

Abstract
Introduction
Alzheimer's disease (AD) is a devastating condition with no effective treatments, with promising findings in rodents failing to translate into successful therapies for patients.
Methods
Targeting the vulnerable entorhinal cortex (ERC), rhesus monkeys received two injections of an adeno‐associated virus expressing a double tau mutation (AAV‐P301L/S320F) in the left hemisphere, and control AAV‐green fluorescent protein in the right ERC. Noninjected aged‐matched monkeys served as additional controls.
Results
Within 3 months we observed evidence of misfolded tau propagation, similar to what is hypothesized to occur in humans. Viral delivery of human 4R‐tau also coaptates monkey 3R‐tau via permissive templating. Tau spreading is accompanied by robust neuroinflammatory response driven by TREM2+ microglia, with biomarkers of inflammation and neuronal loss in the cerebrospinal fluid and plasma.
Discussion
These results highlight the initial stages of tau seeding and propagation in a primate model, a more powerful translational approach for the development of new therapies for AD.
Keywords: Alzheimer's Disease, biomarkers, inflammation, microglia, rhesus monkey, tau
1. BACKGROUND
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that affects 1 in 10 people age 65 and older, 1 , 2 becoming more prevalent with the projected increase of the aged population worldwide. Current thinking is that decades may pass between the development of the initial brain lesions—mainly amyloid beta (Aβ) accumulation and misfolded tau propagation—and the development of the first cognitive symptoms, often occurring when the degenerative process is so substantial that no current therapies can restrain progression of the disease. Our understanding of the pathological events underlying AD have advanced vastly in recent years but successful translation from rodent models into efficient therapies for humans has been extremely limited. More than 400 clinical trials have been conducted in patients after promising discoveries in rodent models, but so far no new drugs have been brought to the market, and these disappointments have led several pharmaceutical companies to downsize their AD programs. 1 , 2 , 3 , 4
The search for animal models that better correlate with the disease pathogenesis has led the field to explore other options, including the use of non‐human primates (NHPs), phylogenetically much closer to humans. Macaque monkeys, for example, share >98% homology with humans for the longest form of the tau protein, and 100% homology for the Aβ sequence. 5 Whether NHPs spontaneously develop the full spectrum of AD pathology that results in dementia remains in question. Great apes and macaques develop amyloid plaques with age, in a similar distribution pattern to that observed in humans, but development of end‐stage neurofibrillary tangles (NFT) and the extent of tau hyperphosphorylation in NHPs is still controversial, as we and others have discussed previously. 6 , 7 , 8 Recent reports suggest that chimpanzees can develop tangles later in life, 9 as well as cynomolgus macaques 10 and rhesus monkeys, 11 but these findings were reported only among the oldest‐old animals, limiting the usefulness of naturally occurring pathology for development of therapeutics.
Taking advantage of the high translational power of NHPs due to their physiological and genetic similarities to humans, we describe here the initial steps of the development of a monkey model of AD with accelerated tau pathology and neuroinflammation. Multiple tau‐based approaches have proven to be promising in mouse models, which gives us a strong foundation for moving to an NHP model. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Four adult rhesus monkeys received a one‐time double infusion of recombinant adeno‐associated virus capsid 1 (AAV1) carrying two tauopathy‐related mutations (P301L/S320F) in the entorhinal cortex (ERC). 22 Three months after the gene delivery, all animals exhibited abnormal hyperphosphorylated tau and extensive misfolded tau propagation with frank NFTs in directly connected hippocampal and neocortical areas. Importantly, an extensive neuroinflammatory response driven by TREM2+ microglia was observed in these areas, with the mutant isoform 4R‐tau injected coaptating monkey 3R‐tau isoform, accelerating tau aggregation and spreading. Longitudinal analysis of cerebrospinal fluid (CSF) and blood indicated substantial axonal damage and neuroinflammation in tau‐expressing animals compared to control animals. These results highlight the initial stages of a robust accelerated tau pathology and propagation in the monkey brain and support the potential of a NHP model of AD, with natural full expression of tau isoforms, allowing advanced testing for therapeutic interventions over a short time frame.
RESEARCH IN CONTEXT
Systematic review: Using PubMed, the authors searched for nonhuman primate models of Alzheimer's disease (AD), more translatable to humans. Studies have documented the natural occurrence of AD‐like tau pathology in monkeys, but only among the oldest‐old animals, limiting the usefulness of such models for the development of therapeutics.
Interpretation: Our results indicate that genetic delivery of dual tau mutations in young adult rhesus monkey generated profound tau‐based neuropathology and intense neuroinflammation, with reflections in the cerebrospinal fluid and plasma.
Future directions: We describe here the initial steps for the development of a monkey model of AD with accelerated tau pathology propagation and neuroinflammation. Assessment of several human AD‐typical biomarkers also suggests clear similarities between our monkey model and human disease progression. These results highlight the initial stages of misfolded tau propagation in a nonhuman primate model, with natural full expression of tau isoforms, allowing advanced testing for therapeutic interventions over a short timeframe.
HIGHLIGHTS
Genetic delivery of mutated tau in the monkey brain induce misfolded tau propagation.
Tau spreading is followed by neuroinflammatory response driven by TREM2+ microglia.
Injected human tau coaptates monkey tau via permissive templating to spread pathology.
Treated monkeys developed robust alterations in AD core biomarkers in cerebrospinal fluid and blood.
These results lend support for the use of monkeys as a platform to develop AD therapies.
2. METHODS
2.1. Animals and neurosurgery
Four adult female rhesus macaques (10 to 15 years old) had bilateral stereotaxic injections of the adeno‐associated virus expressing a double tau mutation in the left ERC (AAV‐P301L/S320F, 1.176 × 10ˆ13 genomic copies/mL), and the control injection (AAV‐green fluorescent protein [GFP], 1.6 × 10ˆ13 genomic copies/mL) in the right ERC. Four additional healthy non‐injected age‐matched females (10 to 14 years old) and four aged female animals (21 to 25 years old) were also used as controls. All of the 10‐ to 15‐year‐old females were cycling, premenopausal monkeys. All experiments were approved by the Institutional Animal Care and Use Committee at the University of California–Davis (protocol no. 20752).
2.2. AAV preparation
Human 0N/4R tau carrying two mutations (P301L/S320F) was expressed under the control of the hybrid cytomegalovirus enhancer/chicken β‐actin (CMV/CBA) promoter, a CBA intron (first intron of chicken β‐actin gene plus the splice acceptor of the rabbit β‐globin gene), woodchuck hepatitis virus post‐transcriptional regulatory element (WPRE), and bovine polyA, as previously described. 22 Recombinant AAVs containing the P301L/S320F tau gene or an empty vector were packaged in capsid 1 and purified under good laboratory practices at the Penn Vector Core (University of Pennsylvania). The AAV preparations were tested for endotoxins (< 5 endotoxin units per mL), tested for purity using the sodium dodecyl sulphate—polyacrylamide gel electrophoresis method and titered using three rounds of digital polymerase chain reaction. The AAVs were aliquoted and stored at –80○C until further use.
2.3. CSF and plasma collection
CSF and plasma were collected prior to surgery, once per moth post‐surgery, and immediately prior to necropsy (totaling four collections). For CSF, animals were sedated with ketamine (5 to 30 mg/kg, IM) and dexmedetomidine (0.0075 to 0.015 mg/kg, IM). A 23‐gauge spinal needle was inserted into the subarachnoid space of the cisterna magna and 1 to 2 mL of CSF aspirated and transferred to cryotube and stored at –80°C. For plasma, ≈30 mL of whole blood was collected in EDTA‐containing tubes and immediately centrifuged at 1500 x g for 15 minutes and apportioned into 0.5 mL aliquots and stored at –80°C until use.
2.4. Perfusion and tissue preparation
All monkeys were deeply anesthetized with ketamine (25 mg/kg) and intubated. Perfusion was performed following the same protocol described recently. 7 After perfusion, samples for microscopy were immersed in fixative (4% paraformaldehyde with 0.125% glutaraldehyde in phosphate buffer, pH 7.4) at 4°C for 48 hours under agitation and stored in phosphate‐buffered saline (PBS) with 0.1% sodium azide at 4°C until processed.
2.5. Human CSF and plasma samples
CSF and plasma from four patients confirmed with AD (Braak IV‐V) and four age‐matched controls were obtained from the University of California Davis Alzheimer's Disease Center, grant: P30 AG010129, provided by Dr. Lee‐Way Jin.
2.6. Immunohistochemistry
50 μm‐thick free‐floating sections were incubated in an antigen retrieval solution (Wako, S1700) at 60°C for 30 minutes. After that, sections were incubated in blocking solution: 5% donkey serum, 5% goat serum, 5% bovine serum albumin in PBS 0.3% triton for 2 hours at room temperature under agitation. Sections were then incubated for 48 hours with the following antibodies: IBA‐1 (234‐006, Synaptic Systems, 1:1000), AT8 (MN1020, Invitrogen, 1:500), AT100 (MN1060, Invitrogen, 1:1000), pTau 422 (ab79415, Abcam, 1:1000), NeuN (266004, Syn. Systems, 1:1000), TREM2 (AF1828, R&D systems, 1:500), MAP2 (188004, Syn, Systems, 1:1000), Tau 3R (2A1‐1F4, Wako, 1:400), Tau 4R (ab218314, Abcam, 1:500), CP3, MC1, and PHF1 (provided by Dr. Peter Davies, 1:400). TOC1 antibody for tau oligomers was provided by Dr. Nicholas Kanaan and used at 1:1000. Tissue was washed thoroughly with PBS and incubated with Alexa Fluor secondary antibodies (Invitrogen, 1:500) for 2 hours, at room temperature. True Black solution (Biotium) was used for 30 minutes to eliminate lipofuscin autofluorescence. Slides were then mounted with Prolong Diamond Antifade with Dapi (Invitrogen).
2.7. Microscopy and image analysis
All images were acquired in 3D (z‐stack variating between 25 and 50 μm) using a Zeiss LSM 800 microscope with Airyscan. For NeuN/AT8/ThioS colocalization analysis, three 20x images were acquired from each hippocampal region analyzed, with at least 240 NeuN+ cells analyzed per region, per animal. Similar analysis was performed for microglia morphological quantification, with at least 20 cells acquired at 63 x per hemisphere, per animal. These images were exported and analyzed in 3D in Imaris software (Bitplane) using the filament tracing tool. Finally, three 20x images were acquired from the CA3/hilus region and layer II of ERC for quantification of AT8/3R and 4R tau colocalization. Microglia engulfment of 3R and 4R tau particles was performed as described previously, 7 with the total volume of internalized puncta (μm3) being divided by the total volume of microglial cells (μm3).
2.8. Enzyme‐linked immunosorbent assays (ELISAs)
Validation of human ELISAs using rhesus monkey samples were validated in our previous study 7 . Aβ1‐42, Aβ1‐40, t‐tau, p‐tau Ser199, p‐tau Thr231, p‐tau Ser396 (Invitrogen), TNF‐α, IL‐6, TREM2, BDNF (Abcam), TDP‐43 (Proteintech), and neurofilament‐light (Uman Diagnostics) ELISA assays were performed according to each kit manufacturer's instructions, after sample dilution optimization.
2.9. Statistical analysis
All analyses were performed with GraphPad Prism 6, and datasets were assessed for normality parameters prior to significance determination. Values are expressed as means ± standard error of the mean. Statistical tests and p values are indicated in each figure legend.
3. RESULTS
3.1. Injected monkeys develop AD core biomarkers in CSF and plasma
To develop transcellular propagation of tau aggregates, we took advantage of a recently established AAV‐mediated gene targeting expressing two tau variants, known to cause frontal‐temporal dementia (FTD) and Pick's disease (PiD) in humans, P301L and S320F mutant tau, respectively (i.e., AAV‐2xTau). The combination of both mutations resulted in a synergistic effect on the development of tau‐related pathology in mice, including synaptic alterations and cognitive impairment. 22 Notably, the AAV used in this study express full length human 0N/4R tau isoform (0N meaning without inserts near the N‐terminal region of the gene). The injections targeted the ERC, using a surgical stereotaxic navigation system for the AAV delivery (Figure 1A, Medtronic StealthStation). We opted for only using female monkeys in this study, because AD generally affects substantially more women than men, 1 and to correlate with previous data from our lab on age‐related cognitive decline and neurodegeneration in female rhesus monkeys, especially after menopause. 23 , 24 , 25
FIGURE 1.

Viral delivery of mutated tau in the monkey entorhinal cortex (ERC) led to accelerated Alzheimer's disease (AD) pathology as reflected in fluid biomarkers. Four adult rhesus monkeys (10 to 14 years old) received a single time viral injection expressing a double human tau mutation (AAV‐2xTau) in the left ERC, and control AAV‐GFP in the right ERC, as shown in (A). Cerebrospinal fluid (CSF) and plasma were collected prior to surgery and every 30 days until necropsy (B). A panel of several biomarkers relevant to AD patients were analyzed, and divided into four major categories, as indicated in (C). A robust increase in neurofilament light levels (NfL, D) and total tau (E) were observed in the CSF and plasma of injected animals compared to young and aged non‐injected monkeys. A progressive reduction in BDNF levels in the CSF of AAV‐2xTau injected animals was also observed (F), but no changes were detected in TDP‐43 levels (G). Notably, all three biomarkers for neuroinflammation analyzed were increased in treated animals compared to controls, both in CSF and plasma: sTREM2 (H) and the proinflammatory cytokines TNFα (I) and IL‐6 (J). Substantial increase in tau phosphorylation in different epitopes was also observed in treated monkeys (K, L, M), possibly reflecting neurofibrillary tangle formation in the brain and accelerated progression of AD pathology. On the other hand, no changes were observed in the total levels of amyloid beta (Aβ)40 (N) and Aβ42 (O) in the CSF of treated monkeys. *P < .05, **P < .01, ***P < .001; one‐way analysis of variance, Tukey's post hoc test
We recently reported that rhesus monkeys normally present similar levels of several AD‐related proteins in fluid samples to that seen in humans. 7 To deepen the investigation of AD‐relevant biomarkers in the monkeys, CSF and blood samples were collected prior to and after the AAV‐2xTau gene delivery, as indicated in Figure 1B. Twelve biomarkers were investigated, being selected for their relevance to the AD progression in patients, 26 , 27 , 28 , 29 , 30 , 31 , 32 and divided into four major categories, as shown in Figure 1C. CSF and plasma from young (10 to 16 years old) and aged (19 to 26 years old) non‐treated monkeys were used as controls, as well as samples from patients with confirmed AD diagnoses (Figure S1 in supporting information). Among the markers analyzed for neuronal damage, neurofilament light (NfL) had the highest increase in CSF and plasma in injected monkeys (Figure 1D and Figure S1A‐B), as well as total tau in the CSF (Figure 1E and Figure S1D‐E). There was also a temporal reduction of BDNF levels (Figure 1F), but no changes in TDP‐43 (Figure 1G) in CSF of treated animals. To investigate possible changes in inflammatory markers associated with tau pathology, soluble TREM2 (sTREM2, Figure 1H and Figures S1K‐L) and two inflammatory cytokines, TNFα (Figure 1I, Figures S1M‐N) and interleukin 6 (IL6, Figure 1J, Figures S1O‐P) were measured. The profile of changes in all three proteins were very similar among treated animals and across CSF and plasma, with a robust increase occurring 2 months after infusions, which would be consistent with AAV‐mediated gene expression timeline (3 weeks after brain infusion). In the human samples analyzed, consistent increases in sTREM2 levels across plasma and CSF were found compared to age‐matched samples (Figure S1K‐L), but no detectable differences were observed in the TNFα and IL‐6 levels between human samples (Figure S1M‐P).
In AD, p‐tau is the main component of the NFTs, and its detection in the CSF reflects the disease's progression in the brain. We observed a high increase of p‐tau levels in three different epitopes related to NFT pathogenesis in the CSF of treated monkeys and AD controls: tau phosphorylation at Ser 199 (Figure 1K, Figure S1G), at Thr 231 (Figure 1L, Figure S1H) and at Ser 396 (Figure 1M, Figure S1F). Increases in all three occurred quickly, detected in the first month after injections, and they were sustained at high levels in the CSF during subsequent months. All these sites of phosphorylation are known to cause a structural impact in tau interaction with microtubules and are reflections of the disease's progression. Finally, no changes were observed in Aβ42 (Figure 1N) and Aβ40 (Figure 1O) levels, suggesting the AAV‐2xTau infusion did not trigger amyloid pathology in this model. Notably, increases in amyloid levels were observed among the human samples (Figures S1I‐J).
3.2. AAV‐2xTau injection in the ERC induce extensive tau pathology in the hippocampus
Initial analysis of the hippocampus from injected monkeys revealed substantial reactivity for different markers for tau pathology, including AT8 (Ser 202/Thr 205) and ThioS (Figure 2A‐D). Other markers analyzed were AT100 (Thr212/214), tau oligomers, pTau Ser 262, pTau Ser 422, pTau Thr 231, pTau 214, PHF1 (p‐Tau Ser 396/Ser 404) and tau conformational dependent antibody MC1 (representative images for all additional markers can be found in Figure S2A‐T in supporting information). Investigation of the hippocampal subareas from the injected monkeys was performed by combining triple staining and 3D confocal microscopy, allowing the precise quantification of colocalization between neuronal marker NeuN with p‐tau marker (AT8) and NFTs (ThioS), as highlighted in Figure 2E‐J. Although most of the hippocampal subfields receive direct projections from ERC (Figure 2K), intrinsic variability in the pattern of misfolded tau deposition was observed, with the majority of the tau tangles being present in the CA3 region (Figure 2L‐M), followed by CA1 (Figure 2N‐O). ThioS‐positive cells in these regions often no longer express the neuronal marker NeuN, indicating these are mostly ghost tangles, similar to what is seen in AD. Other hippocampal areas heavily affected by tau pathology include the subiculum (Figure 2P‐Q) and dentate gyrus (Figure 2R‐S). Pathology was also observed in layer 2 of the ERC (Figure 2T‐U), known also to be highly vulnerable in patients with AD, 33 , 34 and the site of injection, with a minor expansion to the right ERC (Figure 2V‐X). Notably, although the contralateral hemisphere did not present markers for fibrillary tau pathology as did the left hemisphere, expression of early tau pathological events such as phosphorylation in Ser 262 and Thr 231 were detected in the CA1, CA3, and DG of the right hippocampus. The expression of these markers suggest the presence of events related to decreased binding of tau to microtubules in this region (Figure S3A‐T in supporting information), similar to early events observed in the hippocampus of AD patients. 35 , 36 Our assumption is that the early pathology in contralateral hippocampus reflects the involvement of the contralateral projection of the ERC connectome.
FIGURE 2.
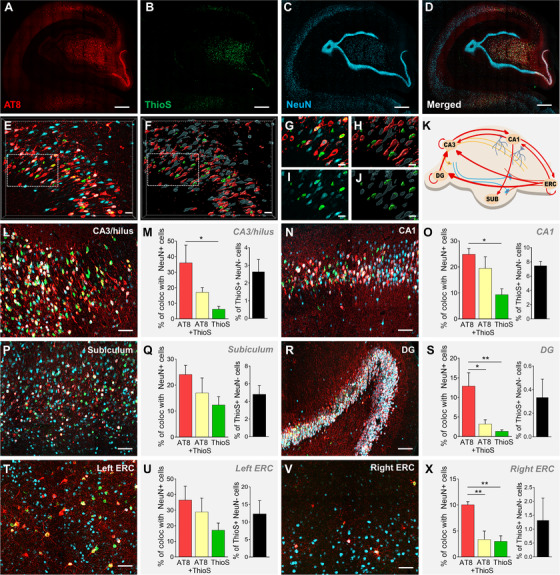
AAV‐2xTau injection in the entorhinal cortex (ERC) induces robust misfolded tau propagation in the left hippocampus of rhesus monkeys. Three months after the viral delivery of dual tau mutation in the ERC, p‐Tau (red, AT8) and ThioS (green) were used as markers for pathological tau spreading and tangle formation. First panel (A‐D) shows the spatiotemporal progression of tau accumulation in the monkey hippocampus, colocalizing also with NeuN (blue). Scale bar: 500 μm. Note that AT8 labeling in the granule cell layer is limited to the lower right corner at this stage, reflecting the topographic organization of the ERC/DG projection. High‐resolution confocal microscopy images were exported and analyzed in Imaris software by creating a 3D volume rendering for each marker. This allowed a precise quantitative measurement of protein‐protein colocalization, as exemplified in the CA1 region in (E, G, I; original and zoom images, respectively), and (F, H, J; 3D surface rendering and zoom regions, respectively). Scale bar: 50 μm. Anatomic organization of neural circuits connecting ERC and hippocampus analyzed in this study are shown in (K) and determined the pattern of misfolded tau propagation and deposition in injected monkeys (L‐X). Quantification of tau pathology progression in the left hippocampus demonstrated that the major areas affected are CA3‐Hilus (L‐M), CA1 (N‐O), subiculum (P‐Q), and DG (R‐S). Injection of AAV‐2xTau in the left ERC induced robust tau pathology in pyramidal neurons in layer 2 (T‐U). The right ERC injected with AAV‐GFP displayed a very low‐level p‐Tau and ThioS labeling compared to the other regions analyzed, which likely reflects propagation through interhemispheric ERC connectivity (V‐X). Scale bar: 50 μm. *P < .05, **P < .01; One‐way analysis of variance, Tukey's post hoc test
3.3. Misfolded tau propagation in the hippocampus induces extensive microglial response
Accumulating evidence suggests a major role for glial cells and neuroinflammation in different neurodegenerative diseases, including AD. 37 , 38 Healthy microglia and astrocytes are normally involved in the clearance of pathological proteins, including Aβ and soluble and insoluble forms of tau, 39 , 40 but when activated these cells can actually facilitate spreading of pathological proteins and exacerbate the degenerative process. 40 To investigate a potential role of reactive microglia in the tau propagation observed in the AAV‐2xTau monkeys, we evaluated 3D morphological changes in microglia between left and right hemispheres (Figure 3A‐C), and quantification of these cells demonstrated the increase of reactive microglia in the left hippocampus (Figure 3D‐G). Interaction between microglia and AT8+ neurons is highlighted in Figure 3H‐O (see also movie S1 in supporting information). Finally, quantification of the colocalization between microglia general marker IBA1 and triggering receptor expressed on myeloid cells 2 (TREM2), a key marker for pathological microglia in AD, 41 demonstrated that TREM2 activation is robust in the presence of tau pathology, but not in the control hemisphere (Figure 3P‐Z). Reactive astrocytes were also observed in response to tau pathology, but they do not closely interact with p‐tau–positive cells the same way as microglia, as shown in Figure S4 in supporting information.
FIGURE 3.

Robust inflammatory response in the hippocampus is modulated by reactive TREM2+ microglia. AAV‐2XTau injection in the left entorhinal cortex produced substantial microgliosis in the left hippocampus (A), but not right side (B). Representative micrographs and the 3D surface rendering created to analyze microglia morphological complexity are highlighted in (C). Reactive microglia are abundant in the left hippocampus, but not on the right side (D). Similarly, other parameters analyzed including number (E) and length of segments (F), and soma volume (G), suggest microglia shifted from a physiological to a reactive state in the presence of tau pathology, ultimately leading to an increase in the neuroinflammatory process. Representative images from panel (H‐J) show intense distribution of activated microglia (green, IBA1) in direct response to the presence of p‐Tau (AT8) in the hippocampus. Scale bar: 500 μm. Panel K‐N and the 3D surface rendering of the image highlighted in (O), demonstrate the close interaction between reactive microglia and p‐Tau in the left hippocampus of treated animals. Scale bar: 10 μm. Finally, representative images shown in panels (P‐S; left hippocampus) and (T‐X; right hippocampus) revealed a high degree of colocalization between IBA and TREM2 in the left but not in the right hippocampus, as demonstrated in (Z). These results highlight a unique profile of reactive microglia TREM2+ directly interacting with affected neurons, similar to what is observed in the brain of Alzheimer's disease patients. Scale bar: 20 μm. *P < .05, Unpaired t test
FIGURE 4.

Injected tau isoform 4R coaptates monkey 3R‐tau to spread pathology in a prion‐like manner. The first panel (A‐E) shows that in the entorhinal cortex (ERC), most of the p‐tau (green, AT8) colocalizes with 4R‐tau (blue) to a much greater extent than with 3R‐tau (red). The opposite is observed in the hippocampus, with a higher degree of colocalization occurring between 3R‐tau (red) and AT8, as shown in (F‐J). Scale bar: 50 μm and for the zoom area: 10 μm. Reactive microglia surrounding affected neurons were stained for IBA1 (white, K, P), and as shown in (K‐O) for ERC and (P‐S) for hippocampus; microglia are actively taking up 3R and 4R‐tau from neurons. The volume of internalized 3R and 4R‐tau particles (μm3) was divided by the total volume of microglia cells analyzed (μm3) to generate the total percentage of engulfment, calculated in (O; ERC) and (T; hippocampus)
3.4. Presence of 4R human tau templates 3R monkey tau to induce propagation
Abnormal splicing affecting only one tau isoform without altering the amino acid sequence of the protein is enough to cause tauopathies such as Pick's disease (3R), corticobasal degeneration (4R), and progressive supranuclear palsy (4R). 42 AD involves both tau isoforms equally (3R + 4R), while FTD can be either 3R or 4R, or also involve both. 43 Our knowledge about how these isoform profiles vary during the different stages of tau pathology in AD is largely unknown and difficult to resolve in rodent studies, because mice and rats do not express the 3R tau isoform in the adult brain. 44 Rhesus monkeys offer a unique opportunity, because this species expresses both tau isoforms in the brain, similar to humans, but different from rodents and also even from marmosets. 45
Taking advantage of the use of an AAV construct expressing only human 4R tau, we were able to explore the role of the monkey 3R tau in the misfolded tau propagation. As shown in Figure 4A‐E for ERC and Figure 4F‐J for hippocampus, AT8 colocalizes mostly with 4R tau in the injected ERC, but in the hippocampus, most AT8+ neurons colocalize with monkey 3R tau. These results suggest the human 4R tau delivered in the ERC is permissively templating monkey 3R tau in the hippocampus. Analysis of microglia in both regions revealed a similar profile of isoform distribution, with microglia in the ERC taking up mostly 4R tau fragments (Figure 4K‐O) and more 3R tau fragments in the hippocampus (Figure 4P‐T). Importantly, the tau transgenes used in this study are driven by a hybrid chicken β‐actin (hCBA) promoter, and the AAV1 used is neurotropic. 22 This indicates that the AAV‐2xTau injected could not induce microglial expression 3R and 4R tau isoforms, but rather tau presence inside these cells is due to phagocytic engulfment of neuronal particles.
3.5. Tau seeds propagate outside ERC‐hippocampus in a prion‐like manner
We also explored misfolded tau propagation after AAV‐2xTau delivery in sites beyond the ERC and hippocampus. Given the well‐defined corticocortical circuitry in the visual system, we initially chose to determine the degree to which tau pathology could be linked to key interconnected visual regions. Interestingly, we found a unique pattern of tau propagation distant from the injection site, reaching areas in the occipital lobe and V4. A proposed scheme of the affected neurocircuitry is presented in Figure 5A. Major areas affected by misfolded tau propagation include the previously described ERC and CA1 (Figure 5B‐E). However, the retrosplenial cortex (RSC) was also affected, presenting intense AT8 staining and several NFTs in all treated monkeys. Two other major sites were identified with misfolded tau accumulation, the visual regions V4 and V1 (Figure 5H‐K). Interestingly, V4 and V1 are a minimum of two and three synapses from ERC, respectively, which likely explains the pattern of misfolded tau propagation we observed in these areas. V4 and V1 presented only AT8 staining but no tangles with VI having only AT8‐positive axons, suggesting that these areas were affected later in the spatiotemporal progression of the pathology.
FIGURE 5.

Spatiotemporal progression of misfolded tau propagation in the rhesus monkey brain. Hippocampal and neocortical circuits linked to observations in (B‐K) shown in (A). Injected entorhinal cortex (ERC) presented strong staining and colocalization of both markers for tau pathology, p‐Tau (red, AT8) and ThioS (green), as shown in (B) and highlighted in (C). CA1 is also highly affected, as shown in (D) and (E). The retrosplenial cortex (RSC), a region directly connected to ERC, is also affected, with AT8 immunoreactivity present in several cortical layers, but fewer neurons being ThioS+ (F; and zoom in G). Two connected areas of the occipital lobe were also affected, V4 (H‐I) and V1 (J‐K), but the AT8 labeling was prevalent in processes and there was no ThioS+ labeling at 3 months in these neocortical regions, suggesting that the progression toward degeneration is at an earlier stage in these more distant sites. Scale bar: (B‐J): 50 μm and (C‐K): 20 μm
4. DISCUSSION
As discussed above, preclinical animal work in AD is heavily invested in the use of genetically altered rodent models, with >170 transgenic mice being developed exclusively to investigate AD pathology. One route to close the translational gap between the mouse model and human clinical data is to develop NHP models of AD. Rhesus macaques and marmosets are among the options, and although they do not develop AD pathology identical to humans, their brains undergo structural and biochemical changes that mirror those seen in people, including Aβ deposition and cognitive decline. 4
Recently we described a NHP model of early AD by injecting Aβ oligomers (AβOs) in the lateral ventricle of the female monkey brain. 7 AβO infusions lead to early stages of synapse dysfunction in the prefrontal cortex and hippocampus, mimicking the early synaptic stage of AD pathology in humans. Here we present a more aggressive model recapitulating the degenerative phase of AD. Using AAV‐mediated gene delivery of a dual tau mutation into the ERC, we were able to demonstrate the development of end‐stage tangles in AD‐vulnerable areas such as the ERC and hippocampus, as well as tau hyperphosphorylation in regions several synaptic connections from the injection site, such as visual areas in the occipital lobe, 3 months after injection. In vivo reflections of disease progression were reflected in monthly samples of CSF and plasma from treated animals. The profile of the 12 biomarkers analyzed revealed an immediate robust increase in NF‐L, total tau, and p‐tau, and a later increase in inflammatory markers such as sTREM2 and proinflammatory cytokines TNFα and IL‐6. Most importantly, we were able to confirm the ability of the human 4R tau injected to self‐propagate inside the monkey brain via permissive templating of endogenous 3R tau. Besides not being expressed in the adult mouse brain, tau 3R isoform is also not expressed in marmosets, as reported recently, 45 highlighting the translational power of using rhesus monkey to develop a platform to test therapies for AD drug discovery.
Abnormal tau phosphorylation is a key feature in AD and other related dementias. 46 , 47 Events that decrease tau's capacity to interact with microtubules, such as phosphorylation in Ser 262 and Thr 231, are thought to be involved in the early stages of AD pathology, before an abnormal increase in tau's ability to self‐associate. 35 , 48 , 49 An increase in both Ser 262 and Thr231 was detected in the contralateral hemisphere of the AAV‐2xTau injected monkeys, but not late‐stage markers that are thought to be involved in tau polymerization into filaments, such as AT8 or ThioS. 35 , 48 The presence of these events in the contralateral hemisphere suggests that the mutant tau injection is affecting intrinsic interhemispheric connections in the ERC and/or hippocampus.
Several AD studies have used AT8 antibodis to perform neuropathological examination of human post mortem samples and mouse models of the disease. 50 , 51 Although AT8 (Ser 202/Thr 205) is an important marker for AD pathology, it is important to highlight that it detects hyperphosphorylated tau, but not end‐stage tangles or ghost tangles. 52 Here, we were able to detect that, in several areas of the brain, including the DG in the hippocampus and V1 and V4 in the occipital lobe, immunoreactivity for AT8 was strong in the neuronal cell bodies or axons, but many of these cells did not also label for ThioS reactivity. ThioS binds to beta sheet‐rich structures that compose misfolded protein aggregates such as amyloid plaques and tau tangles. AT8 and ThioS colocalize inside the same cells in some regions like ERC, CA3, and CA1, but not in the DG or V1 and V4, where only AT8 immunoreactivity was detected. This is a key observation regarding the progression to NFT formation, because the conformational changes detected by ThioS are essentially irreversible, ultimately labelling dying neurons. On the other hand, neurons that are solely affected by hyperphosphorylated tau accumulation did not enter an apoptotic route, and therefore therapeutic approaches that can rescue these neurons from cell death may be successful. In addition, this suggests that AT8 is not a dependable label for NFTs that represent dying or dead neurons.
The pathogenetic progression of events that lead to AD progression is still not fully understood, but selective vulnerability of pyramidal neurons in the hippocampus and other neocortical regions has been described by us and others. 53 , 54 , 55 Layer 2 of the ERC, the subiculum and the CA1, are the regions that are known to be particularly vulnerable to NFT formation, especially pyramidal cells. 53 , 56 Interestingly, late‐stage markers of tau pathology were found mostly in the CA3, DG, and subiculum of treated monkeys, with fewer present in CA1. One hypothesis is that, at the timepoint analyzed in this study, CA1 neurons were already extensively affected, and would only be counted as ghost‐tangles, no longer expressing NeuN and AT8. This would explain why after the left ERC, CA1 is the region with higher expression of ghost tangles (ThioS+ NeuN–), but lower expression of AT8 compared to CA3. The balance of pathology between these two regions in our NHP model is likely to reflect the degree of spreading of pathology between circuits intrinsic to the hippocampus (e.g., CA3 to CA1, Schaffer collaterals), but it is also worth noting that DG, CA3/Hilus, and CA1 all receive direct projections from ERC, though the laminar origins differ in ERC with the projections to CA1 and CA3 coming largely from deep layers and the projection to DG largely from Layer 2. 57 Thus, the slight variations in laminar location of the ERC injections could affect the relative distribution of pathology at this early time point. Interestingly, early events in AD progression associated with reduced tau binding to microtubules were found in higher expression in CA1 compared to CA3 and DG in the contralateral hippocampus. The differences in the temporal progression of tau pathology between the left and right hippocampus might indicate that CA1, together with subiculum and ERC layer 2, are regions primary involved in the spread of pathological tau, similar to what is observed in AD patients.
Another important finding from this study is the extensive neuroinflammatory response after the AAV‐mediated gene delivery. The complexity of monkey microglia was described by us and others 7 , 58 and microglia analyzed in this study were found to develop a TREM2+ profile, suggesting its transition from a physiological healthy state to a disease‐related reactive state, contributing to the disease by spreading bioactive tau seeds across different brain regions. In addition, by seeing a change in the tau isoforms inside microglia between the injection site and a terminal projection site (ERC‐hippocampus) we were able to demonstrate the highly dynamic profile of these cells and their contribution to accelerating the propagation of tau seeds in different brain areas.
5. CONCLUSIONS AND FUTURE DIRECTIONS
Even though our knowledge of the biology underlying AD pathology has progressed greatly in the last three decades, translation to tau‐based therapeutics for humans has been disappointing, pointing to the difficulty in translating findings from rodents into effective therapies for humans. Although we and others have demonstrated that rhesus monkeys represent valuable models for studying aging and age‐related neurodegenerative diseases, 3 , 7 , 11 , 59 it is still to be determined if NHPs also present a similar cognitive and memory dysfunction profile that is frequently observed in AD patients. After the initial steps of biochemical validation of the tau pathology in NHPs, longitudinal cognitive assessment using treated monkeys will be necessary, especially comparing animals across different sex and ages, and with the presence of comorbidities such as diabetes and hypertension that can increase the risk of AD. In the present study, injections of the AAV‐2xTau were performed in the left ERC, and AAV‐GFP in the right ERC. Unilateral treatment was chosen for this project to maximize the power of each monkey and to provide a validation of the biochemical progression of the pathology across hemispheres. For future behavioral studies, bilateral injection of AAV‐2xTau will be essential, especially for studies aimed at demonstrating cognitive decline.
In summary, we report here the initial steps of the development of a tau‐based model of AD in rhesus monkeys. While it is still unknown whether the animals treated with AAV‐2xTau will present the full spectrum of AD, including severe cognitive impairment, these initial observations set the stage for the next steps in testing tau‐based therapeutics for AD patients.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Danielle Beckman, Jeffrey H. Kordower, and John H. Morrison designed the research. All authors performed the research. Danielle Beckman, Jeffrey H. Kordower, and John H. Morrison wrote the paper.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We want to thank Dr. Benoit I. Giasson for providing the original plasmid expressing the dual tau mutation, Dr. Peter Davies for the tau antibodies, and Dr. Nicholas Kanaan for the tau oligomer antibody. We also thank the UC Davis Alzheimer's Disease Center (P30‐AG010129) for providing human CSF and plasma samples and the Bakalar Family. This project was supported by NIH grants P51‐OD011107‐57S3 and P51‐OD011107‐58S2. D. Beckman is an Alzheimer's Association Postdoctoral Fellow, AARF‐20‐685431. The California National Primate Research Center is supported by NIH Office of the Director award P51‐OD011107.
Beckman D, Chakrabarty P, Ott S, et al. A novel tau‐based rhesus monkey model of Alzheimer's pathogenesis. Alzheimer's Dement. 2021;17:933–945. 10.1002/alz.12318
[The copyright line for this article was changed on April 20, 2021 after original online publication.]
DATA AVAILABILITY STATEMENT
All data associated with this study are present in this paper. Raw data used to generate images and graphs are available from the corresponding author, J.H.M., upon request.
REFERENCES
- 1. 2020 Alzheimer's disease facts and figures. Alzheimer's Dement 2020;16:391‐460. [DOI] [PubMed] [Google Scholar]
- 2. Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimer's Dement. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beckman D, Baxter MG, Morrison JH. Future directions in animal models of Alzheimer's disease. J Neurosci Res. 2018;96:1829‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. King A. The search for better animal models of Alzheimer's disease. Nature. 2018;559:S13‐5. [DOI] [PubMed] [Google Scholar]
- 5. Drummond E, Wisniewski T. Alzheimer's disease: experimental models and reality. Acta Neuropathol. 2017;133(2):155‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker LC, Jucker M. The exceptional vulnerability of humans to Alzheimer's disease. Trends Mol Med. 2017;23(6):534‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beckman D, Ott S, Donis‐Cox K, et al. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc Natl Acad Sci U S A. 2019;116(52):26239‐26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Latimer CS, Shively CA, Keene CD, et al. A nonhuman primate model of early Alzheimer's disease pathologic change: Implications for disease pathogenesis. Alzheimer's. Dement. 2019;15:93‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edler MK, Sherwood CC, Meindl RS, et al. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer's disease. Neurobiol Aging. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oikawa N, Kimura N, Yanagisawa K. Alzheimer‐type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res. 2010;1315:137‐149. [DOI] [PubMed] [Google Scholar]
- 11. Paspalas CD, Carlyle BC, Leslie S, et al. The aged rhesus macaque manifests Braak stage III/IV Alzheimer's‐like pathology. Alzheimer's Dement. 2018:680‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Götz J, Deters N, Doldissen A, et al. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007;17:91‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu JW, Hussaini SA, Bastille IM, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19:1085‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pooler AM, Polydoro M, Wegmann S. Propagation of tau pathology in Alzheimer's disease: identification of novel therapeutic targets. Alzheimers Res Ther. 2013;5:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harris JA, Koyama A, Maeda S, et al. Human P301L‐mutant tau expression in mouse entorhinal‐hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One. 2012;7:e45881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu L, Drouet V, Wu JW, et al. Trans‐synaptic spread of tau pathology in vivo. PLoS One. 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Calignon A, Polydoro M, Surez‐Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Narasimhan S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in non‐transgenic mouse brain. J Neurosci. 2017:1230‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell‐to‐cell transmission of pathological tau: a review. JAMA Neurol. 2019;76(1):101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mudher A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion‐like propagation?. Acta Neuropathol Commun. 2017;5(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu H, Rodriguez GA, Herman M, et al. Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer's disease. Neuron. 2017;93(3):533‐541.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koller EJ, Gonzalez De La, Cruz E, et al. Combining P301L and S320F tau variants produces a novel accelerated model of tauopathy. Hum Mol Genet. 2019;28(19):3255‐3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morrison JH, Brinton RD, Schmidt PJ, Gore AC. Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J Neurosci. 2006;26(41):10332‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hara Y, Waters EM, McEwen BS, Morrison JH. Estrogen effects on cognitive and synaptic health over the lifecourse. Physiol Rev. 2015;95:785‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dumitriu D, Rapp PR, McEwen BS, Morrison JH. Estrogen and the aging brain: an elixir for the weary cortical network. Ann N Y Acad Sci. 2010;1204:104‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang WY, Tan MS, Yu JT, Tan L. Role of pro‐inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. 2015;3(10):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age‐related diseases. Nat Rev Endocrinol. 2018;14(10):576‐590. [DOI] [PubMed] [Google Scholar]
- 28. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131‐144. [DOI] [PubMed] [Google Scholar]
- 29. Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004;61:95‐102. [DOI] [PubMed] [Google Scholar]
- 30. Tapia‐Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59(1):201‐20. [DOI] [PubMed] [Google Scholar]
- 31. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130‐133. [DOI] [PubMed] [Google Scholar]
- 32. Mrak RE, Griffin WST. Potential Inflammatory biomarkers in Alzheimer's disease. J Alzheimer's Dis. 2005;21(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 33. van Hoesen GW, Hyman BT, Damasio AR. Entorhinal cortex pathology in Alzheimer's disease. Hippocampus. 1991;1(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 34. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 35. Johnson GVW, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117(Pt 24):5721‐9. [DOI] [PubMed] [Google Scholar]
- 36. Despres C, Byrne C, Qi H, et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci. 2017:201708448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14(4):388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777‐783. [DOI] [PubMed] [Google Scholar]
- 39. Bolós M, Llorens‐Martín M, Jurado‐Arjona J, Hernández F, Rábano A, Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimer's Dis. 2016;50(1):77‐87. [DOI] [PubMed] [Google Scholar]
- 40. Fakhoury M. Microglia and astrocytes in Alzheimer's disease: implications for therapy. Curr Neuropharmacol. 2017;16(5):508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ulland TK, Colonna M. TREM2 — a key player in microglial biology and Alzheimer's disease. Nat Rev Neurol. 2018;14(11):667‐675. [DOI] [PubMed] [Google Scholar]
- 42. Lee VM‐Y, Goedert M, Trojanowski JQ, Neurodegenerative Tauopathies. Annu Rev Neurosci. 2001;24:1121‐59. [DOI] [PubMed] [Google Scholar]
- 43. Iqbal K, Liu F, Gong C‐X, Grundke‐Iqbal I. Tau in Alzheimer's disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uemura N, Uemura MT, Luk KC, Lee VM‐Y, Trojanowski JQ. Cell‐to‐cell transmission of tau and α‐Synuclein. Trends Mol Med. 2020;26(10):936‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma G, Huo A, Kimura T, et al. Tau isoform expression and phosphorylation in marmoset brains. J Biol Chem. 2019;294(30):11433‐11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steinhilb ML, Dias‐Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007;18(12):5060‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goedert M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018;592(14):2383‐2391. [DOI] [PubMed] [Google Scholar]
- 48. Gong CX, Liu F, Grundke‐Iqbal I, Iqbal K. Post‐translational modifications of tau protein in Alzheimer's disease. J Neural Transm. 2005;112(6):813‐38. [DOI] [PubMed] [Google Scholar]
- 49. Fischer D, Mukrasch MD, Biernat J, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48(42):10047‐55. [DOI] [PubMed] [Google Scholar]
- 50. Gandhi NS, Landrieu I, Byrne C, et al. A phosphorylation‐induced turn defines the Alzheimer's disease AT8 antibody epitope on the tau protein. Angew Chemie ‐ Int Ed 2015;54(23):6819‐23. [DOI] [PubMed] [Google Scholar]
- 51. Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167‐170. [DOI] [PubMed] [Google Scholar]
- 52. Rapp MA, Schnaider‐Beeri M, Purohit DP, Perl DP, Haroutunian V, Sano M. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry. 2008;16:168‐174. [DOI] [PubMed] [Google Scholar]
- 53. Morrison JH, Hof PR. Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer's disease. Prog. Brain Res. 2002;136:467‐86. [DOI] [PubMed] [Google Scholar]
- 54. Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer's disease. Science. 1985;230(4730):1179‐1181. [DOI] [PubMed] [Google Scholar]
- 55. Geula C. Abnormalities of neural circuitry in Alzheimer's disease: hippocampus and cortical cholinergic innervation. Neurology. 1998;51(1 Suppl 1):S18‐29. [DOI] [PubMed] [Google Scholar]
- 56. Hof PR, Morrison JH. The aging brain: morphomolecular senescence of cortical circuits. Trends Neurosci. 2004;27:607‐613. [DOI] [PubMed] [Google Scholar]
- 57. Kondo H, Lavenex P, Amaral DG. Intrinsic connections of the macaque monkey hippocampal formation: iI. CA3 connections. J Comp Neurol. 2009;515(3):349‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Robillard KN, Lee KM, Chiu KB, MacLean AG. Glial cell morphological and density changes through the lifespan of rhesus macaques. Brain Behav Immun. 2016;55:60‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Latimer CS, Shively CA, Keene CD, et al. A nonhuman primate model of early Alzheimer's disease pathologic change: implications for disease pathogenesis. Alzheimer's Dement. 2019;15(1):93‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
All data associated with this study are present in this paper. Raw data used to generate images and graphs are available from the corresponding author, J.H.M., upon request.