

Abstract
Nature segregates fundamental tasks such as information storage/transmission and catalysis between two different compound classes (e.g. polynucleotides for replication and folded polyamides for catalysis). This division of labor is likely a product of evolution, raising the question of how simpler systems in which replicators and folded macromolecules co‐exist may emerge in the transition from chemistry to biology. In synthetic systems, achieving co‐existence of replicators and foldamers in a single molecular network remains an unsolved problem. Previous work on dynamic molecular networks has given rise to either self‐replicating fibers or well‐defined foldamer structures (or completely un‐sorted complex systems). We report a system in which two cross‐reactive dithiol (nucleobase‐ and peptide‐based) building blocks self‐sort into a replicator fiber and foldamer that both emerge spontaneously and co‐exist. The self‐sorting behavior remains prevalent across different building block ratios as two phases of emergence occur: replicator growth followed by foldamer formation. This is attributed to the autocatalytic formation of the replicator fiber, followed by enrichment of the system in the remaining building block, which is subsequently incorporated into a foldamer.
Keywords: dynamic combinatorial chemistry, foldamers, self-assembly, self-replicators, self-sorting
Two different dithiol building blocks interconvert through reversible covalent bonds to initially form a dynamic combinatorial library of macrocycles of various size. The self‐sorting process starts by the amplification of a single library member to form self‐replicating fibers. This step is followed by enrichment of the system in the remaining building block, which forms a large macrocyclic foldamer.
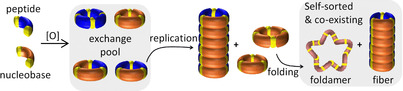
Introduction
Life heavily relies on the synthesis of different sets of macromolecules and their ability to function orthogonally yet harmonically. Nature accomplishes this by segregating the synthesis of these macromolecules based on compound class by using different connecting chemistries. For example, nucleotides are synthetically linked by phosphodiester bonds to form a polynucleotide that stores and transmits genetic information. Nucleic acids are orthogonal to proteins, which have a folded polyamide backbones, bind biomolecules and catalyze reactions. The building blocks of these two classes of biomacromolecules (nucleotides and amino acids) mostly do not cross‐react (except in t‐RNA‐amino acid conjugates). This raises the question of how this division of labor over two compound classes can come about during the emergence and evolution of life and whether it is necessary to have two different connecting chemistries, or whether simpler systems can form based on a single connecting chemistry in which replicators and folded macromolecules co‐exist. The “RNA world hypothesis” attempts to address these questions by assuming that RNA can both store/transmit information and catalyze reactions. [1] These functions are accomplished by adopting significantly different conformations. For example, RNA‐based enzymes or ribozymes exhibit three‐dimensional folded structures, just like modern proteins require folding for function, yet in order to be replicated RNA needs to adopt an unfolded structure. [2] These different conformational requirements are difficult to reconcile and limits the ability of RNA to self‐replicate and have ribozymal activity at the same time. [3]
We reasoned that it should be possible to achieve the parallel emergence and co‐existence of self‐replicators and folded structures through a self‐sorting process between compound classes in a single molecular network using a single connecting chemistry. Self‐sorting of mixtures of interacting monomers is a relatively well‐established method for achieving the co‐existence of complex assemblies in both organic solvents and aqueous medium. [4] Yet such sorting tends to be limited to different assemblies within a single class of structures, such as coordination cages, [5] foldamers, [6] nanofibers [7] or knots. [8] Self‐sorting of complex structures into two dissimilar structure classes is still a largely unmet challenge in systems chemistry.
We also reasoned that dynamic combinatorial chemistry (DCC) should allow access to the desired self‐sorting behavior. [9] Dynamic combinatorial libraries (DCLs) are generated upon mixing building blocks that react with one another through reversible covalent bonds and library members continuously interconvert by exchange of these building blocks. Although the possible number of library members can be vast, molecular recognition events involving added molecules (templates) or between library members can direct the product distribution towards a specific thermodynamically stable system, [10] including self‐replicating molecules and foldamers. [11]
Specifically, we have shown that oxidation of aryl dithiol building block 1 a, functionalized with a pentapeptide sequence, gives rise to a library of macrocycles, from which a specific ring size was amplified as a result of assembling into fibers stabilized by β‐sheet interactions. [12] This assembly process is autocatalytic so the assembling macrocycles can be considered self‐replicators. In other work, the same aryl dithiol motif, functionalized with different moieties (nucleobases as in building block 2 or short peptides), were found to form specific, unexpectedly large macrocycles (ranging from a 9mer to a 23mer).[ 11b , 13 ] These structures form by virtue of folding into well‐defined three‐dimensional conformations, akin to the folding exhibited by proteins.
Our first attempts at achieving the parallel emergence of foldamers and self‐replicators, by mixing the nucleobase building blocks that produced foldamers with the peptide building blocks that produced replicators, failed as a result of the dominance of self‐replicators that incorporated both building block types at a specific building block ratio and the absence of self‐sorting when varying building block ratios. A representative example of the observed diversity in products obtained by mixing equimolar amounts of building blocks 1 a and 2 is shown in Figure 1 a. [14]
Figure 1.
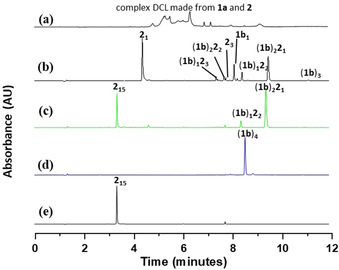
UPLC analysis reveals a) a complex, mixed DCL made from building blocks 1 a (1.0 mM) and 2 (1.0 mM), published previously; [14] b) a DCL of macrocycles after 36 hours upon combining 1 b (1.0 mM) and 2 (1.0 mM); c) self‐sorting into two distinct species (1 b)2 21 and 215, after the DCL in b was incubated for 7 days; d) 1 b (2.0 mM) selectively forming (1 b)4 and e) 2 (2.0 mM) selectively forming 215 after stirring for 10 days in Na2B4O7 buffer (50 mM, pH 8.0, 1.0 M NaCl).
We now report a system in which two cross‐reactive building blocks self‐sort into a replicator fiber and a foldamer that both emerge spontaneously and stably co‐exist subsequently. The two structures rely on reversible disulfide chemistry and in the self‐sorting process replicator fiber formation is followed by the emergence of the folded structure. These results demonstrate that a mixture of nucleobase and peptide building blocks can autonomously self‐sort.
Results and Discussion
In the previously reported system [14] based on peptide‐based building blocks 1 a and nucleobase‐containing building block 2 self‐sorting did not occur, despite the fact that 1 a and 2, individually, can give rise to replicators [12] and foldamers, [11b] respectively. In order to favor self‐sorting we choose to replace 1 a with the somewhat more hydrophobic building block 1 b (Scheme 1), featuring a β‐alanine instead of a glycine residue, which should enhance the stability of the stacks of replicators and facilitate their emergence across a wider range of building block stoichiometries. When 1 b would still form a replicator also in the presence of excess 2 then foldamer formation from the latter would be possible at the same time.
Scheme 1.
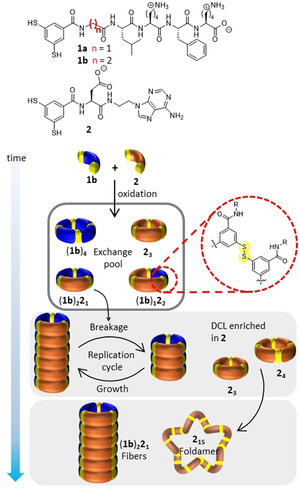
Cartoon illustration of the self‐sorting process. A DCL made from building blocks 1 b and 2 first forms autocatalytic (1 b)2 21 fibers, facilitating the formation of a single building block foldamer, 215, by enriching the remaining products in 2.
Indeed, two distinct species emerge from a DCL made from an equimolar solution of 1 b and 2 ([1 b]=[2]=1.0 mM) in aqueous borate buffer (50 mM Na2B4O7, pH 8.0, 1.0 M NaCl) upon stirring at room temperature in the presence of air. The reaction progress was monitored by UPLC‐MS at a wavelength where building blocks 1 b and 2 have comparable molar absorptivities. Control experiments showed that these absorptivities are independent of the macrocycles in which the building blocks reside (see Figure S6), so that peak areas directly reflect concentrations (in terms of units of building block). The first 24 hours revealed a mixture of differently sized mixed building block macrocycles (Figure 1 b), however, after 7 days, the DCL was dominated by two major species: cyclic trimer (1 b)2 21 and foldamer 215, accompanied by a small amount of another cyclic trimer (1 b)1 22 (Figure 1 c). This contrasts with the results of a DCL prepared from 1 b alone which forms only the tetramer (1 b)4 (Figure 1 d). Interestingly, 215 was the only foldamer that emerged from a plethora of foldamer possibilities and the ring size was preserved as 2 alone also generates a foldamer consisting of 215 (Figure 1 e). [11b]
Self‐sorting was found to be robust to changes in pH in the analyzed range of pH 6–8, while high ionic strength (0.5–1.0 M NaCl) promoted foldamer formation (see Figure S22).
The self‐sorting behavior was monitored over time at different building block ratios and revealed two phases of emergence. A series of libraries was set up with a total concentration of 2.0 mM with different molar fractions of 1 b (100 %−0 %) in borate buffer and monitored by UPLC. In cases where 1 b was present at a molar fraction of less than 60 %, the kinetic traces revealed two phases of emergence: the immediate formation of (1 b)2 21 until all of building block 1 b was consumed, which was followed by the second phase of 215 growth until finally both species co‐exist. The time of completion of (1 b)2 21 formation (and the subsequent initiation of foldamer formation) is dependent on the concentration of building block 1 b available in the library (Figure S1, S2). For example, at 50 % in 1 b, the (1 b)2 21 species forms in 96 h (Figure 2 a, shaded area) corresponding to the onset of the formation of 215, whereas at 30 % in 1 b (Figure 2 b, shaded area), the (1 b)2 21 species forms in 48 h, thereby promoting the early formation of 215. This suggests that (1 b)2 21 indirectly promotes the formation of only a single foldamer in the two‐component system by removing 1 b from the exchange pool, reminiscent of the behavior observed previously for coordination cages. [5a] The self‐sorting of the system into (1 b)2 21 and 215 is preserved across a wide range of building block ratios. Only when the mole fraction of building block exceeds 70 % in 1 b foldamer formation is no longer observed, as all 2 is now used for replicator formation (Figure 2 c, S1 and S2). We were thus interested in characterizing (1 b)2 21 given its important role in the self‐sorting process.
Figure 2.
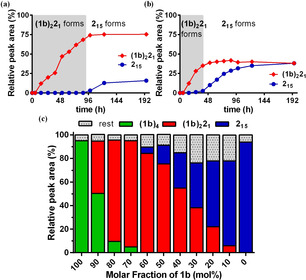
a) Kinetics of emergence of replicator (1 b)2 21 and foldamer 215 from an equimolar solution of 1 b (1.0 mM) and 2 (1.0 mM) show that 215 emerges only after formation of (1 b)2 21 is near complete (shaded area, 96 h). b) Earlier emergence of 215 is observed in an analogous experiment starting from a different building block ratio (30 % in 1 b), as (1 b)2 21 growth is complete early at 48 h (shaded area). c) Experiments starting from different ratios of 1 b and 2 show that replicators and foldamers dominate the DCL.
Seeding experiments reveal (1 b)2 21 and (1 b)4 are both self‐replicators that form fibers. The addition of 6 mol % of preformed (1 b)2 21 (seed) into a fresh DCL made from 1 b (1.0 mM) and 2 (0.50 mM) accelerated the formation of (1 b)2 21 in comparison to the non‐seeded library, which is a hallmark of autocatalytic self‐replication (Figure 3 a). [11a] Acceleration of (1 b)4 formation was also observed upon the addition of (1 b)4 seed to a fresh solution of 1 b (Figure 3 b). Previous reports by our group show that peptide‐based replicators owe their autocatalytic behavior to the formation and breakage of fibrous structures formed by β‐sheet stacking. [15] Note that, as expected, the formation of foldamer 2 15 did not show any signs of autocatalysis (Figure S24).
Figure 3.
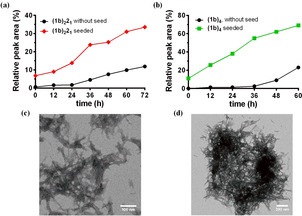
Seeding experiments reveal accelerated formation of (1 b)2 21 or (1 b)4 indicative of self‐replication when a) (1 b)2 21 (6 mol %) seed is added to a solution of 1 b and 2 ([1 b] 1.0 mM, [2]=0.5 mM, red trace) and when b) (1 b)4 (10 mol %) seed is added to 1 b alone ([1 b]=1.0 mM, green trace) in comparison to non‐seeded samples (black traces). TEM images reveal bundled fibers from samples of c) (1 b)2 21 and d) (1 b)4.
Negative staining transmission electron microscopy (TEM) micrographs of the libraries dominated by replicators (1 b)2 21 and (1 b)4 showed bundled fibrous nanostructures (Figures 3 c and d). β‐sheet stacking of the pentapeptide sequence contributes to the formation of these fibers as evident from both Circular Dichroism (CD) spectroscopy and Thioflavin T fluorescence (Figure S3a). The CD spectra of replicator (1 b)4 showed strong positive helicity at 206 nm and negative helicity around 220 nm, indicative of the presence of β‐sheet structure [12] (Figure S3b). Similar yet weaker helicities were observed for replicator (1 b)2 21.
The emergence of self‐replicating fibers, such as (1 b)2 21, operates via a fiber elongation/breakage mechanism and is heavily affected by mechanical agitation. [16] In the absence of mechanical agitation, a fresh library was set up by dissolving equimolar building blocks 1 b and 2. UPLC analysis revealed that self‐sorting was less efficient in the absence of agitation (Figure S4). A range of different small rings composed of building block 1 b and 2 formed. 15mer rings formed as well but incorporated one or two units of building block 1 b (e.g. (1 b)1 214). We conclude that, in the absence of efficient self‐replication by (1 b)2 21, the degree of self‐sorting is diminished. Furthermore, the autocatalytic (1 b)2 21 fiber is more stable than a mixture of 215 and (1 b)4. The addition of 1 b (1.0 mM) to a solution containing 215 (1.0 mM) promoted the partial degradation of the foldamer and, following the transient formation of (1 b)4, the dominant species was (1 b)2 21, consuming both 215 and (1 b)4 (Figure S5).
The self‐sorting observed in the 1 b/2 system was absent in the 1 a/2 system. The difference between the two systems is relatively small: 1 b contains an additional methylene group. Yet this difference makes 1 b more hydrophobic and more efficient at assembling as compared to 1 a, as evident from the fact that cyclic oligomers of 1 b are capable of self‐assembling at a smaller ring size than those of 1 a (tetramer vs. hexamer; we know from previous work that the minimal ring size required for assembly is inversely correlated to building block hydrophobicity). [12] We speculate that this enhanced tendency for 1 b‐containing rings to self‐assemble translates into (1 b)2 2 assemblies being more stable than the corresponding (1 a)2 2 assemblies. This notion is supported by the fact that (1 b)2 2 assemblies are more robust to changes in building block stoichiometry than the corresponding (1 a)2 2 assemblies, as evident from Figure S23.
Conclusion
We have shown how a DCL made from a nucleobase‐containing and a peptide‐based building block self‐sorts into a foldamer incorporating 15 units of the nucleobase building block and a self‐replicator that contains both building block types. Intriguingly, the foldamer is formed from the nucleobase building block while the peptide building block is fully incorporated into the self‐replicator, which is opposite to the distribution of these compound classes in Nature, where peptides make folded proteins and nucleic acid oligomers are replicated. The emergence of the self‐replicator promotes self‐sorting and indirectly facilitates foldamer formation by homogenizing library composition: by taking the peptide building block out of the exchange pool the latter becomes relatively enriched in the remaining nucleobase building block, which is then more readily incorporated into the foldamer. These results show for the first time that an initially diverse mixture of compounds can transform spontaneously and selectively into discrete and stably co‐existing foldamers and self‐replicators. Since self‐sorting occurs also when the system is closed (i.e. not agitated; cf. Figure S4), albeit less efficiently, the sorting process is thermodynamically favorable, indicating that the strength of the non‐covalent interactions in the folded and self‐assembled structures are sufficient to overcome the entropic cost that is inevitably associated with sorting. These findings show that assembly processes are a powerful force to counteract the inherently divergent nature of chemistry and push chemical systems to the formation of a few specific structures where many alternatives are also potentially accessible. Such changeover from divergence to specificity is key to the transition from chemistry to biology in the contexts of the origin and de‐novo synthesis of life.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the EU (ERC AdG 741774, MCIF 745805‐DSR; MCIF 786350‐PSR), NWO (VICI grant) and the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035).
B. Liu, M. A. Beatty, C. G. Pappas, K. Liu, J. Ottelé, S. Otto, Angew. Chem. Int. Ed. 2021, 60, 13569.
References
- 1.
- 1a. Crick F. H. C., J. Mol. Biol. 1968, 38, 367–379; [DOI] [PubMed] [Google Scholar]
- 1b. Orgel L. E., J. Mol. Biol. 1968, 38, 381–393; [DOI] [PubMed] [Google Scholar]
- 1c. Gilbert W., Nature 1986, 319, 618–618; [Google Scholar]
- 1d. Szilágyi A., Könnyű B., Czárán T., Sci. Rep. 2020, 10, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kun Á., Szilágyi A., Könnyű B., Boza G., Zachar I., Szathmáry E., Ann. N. Y. Acad. Sci. 2015, 1341, 75–95. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Robertson M. P., Joyce G. F., Chem. Biol. 2014, 21, 238–245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Lincoln T. A., Joyce G. F., Science 2009, 323, 1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Safont-Sempere M. M., Fernández G., Würthner F., Chem. Rev. 2011, 111, 5784–5814; [DOI] [PubMed] [Google Scholar]
- 4b. Mukhopadhyay P., Wu A., Isaacs L., J. Org. Chem. 2004, 69, 6157–6164; [DOI] [PubMed] [Google Scholar]
- 4c. Ghosh S., Isaacs L., Complex self-sorting systems, Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 4d. Aratsu K., Takeya R., Pauw B. R., Hollamby M. J., Kitamoto Y., Shimizu N., Takagi H., Haruki R., Adachi S.-i., Yagai S., Nat. Commun. 2020, 11, 1623; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Ayme J.-F., Dhers S., Lehn J.-M., Angew. Chem. Int. Ed. 2020, 59, 12484–12492; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12584–12592; [Google Scholar]
- 4f. Ghosh A., Schmittel M., Beilstein J. Org. Chem. 2020, 16, 2831–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Rizzuto F. J., Nitschke J. R., J. Am. Chem. Soc. 2020, 142, 7749–7753; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Lei Y., Chen Q., Liu P., Wang L., Wang H., Li B., Lu X., Chen Z., Pan Y., Huang F., Li H., Angew. Chem. Int. Ed. 2021, 60, 4705–4711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4755–4761. [Google Scholar]
- 6. Tsiamantas C., de Hatten X., Douat C., Kauffmann B., Maurizot V., Ihara H., Takafuji M., Metzler-Nolte N., Huc I., Angew. Chem. Int. Ed. 2016, 55, 6848–6852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6962–6966. [Google Scholar]
- 7. Kubota R., Nagao K., Tanaka W., Matsumura R., Aoyama T., Urayama K., Hamachi I., Nat. Commun. 2020, 11, 4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhong J., Zhang L., August D. P., Whitehead G. F. S., Leigh D. A., J. Am. Chem. Soc. 2019, 141, 14249–14256. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hsu C. W., Miljanić O. Š. in Dynamic Covalent Chemistry (Ed.: Zhang W., Jin Y.), John Wiley & Sons Ltd, Chichester, UK, 2017, pp. 253–286; [Google Scholar]
- 9b. Miljanić O. Š., Chem 2017, 2, 502–524; [Google Scholar]
- 9c. Herrmann A., Chem. Soc. Rev. 2014, 43, 1899–1933; [DOI] [PubMed] [Google Scholar]
- 9d. Kołodziejski M., Stefankiewicz A. R., Lehn J.-M., Chem. Sci. 2019, 10, 1836–1843; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Klotzbach S., Beuerle F., Angew. Chem. Int. Ed. 2015, 54, 10356–10360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10497–10502. [Google Scholar]
- 10.
- 10a. Frei P., Hevey R., Ernst B., Chem. Eur. J. 2019, 25, 60–73; [DOI] [PubMed] [Google Scholar]
- 10b. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Carnall J. M. A., Waudby C. A., Belenguer A. M., Stuart M. C. A., Peyralans J. J.-P., Otto S., Science 2010, 327, 1502–1506; [DOI] [PubMed] [Google Scholar]
- 11b. Liu B., Pappas C. G., Zangrando E., Demitri N., Chmielewski P. J., Otto S., J. Am. Chem. Soc. 2019, 141, 1685–1689; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Kosikova T., Philp D., J. Am. Chem. Soc. 2019, 141, 3059–3072; [DOI] [PubMed] [Google Scholar]
- 11d. Kosikova T., Philp D., Chem. Soc. Rev. 2017, 46, 7274–7305. [DOI] [PubMed] [Google Scholar]
- 12. Malakoutikhah M., Peyralans J. J., Colomb-Delsuc M., Fanlo-Virgos H., Stuart M. C., Otto S., J. Am. Chem. Soc. 2013, 135, 18406–18417. [DOI] [PubMed] [Google Scholar]
- 13. Pappas C. G., Mandal P. K., Liu B., Kauffmann B., Miao X., Komáromy D., Hoffmann W., Manz C., Chang R., Liu K., Pagel K., Huc I., Otto S., Nat. Chem. 2020, 12, 1180–1186. [DOI] [PubMed] [Google Scholar]
- 14. Liu B., Pappas C. G., Ottelé J., Schaeffer G., Jurissek C., Pieters P. F., Altay M., Marić I., Stuart M. C. A., Otto S., J. Am. Chem. Soc. 2020, 142, 4184–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frederix P. W. J. M., Idé J., Altay Y., Schaeffer G., Surin M., Beljonne D., Bondarenko A. S., Jansen T. L. C., Otto S., Marrink S. J., ACS Nano 2017, 11, 7858–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colomb-Delsuc M., Mattia E., Sadownik J. W., Otto S., Nat. Commun. 2015, 6, 7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary