

Abstract
Background
Isatuximab (Isa), an anti‐CD38 monoclonal antibody, and carfilzomib (K), a next‐generation proteasome inhibitor (PI), both have potent single‐agent activity in relapsed and refractory multiple myeloma (RRMM).
Methods
This phase 1b study evaluated the combination of Isa and K in 33 patients with RRMM. Isa was administered by intravenous infusion in 3 dosing cohorts: dose level 1 (Isa at 10 mg/kg biweekly), dose level 2 (DL2; Isa at 10 mg/kg weekly for 4 doses and then biweekly), and dose level 3 (Isa at 20 mg/kg weekly for 4 doses and then biweekly) and all patients received K (20 mg/m2 intravenously for cycle 1, days 1 and 2, and then 27 mg/m2 for all subsequent doses). A standard 3+3 dose‐escalation design was used, no dose‐limiting toxicity was observed, and the maximum tolerated dose was not reached. An expansion cohort of 18 patients was enrolled at DL2 to further evaluate safety and efficacy. Responses were assessed with the International Myeloma Working Group response criteria, and patients continued treatment until disease progression or unacceptable toxicity.
Results
With a median follow‐up of 26.7 months, in this heavily pretreated population with a median of 3 prior lines (refractory to PIs and immunomodulatory drugs, 76%; refractory to K, 27%), the overall response rate was 70% (stringent complete response/complete response, 4; very good partial response, 8; partial response, 11). The median progression‐free survival was 10.1 months, and the 2‐year survival probability was 76%. The most common treatment‐related adverse events (grade 2 or higher) were anemia, leukopenia, neutropenia, thrombocytopenia, hypertension, and infection. Infusion reactions were common (55%) but did not limit dosing.
Conclusions
Treatment with Isa plus K was well tolerated with no unexpected toxicity. The combination was effective despite the enrollment of heavily pretreated patients with RRMM.
Lay Summary
This phase 1b study was designed to assess the safety, pharmacokinetics, and preliminary efficacy of isatuximab and carfilzomib in patients with relapsed and refractory multiple myeloma.
Thirty‐three patients were treated: 15 in dose escalation and 18 in dose expansion. Patients received an average of 10 cycles.
The treatment was safe and effective. No unexpected toxicity or drug‐drug interactions were noted. Seventy percent of the subjects responded to therapy, and the progression‐free survival was 10.1 months.
Keywords: carfilzomib, isatuximab, immunomodulatory, monoclonal antibody, pharmacokinetics, proteasome
Short abstract
The combination of isatuximab and carfilzomib is safe with low levels of grade 3/4 hematologic and nonhematologic toxicities and no unexpected drug‐drug interactions. This treatment combination appears effective with an overall response rate of 70% and progression‐free survival of 10.1 months in patients with highly refractory multiple myeloma.
Introduction
Over the past decade, there have been more than 10 new drug approvals for the treatment of multiple myeloma (MM). The majority of these agents have been approved for the treatment of patients with relapsed and/or refractory disease. 1 Despite these advances, the disease is considered incurable, and additional agents as well as novel combinations are needed. There are now 5 classes of drugs that have demonstrated potent single‐agent activity for the treatment of MM, including proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), monoclonal antibodies (mAbs), a selective inhibitor of nuclear export, and a B‐cell maturation antigen–targeted antibody‐drug conjugate. 1 , 2 Numerous studies have evaluated combinations of these classes of drugs in patients with relapsed and refractory multiple myeloma (RRMM) as well as patients with newly diagnosed multiple myeloma (NDMM). Perhaps most encouraging have been the results from trials using the novel anti‐CD38 antibodies daratumumab (Dara) and isatuximab (Isa). These agents have demonstrated single‐agent activity but even greater activity when used in combination with IMiDs and PIs for patients with NDMM as well as patients with RRMM. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 Dara is currently approved for 7 indications, including the treatment of RRMM (5 different regimens) and the treatment of NDMM (3 multidrug combinations). Isa recently received approval from the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) on the basis of the phase 3 ICARIA study. 6 This study evaluated the combination of isatuximab, pomalidomide, and dexamethasone (IsaPd) versus pomalidomide and dexamethasone in a more heavily pretreated population with RRMM, and it showed significantly longer progression‐free survival (PFS) in the IsaPd arm (11.5 vs 6.5 months). There have been many other in vitro and clinical studies showing synergistic effects between IMiDs and mAbs in the relapsed setting and in NDMM. 11 , 12 , 13 , 14 Overall, IMiDs have been the preferred partner for mAbs because of their immunostimulatory effects.
Until recently, there was less enthusiasm for combining the CD38 mAbs with PIs. PIs can induce immunosuppression, and thus it was believed that CD38 antibodies with PIs would be less effective than combinations with IMiDs. The recent positive results from the phase 3 CANDOR study, which combined Dara with carfilzomib and dexamethasone (Kd), and the phase 3 IKEMA trial, which combined Isa with K and dexamethasone, have demonstrated that CD38 antibodies are quite effective when combined with K and dexamethasone. 15 , 16 The combination of Dara with K and dexamethasone showed an overall response rate (ORR) of 84% and is now FDA approved in the early RRMM setting.
In 2015, when this study was initiated, there were limited data from in vitro and xenograft studies that demonstrated at least additive effects when either Dara or Isa was combined with bortezomib or K 11 , 17 (B. Aftab, PhD and B. C. Hann, PhD, unpublished data, 2013). Because Isa was selected for development on account of its ability to induce cellular apoptosis without crosslinking, we hypothesized that Isa might have synergistic effects with K. Thus, we initiated this phase 1 study combining Isa and K, a doublet, in patients with RRMM who had received 2 or more prior lines of therapy. Early results from this study led to the phase 3 IKEMA trial.
Materials and Methods
Study Design and Objectives
This open‐label, multicenter, phase 1b, dose‐escalation study was designed to assess the safety, tolerability, pharmacokinetics, and preliminary efficacy of Isa and K in patients with RRMM. Patients were enrolled at 3 sites within the United States. The study was approved by the institutional review board at each site and was conducted in accordance with Guidelines for Good Clinical Practice and with provisions of the Declaration of Helsinki. All patients provided written informed consent before initiating any study procedures. The study is registered at ClinicalTrials.gov (NCT02332850). This is a Multiple Myeloma Research Consortium study supported by the corporations Sanofi and Amgen.
The primary objective of this study was to assess safety and define the maximum tolerated dose of Isa administered intravenously in combination with standard‐dose K (27 mg/m2 given twice weekly). Secondary objectives included an assessment of the safety/tolerability, immunogenicity, pharmacokinetics, and preliminary efficacy, including ORR, PFS, and overall survival (OS), of this combination.
Study Design
A standard phase 1, 3+3 dose‐escalation design was used, and 3 dose levels were evaluated: dose level 1 (DL1; Isa at 10 mg/kg intravenously on days 1 and 15 of every cycle), dose level 2 (DL2; Isa at 10 mg/kg intravenously on days 1, 8, 15, and 22 of cycle 1 and then on days 1 and 15 of all subsequent 28‐day cycles), and dose level 3 (DL3; Isa at 20 mg/kg on days 1, 8, 15, and 22 of cycle 1 and then on days 1 and 15 of all subsequent 28‐day cycles). In addition, all patients received standard‐dose K (20 mg/m2 intravenously for cycle 1, days 1 and 2, and then 27 mg/m2 intravenously on days 8, 9, 15, and 16 and for all subsequent doses). After cycle 8, patients were allowed to decrease the K frequency to days 1, 2, 15, and 16 per cycle while maintaining biweekly Isa per investigator and patient choice. Dexamethasone was not considered part of the treatment regimen but was given to prevent infusion reactions. The maximum tolerated dose was defined as 1 dose level below the dose at which dose‐limiting toxicity (DLT) was observed in >33% of the participants. An expansion cohort was planned when 6 patients were treated without safety concerns at the selected dose level. DLT was defined as 1 or more of the following (which were at least possibly related to study therapy and occurred in the first 28 days): any grade 3 or higher nonhematologic toxicity except for reversible electrolyte abnormalities; grade 3 fatigue and hypertension; grade 3 nausea, vomiting, and diarrhea that responded to medical management within 72 hours; grade 3/4 or higher hypersensitivity to Isa or K; any severe hematologic toxicity (defined as grade 4 neutropenia lasting ≥7 days, grade 3/4 neutropenia with fever or infection, or grade 3/4 thrombocytopenia with life‐threatening bleeding); and any treatment delay for >2 weeks or dose modification required between day 1 and day 28 of cycle 1. For each dosing cohort, an independent data monitoring committee at the University of California San Francisco was convened to review data, determine safety, and allow dose escalation. The selected Isa dose for the expansion cohort was determined on the basis of the assessment of safety, early efficacy (ORR), and pharmacokinetic data from this study as well as data generated from other Isa trials.
All patients received standard prophylactic medications to prevent infusion reactions. Before Isa in cycle 1, patients received dexamethasone at 20 mg intravenously, diphenhydramine at 25 to 50 mg intravenously, ranitidine at 50 mg intravenously, and acetaminophen at 650 to 1000 mg orally within 15 to 60 minutes of dosing. Before K dosing in cycle 1, patients received dexamethasone at 4 mg orally. For cycle 2 and beyond, premedications for Isa and K were determined by the investigator. Antiviral prophylaxis for herpes zoster was recommended. Patients continued the study therapy until disease progression, unacceptable toxicity, death, or patient or physician choice.
Study Population
The study enrolled patients 18 years old or older with RRMM who previously had been treated with at least 2 prior lines of therapy and had confirmed progression or refractoriness to their immediate prior line of therapy. Prior K exposure was allowed, but patients had to be >4 weeks from their last K dose. Prior anti‐CD38 therapy was not allowed. Patients were required to have measurable disease (serum M‐protein ≥ 0.5 g/dL, urinary M‐protein ≥ 200 mg/24 h, involved free light chain ≥ 10 mg/dL [with an abnormal free light chain ratio], or quantitative serum immunoglobulin A or D level > 0.5 g/dL by nephelometry), adequate marrow function (absolute neutrophil count > 1000/µL, platelets >50,000/µL, and hemoglobin ≥ 8 g/dL), adequate renal and hepatic function (creatinine clearance ≥ 30 mL/min and bilirubin level ≤ 1.5 × upper limit of normal), and an Eastern Cooperative Oncology Group performance status of 0 to 2. Patients with an active or uncontrolled infection, significant cardiac disease (including second/third‐degree heart block), significant ischemic heart disease, a QTc interval > 450 milliseconds at the baseline, New York Heart Association class II or worse congestive heart failure, uncontrolled hypertension or hyperglycemia, grade 2 or higher painful neuropathy, or evidence of amyloidosis were excluded. Additional exclusion criteria were prior autologous transplantation within 12 weeks of study therapy, prior anticancer treatment within 14 days or investigational therapy within 21 days (or 4 half‐lives) of study initiation, prior malignancy (except for adequately treated basal cell carcinoma/squamous cell carcinoma, cervical cancer in situ, or other malignancies from which the subject had been disease‐free for at least 3 years), a daily requirement for corticosteroids (>10 mg of prednisone or the equivalent), evidence of mucosal or internal bleeding, and a major surgical procedure within 4 weeks of study therapy.
End Points and Statistical Methods
The primary end point was safety, and it was evaluated continuously from the day of consent. Patients who received at least 1 dose of the study therapy were included in the DLT evaluation for dose escalation and in the assessment of adverse events (AEs). Events were documented by history, physical examinations, and laboratory tests and were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (version 4.03). Secondary end points included ORR, duration of response (DOR), PFS, and OS. Responses were assessed by the investigators using the International Myeloma Working Group uniform response criteria 18 with incorporation of the minimal response per the European Group for Blood and Marrow Transplantation criteria. The clinical benefit rate was calculated as the number of responders (per the International Myeloma Working Group) plus those achieving a minimal response divided by the number of evaluable patients. Patients were considered evaluable for response if they had baseline data available, had received at least 1 cycle of therapy, and had their disease re‐evaluated or had evidence for disease progression.
PFS was defined as the time interval from the date of the first dose of the study drug to the first documented disease relapse, progression, or death from any cause, whichever occurred first. OS was defined as the time interval from the date of the first dose of the study drug to the date of death from any cause. DOR was defined as the time interval from the date of the first documented response (stringent complete response [sCR], complete response [CR], very good partial response [VGPR], or partial response [PR]) to documented disease relapse, progression, or death, whichever occurred first. OS was censored with the date of last known contact for those alive at the time of analysis. For patients with no documented date of progression or death, PFS and DOR were censored at the date of the last adequate assessment.
Continuous data for the entire treated population were summarized with descriptive statistics, including means, standard deviations, medians, and ranges, as appropriate. Categorical and ordinal data were summarized with numbers and percentages along with the binomial exact 95% confidence intervals (CIs) for each dose level and overall. Time to response, DOR, and OS were estimated via Kaplan‐Meier methods. The median time to event and event‐free probabilities are reported along with 95% CIs.
Pharmacokinetic Assessments
Intensive pharmacokinetic blood sampling was performed for Isa on cycle 1, day 1, and on cycle 3, day 1. Samples were obtained before the Isa infusion, in the middle of the infusion, at the end of the infusion, and then 3, 24, 48, 168, and 336 hours after the infusion. Samples were also collected intermittently just before infusion on day 1 from cycle 2 through 8. Concentrations were assessed in plasma by an enzyme‐linked immunoadsorption assay using a lower limit of quantification of 0.500 ng/mL. Noncompartmental analysis was performed with PKDMS software (version 3; Pharsight) with Model 202 (constant infusion). Pharmacokinetic parameters included the area under the curve over the dosing interval (1 week in cycle 1, day 1, and 2 weeks in cycle 3, day 1); the area under the curve until the last quantifiable time point; the maximum plasma concentration; the time to reach the maximum plasma concentration; the time corresponding to the last observed concentration above the lower limit of quantification; the accumulation ratio for the maximum plasma concentration from cycle 3 to cycle 1; and the accumulation ratio for the area under the curve over the dosing interval (per week intervals) from cycle 3 to cycle 1.
Intensive pharmacokinetic blood sampling was also performed for K at cycle 1, day 1, and cycle 3, day 1, with samples obtained before the infusion and then 10 minutes, 30 minutes, 1 hour, 2 hours, and 3 hours after the end of the infusion. Concentrations were assessed in plasma by liquid chromatography with tandem mass spectrometry with a lower limit of quantification of 0.250 ng/mL. Mean concentrations (and standard deviations) over time were plotted.
Results
Patient Characteristics
A total of 33 patients were enrolled from January 2015 through February 2018: 15 in dose escalation (3 at DL1, 6 at DL2, and 6 at DL3) and 18 in dose expansion (at DL2). All patients were considered evaluable for safety and efficacy. This was a heavily pretreated population with a median time since MM diagnosis of 6 years (range, 1‐14 years), and subjects had received a median of 3 prior lines of therapy (range, 2‐8). The majority were male (73%), and 30% were older than 65 years. Seventy‐three percent were refractory to lenalidomide (Len), 48% were refractory to pomalidomide, 70% were refractory to bortezomib, 27% were refractory to K, and 76% were dual‐refractory to PIs and IMiDs. Six patients (18%) had high‐risk (HR) cytogenetic features: 1 with t(4;14), 1 with t(14;16), and 4 with del 17p. The most common fluorescence in situ hybridization (FISH) abnormality was a 1q21 gain, which was present in the absence of other HR FISH findings in 42% of the patients. Patient demographics and baseline disease characteristics are summarized in Table 1.
TABLE 1.
Patient Demographics and Baseline Characteristics
| Characteristic | Isatuximab Dose | Overall (n = 33) | ||
|---|---|---|---|---|
| 10 mg/kg Q2W (n = 3) | 10 mg/kg QW/Q2W (n = 24) | 20 mg/kg QW/Q2W (n = 6) | ||
| Age, median (range), y | 59 (38‐63) | 61 (53‐74) | 64 (55‐78) | 61 (38‐78) |
| Male/female, No. | 2/1 | 16/8 | 6/0 | 24/9 |
| Race, No. (%) | ||||
| White | 3 | 13 | 5 | 21 (64) |
| Asian | 5 | 5 (15) | ||
| Hispanic | 4 | 1 | 5 (15) | |
| Other | 2 | 2 (6) | ||
| Time since diagnosis, median (range), y | 6 (2.5‐10.6) | 5.5 (1‐13.8) | 5.5 (1.8‐14.3) | 6.2 (1‐14.3) |
| Myeloma type at diagnosis, No. (%) | ||||
| IgG | 1 (33) | 14 (58) | 2 (33) | 17 (52) |
| IgA | 1 (33) | 5 (21) | 2 (33) | 8 (24) |
| Light chain only | 1 (33) | 5 (21) | 2 (33) | 8 (24) |
| Prior lines of therapy, median (range), No. | 7 (2‐8) | 2.5 (2‐6) | 3.5 (2‐6) | 3 (2‐8) |
| Previous stem cell transplant, No. (%) | 3 (100) | 18 (75) | 6 (100) | 27 (82) |
| Refractoriness to therapy | ||||
| IMiDs, No. | ||||
| Len, exp/refr | 3/2 | 24/17 | 6/5 | 33/24 |
| Pom, exp/refr | 3/2 | 8/8 | 6/6 | 17/16 |
| PIs, No. | ||||
| Bort, exp/refr | 3/2 | 24/17 | 6/4 | 33/23 |
| K, exp/refr | 3/2 | 6/3 | 5/4 | 14/9 |
| Dual refr (IMiD + PI), % | 67 | 71 | 83 | 76 |
| Bone marrow plasma cells, median (range) | 60 (35‐85) | 26 (1‐80) | 22.5 (1‐30) | 30 (1‐85) |
| Cytogenetics/FISH at study entry, No. (%) | ||||
| HR a | 0 | 4 (18) | 2 (33) | 6 (19) |
| 1q gain (without HR abnl) | 2 (67) | 9 (41) | 2 (33) | 14 (42) |
Abbreviations: abnl, abnormality; Bort, bortezomib; exp, exposed; FISH, fluorescence in situ hybridization; HR, high risk; IgA, immunoglobulin A; IgG, immunoglobulin G; IMiD, immunomodulatory drug; K, carfilzomib; Len, lenalidomide; PI, proteasome inhibitor; Pom, pomalidomide; Q2W, every 2 weeks; QW, weekly; refr, refractory.
Includes del(17p), t(4;14), and t(14;16).
The median follow‐up was 26.7 months (range, 13.3‐61 months) with a data cutoff date of March 20, 2020. The median number of cycles administered was 10 (range, 2‐34). At the data cutoff, 28 patients experienced disease progression, 1 patient opted to proceed to autologous stem cell transplantation in response, and 4 patients remained on therapy. Eleven patients died of MM, but there were no deaths or treatment discontinuations due to toxicity. One patient withdrew consent after disease progression.
DLT/Selected Dose for Expansion
The combination of Isa and K was very well tolerated, and no DLT was encountered at any dose level. Three patients were initially treated at DL1, DL2, and DL3, and then 3 additional patients were treated at DL2 and DL3 before dose expansion. Safety and response data from DL2 and DL3 were both favorable, with longer infusion times noted in DL3. These data combined with additional response and pharmacokinetic data from other Isa studies identified DL2 (Isa at 10 mg/kg weekly for 4 doses and then biweekly) as the optimal biologic dose for Isa in combination with other agents. Thus, DL2 was selected for the expansion cohort of 18 patients.
Safety/AEs
AEs of any grade considered at least possibly related to the study treatment were seen in 100% of the subjects. There were no treatment‐related deaths or treatment discontinuations due to an AE. Grade 3 or 4 AEs considered at least possibly related to the study therapy were observed in 16 patients (48%), with hypertension (15%), diarrhea (9%), anemia (9%), and neutropenia (9%) being most common. Notably, lymphopenia (grades 1‐4, 94%; grades 3 and 4, 55%) was frequent but was excluded from analyses because this was an expected AE. The most common grade 2 or higher treatment‐related AEs were anemia (48%), leukopenia (48%), neutropenia (24%), upper respiratory infections (24%), thrombocytopenia (21%), and hypertension (21%). Select treatment‐related AEs are shown in Table 2. One patient experienced a grade 3 deep vein thrombosis, but there were no other severe hematologic, vascular, or cardiac AEs.
TABLE 2.
Treatment‐Related AEs by Dose Level
| Isatuximab Dose | Overall | |||||||
|---|---|---|---|---|---|---|---|---|
| 10 mg/kg Q2W | 10 mg/kg QW/Q2W | 20 mg/kg QW/Q2W | ||||||
| No. of patients | 3 | 24 | 6 | 33 | ||||
| Toxicity grade | 1‐4 | ≥3 | 1‐4 | ≥3 | 1‐4 | ≥3 | 1‐4 | ≥3 |
| Hematologic AEs, No. (%) | ||||||||
| Neutropenia | 2 (67) | 1 (33) | 8 (33) | 2 (8) | 0 | 0 | 10 (30) | 3 (9) |
| Anemia | 1 (33) | 1 (33) | 18 (75) | 1 (4) | 5 (83) | 1 (17) | 19 (73) | 3 (9) |
| Thrombocytopenia | 2 (67) | 0 | 20 (83) | 1 (4) | 6 (100) | 0 | 26 (85) | 1 (3) |
| Leukopenia | 1 (33) | 1 (33) | 17 (71) | 1 (4) | 1 (17) | 0 | 19 (73) | 2 (6) |
| Lymphopenia | 1 (33) | 1 (33) | 24 (100) | 14 (58) | 6 (100) | 3 (50) | 31 (94) | 18 (55) |
| Nonhematologic AEs, No. (%) | ||||||||
| Fatigue | 2 (67) | 0 | 8 (33) | 0 | 1 (17) | 0 | 11 (33) | 0 |
| Nausea | 2 (67) | 0 | 6 (25) | 0 | 3 (50) | 0 | 11 (33) | 0 |
| Diarrhea | 2 (67) | 1 (33) | 9 (38) | 2 | 0 | 0 | 11 (33) | 3 (9) |
| Constipation | 2 (67) | 0 | 7 (29) | 0 | 3 (50) | 0 | 12 (36) | 0 |
| Hypertension | 0 | 0 | 9 (38) | 3 (13) | 2 (33) | 2 (33) | 11 (33) | 5 (15) |
| Headache | 1 (33) | 0 | 12 (50) | 0 | 2 (33) | 0 | 15 (45) | 0 |
| Muscle cramps | 0 | 0 | 7 (29) | 0 | 4 (67) | 0 | 11 (33) | 0 |
| Insomnia | 0 | 0 | 5 (21) | 2 (8) | 2 (33) | 0 | 7 (21) | 2 (6) |
| Dyspnea | 1 (33) | 0 | 7 (29) | 0 | 1 (33) | 0 | 9 (27) | 0 |
| URI | 1 (33) | 0 | 7 (29) | 0 | 0 | 0 | 8 (24) | 0 |
Abbreviations: AE, adverse event; Q2W, every 2 weeks; QW, weekly; URI, upper respiratory infection.
Infusion reactions were considered AEs of special interest and were scored separately. Infusion reactions were common and occurred in 18 patients (55%), with the majority occurring during the first infusion (17 of 18) and most attributed to Isa (17 of 18). These AEs were manageable and mostly grade 1 or 2 (only 1 grade 3 AE with Isa), and they did not lead to treatment discontinuation in any patient (Fig. 1). Also of interest, treatment‐induced peripheral neuropathy was infrequent (9%) and mild (all grade 1) and was associated with the only dose reduction in the study attributed to K. Missed or delayed dosing of Isa and K was infrequent and was most commonly related to an upper respiratory infection or a planned vacation. Overall, more than 96% of the study therapy was administered. Antidrug antibodies were detected in 2 patients for whom there were no clinical sequalae.
Figure 1.
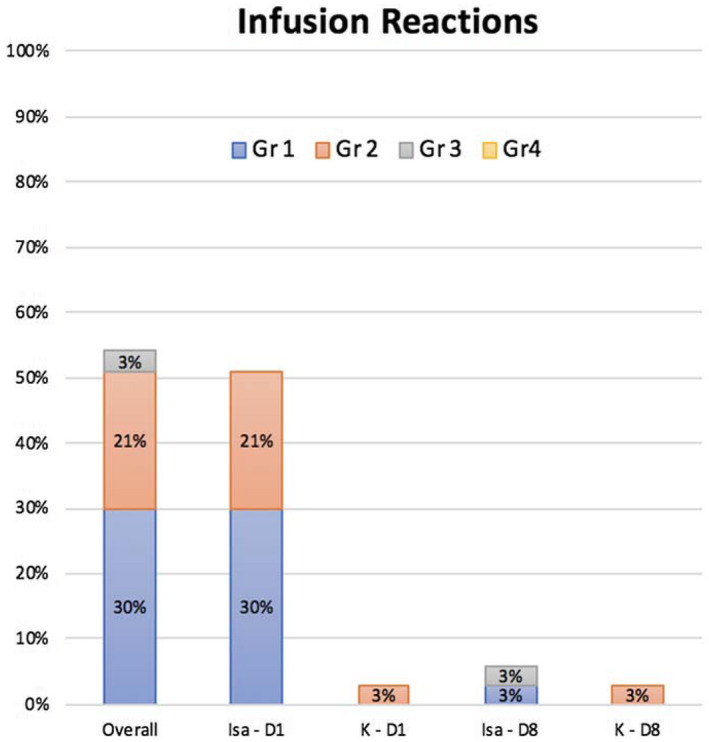
Infusion reactions in cycle 1 by drug, grade, and infusion day. No reactions were seen after cycle 1, D8. D indicates day; Gr, grade, Isa, isatuximab; K, carfilzomib.
Ten patients experienced 12 severe AEs, with all requiring hospitalization. These were mostly due to infection: upper respiratory infection (6), gastroenteritis (2), pneumonia (2), febrile neutropenia (1), and deep vein thrombosis (1). All severe AEs resolved with appropriate treatment, and all patients resumed the study therapy.
Pharmacokinetics
All patients were exposed to Isa and K. Isa pharmacokinetic parameters are provided in Table 3. No major deviation from dose proportionality was observed between 10 and 20 mg/kg. Both the maximum plasma concentration and the area under the curve over the dosing interval increased by ∼1.5‐fold and ∼1.9‐fold (geometric mean ratio) for a 2‐fold increase in the dose after cycle 1 and cycle 3, respectively. The accumulation at cycle 3 (seventh administration) versus the first administration was ∼2 to 3.4. Overall, the Isa pharmacokinetics mirrored single‐agent Isa studies, and there were no apparent drug‐drug interactions or unexpected changes in pharmacokinetics for either Isa or K.
TABLE 3.
Pharmacokinetic Parameters of Isatuximab
| Isatuximab Dosing Regimen | |||
|---|---|---|---|
| 10 mg/kg Q2W | 10 mg/kg QW/Q2W | 20 mg/kg QW/Q2W | |
| Cycle 1, day 1 | |||
| No. | 3 | 20 | 6 |
| tmax, median (range), h | 6.30 (5.37‐24.00) | 5.17 (2.02‐24.18) | 8.83 (5.58‐11.12) |
| Cmax, μg/mL | 155 ± 32.1 (153) [21] | 174 ± 46.8 (169) [27] | 263 ± 59.9 (258) [23] |
| AUC1week, μg h/mL | 20,200 ± 5400 (19,700) [27] | 15,100 ± 3850 (14,600) [26] | 24,600 ± 8030 (23,600) [33] |
| AUC2weeks, μg h/mL | 33,300 ± 11,500 (31,900) [34] | — | — |
| Cycle 3, day 1 | |||
| No. | 16 | 6 | |
| tmax, median (range), h | — | 5.34 (2.07‐48.00) | 7.74 (4.23‐8.98) |
| Cmax, μg/mL | — | 367 ± 127 (346) [35] | 643 ± 173 (623) [27] |
| AUC2weeks, μg h/mL | — | 78,900 ± 37,600 (70,500) [48] | 151,000 ± 60,300 (140,000) [40] |
| Rac Cmax | — | 2.13 ± 0.639 (2.03) [30] | 2.45 ± 0.449 (2.41) [18] |
| Rac AUC1week | — | 3.09 ± 0.971 (2.95) [31] | 3.45 ± 0.872 (3.36) [25] |
Abbreviations: AUC1week, area under the curve over the dosing interval (1 week); AUC2weeks, area under the curve over the dosing interval (2 weeks); Cmax, maximum plasma concentration; CV, coefficient of variation; Q2W, every 2 weeks; QW, weekly; Rac AUC1week, cycle 3 to cycle 1 accumulation ratio of AUC1week; Rac Cmax, cycle 3 to cycle 1 accumulation ratio for Cmax; SD, standard deviation; tmax, time to reach maximum plasma concentration.
Continuous variables are summarized as mean ± SD (geometric mean) [CV %] unless otherwise stated.
Efficacy
All 33 patients were evaluable for a response. The ORR for the entire study population was 70% (23 of 33; 95% CI, 51%‐84%), with 4 patients (12%) achieving a CR (sCR, n = 3; CR, n = 1), 8 patients (24%) achieving a VGPR, 11 patients (33%) achieving a PR, and an additional 5 patients (15%) experiencing a minimal response (clinical benefit rate, 85%; Fig. 2).
Figure 2.
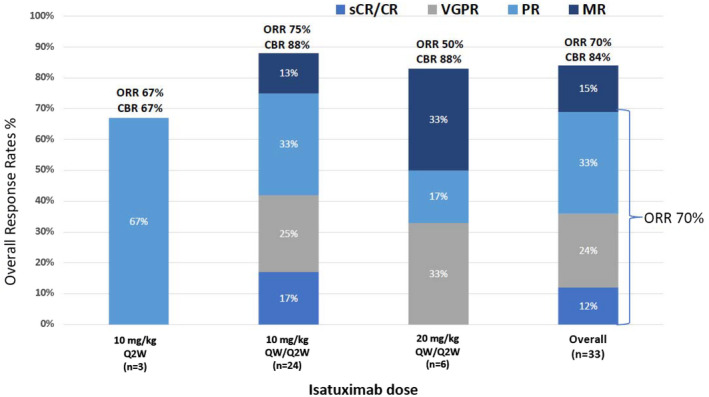
ORRs by dose level and overall. CBR indicates clinical benefit rate; MR, minor response; ORR, overall response rate; PR, partial response; Q2W, every 2 weeks; QW, weekly; sCR/CR, stringent complete response/complete response; VGPR, very good partial response.
At a median follow‐up of 26.7 months, the median PFS was 10.1 months (95% CI, 6.47‐16.4 months; Fig. 3). The 2‐year OS rate was 76% (95% CI, 63%‐92%), and the median OS was not reached (Fig. 4). Among responders, the median time to first response (≥PR) was 4 weeks (4‐48 weeks) with a median time to best response of 12 weeks (4‐96 weeks). Six patients had a deepening of response even after cycle 4 at a median of 38 weeks of therapy (16‐96 weeks) with an improvement to an sCR/CR (n = 2), VGPR (n = 2), or PR (n = 2).
Figure 3.
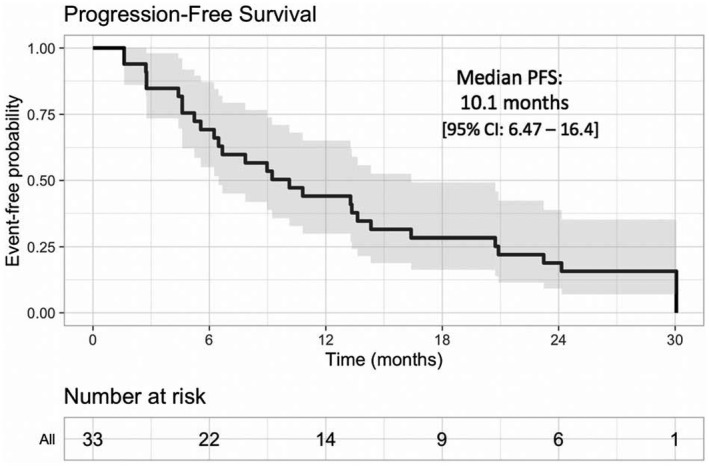
Kaplan‐Meier analysis of PFS. The median PFS (along with the 95% CI) is plotted for all treated patients (isatuximab plus carfilzomib). CI indicates confidence interval; PFS, progression‐free survival.
Figure 4.
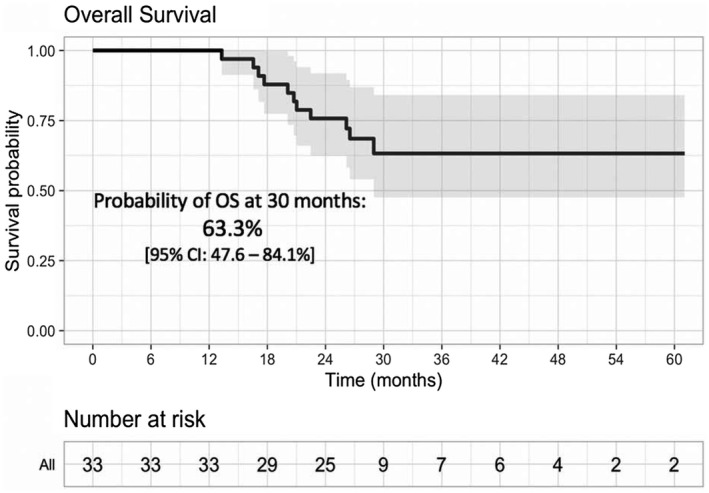
Kaplan‐Meier analysis of OS. OS is plotted for the total population treated with isatuximab plus carfilzomib. CI indicates confidence interval; OS, overall survival.
Responses were consistently observed in all subgroups. For patients who were refractory to Len (n = 24 [the majority receiving a Len doublet or triplet]), any IMiD (n = 28), or PIs (n = 25), the ORRs were 54%, 68%, and 72%, respectively. For dual‐refractory patients (IMiDs and PIs; n = 25), the ORR was similar (68%). For the patients with prior K exposure (n = 14) and the K‐refractory patients (n = 9), the ORRs were 62% and 60%, respectively. In a subgroup analysis based on cytogenetic/FISH results, the HR subgroup (n = 6) had an ORR of 83% (3 VGPRs and 2 PRs), and the 14 patients with a gain (1q21) had an ORR of 54% (combined HR and 1q gain ORR, 65% [n = 20]). Age was not significantly associated with response; the ORR for an age < 65 years was 73%, and the ORR for an age ≥ 65 years was 64%. Responses were durable with a median DOR of 10 months (≥PR; range, 1.9‐29.4 months), and more than one‐third of the responders achieved a remission duration longer than 18 months.
Discussion
The treatment landscape for RRMM has changed since mAbs gained FDA approval. Dara was initially approved in 2015 as monotherapy for the treatment of IMiD and PI–refractory MM, and since then, anti‐CD38 antibodies have been approved for use in 9 additional combinations and indications. 3 , 6 , 7 , 8 , 9 , 10 , 14 In general, these combinations have shown improved efficacy with nonoverlapping toxicity. 19 , 20 This study was originally designed to assess the safety and efficacy of Isa plus K, a novel combination, in a more heavily pretreated population with RRMM. This study was initiated before the approvals of high‐dose Kd (K 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 plus d 40 mg weekly) and weekly Kd (K 70 mg/m2 on days 1, 8, and 15 plus d 40 mg weekly) for RRMM and thus used K at 27 mg/m2 on days 1, 2, 8, 9, 15, and 16. The early results from this trial led to the phase 3 IKEMA trial (Isa plus K56d vs K56d), which may lead to FDA approval for Isa plus Kd.
This phase 1b study demonstrated excellent safety; the combination of Isa and K27 was well tolerated, and there were very few treatment‐related grade 3 or higher events. The most common grade 3 events were hypertension (15%), diarrhea (9%), anemia (9%), and neutropenia (9%), and this is in line with the expected toxicity of these agents. No patient discontinued treatment because of toxicity, and only 1 patient required a dose reduction of K due to neuropathy. Importantly, no additional significant cardiovascular AEs were seen other than the expected hypertension and 1 episode of deep vein thrombosis. Overall, the levels of hematologic, cardiovascular, and nonhematologic toxicity compared favorably with other doublet and triplet combinations used in the treatment of RRMM, and patients were able to receive >96% of the intended therapy.
This combination also demonstrated promising efficacy with an ORR of 70%. Responses were noted in dual‐refractory patients (refractory to IMiDs and PIs; ORR, 68%), Len‐refractory patients (ORR, 54%), and K‐refractory patients (ORR, 60%). The PFS was 10.1 months, and the 2‐year OS rate was 76%. These data compare favorably with the ICARIA study, which included similarly refractory patients and noted an ORR of 60.4%, PFS of 11.5 months, and 1‐year OS of 72% in IsaPd‐treated patients. 6 It is worth noting that the majority of IMiD‐refractory patients in this study became refractory on therapeutic Len or Pom as part of a doublet or triplet, and very few became refractory solely on Len maintenance. Kumar et al 21 published on PI and IMiD–refractory patients unexposed to CD38 antibodies, who showed expected median PFS and OS of 5 and 15.2 months, respectively, on subsequent non–CD38‐containing therapy. Although cross‐trial comparisons have limitations, another trial that enrolled similarly exposed patients (median number of prior lines, 3; Len‐refractory, 90%; dual‐refractory, 68%) was the Eloquent‐3 trial, a randomized phase 3 which evaluated the triplet of elotuzumab, pomalidomide, and dexamethasone versus the doublet Pd and showed an advantage for EloPd with an ORR of 53%, PFS of 10.3 months, and OS at 18 months of 68%. 22 Collectively, these comparisons show that Isa plus K27 is an active regimen, with favorable responses noted in heavily pretreated, Len‐refractory, dual‐refractory, and K‐refractory patients with MM.
These data also support our hypothesis of potential synergy between Isa and K through the induction of cellular apoptosis. K can induce apoptosis through multiple mechanisms, including induction of the unfolded protein response, activation of c‐Jun NH2‐terminal kinase (JNK) and p53, and prevention of degradation of proapoptotic family members such as Bim, Bid, and NOXA. 23 , 24 Isa triggers both the caspase‐dependent apoptotic pathway and the lysosome‐mediated cell death pathway, and these direct effects are independent of Fc fragment binding. 25 Although the direct mechanism for this synergy remains unclear, the early clinical results from Isa plus K(27), including the high ORR of 60% in K‐refractory patients, support further development of Isa plus K.
Recently, data from the phase 3 IKEMA trial, which evaluated Isa plus K56d versus K56d, showed significantly improved ORR (87% vs 83%) and PFS (not reached at 20.7 months of follow‐up vs 19.2 months) in patients receiving IsaKd versus Kd. 16 Also supporting the combination of CD38 antibodies and K is the phase 3 CANDOR trial, which reported significantly improved ORR (84% vs 75%) and PFS (not reached vs 15.8 months) in patients receiving DaraK56d versus K56d. 15 A phase 1b study evaluated Dara with weekly K70d and demonstrated an ORR of 84% and a PFS rate of 75% at 12 months of follow‐up. 26 Full FDA approval has been granted for both Dara plus Kd regimens. Although the ORR and PFS in our study (Isa plus K27) are significantly lower than those in CANDOR (Dara plus K56d) and IKEMA (Isa plus K56d), the patient populations and K dosing were significantly different. Both CANDOR and IKEMA enrolled less heavily pretreated patients (1‐3 prior lines), with only a minority of patients refractory to Len or PIs. In our study, more than three‐quarters of the patients were dual‐refractory. Overall, the favorable responses seen in all these studies and especially in refractory patients support the use of the combination of CD38 antibodies with K.
The selected Isa dosing of 10 mg/kg weekly for 4 doses and then every other week as the optimal dose was based on multiple factors, including receptor occupancy, pharmacokinetics, and response data from all Isa studies. Importantly, Isa exposure in this study (area under the curve over the dosing interval (1 week) [AUC1week], 15,100 µg h/mL; coefficient of variation, 26%) was comparable to that observed with Isa monotherapy (mean AUC1week; 17,000 μg h/mL). 4 The pharmacokinetic parameters of Isa were unaffected by coadministration with K, and K concentration‐time profiles were in accordance with those reported in the literature. 27 These results suggest that there is no interaction between Isa and K when they are given in combination. We saw no overlapping or unexpected toxicity from this combination, and this also supports the use of this combination.
Overall, the results of this study support the use of a CD38 antibody plus K in Len‐, PI‐, and/or dual‐refractory patients. In the United States, where many patients are treated with Len maintenance after frontline therapy, the switch in drug classes to a CD38 antibody plus a PI (K) upon progression would be strongly supported by these data. Although the approved K dosing with anti‐CD38 therapy in RRMM (1‐3 prior lines) is 56 mg/m2 twice weekly, in the real‐world setting, treatment approaches are often tailored to individual patients, 28 and K dosing at 27 mg/m2 for those unable to tolerate higher doses would be supported by these data. An ongoing expansion cohort is evaluating Isa with weekly dosing of K.
In summary, the results of this phase 1b study demonstrate that Isa in combination with standard‐dose K (27 mg/m2) is well tolerated and active in patients with RRMM. No DLTs were observed, and the maximum tolerated dose was not reached. Objective responses were noted in the majority of the patients (ORR, 70%) even though more than three‐fourths of the patients were dual‐refractory to PIs and IMiDs, and 25% were K‐refractory before study entry. The results of this study and other Isa combination trials support the clinical use of Isa at 10 mg/kg weekly/every 2 weeks as the preferred dosing regimen. Overall, this combination has great potential for the treatment of patients with IMiD and PI–refractory disease.
Funding Support
This was a Multiple Myeloma Research Consortium study supported by research funding from Sanofi and Amgen. All study medications (isatuximab and carfilzomib) were provided from an investigational supply.
Conflict of Interest Disclosures
Thomas G. Martin reports research funding (institutional) from Sanofi, Amgen, Janssen, and Seattle Genetics and consultancy for Roche and GlaxoSmithKline. Nina Shah reports research funding (institutional) from Bluebird Bio, Janssen, Celgene/Bristol‐Myers Squibb, Sutro Biopharma, TeneoBio, Poseida, and Nektar and consultancy for and honoraria from Celgene, Janssen, Bluebird Bio, Sutro Biopharma, Genentech, Seattle Genetics, Oncopeptides, Karyopharm, Surface Oncology, Precision Biosciences, GlaxoSmithKline, Nektar, Amgen, Indapta Therapeutics, Bristol‐Myers Squibb, CareDx, Kite, Karyopharm, and Sanofi. Joshua Richter reports consultancy and membership on an advisory board for Sanofi. David H. Vesole reports research funding (institutional) from Sanofi and Amgen; stock and other ownership interests in Amgen, AbbVie, Biogen, Celgene/Bristol‐Myers Squibb, Gilead Sciences, Johnson & Johnson, Lilly, and Novartis; honoraria from or consultancy for Amgen, Takeda, Celgene, and Pfizer; and membership in speakers’ bureaus for Amgen, Celgene/Bristol‐Myers Squibb, Janssen Oncology, GlaxoSmithKline, and Takeda. Sandy W. Wong reports research funding (institutional) from Bristol‐Myers Squibb, GlaxoSmithKline, Janssen, Roche/Genentech, and Fortis and consultancy or membership on an advisory committee for Amgen and Sanofi. Deepu Madduri reports research funding (institutional) from Janssen and Regeneron; membership in speakers’ bureaus for Shire, Legend, Kinevant, and GlaxoSmithKline; honoraria from Janssen, Celgene, and AbbVie; and consultancy for Foundation Medicine and Takeda. Sundar Jagannath reports consultancy for Legend Biotech, Takeda, AbbVie, Celgene, Bristol‐Myers Squibb, Karyopharm, Janssen, and Merck and membership on scientific advisory boards for Celgene, Bristol‐Myers Squibb, and Sanofi‐Aventis. David S. Siegel reports research funding (institutional) from Celgene; stock and other ownership interests in Cellularity; consulting or advisory roles with Amgen, Celgene, Takeda, Janssen Oncology, Bristol‐Myers Squibb, Karyopharm Therapeutics, and Merck; and membership in speakers’ bureaus for Amgen, Celgene, Takeda, Janssen Oncology, and Bristol‐Myers Squibb. Noa Biran reports research funding (institutional) from Merck, Karyopharm Therapeutics, and Bristol‐Myers Squibb; honoraria from or consultancy for Bristol‐Myers Squibb, Amgen, Celgene, Takeda, and Janssen Oncology; and membership in speakers’ bureaus for Takeda, Celgene, Amgen, Janssen, and Bristol‐Myers Squibb. Jeffrey L. Wolf reports consultancy for Adaptive Biotech, TeneoBio, Amgen, Bristol‐Myers Squibb, Takeda, and Janssen. Samir Parekh reports consultancy for Foundation Medicine and research funding (institutional) from Celgene, Pfizer, and Karyopharm. Hearn J. Cho is employed by the Multiple Myeloma Research Consortium and reports research funding from Takeda, Celgene, and Genentech. Samira Ziti‐Ljajic is an employee of Sanofi. Ajai Chari reports consultancy for Secura Bio, Novartis, Amgen, Bristol‐Myers Squibb, Celgene, Antengene, Takeda, Janssen, and Karyopharm; has received research funding from Amgen, Array Biopharma, Celgene, GlaxoSmithKline, Janssen, Takeda, Novartis, Oncopeptides, Pharmacyclics, and Seattle Genetics; and is an advisory board member for Amgen, Celgene, Millennium/Takeda, Novartis, Janssen, Karyopharm, Sanofi, GlaxoSmithKline, Secura Bio, and Seattle Genetics. The other authors made no disclosures.
Author Contributions
Thomas G. Martin: Research design, writing of the protocol, treatment of patients, data acquisition, analysis and interpretation of the data, and writing of the manuscript. Nina Shah: Treatment of patients, data acquisition, analysis and interpretation of the data, and writing of the manuscript. Joshua Richter: Treatment of patients, data acquisition, and analysis and interpretation of the data. David H. Vesole: Treatment of patients, data acquisition, analysis and interpretation of the data, and writing of the manuscript. Sandy W. Wong: Treatment of patients, data acquisition, and analysis and interpretation of the data. Chiung‐Yu Huang: Analysis and interpretation of the data. Deepu Madduri: Treatment of patients and data acquisition. Sundar Jagannath: Treatment of patients and data acquisition. David S. Siegel: Treatment of patients and data acquisition. Noa Biran: Treatment of patients and data acquisition. Jeffrey L. Wolf: Research design, treatment of patients, and data acquisition. Samir Parekh: Treatment of patients and data acquisition. Hearn J. Cho: Treatment of patients and data acquisition. Pamela Munster: Research design. Shambavi Richard: Treatment of patients and data acquisition. Samira Ziti‐Ljajic: Vital analytical tools. Ajai Chari: Treatment of patients, data acquisition, analysis and interpretation of the data, and writing of the manuscript. All authors reviewed the manuscript, made edits, and approved the final product.
Martin TG, Shah N, Richter J, Vesole DH, Wong SW, Huang C‐Y, Madduri D, Jagannath S, Siegel DS, Biran N, Wolf JL, Parekh S, Cho HJ, Munster P, Richard S, Ziti‐Ljajic S, Chari A. Phase 1b trial of isatuximab, an anti‐CD38 monoclonal antibody, in combination with carfilzomib as treatment of relapsed/refractory multiple myeloma. Cancer. 2021. 10.1002/cncr.33448
References
- 1. Durer C, Durer S, Lee S, et al. Treatment of relapsed multiple myeloma: evidence‐based recommendations. Blood Rev. 2020;39:100616. [DOI] [PubMed] [Google Scholar]
- 2. Demel I, Bago JR, Hajek R, Jelinek T. Focus on monoclonal antibodies targeting B‐cell maturation antigen (BCMA) in multiple myeloma: update 2020. Br J Haematol. Published online November 20, 2020. doi: 10.1111/bjh.17235 [DOI] [PubMed] [Google Scholar]
- 3. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment‐refractory multiple myeloma (SIRIUS): an open‐label, randomised, phase 2 trial. Lancet. 2016;387:1551‐1560. [DOI] [PubMed] [Google Scholar]
- 4. Martin T, Strickland S, Glenn M, et al. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin T, Baz R, Benson DM, et al. A phase Ib study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;129:3294‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low‐dose dexamethasone versus pomalidomide and low‐dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA‐MM): a randomised, multicenter, open‐label, phase 3 study. Lancet. 2019;37:2096‐2107. [DOI] [PubMed] [Google Scholar]
- 7. Palumbo A, Chanan‐Khan A, Weisel K, et al; CASTOR Investigators . Daratumumab, bortezomib and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754‐766. [DOI] [PubMed] [Google Scholar]
- 8. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319‐1331. [DOI] [PubMed] [Google Scholar]
- 9. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380:2104‐2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mateos MV, Cavo M, Blade J, et al. Overall survival with daratumumab, bortezomib, melphalan and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open‐label, phase 3 trial. Lancet. 2020;395:132‐141. [DOI] [PubMed] [Google Scholar]
- 11. Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38‐targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20:4574‐4583. [DOI] [PubMed] [Google Scholar]
- 12. van de Donk NWCJ, Usmani SZ. CD38 antibodies in multiple myeloma: mechanism of action and modes of resistance. Front Immunol. 2018;9:2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lonial S, Dimopoulos M, Palumbo A, et al; ELOQUENT‐2 Investigators . Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621‐631. [DOI] [PubMed] [Google Scholar]
- 14. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130:974‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomized, multicenter, open‐label, phase 3 study. Lancet. 2020;396:186‐197. [DOI] [PubMed] [Google Scholar]
- 16. Moreau P, Dimopoulos MA, Mikhael J, et al.Isatuximab plus carfilzomib and dexamethasone vscarfilzomib and dexamethasone in relapsed/refractory multiple myeloma (IKEMA): interim analysis of a phase 3 randomized, open‐label study [abstract LB2603]. Abstract presented at 25th European Hematology Association Annual Congress; June 11‐21, 2020.
- 17. Nijhof IS, Groen RW, Noort WA, et al. Preclinical evidence for the therapeutic potential of CD38‐targeted immune‐chemotherapy in multiple myeloma patients refractory to lenalidomide and bortezomib. Clin Cancer Res. 2015;21:2802‐2810. [DOI] [PubMed] [Google Scholar]
- 18. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328‐e346. [DOI] [PubMed] [Google Scholar]
- 19. Sun Z, Zheng F, Wu S, Liu Y, Guo H, Liu Y. Triplet versus doublet combination regimens for the treatment of relapsed or refractory multiple myeloma: a meta‐analysis of phase III randomized controlled trials. Crit Rev Oncol Hematol. 2017;113:249‐255. [DOI] [PubMed] [Google Scholar]
- 20. van de Donk NWCJ, Moreau P, Plesner T, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127:681‐695. [DOI] [PubMed] [Google Scholar]
- 21. Kumar S, Dimopoulos M, Kastritis E, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31:2443‐2448. [DOI] [PubMed] [Google Scholar]
- 22. Dimopoulos M, Dytfeld D, Gropsiki S, et al. Elotuzumab plus pomalidomide and dexamethasone for relapsed/refractory multiple myeloma: efficacy results after additional follow‐up of the phase 2, randomized ELOQUENT‐3 study [abstract PS1370]. Poster presented at: 24th European Hematology Association Annual Congress; June 15, 2019; Amsterdam, the Netherlands.
- 23. Chhabra S. Novel proteasome inhibitors and histone deacetylase inhibitors: progress in myeloma therapeutics. Pharmaceuticals (Basel). 2017;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA‐mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282‐6293. [DOI] [PubMed] [Google Scholar]
- 25. Martin T, Corzo K, Chiron M, et al. Therapeutic opportunities with pharmacologic inhibition of Cd38 with isatuximab. Cells. 2019;8:1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chari A, Martinez‐Lopez J, Mateos MV, et al. Daratumumab plus carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood. 2019;134:421‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z, Yang J, Kirk C, et al. Clinical pharmacokinetics, metabolism, and drug‐drug interactions of carfilzomib. Drug Metab Dispos. 2013;41:230‐237. [DOI] [PubMed] [Google Scholar]
- 28. Richardson PG, San Miguel JF, Moreau P, et al. Interpreting clinical trial data in multiple myeloma: translating findings to the real‐world setting. Blood Cancer J. 2018;8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]