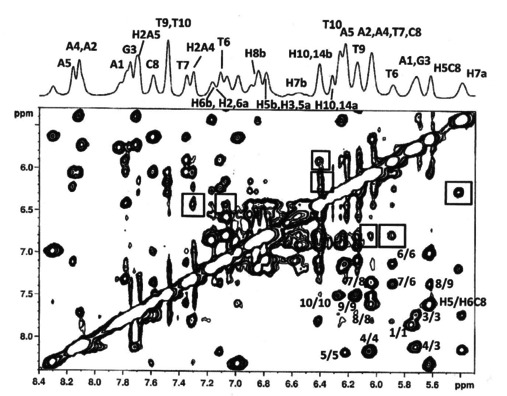
Selected region of 2D‐NOESY spectrum of ds10 duplex treated with (±)‐trans‐δ‐viniferin at 15 °C in H2O/D2O (9 : 1), 100 mM NaCl, 10 mM sodium phosphate buffer (pH 7.0), at R=[(±)‐trans‐δ‐viniferin]/[DNA]=2.0. The numbers indicate intra‐ and inter‐residue NOE interactions. The boxes indicate the NOE contacts between the ligand and ds10 duplex (see Table 2).
Table 2.
Intermolecular NOE interactions between (±)‐trans‐δ‐viniferin and ds10 duplex.[a,b]
|
(±)‐trans‐δ‐viniferin protons [ppm] |
ds10 protons |
|---|---|
|
H3,5a (6.79) |
H1’T6 |
|
H3,5a (6.79) |
H1’T7 |
|
H3,5a (6.79) |
H6T6 |
|
H7a (5.42) |
H1’A5 |
|
H10,14b (6.42) |
H1’T6 |
|
H10,14b (6.42) |
H6T6 |
|
H10,14b (6.42) |
H1’T7 |
|
H10,14b (6.42) |
H1’A4 |
|
H10,14b (6.42) |
H2 A4 |
[a] Measured at 15 °C in H2O/D2O (9 : 1), 100 mM NaCl, 10 mM sodium phosphate buffer (pH 7.0) and R=2.0. [b] Measured in ppm from external DSS.
Overall, these findings allowed excluding an intercalative binding mode in the interaction of (±)‐trans‐δ‐viniferin with the two investigated duplex models. On the other hand, binding to both minor and major grooves appeared to be the preferential binding mode of (±)‐trans‐δ‐viniferin to ds10 duplex, while the line broadening of 1H NMR signals together with the absence of NOE interactions did not allow to get detailed information about the preferred binding sites of (±)‐trans‐δ‐viniferin on the ds6 duplex.
1H NMR experiments of (±)‐trans‐δ‐viniferin with a G‐quadruplex model
The imino proton signals of G‐quadruplex‐forming oligonucleotides are correlated with their well‐defined global structure. The parallel G‐quadruplex Pu22T14T23, originating from c‐myc oncogene promoter of sequence d(TGAGGGTGGGTAGGGTGGGTAA), comprising two G to T mutations, was chosen as a target model because it gave higher quality spectra in K+ solution in comparison to the wild‐type oligonucleotide (Figure 4A). Notably, Pu22T14T23 has already proved to be a good model to study G‐quadruplex/ligand interactions.[ 23 , 40 ] Twelve imino proton signals were observed and assigned (Figure 4B, bottom), [23] consistent with the formation of a G‐quadruplex structure composed of three stacked quartets. [40]
Figure 4.

A) Schematic representation of Pu22T14T23 G‐quadruplex. B) Imino protons region of the 1H NMR titration spectra of Pu22T14T23 G‐quadruplex with (±)‐trans‐δ‐viniferin at 25 °C in H2O/D2O (9 : 1), 70 mM KCl, 25 mM potassium phosphate buffer (pH 6.9), at different ratios R=[(±)‐trans‐δ‐viniferin]/[DNA].
1H NMR titration experiments were performed by adding increasing amounts of (±)‐trans‐δ‐viniferin to the G‐quadruplex solution with ratios R=[(±)‐trans‐δ‐viniferin]/[DNA] ranging from 0 to 2.0. A single new set of only nine imino proton signals, because of some overlapping, was observed at R=2.0 (Figure 4B). This suggested that well‐defined DNA/ligand complexes were present in our conditions and allowed assigning imino and aromatic protons by 2D‐NOESY spectra (Table S3). In the NOESY spectrum, the sequential and imino proton inter‐residue NOE interactions of Pu22T14T23 G‐quadruplex in the complex with (±)‐trans‐δ‐viniferin were detected (Table S4 and Figures S5 and S6). The addition of (±)‐trans‐δ‐viniferin caused perturbations to the chemical shifts of the imino protons (Figure 4B and Table S3), giving a first indication that an interaction between (±)‐trans‐δ‐viniferin and Pu22T14T23 G‐quadruplex occurred. In particular, signals belonging to both 3’‐end (G9, G18 and G22) and 5’‐end (G16) quartets, as well as to G17 in the middle quartet, were the most shifted ones. In detail, upfield shift variations were found for the imino protons of G16, G17 and G18 in the complex with (±)‐trans‐δ‐viniferin, whereas for G9 and G22 downfield shift variations were observed (Figure 4B and Table S3). Considering that typical end‐stackers have been reported to induce upfield shifts in the guanine residues of quartets in the G‐quadruplex NMR spectra,[ 23 , 40 ] the chemical shift changes induced by (±)‐trans‐δ‐viniferin reflect the complexity of the ligand interaction with the G‐quadruplex (see fluorescence spectroscopy experiments and molecular modelling sections).
The 5’‐ and 3’‐end flanking residues did not present NOE contacts with the ligand, whereas some chemical shift variations were observed for 3’‐end flanking residues, suggesting a rearrangement of this segment induced by the ligand. In detail, while no chemical shift variations were observed for protons of A6 at 5’‐end, thus indicating that A6 was still stacked on G7, [41] T23 methyl protons underwent a downfield shift variation, proving a partial de‐stacking of T23 upon addition of the ligand. Additionally, the downfield shift observed for A24 and G9 protons suggested that A24 was no more involved in stacking with the T23:A25 base pair, and A25 was no more folded over the G9 aromatic moiety, as observed in the free Pu22T14T23 G‐quadruplex structure. [41] Conversely, no relevant chemical shift changes were detected for the residues located in the loops of the G‐quadruplex, i. e. T10, T14, A15 and T19 (Figures S7 and S8 and Table S3). Moreover, new broad peaks were detected upon ligand addition in the range of 5.5–6.8 ppm that were attributed to the protons of (±)‐trans‐δ‐viniferin. Their broad shape indicated that the ligand could either move in the binding site or bind to different binding sites of the target (Figure 5). Unfortunately, the overlap between the signals of G‐quadruplex deoxyribose moieties protons and (±)‐trans‐δ‐viniferin protons prevented the unambiguous assignment of the protons belonging to the ligand. Nevertheless, few intermolecular NOE contacts with imino protons of G9, G13, G16 and G18 were detected (Figure S9). These NOEs were in agreement with the relatively large chemical shift perturbation observed in 1H NMR spectra for these residues.
Figure 5.
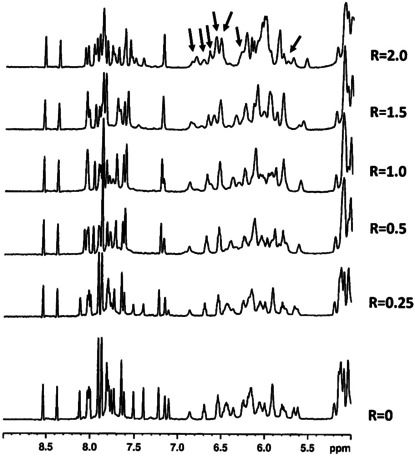
Aromatic and anomeric protons region of the 1H NMR titration spectra of Pu22T14T23 G‐quadruplex with (±)‐trans‐δ‐viniferin at 25 °C in H2O/D2O (9 : 1), 70 mM KCl, 25 mM potassium phosphate buffer (pH 6.9), at different ratios R=[(±)‐trans‐δ‐viniferin]/[DNA]. The arrows indicate the (±)‐trans‐δ‐viniferin signals.
Overall, these findings suggested that the binding of (±)‐trans‐δ‐viniferin did not disrupt the original parallel fold of the Pu22T14T23 G‐quadruplex. However, rearrangements of the flanking residues covering the external quartets were observed as a consequence of the ligand binding at both the 3’‐ and 5’‐end quartets through stacking mode.
Circular dichroism experiments
Circular dichroism experiments were performed on ds6 and ds10 duplexes and Pu22T14T23 G‐quadruplex in the absence and presence of (±)‐trans‐δ‐viniferin, trans‐resveratrol, trans‐pterostilbene and (±)‐pterostilbene‐trans‐dihydrodimer to assess the effects of these ligands on different DNA secondary structures. The pure enantiomers (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin were also studied in order to investigate the role of these two different configurations of trans‐δ‐viniferin in the DNA binding ability. First, CD spectra and CD‐melting experiments were carried out on ds6 and ds10 duplexes and Pu22T14T23 G‐quadruplex alone to characterize the CD behaviour of the three DNA models of choice. The analysis of CD spectra indicated that ds6 and ds10 folded into duplexes adopting B‐conformation, featured by a maximum at 279 or 278 nm and a minimum at 257 or 248 nm respectively (Figure S10A, B), while Pu22T14T23 folded into a parallel G‐quadruplex, featured by a maximum at 265 nm and a minimum at 244 nm (Figure S10C, black line), confirming the expected conformations for these duplex and G‐quadruplex structures under the here used conditions,[ 42 , 43 , 44 ] i. e. 110 mM Na+‐ or 95 mM K+‐containing buffer for duplexes or G‐quadruplex, respectively. The CD‐melting experiments, recorded by following the CD changes at the wavelength of intensity minimum or maximum characteristic of each DNA secondary structure, allowed determining Tm values of 18, 22 and ca. 92 (±1) °C for ds6, ds10 duplexes and Pu22T14T23 G‐quadruplex, respectively (Figure S11A−C, black lines). The very high stability found for Pu22T14T23 G‐quadruplex in the above conditions prevented the observation of a full sigmoidal melting curve and the consequent determination of its melting temperature with good accuracy. To overcome this drawback, Pu22T14T23 G‐quadruplex was studied alone and in its interaction with the different ligands in a buffer containing a lower concentration of potassium ions, i. e. 10 mM. Under these conditions, Pu22T14T23 still folded in a parallel G‐quadruplex structure showing the same CD signature as observed at higher K+ concentration (Figure S10C, red line), but with the remarkably lower Tm of 75 (±1) °C (Figure S11C, red line). Then, solutions of ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex were titrated with increasing amounts of each ligand and CD spectra recorded after each addition (Figures 6 and S12‐S16). The contribution of the chiral ligands (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin, even if of low intensity (data not shown), was taken into consideration and subtracted at each titration point for all the systems containing the two pure enantiomers (Figures S12 and S13). No significant variation of the CD profiles was observed for any of the investigated systems (Figures 6 and S12‐S16), thus proving that the overall folds of the here studied duplexes and G‐quadruplex were preserved even upon ligand binding,[ 45 , 46 , 47 ] consistently with the NMR results. Moreover, no induced CD signal was observed for all the investigated systems (data not shown).
Figure 6.
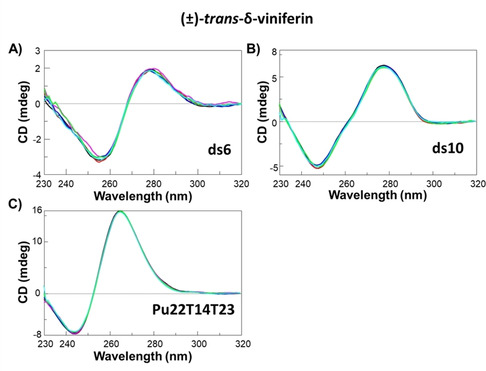
CD spectra of solutions of: A) ds6 duplex, B) ds10 duplex and C) Pu22T14T23 G‐quadruplex at 2 μM DNA concentration at A), B) 100 mM NaCl, 10 mM sodium phosphate buffer (pH 7.0) and 15 °C or C) 5 mM KCl, 5 mM potassium phosphate buffer (pH 6.9) and 25 °C, in the absence (black lines) and presence of 0.5 (red lines), 1 (blue lines), 2 (pink lines), 3 (green lines) or 4 (light blue lines) molar equivalents of (±)‐trans‐δ‐viniferin.
CD‐melting experiments were also performed to evaluate the effects on the ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex thermal stability upon incubation with each of the investigated ligands. CD‐melting curves were recorded in the presence of each ligand at 1 : 4 DNA:ligand ratio (Figures S17 and S18). The Tm and ΔTm values for each of the investigated systems are reported in Table 3. No significant stabilizing effects were observed on ds6 duplex, with the only exception of (R,R)‐trans‐δ‐viniferin (ΔTm=+2 °C). On the other hand, very slight stabilizing effects were induced by (±)‐δ‐trans‐viniferin, (R,R)‐trans‐δ‐viniferin, (S,S)‐trans‐δ‐viniferin and trans‐resveratrol on Pu22T14T23 G‐quadruplex (ΔTm=+1 °C), while trans‐pterostilbene or (±)‐pterostilbene‐trans‐dihydrodimer induced no stabilization of Pu22T14T23 G‐quadruplex. Notably, the highest stabilizing effects were detected for all the ligands on ds10 duplex. Interestingly, (±)‐trans‐δ‐viniferin was able to stabilize ds10 duplex slightly more (ΔTm=+4 °C) than the single pure enantiomers (R,R)‐trans‐δ‐viniferin (ΔTm=+2 °C) and (S,S)‐trans‐δ‐viniferin (ΔTm=+3 °C). A similar stabilization as found for (±)‐δ‐trans‐viniferin was observed for trans‐resveratrol and trans‐pterostilbene (ΔTm=+4 °C), while (±)‐pterostilbene‐trans‐dihydrodimer produced only a slight increase of Tm of ds10 duplex (ΔTm=+2 °C), revealing minor stabilizing effects on this DNA sequence.
Table 3.
Melting temperatures (Tm) for solutions of ds6 and ds10 duplexes and Pu22T14T23 G‐quadruplex in the absence and presence of 4 molar equivalents of each ligand. Each ΔTm was calculated as the difference between Tm of 1 : 4 DNA/ligand ratio system and Tm of DNA alone.
|
|
Tm [°C] (±1) |
ΔTm [°C] |
||||
|---|---|---|---|---|---|---|
|
Ligand/DNA target |
ds6 |
ds10 |
Pu22T14T23 |
ds6 |
ds10 |
Pu22T14T23 |
|
No ligand |
18 |
22 |
75 |
– |
– |
– |
|
(±)‐trans‐δ‐ viniferin |
18 |
26 |
76 |
0 |
+4 |
+1 |
|
(R,R)‐trans‐δ‐ viniferin |
20 |
24 |
76 |
+2 |
+2 |
+1 |
|
(S,S)‐trans‐δ‐ viniferin |
18 |
25 |
76 |
0 |
+3 |
+1 |
|
trans‐resveratrol |
18 |
26 |
76 |
0 |
+4 |
+1 |
|
trans‐pterostilbene |
18 |
26 |
75 |
0 |
+4 |
0 |
|
(±)‐pterostilbene‐trans‐ dihydrodimer |
18 |
24 |
75 |
0 |
+2 |
0 |
Fluorescence spectroscopy experiments
To get information about the binding stoichiometry and constants for the complexes formed between trans‐δ‐viniferin, trans‐resveratrol, trans‐pterostilbene and (±)‐pterostilbene‐trans‐dihydrodimer and ds6, ds10 duplexes or Pu22T14T23 G‐quadruplex, titration fluorescence experiments were performed. In detail, stilbenoid solutions were titrated with increasing amounts of ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex solutions, and, after each addition, the corresponding fluorescence spectra were recorded. A significant fluorescence enhancement was observed in all titration experiments (Figures S19–S23), except for titrations of (±)‐pterostilbene‐trans‐dihydrodimer with both the investigated duplex and G‐quadruplex structures, which resulted in fluorescence quenching (Figure S24). These data further corroborated the effective interaction between the here studied ligands and the duplex/G‐quadruplex structures of choice. The fraction of bound ligand was then calculated from the obtained fluorescence intensity values and reported as a function of the DNA concentration. These experimental data were fitted with an independent and equivalent‐sites model, [48] thus providing binding stoichiometries and constants for all DNA/ligand systems (Figures S25–S28 and Table 4). A binding stoichiometry of 1 : 3 DNA/ligand was found for the complexes between (±)‐trans‐δ‐viniferin, (R,R)‐trans‐δ‐viniferin, (S,S)‐trans‐δ‐viniferin and ds6 and ds10 duplexes, as well as for the complexes between (±)‐trans‐δ‐viniferin and Pu22T14T23 G‐quadruplex, whereas a binding stoichiometry of 1 : 2 DNA/ligand was observed for (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin in their interaction with Pu22T14T23 G‐quadruplex. Apparent binding constants of (6.1±6.0) ×106, (1.1±1.9) ×107 or (1.3±0.9)×107 M−1 were found for the complexes between (±)‐trans‐δ‐viniferin and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively. On the other hand, apparent binding constants of (5.7±5.3) ×106, (4.4±6.3)×106 or (1.7±1.5)×106 M−1 were obtained for the complexes between (R,R)‐trans‐δ‐viniferin and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively, while apparent binding constants of (1.0±1.4) ×107, (1.7±1.0)×106 or (0.5±0.7)×106 M−1 were obtained for the complexes between (S,S)‐trans‐δ‐viniferin and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively.
Table 4.
Binding constant (K b) and stoichiometry (n) values for complexes of each ligand with ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, obtained by fitting fluorescence data with an independent and equivalent‐sites model. The errors associated to the binding constant and stoichiometry values are based on the fit.
|
Ligand/DNA target |
ds6 |
ds10 |
Pu22T14T23 |
|
|---|---|---|---|---|
|
(±)‐trans‐δ‐viniferin |
K b (M−1) |
(6.1±6.0)×106 |
(1.1±1.9)×107 |
(1.3±0.9)×107 |
|
n |
3.3±0.6 |
3.1±0.7 |
2.8±0.2 |
|
|
(R,R)‐trans‐δ‐viniferin |
K b (M−1) |
(5.7±5.3)×106 |
(4.4±6.3)×106 |
(1.7±1.5)×106 |
|
n |
3.5±0.6 |
3.3±1.4 |
2.2±0.9 |
|
|
(S,S)‐trans‐δ‐viniferin |
K b (M−1) |
(1.0±1.4)×107 |
(1.7±1.0)×106 |
(0.5±0.7)×106 |
|
n |
3.4±0.6 |
2.8±0.8 |
1.8±1.7 |
|
|
trans‐resveratrol |
K b (M−1) |
(1.6±1.4)×106 |
(1.6±1.3)×106 |
(1.2±0.6)×106 |
|
n |
1.2±0.5 |
1.3±0.5 |
0.9±0.3 |
|
|
trans‐pterostilbene |
K b (M−1) |
(1.2±0.5)×106 |
(1.6±0.9)×106 |
(3.9±0.9)×106 |
|
n |
0.9±0.2 |
1.2±0.3 |
0.8±0.1 |
|
|
(±)‐pterostilbene‐trans‐dihydrodimer |
K b (M−1) |
(1.8±1.8)×106 |
(0.9±1.3)×106 |
(1.9±1.5)×106 |
|
n |
2.1±0.9 |
2.0±0.8 |
2.2±0.8 |
|
A binding stoichiometry of 1 : 1 and binding constants of (1.6±1.4)×106, (1.6±1.3) ×106 or (1.2±0.6) ×106 M−1 were found for the complexes between trans‐resveratrol and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively. The same binding stoichiometry as trans‐resveratrol and binding constants of (1.2±0.5)×106, (1.6±0.9)×106 or (3.9±0.9)×106 M−1 were obtained for the complexes between trans‐pterostilbene and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively.
On the other hand, a binding stoichiometry of 1 : 2 was found for the complexes between (±)‐pterostilbene‐trans‐dihydrodimer and all the investigated DNA oligonucleotides. Apparent binding constants of (1.8±1.8)×106, (0.9±1.3)×106 and (1.9±1.5)×106 were found for the complexes between (±)‐pterostilbene‐trans‐dihydrodimer and ds6 and ds10 duplexes or Pu22T14T23 G‐quadruplex, respectively.
In excellent agreement with NMR data for the G‐quadruplex model here investigated, the binding stoichiometries observed for the (±)‐trans‐δ‐viniferin/(R,R)‐trans‐δ‐viniferin/(S,S)‐trans‐δ‐viniferin:Pu22T14T23 G‐quadruplex complexes – including 1 : 2 up to 1 : 3 DNA/ligand complexes – provided an additional and independent evidence in support of a stacking binding mode at both the outer Pu22T14T23 G‐quadruplex quartets. Notably, the 1 : 2 DNA/ligand binding stoichiometry found for the (±)‐pterostilbene‐trans‐dihydrodimer:Pu22T14T23 G‐quadruplex complexes suggested a similar binding mode, as expected considering its structural similarity with (±)‐trans‐δ‐viniferin.
The binding stoichiometry found for the (±)‐trans‐δ‐viniferin/(R,R)‐trans‐δ‐viniferin/(S,S)‐trans‐δ‐viniferin:ds10 duplex complexes was also plausible considering the interaction of the ligands at both the minor and major grooves of ds10 duplex inferred by the NMR analysis.
Moreover, the 1 : 1 binding stoichiometries observed for both trans‐resveratrol and trans‐pterostilbene with all the investigated DNA sequences suggested a similar binding mode for these ligands to the duplex or G‐quadruplex structures as a consequence of their structural similarity.
For the here studied systems featured by 1 : 2 or 1 : 3 DNA/ligand binding stoichiometries, the obtained binding constants were considered only as apparent constants. Indeed, in the case of multiple binding events which, appearing unresolved when following spectroscopic changes, can be represented only by a simplified model based on independent and equivalent sites, as is our case, only averaged constant values over all the occurring binding events can be extrapolated.[ 49 , 50 ] Hence, the here obtained data allow only a rough estimation of the real binding constants, which still represent useful indications in the overall understanding of the systems under investigation.
Altogether these data proved a tight interaction between all the explored ligands and both duplex‐ and G‐quadruplex‐forming DNA oligonucleotides, with no relevant marked preference for a specific DNA sequence/secondary structure.
Docking studies
To get a deeper insight into the binding mode of trans‐δ‐viniferin to duplex and G‐quadruplex structures, molecular docking studies were performed for (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin using as targets both ds10 duplex and Pu22T14T23 G‐quadruplex. trans‐Resveratrol was also studied in the complexes with the same DNA targets in order to compare the binding behaviour of trans‐δ‐viniferin with its parent compound. On the basis of the results of the fluorescence spectroscopy experiments, 1 : 2 or 1 : 1 DNA/ligand models were built for DNA/trans‐δ‐viniferin or DNA/trans‐resveratrol complexes, respectively. In detail, the lowest energy docked conformations of the first or both the first and second most populated clusters were selected for 1 : 1 or 1 : 2 DNA/ligand systems, respectively.
As far as the interaction of (R,R)‐trans‐δ‐viniferin with ds10 duplex is concerned, the most populated cluster showed poses involving the binding of the ligand in the middle region of the minor groove, while in the second most populated cluster the ligand was located in the middle region of the major groove of the duplex model. Binding energies of −7.9 and −7.7 kcal/mol were calculated for the first and second binding poses, respectively. Moreover, one hydrogen bond between O2T6 and 11,13b‐OH was detected for (R,R)‐trans‐δ‐viniferin bound to the middle region of the minor groove, while one hydrogen bond was found between O6G3 and 11,13a‐OH for (R,R)‐trans‐δ‐viniferin bound to the middle region of the major groove (Figures 7A and S29 A, B). Conversely, the most populated cluster found for (S,S)‐trans‐δ‐viniferin involved the binding of the ligand in the middle region of the major groove of ds10 duplex, while in the second most populated cluster the ligand was bound in the middle region of the minor groove of the duplex model. Binding energies of −8.0 and −8.1 kcal/mol were obtained for the first and second binding poses, respectively. Additionally, one hydrogen bond between O5’G3 and 11,13a‐OH was found for (S,S)‐trans‐δ‐viniferin bound to the middle region of the major groove, while three hydrogen bonds were found between O2T7 and 11,13b‐OH, between O3’T6 and 11,13b‐OH and between O3’A5 and 11,13b‐OH for (S,S)‐trans‐δ‐viniferin bound to the middle region of the minor groove (Figures 7B and S29 C, D).
Figure 7.

Binding mode of (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin when docked into ds10 duplex. The four panels represent the four possible 1 : 2 DNA/ligand complexes which can be formed between ds10 duplex and the two enantiomers targeting the minor and/or major grooves as follows: A) minor groove: (R,R)‐trans‐δ‐viniferin, major groove: (R,R)‐trans‐δ‐viniferin, B) minor groove: (S,S)‐trans‐δ‐viniferin, major groove: (S,S)‐trans‐δ‐viniferin, C) minor groove: (R,R)‐trans‐δ‐viniferin, major groove: (S,S)‐trans‐δ‐viniferin and D) minor groove: (S,S)‐trans‐δ‐viniferin, major groove: (R,R)‐trans‐δ‐viniferin. (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin are represented as cyan and green sticks, respectively.
As far as the interaction of the pure trans‐δ‐viniferin enantiomers with Pu22T14T23 G‐quadruplex is concerned, in both cases the most populated cluster involved the binding of the ligand to the 3’‐end quartet, while in the second cluster the ligand was bound at the 5’‐end quartet. Binding energies of −8.6 and −7.7 kcal/mol were calculated for the first and second binding poses of (R,R)‐trans‐δ‐viniferin onto Pu22T14T23 G‐quadruplex, respectively, while binding energies of −8.7 and −7.8 kcal/mol were found for the first and second binding poses of (S,S)‐trans‐δ‐viniferin onto Pu22T14T23 G‐quadruplex, respectively. π‐π Stacking interactions of G13 and G18 with A2 ring of the ligand were detected for (R,R)‐trans‐δ‐viniferin bound to the 3’‐end quartet, while π‐π stacking of G16 with A2 ring of the ligand and two hydrogen bonds between phosphate oxygen of G20 and 11,13b‐OH and between O6G7 and 11,13a‐OH were found for (R,R)‐trans‐δ‐viniferin bound to the 5’‐end quartet (Figures 8A and S30 A,B). In turn, π‐π stacking of G13 with A2 ring of the ligand and two hydrogen bonds between phosphate oxygen of T10 and 11,13b‐OH and between O4T23 and 4a‐OH were found for (S,S)‐trans‐δ‐viniferin bound to the 3’‐end quartet, while π‐π stacking of G16 with B2 ring of the ligand and one hydrogen bond between the exocyclic amino group of A6 and 11,13a‐OH were found for (S,S)‐trans‐δ‐viniferin bound to 5’‐end quartet (Figures 8B and S30 C,D).
Figure 8.
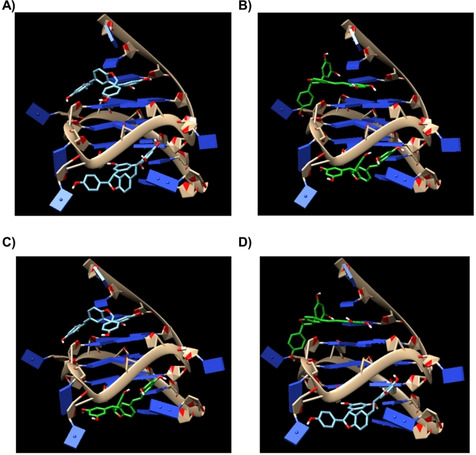
Binding mode of (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin when docked into Pu22T14T23 G‐quadruplex. The four panels represent the four possible 1 : 2 DNA/ligand complexes which can be formed between Pu22T14T23 G‐quadruplex and the two enantiomers targeting the 5’‐end quartet (top) and/or the 3’‐end quartet (bottom) as follows: A) 5’‐end quartet: (R,R)‐trans‐δ‐viniferin, 3’‐end quartet: (R,R)‐trans‐δ‐viniferin, B) 5’‐end quartet: (S,S)‐trans‐δ‐viniferin, 3’‐end quartet: (S,S)‐trans‐δ‐viniferin, C) 5’‐end quartet: (R,R)‐trans‐δ‐viniferin, 3’‐end quartet: (S,S)‐trans‐δ‐viniferin and D) 5’‐end quartet: (S,S)‐trans‐δ‐viniferin, 3’‐end quartet: (R,R)‐trans‐δ‐viniferin. (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin are represented as cyan and green sticks, respectively.
While in the ds10 duplex/trans‐δ‐viniferin systems no significant variations were detected in terms of binding energies between the two best poses at minor and major grooves, in the case of Pu22T14T23 G‐quadruplex/trans‐δ‐viniferin systems a more relevant difference between binding energies for the interaction at the 3’‐ and 5’‐end quartets was found, suggesting that the 3’‐end quartet could be the preferential binding site on Pu22T14T23 G‐quadruplex for both (R,R)‐trans‐δ‐viniferin and (S,S)‐trans‐δ‐viniferin.
Overall, docking results were fully consistent with binding modes inferred by NMR data for both ds10 duplex/trans‐δ‐viniferin and Pu22T14T23 G‐quadruplex/trans‐δ‐viniferin complexes. Binding energies averaged over the two binding events for each DNA/trans‐δ‐viniferin system were also consistent with fluorescence‐derived binding constants, showing no relevant preference of both (R,R) and (S,S) enantiomers of trans‐δ‐viniferin for the duplex or G‐quadruplex targets, as well as no enantioselective binding.
Combining together the findings from NMR‐derived NOE contacts, binding stoichiometries and constants derived from fluorescence spectroscopy and best docking poses and energies, four different models appear plausible for the ds10 duplex/trans‐δ‐viniferin and Pu22T14T23 G‐quadruplex/trans‐δ‐viniferin complexes (Figures 7A−D and 8A−D, respectively).
As far as the trans‐resveratrol/ds10 duplex system is concerned, the best binding pose of the ligand was in the middle region of the duplex minor groove, in analogy with recent studies,[ 30 , 51 ] and was featured by a binding energy of −7.0 kcal/mol and forming one hydrogen bond with the duplex involving O3’T6 and 3,5‐OH (Figure S31A,B). On the other hand, the best binding pose onto Pu22T14T23 G‐quadruplex was at the 3’‐end quartet. A binding energy of −6.1 kcal/mol was calculated for the Pu22T14T23 G‐quadruplex/trans‐resveratrol complex, stabilized by π‐π stacking of G9 and G13 with the 1,3‐benzenediol ring of the ligand along with one hydrogen bond between O4T23 and 3,5‐OH (Figure S31C,D). Notably, similar binding modes were observed comparing trans‐δ‐viniferin and its parent monomer trans‐resveratrol. In full agreement with fluorescence‐derived binding constants, lower binding energies were found with both targets for trans‐resveratrol compared to trans‐δ‐viniferin.
Antiproliferative activity evaluation
(±)‐trans‐δ‐Viniferin, trans‐resveratrol, trans‐pterostilbene and (±)‐pterostilbene‐trans‐dihydrodimer were tested on a panel of human tumour cell lines, i. e. melanoma A375, non‐small cell lung cancer H460 and prostate cancer PC3, as well as on human normal skin WS1 fibroblasts. The antiproliferative potency of the compounds was evaluated after 48 h exposure using the MTS cell proliferation assay, and the IC50 values were determined as the concentrations of compound causing 50 % cell growth inhibition.
Among the tumour cell lines considered, H460 cells were overall the most sensitive ones. Conversely, PC3 cells resulted the least sensitive cells (Table 5). The most active compound on tumour cell lines was (±)‐pterostilbene‐trans‐dihydrodimer. The compound was 1.75‐ and 1.3‐fold more active than trans‐resveratrol and trans‐pterostilbene, respectively, on A375, about 2.3‐fold more active than trans‐resveratrol and trans‐pterostilbene on PC3 cells, and showed cytotoxic potency similar to trans‐resveratrol and trans‐pterostilbene on H460 cells. Comparing the cytotoxic potency produced on WS1 fibroblasts, trans‐resveratrol showed the highest selectivity (IC50>200 μM on WS1) and (±)‐pterostilbene‐trans‐dihydrodimer showed about 2‐fold (PC3) and 3‐fold (A375 and H460) higher cytotoxicity on tumour cells. Conversely, (±)‐trans‐δ‐viniferin showed a similar cytotoxic profile on all the tested cell lines (IC50 in the range 69–120 μM).
Table 5.
Cytotoxic activity of the here investigated natural stilbenoids.[a]
|
Compound |
A375 |
H460 |
PC3 |
WS1 |
|---|---|---|---|---|
|
IC50 (μM)[b] |
|
|
|
|
|
(±)‐trans‐δ‐viniferin |
95±7 |
81±4 |
120±7.8 |
69±5.6 |
|
trans‐resveratrol |
44.5±3.5 |
25±0.77 |
>100 |
>200 |
|
trans‐pterostilbene |
33±0.7 |
25±0.4 |
97±4.9 |
57±10 |
|
(±)‐pterostilbene‐trans‐dihydrodimer |
25.5±2.12 |
24.7±0.35 |
42.7±0.5 |
82.7±1.06 |
[a] 24 h after seeding, cells were exposed for 48 h to the compounds and cytotoxicity was measured using MTS assay. Data represent mean values ±SD of three independent experiments. [b] IC50 is defined as the concentration of compound causing 50 % cell growth inhibition.
To evaluate the ability of the selected molecules to produce DNA damage, the expression of γ‐H2AX after compound exposure was evaluated in all the cell lines. Chromatin is composed by DNA wrapping around histone octamers and forming the nucleosome. Among the histones, H2AX plays a pivotal role in nucleosome formation, chromatin remodelling and DNA repair. Phosphorylation of the Ser‐139 residue of the histone H2AX, leading to γ‐H2AX, is an early cellular response useful in monitoring the induction of DNA double‐strand breaks. The detection and measure of γ‐H2AX levels emerged as a highly specific and sensitive molecular marker for monitoring DNA breaks induced by γ‐radiation and antitumor drugs. [52] Cells were exposed for 48 h to the studied compounds at a concentration corresponding to their respective IC50 and the level of γ‐H2AX was evaluated by Western blot assay (Figure 9A). The level of DNA damage was defined by densitometric analysis. Actin/β‐tubulin was used to define the relative expression levels by measuring the ratio between the band intensity of γ‐H2AX and the corresponding band intensity of actin/β‐tubulin (Figure 9B). Low level of DNA damage was evidenced in normal WS1 fibroblasts exposed to 200 μM trans‐resveratrol. This behaviour confirms a previous work demonstrating that trans‐resveratrol employed at similar doses on normal or cancer cells has no effects on the former ones. [30] (±)‐trans‐δ‐Viniferin, trans‐pterostilbene, as well as (±)‐pterostilbene‐trans‐dihydrodimer produced DNA damage in all the considered cell lines, with (±)‐pterostilbene‐trans‐dihydrodimer being the most active DNA damaging compound of the series. These results are in agreement with the above reported NMR and fluorescence spectroscopy experiments, which proved a tight interaction between the explored ligands and both duplex‐ and G‐quadruplex‐forming DNA oligonucleotides. The lack of linear correlations between IC50 values and observed DNA damage is not surprising, considering that multiple events, involving compounds metabolism and the recently reported repair mechanisms triggered by stilbenoids themselves, can in parallel occur in cell. This complex interplay of factors can also explain the overall moderate cytotoxicity found for these compounds. [28]
Figure 9.
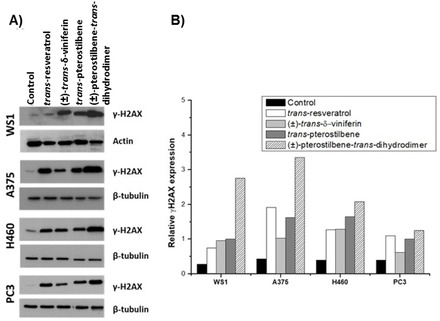
Compound‐mediated DNA damage. A) The capability of the compounds to produce DNA damage was evaluated in Western blot assay by measuring the level of γ‐H2AX before and after compound exposure. After seeding, cells were exposed to compounds for 48 h at concentrations corresponding to their IC50 values. Cells were harvested for protein extraction and cell lysates were fractionated on SDS‐PAGE. Actin or β‐tubulin were used as control for loading. B) The level of DNA damage was measured by densitometric analysis. The band intensity quantitation of the γ‐H2AX and actin/β‐tubulin were used to define the relative expression levels (ratio between the band intensity of γ‐H2AX and the corresponding band intensity of actin/β‐tubulin).
Discussion
DNA is one of the main targets for anticancer drugs. Indeed, the possibility of finely modulating typical mechanisms of cancer cells acting at the genome level represents a fascinating therapeutic strategy, offering several effective precedents described in the literature.[ 53 , 54 , 55 , 56 , 57 ] Several DNA duplex and/or G‐quadruplex ligands, mostly synthetic compounds, have been thus analysed in preclinical and clinical studies.[ 58 , 59 , 60 , 61 , 62 , 63 ] Conversely, relatively few studies are reported for naturally occurring compounds.[ 29 , 30 , 64 ] In this frame, the ability of trans‐resveratrol to interact with DNA duplex and G‐quadruplex structures was recently investigated.[ 29 , 30 ] Notably, the ability to interact with DNA observed for this compound proved to be one of the mechanisms involved in its anticancer activity.
Based on these findings, we investigated the DNA targeting ability and the cytotoxic activity on cancer and normal cells of a trans‐resveratrol dimeric analogue, i. e. (±)‐trans‐δ‐viniferin. Dimeric analogues, compared to their monomeric counterpart, can represent precious scaffolds of major interest due to their more extended surface and higher number of functional moieties, which can confer higher selectivity in the recognition of biologically relevant macromolecules.
NMR analysis along with docking studies allowed building molecular models for the complexes formed between the duplex/G‐quadruplex target of choice and (±)‐trans‐δ‐viniferin. Intercalative binding mode could be fully excluded in its interaction with the here investigated duplex models (ds6 and ds10); analogously, also groove binding mode could be ruled out as possible interaction mode of (±)‐trans‐δ‐viniferin with the G‐quadruplex model (Pu22T14T23). The ligand was located in the middle regions of both minor and major grooves of the ds6 and ds10 duplexes, and the interaction was mainly stabilized by hydrogen bonds. As far as the Pu22T14T23 G‐quadruplex is concerned, the binding of (±)‐trans‐δ‐viniferin did not disrupt its original fold, even if rearrangements of its flanking residues covering the external quartets were observed as a consequence of the ligand binding at both the 3’‐ and 5’‐end quartets through stacking mode. In detail, the binding of (±)‐trans‐δ‐viniferin at the 5’‐end occurred through stacking interactions on the most accessible guanine of the upper quartet, i. e. G16, and no relevant rearrangement of the 5’‐end flanking residues was necessary to stably accommodate the ligand onto this quartet. This was proved by the absence of significant chemical shift variations for the flanking residue A6 as well as for the upper quartet residues G7, G11 and G20. Conversely, the upfield shift for G16 was fully consistent with additional stacking interactions provided by the bound ligand, absent on the uncovered G16 of the free G‐quadruplex. On the other hand, the binding of (±)‐trans‐δ‐viniferin at the 3’‐end occurred by stacking on G9, G13, G18 and G22 accompanied by a rearrangement of T23, A24 and A25. In particular, upon addition of the ligand, the downfield shifts observed for G9, G13, G22, T23, A24 and A25 revealed their partial de‐stacking, whereas the upfield shift of G18 indicated additional stacking interactions, absent in the free G‐quadruplex. These findings indicated that the ligand was located over G9, G13, G18 and G22, thus covering the previously uncovered G18 and hampering the original direct stacking of T23 on G22 and A25 on G9. Notably, the binding of (±)‐trans‐δ‐viniferin at the 3’‐end of the G‐quadruplex model was found to be energetically preferred over the 5’‐binding, as suggested by molecular docking. This could be probably due to the favourable rearrangement of the 3’‐end flanking residues which together with the lower quartet might form a peculiar pocket for the binding of the ligand. On the other hand, perturbations of 1H NMR signals of the residues forming the middle quartet could be the result of a slight modification of the original stacking geometry of both the upper and lower quartets on the middle one upon ligand binding.
CD analysis confirmed that the folds of the three investigated oligonucleotide targets were not perturbed by the ligand, providing a further indication that intercalation did not occur in the duplex structures and that the parallel fold of the G‐quadruplex model was preserved upon ligand binding. As a consequence of the low‐to‐null changes in the overall structure of each DNA target upon ligand interaction, limited stabilizing effects were observed for (±)‐trans‐δ‐viniferin by CD‐melting experiments. This is a further indication that the ligand was perfectly accommodated into the grooves of the duplex model without altering its conformation to achieve a stable binding, while the binding mode typically associated to stabilizing effects on duplexes, i. e. intercalation, did not occur. On the other hand, ligand stacking on the G‐quadruplex target was not associated to stabilizing effects because the ligand insertion onto the outer quartets was possible only after rearrangement of the flanking residues. Therefore, ligand binding to the G‐quadruplex model resulted in a global process with no dramatic effects on the thermal stability of the target since it comprised two equally relevant, but opposite events: the first one inducing destabilizing effects, i. e. the de‐stacking of the flanking residues, and the second one providing stabilizing effects, i. e. the stacking of the ligand onto the outer quartets. Fluorescence analysis demonstrated the high affinity of (±)‐trans‐δ‐viniferin for both duplex and G‐quadruplex structures, with binding constants of the 107 M−1 magnitude order, comparable or even higher than known DNA binders endowed with biological activities both in vitro and in vivo.[ 65 , 66 ] Additionally, fluorescence‐derived binding stoichiometries, along with NMR data, suggested that two ligand molecules were involved in the binding, by targeting both the minor and major grooves of the duplex models, as well as both the 3’‐ and 5’‐end quartets of the G‐quadruplex model. In few cases, binding stoichiometries of 1 : 3 DNA/ligand were observed. These complexes of higher stoichiometries were probably obtained due to a third molecule of (±)‐trans‐δ‐viniferin, not detected by NMR, thus probably binding to different sites on each macromolecule in a weak and unspecific manner. Investigation of pure (R,R) and (S,S) enantiomers of trans‐δ‐viniferin did not show relevant differences in targeting duplex and G‐quadruplex structures with respect to the racemic mixture.
Interestingly, comparing (±)‐trans‐δ‐viniferin with its parent compound trans‐resveratrol, it emerged that the dimeric stilbenoid had higher affinity towards the DNA targets than the monomeric species. Similar binding modes were observed for both ligands in the interaction with duplex and G‐quadruplex structures, though 1 : 1 DNA/ligand complexes were consistently formed by trans‐resveratrol.
The methoxylated analogues of (±)‐trans‐δ‐viniferin and trans‐resveratrol, i. e. (±)‐pterostilbene‐trans‐dihydrodimer and trans‐pterostilbene, respectively, showed similar binding modes, affinities and stoichiometries with all the DNA targets compared to their parent analogues, in line with their structural similarities.
All compounds were cytotoxic in the μM concentration range on a panel of cancer cell lines and produced DNA damage, consistent with their interaction with both DNA duplex and G‐quadruplex structures. Thus, interaction with DNA could be considered one of the anticancer mechanisms of action of these stilbenoids, even if additional mechanisms involving other targets cannot be excluded, in accordance with previous reports in the literature.[ 29 , 30 ]
With the aim of synthesizing more active and selective DNA‐binding ligands based on natural stilbenoids, the design of novel dimeric and monomeric analogues has been now actively undertaken in our laboratories. The molecular models here built for the complexes of (±)‐trans‐δ‐viniferin and trans‐resveratrol with DNA duplex and G‐quadruplex structures are indeed a precious source of inspiration for an effective optimization process, taking into account the unique features of the ligands in their bound conformations.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
C.P. was supported by a FIRC‐AIRC fellowship for Italy. D.M. thanks AIRC – Associazione Italiana per la Ricerca sul Cancro (grant IG2020 n. 25046) for financial support.
C. Platella, S. Mazzini, E. Napolitano, L. M. Mattio, G. L. Beretta, N. Zaffaroni, A. Pinto, D. Montesarchio, S. Dallavalle, Chem. Eur. J. 2021, 27, 8832.
References
- 1.C. M. Galanakis, Polyphenols: Properties, Recovery, and Applications, Woodhead, Cambridge, 2018.
- 2. Quideau S., Deffieux D., Douat-Casassus C., Pouységu L., Angew. Chem. Int. Ed. 2011, 50, 586–621; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 610–646. [Google Scholar]
- 3. Tsao R., Nutrients 2010, 2, 1231–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niesen D. B., Hessler C., Seeram N. P., J. Berry Res. 2013, 3, 181–196. [Google Scholar]
- 5. Jeandet P., Delaunois B., Conreux A., Donnez D., Nuzzo V., Cordelier S., Clément C., Courot E., BioFactors 2010, 36, 331–341. [DOI] [PubMed] [Google Scholar]
- 6. Akinwumi B. C., Bordun K. A. M., Anderson H. D., Int. J. Mol. Sci. 2018, 19, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weiskirchen S., Weiskirchen R., Adv. Nutr. An Int. Rev. J. 2016, 7, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gambini J., Inglés M., Olaso G., Lopez-Grueso R., Bonet-Costa V., Gimeno-Mallench L., Mas-Bargues C., Abdelaziz K. M., Gomez-Cabrera M. C., Vina J., Borras C., Oxid. Met. 2015, 2015, 837042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin W.-S., Leland J. V., Ho C.-T., Pan M.-H., J. Agric. Food Chem. 2020, 68, 12788–12799. [DOI] [PubMed] [Google Scholar]
- 10. Liu Y., You Y., Lu J., Chen X., Yang Z., Molecules 2020, 25, 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Penãlver P., Zodio S., Lucas R., De-Paz M. V., Morales J. C., J. Agric. Food Chem. 2020, 68, 1609–1620. [DOI] [PubMed] [Google Scholar]
- 12. Kumar A., Levenson A. S., Epigenetics Cancer Prev. 2019, pp. 169–186. [Google Scholar]
- 13. Singh C. K., Ndiaye M. A., Ahmad N., Biochim. Biophys. Acta 2015, 1852, 1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guthrie A. R., Chow H. H. S., Martinez J. A., Pharmacol. Res. Perspect. 2017, 5, e00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee Y. H., Chen Y. Y., Yeh Y. L., Wang Y. J., Chen R. J., Int. J. Mol. Sci. 2019, 20, 2716. [Google Scholar]
- 16. Aluyen J. K., Ton Q. N., Tran T., Yang A. E., Gottlieb H. B., Bellanger R. A., J. Diet. Suppl. 2012, 9, 45–56. [DOI] [PubMed] [Google Scholar]
- 17. McCormack D., McFadden D., J. Surg. Res. 2012, 173, e53–e61. [DOI] [PubMed] [Google Scholar]
- 18. Fulda S., Drug Discovery Today 2010, 15, 757–765. [DOI] [PubMed] [Google Scholar]
- 19. Rhodes D., Lipps H. J., Nucleic Acids Res. 2015, 43, 8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Asamitsu S., Obata S., Yu Z., Bando T., Sugiyama H., Molecules 2019, 24, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burge S., Parkinson G. N., Hazel P., Todd A. K., Neidle S., Nucleic Acids Res. 2006, 34, 5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siddiqui-Jain A., Grand C. L., Bearss D. J., Hurley L. H., Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Musso L., Mazzini S., Rossini A., Castagnoli L., Scaglioni L., Artali R., Di Nicola M., Zunino F., Dallavalle S., Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 615–629. [DOI] [PubMed] [Google Scholar]
- 24. Agrawal P., Lin C., Mathad R. I., Carver M., Yang D., J. Am. Chem. Soc. 2014, 136, 1750–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lipps H. J., Rhodes D., Trends Cell Biol. 2009, 19, 414–422. [DOI] [PubMed] [Google Scholar]
- 26. Lagunas-Rangel F. A., Bermúdez-Cruz R. M., Front. Oncol. 2020, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Colin D. J., Limagne E., Ragot K., Lizard G., Ghiringhelli F., Solary, Chauffert B., Latruffe N., Delmas D., Cell Death Dis. 2014, 5, e1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsieh T., Huang Y., Wu J. M., Carcinogenesis 2011, 32, 93–101. [DOI] [PubMed] [Google Scholar]
- 29. Platella C., Guida S., Bonmassar L., Aquino A., Bonmassar E., Ravagnan G., Montesarchio D., Roviello G. N., Musumeci D., Fuggetta M. P., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2843–2851. [DOI] [PubMed] [Google Scholar]
- 30. Platella C., Raucci U., Rega N., D'Atri S., Levati L., Roviello G. N., Fuggetta M. P., Musumeci D., Montesarchio D., Int. J. Biol. Macromol. 2020, 151, 1163–1172. [DOI] [PubMed] [Google Scholar]
- 31. Mattio L. M., Dallavalle S., Musso L., Filardi R., Franzetti L., Pellegrino L., D'Incecco P., Mora D., Pinto A., Arioli S., Sci. Rep. 2019, 9, 19525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mattio L. M., Marengo M., Parravicini C., Eberini I., Dallavalle S., Molecules 2019, 24, 3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilkens A., Paulsen J., Wray V., Winterhalter P., J. Agric. Food Chem. 2010, 58, 6754–6761. [DOI] [PubMed] [Google Scholar]
- 34. Mazzini S., Bellucci M. C., Mondelli R., Bioorg. Med. Chem. 2003, 11, 505–514. [DOI] [PubMed] [Google Scholar]
- 35. Delepierre M., Dinh T. H., Roques B. P., Biopolymers 1989, 28, 2115–2142. [DOI] [PubMed] [Google Scholar]
- 36. Lown J. W., Hanstock C. C., Bleackley R. C., Imbach J. L., Rayner B., Vasseur J. J., Nucleic Acids Res. 1984, 12, 2519–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ragg E., Mondell R., Garbesi A., Colonna F. P., Battistini C., Vioglio S., Magn. Reson. Chem. 1989, 27, 640–646. [Google Scholar]
- 38.D. G. Gorenstein, Phosphorus-31 NMR: Principles and Applications, Academic Press, New York, 1984.
- 39. Misiak M., Heldt M., Szeligowska M., Mazzini S., Scaglioni L., Grabe G. J., Serocki M., Lica J., Switalska M., Wietrzyk J., Beretta G. L., Perego P., Zietkowski D., Baginski M., Borowski E., Skladanowski A., Oncotarget 2017, 8, 105137–105154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dai J., Carver M., Hurley L. H., Yang D., J. Am. Chem. Soc. 2011, 133, 17673–17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ambrus A., Chen D., Dai J., Jones R. A., Yang D., Biochemistry 2005, 44, 2048–2058. [DOI] [PubMed] [Google Scholar]
- 42. del Villar-Guerra R., Trent J. O., Chaires J. B., Angew. Chem. Int. Ed. 2018, 57, 7171–7175; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7289–7293. [Google Scholar]
- 43. Kypr J., Kejnovská I., Renčiuk D., Vorlíčková M., Nucleic Acids Res. 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim B. G., Evans H. M., Dubins D. N., Chalikian T. V., Biochemistry 2015, 54, 3420–3430. [DOI] [PubMed] [Google Scholar]
- 45. Šmidlehner T., Piantanida I., Pescitelli G., Beilstein J. Org. Chem. 2017, 14, 84–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paramasivan S., Rujan I., Bolton P. H., Methods 2007, 43, 324–331. [DOI] [PubMed] [Google Scholar]
- 47. Carvalho J., Queiroz J. A., Cruz C., J. Chem. Educ. 2017, 94, 1547–1551. [Google Scholar]
- 48. Giancola C., Pagano B., Top. Curr. Chem. 2013, 330, 2011–242. [DOI] [PubMed] [Google Scholar]
- 49. Platella C., Trajkovski M., Doria F., Freccero M., Plavec J., Montesarchio D., Nucleic Acids Res. 2020, 48, 12380–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buchholz I., Karg B., Dickerhoff J., Sievers-Engler A., Lämmerhofer M., Weisz K., Chem. Eur. J. 2017, 23, 5814–5823. [DOI] [PubMed] [Google Scholar]
- 51. Nair M. S., Shukla A., J. Biomol. Struct. Dyn. 2020, 38, 3087–3097. [DOI] [PubMed] [Google Scholar]
- 52. Mah L. J., El-Osta A., Karagiannis T. C., Leukemia 2010, 24, 679–686. [DOI] [PubMed] [Google Scholar]
- 53. Gavande N. S., Vandervere-Carozza P. S., Hinshaw H. D., Jalal S. I., Sears C. R., Pawelczak K. S., Turchi J. J., Author P. T., Pharmacol. Ther. 2016, 160, 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hurley L. H., Nat. Rev. Cancer 2002, 2, 188–200. [DOI] [PubMed] [Google Scholar]
- 55. Sun J., Wei Q., Zhou Y., Wang J., Liu Q., Xu H., BMC Syst. Biol. 2017, 11, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crisci S., Amitrano F., Saggese M., Muto T., Sarno S., Mele S., Vitale P., Ronga G., Berretta M., Di Francia R., Medicina 2019, 55, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cimino-Reale G., Zaffaroni N., Folini M., Curr. Pharm. Des. 2016, 22, 6612–6624. [DOI] [PubMed] [Google Scholar]
- 58. Monchaud D., Teulade-Fichou M. P., Org. Biomol. Chem. 2008, 6, 627–636. [DOI] [PubMed] [Google Scholar]
- 59. Platella C., Pirota V., Musumeci D., Rizzi F., Iachettini S., Zizza P., Biroccio A., Freccero M., Montesarchio D., Doria F., Int. J. Mol. Sci. 2020, 21, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Amato J., Platella C., Iachettini S., Zizza P., Musumeci D., Cosconati S., Pagano A., Novellino E., Biroccio A., Randazzo A., Pagano B., Montesarchio D., Eur. J. Med. Chem. 2019, 163, 295–306. [DOI] [PubMed] [Google Scholar]
- 61. Musumeci D., Amato J., Zizza P., Platella C., Cosconati S., Cingolani C., Biroccio A., Novellino E., Randazzo A., Giancola C., Pagano B., Montesarchio D., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1341–1352. [DOI] [PubMed] [Google Scholar]
- 62. Pirota V., Platella C., Musumeci D., Benassi A., Amato J., Pagano B., Colombo G., Freccero M., Doria F., Montesarchio D., Int. J. Biol. Macromol. 2021, 166, 1320–1334. [DOI] [PubMed] [Google Scholar]
- 63. O'Hagan M. P., Peñalver P., Gibson R. S. L., Morales J. C., Galan M. C., Chem. Eur. J. 2020, 26, 6224–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carvalho J., Mergny J. L., Salgado G. F., Queiroz J. A., Cruz C., Trends Mol. Med. 2020, 26, 848–861. [DOI] [PubMed] [Google Scholar]
- 65. Platella C., Napolitano E., Riccardi C., Musumeci D., Montesarchio D., J. Med. Chem. 2021, 64, 3578–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pagano B., Fotticchia I., De Tito S., Mattia C. A., Mayol L., Novellino E., Randazzo A., Giancola C., J. Nucleic Acids 2010, 247137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary