

Abstract
The development of new methodologies enabling a facile access to valuable heterocyclic frameworks still is an important subject of research. In this context, we describe a dual catalytic cycle merging C−H alkynylation of phenols and oxy‐alkynylation of the newly introduced triple bond by using a unique redox property and the carbophilic π acidity of gold. Mechanistic studies support the participation of a bimetallic gold–silver species. The one‐pot protocol offers a direct, simple, and regio‐specific approach to 3‐alkynyl benzofurans from readily available phenols. A broad range of substrates, including heterocycles, is transferred with excellent functional group tolerance. Thus, this methodology can be used for the late‐stage incorporation of benzofurans.
Keywords: Alkynylation, Au–Ag bimetallic catalysis, Benzofurans, Phenols
A mild and general synthesis of 3‐alkynylbenzofurans from readily available phenols by Au–Ag bimetallic catalysis has been developed. Importantly, this also advances our mechanistic understanding of the “silver effect” in gold redox chemistry.

Benzofuran, as a privileged structure, can be found in many natural products and biologically active molecules. [1] In addition, highly substituted benzofurans are also prevalent in many leading drug candidates [2] and organic materials. [3] Due to the immense importance of this target, several efficient strategies have already been developed.[ 4 , 5 ] One of the most successful modular strategies, which was established over the past decades, is based on a tandem Sonogashira coupling/5‐endo‐dig cyclization and a subsequent trapping of the intermediate alkenyl–metal species by various electrophiles (Scheme 1 a). [6] The requirement of functionalized precursors, such as ortho‐halo or ‐alkynyl phenols, is a drawback. For example, in 2005, Fürstner and Yamamoto independently reported an elegant example of a platinum‐catalyzed cyclization reaction of ortho‐alkynyl phenols to produce 3‐(α‐alkoxyalkyl)benzofurans.[ 6a , 6g ] In 2014, Blum's group reported an intramolecular gold‐catalyzed alkoxyboration of C−C multiple bonds, an impressive method for the preparation of benzofuran boronic acid derivatives, ideal precursors for downstream transformations. [6b] Recently, Fensterbank et al. developed a dual gold‐ and photo‐induced alkynylative ortho‐cyclization to achieve 3‐alkynyl benzofurans via a photosensitized oxidative addition. [6i] Despite the efficiency and the synthetic potential of these strategies, the requirement of pre‐functionalized substrates is still disadvantageous in these specialized protocols involving multistep procedures. Consequently, there is still a high demand to develop efficient and simple methods for the direct synthesis of functionalized benzofurans.
Scheme 1.

Metal‐catalyzed synthesis of functionalized benzofurans; a) common strategies; b) this work.
Gold catalysis serves as a powerful tool in synthetic organic chemistry, [7] in particular in the fields of heterocyclic chemistry [8] and complex molecule synthesis. [9] Recently, the potential of alkynyl gold(III) species to introduce alkyne moieties as useful synthetic handles for further manipulations was reported. [10] As part of our continuing efforts in gold redox chemistry, we recently developed a ligand‐enabled oxidative addition of alkynylbenziodoxole as key to alkynyl gold(III) complexes. [11] Based on this, we envisioned that an intermolecular tandem process starting from simple phenols might directly afford functionalized benzofurans, enabled by a reactive alkynyl gold(III) species. To the best of our knowledge, the direct synthesis of valuable 3‐alkynyl benzofurans from readily available phenols is yet unknown. Two challenges in a reaction of phenols with alkynyl gold(III) complexes are: a) the sensitivity of phenols towards strong oxidants, which can lead to homocoupling (as known for other transition metals); [12] b) tailoring the conditions for the generation of a reactive alkynyl gold(III) species in a catalytic fashion. Herein, we report the development of an unprecedented direct synthesis of 3‐alkynyl benzofurans by an Au–Ag bimetallic‐catalyzed cascade reaction from readily accessible phenols and alkynylbenziodoxole reagents (Scheme 1 b).
We started our evaluation of the reaction parameters by using the phenol 1 and alkynylbenziodoxole 2 as model substrates to examine the feasibility of the approach and to optimize the reaction conditions (Table 1). After the adjustment of various reaction parameters, the desired 3‐alkynyl benzofuran 4 was isolated in 94 % yield in the presence of 5 mol % Ph3PAuCl/AgNTf2 and 20 mol % Phen in MeCN under open‐flask conditions at 45 °C. Control experiments showed that gold, silver, and Phen all are essential for the reaction. No reaction was observed with pre‐activated Ph3PAuNTf2 (by Celite filtration of the Ph3PAuCl/AgNTf2 mixtures) instead of an in situ activation (Ph3PAuCl/AgNTf2; Table 1, entries 1 and 2). The need for the presence of silver salts indicated a decisive role of silver, it was not only a simple halide scavenger [13] (Table 1, entries 3–6). However, AgCl in combination with a ligand (PPh3, IPr) as additive was not able to promote the reaction, which indicated that free coordination sites at the silver are necessary to coordinate the Phen ligand (Table 1, entries 7 and 8). Various gold(I) and gold(III) complexes were also tested (Table 1, entries 9–11). Among them, AuCl led to formation of 4 in 76 % yield, and the commonly used AuCl3 or IPrAuCl in the presence of AgNTf2 showed no catalytic activity. Other Phen‐type ligands L1 and L2 did not improve the reaction efficiency (Table 1, entries 12–14). When the alkynylbenziodoxolone 3 was used instead of 2, no conversion to 4 was observed (Table 1, entry 15).
Table 1.
Optimization of the reaction conditions.[a]

|
Entry |
Catalytic system |
Yield [%][b] |
|---|---|---|
|
1 |
Ph3PAuCl/AgNTf2/Phen |
94 (0)[c] |
|
2 |
Ph3PAuNTf2/Phen |
0 |
|
3 |
Ph3PAuNTf2/AgCl/Phen |
85 |
|
4 |
Ph3PAuNTf2/AgBr/Phen |
64 |
|
5[d] |
Ph3PAuNTf2/AgI/Phen |
0 |
|
6[d] |
Ph3PAuNTf2/AgNTf2/Phen |
22 |
|
7[d] |
Ph3PAuNTf2/Ph3PAgCl/Phen |
0 |
|
8[d] |
Ph3PAuNTf2/IPrAgCl/Phen |
0 |
|
9 |
AuCl/AgNTf2/Phen |
76 |
|
10 |
IPrAuCl/AgNTf2/Phen |
0 |
|
11 |
AuCl3/AgNTf2/Phen |
0 |
|
12 |
Ph3PAuCl/AgNTf2/bpy |
0 |
|
13 |
Ph3PAuCl/AgNTf2/L1 |
61 |
|
14 |
Ph3PAuCl/AgNTf2/L2 |
82 |
|
15[e] |
Ph3PAuCl/AgNTf2/Phen |
0 |
|
| ||
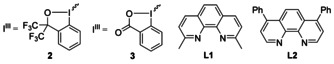
[a] 1 (0.10 mmol), 2 (0.22 mmol), Ph3PAuCl/AgNTf2 (5 mol %), Phen (20 mol %) in CH3CN (2 mL) at 45 °C under open flask conditions. [b] Isolated yields. [c] No Ph3PAuCl, or AgNTf2, or Phen. [d] 24 h for the reaction. [e] 3 instead of 2. Phen=1,10‐phenanthroline.
Under the optimized reaction conditions from Table 1 we investigated the substrate scope of this tandem reaction. First, we explored the scope with regard to the phenol moiety. A variety of phenols smoothly reacted with alkynylbenziodoxole 2 providing the corresponding 3‐alkynyl benzofurans in good to excellent yields. As illustrated in Scheme 2, various types of functional groups, for example, sulfonyl, carboxyl, acyl, formyl, nitro, and cyano as well as F, Br, and Cl substituents, were tolerated (4–10; 13–19). [14] Moreover, disubstituted and trisubstituted phenols smoothly delivered the products in excellent positional selectivity; only for an ortho‐substituted phenol a product mixture of 12 and 12′ was obtained. Naphthols and 9‐phenanthrol also delivered the corresponding π‐extended products in good yield and with very good selectivity (20–23). Notably, hydroxy‐substituted heterocyclic systems were also compatible, providing the products in good to excellent yields (24–28). Only some electron‐rich phenols, such as 4‐methoxyphenol, were not compatible (11); instead, a reductive homocoupling to 1,4‐diphenylbutadiyne was observed. A possible reason is that 4‐methoxyphenol due to the high pK a value (19.1 in DMSO) cannot regenerate the bimetallic catalyst C (Scheme 5), while the lower pK a value of ortho‐C6H4IC(CF3)2OH allows the catalyst regeneration (there is only a literature‐known value for hexafluoro‐2‐propanol, CH(CF3)2OH, which has a pK a of 17.9 in DMSO; due to the H substitutent it should be higher than the pK a of ortho‐C6H4IC(CF3)2OH with a sp2‐C substitutent). And with the electron‐withdrawing effect of two meta‐methoxy groups on the phenol, the pK a value of phenol is increased. Therefore, product 17 was obtained in decent yield. Various complex phenol derivatives of pharmaceutical importance were examined next to prove the synthetic potential of this strategy (29–31). A moderate amount of the desired benzofuran product 30 was obtained from Umbelliferone and 2, without affecting the lactone functionality of the coumarin moiety. Pleasingly, Nitroxoline, an antibiotic that has been used by humans for many years, reacted smoothly with 2 to provide 31 in excellent yield.
Scheme 2.

General substrate scope with respect to phenols and alkynylbenziodoxoles. Reaction conditions: 1 (0.1 mmol), 2 (0.22 mmol), Ph3PAuCl/AgNTf2 (5 mol %), and Phen (20 mol %) were stirred in CH3CN under open flask conditions at 45 °C; Yields refer to isolated product. [a] 4 mmol scale.
Scheme 5.

Proposed mechanism.
The scope with regard to the alkynylbenziodoxoles reagents was studied with both aliphatic and (hetero)aromatic ethynylbenziodoxole derivatives. These could successfully be employed as precursors, providing a variety of 3‐alkynyl benzofurans in good to excellent yield (32–43). [15] In a one‐pot competition experiment of two different alkynylbenziodoxoles, mainly the product 32 was obtained, in addition to a small amount of 38; GC/MS shows only a trace of a heterocoupling.
The obtained benzofurans still possess functional groups, the alkynyl benzofuran was hydrogenated by formic acid under palladium catalysis. Depending on the conditions either the Z‐alkene 44 or the E‐isomer 45 was generated in excellent yield.
Mechanistic evidence for the role of AgCl was gathered by monitoring the conversion rates for different catalyst combinations. As shown in Figure 1, equimolar amounts of Ph3PAuCl/AgNTf2 or Ph3PAuNTf2/AgCl smoothly promoted the reaction, while Ph3PAuNTf2 as single catalyst was inactive. Notably, the addition of an equimolar amount of AgCl to the mixture turned on the catalytic activity again and the reaction proceeded smoothly in a good yield. As the solubility of AgCl in acetonitrile at 25 °C is low (0.05 mg L−1), [16] a coordination by the Phen ligand must take place. Thus, the active catalyst is formed, probably a heterobimetallic complex. The lack of reactivity with AgCl bearing an additional ligand (PPh3 or IPr) can be explained by the missing free coordination site for the Phen ligand in these complexes. This further underlines that a PhenAgCl species is a crucial structural element in the actually operating catalytic species. By ESI‐MS measurements of the Ph3PAuCl/AgNTf2/Phen system, an ion with m/z=781.0009 was detectable. A computational study of different possible isomers originating from these components was carried out. Complex A is the most stable of all calculated species (see the Supporting Information). An analysis of this led us to the proposal of a heterobimetallic species Ph3PAuClAg(Phen) A as active catalytic species (Figure 1, right side). In analogy to previous studies, [11a] we assumed that an alkynyl gold(III) species might be a crucial intermediate in the catalytic cycle as well. To verify this hypothesis, stoichiometric reactions with the preformed (Phen)gold(III) alkynyl species B and 1 were conducted. But neither with nor without AgCl, the desired product was detected (Scheme 3). This indicated that the reaction is not proceeding through intermediate B, which is then activated by the effect of AgCl, but instead via the in situ‐generated heterobimetallic Au/Ag species A (introducing the Ag already at the AuI stage), which serves as the active catalyst.
Figure 1.

1H NMR monitoring of the reaction course with different catalytic systems. Conditions: 1 (0.1 mmol), 2 (0.22 mmol), the catalytic system (5 mol %), and Phen (20 mol %) were stirred in CH3CN under open‐flask conditions at 45 °C.
Scheme 3.

Stoichiometric reactions with alkynyl gold(III) species B.
Next we reacted C3‐unsubstituted benzofuran 47 and 2, but the reaction failed to deliver any 3‐alkynyl benzofuran 5 under the standard reaction conditions (Scheme 4 a). In addition, 2‐alkynyl phenol 48 failed to afford the C3‐unsubstituted benzofuran (Scheme 4 b); no conversion was observed. In contrast, with or without AgCl, the reaction of 2‐alkynyl phenols 48 with 2 afforded the desired 3‐alkynyl benzofuran 5 in excellent yield (Scheme 4 c). This indicates that silver only is essential in the ortho‐alkynylation of phenol, which is the first step of the overall reaction, the step leading to intermediates like 48 (Table 1, entry 2). It also shows nicely that silver is not needed in the oxy‐alkynylation step and not needed in the benzofuran alkynylation. In the absence of silver it is the [PhenAuPPh3]+ complex which is able to undergo oxidative addition of 2, as previously shown. [11a] This strongly indicates that the cascade starts with the alkynylation of the phenol in the first step. It excludes a scenario in which a C3‐unsubstituted benzofuran and gold(I) 3‐benzofuryl complex are first formed which is then alkynylated in the final step;[ 10a , 10b , 10c ] instead, the oxy‐alkynylation is triggered by a bimetallic gold(III)‐acetylide‐Ag complex followed by a subsequent reductive elimination.
Scheme 4.
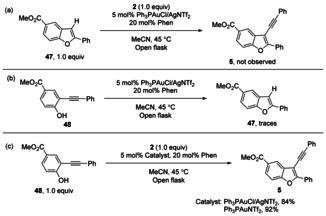
Control experiments.
Then the thermochemistry of the oxidative addition of the used hypervalent iodo species to A was calculated (Figure 2). Two reaction pathways were taken into consideration. On the one hand the two‐step oxidative addition starting from I formed transition state II (ΔH=15.1 kcal mol−1; ΔG=14.8 kcal mol−1). Afterwards species III (ΔH=1.51 kcal mol−1; ΔG=0.93 kcal mol−1) was obtained as an ionic pair of a AuIII‐complex with the alkyne from the hypervalent iodo species and the corresponding benzylic alcoholate anion. Ultimately, species IV (ΔH=−28.3 kcal mol−1; ΔG=−27.6 kcal mol−1) was obtained as the final product of the oxidative addition. Furthermore, a barrier‐free concerted pathway was found, which led to the same product IV. The same reaction was also calculated with Ph3PAuCl. This system did neither undergo cis nor trans oxidative addition of the hypervalent iodo reagent towards a AuIII‐species.
Figure 2.

DFT‐computed free energies of the oxidative addition of the hypervalent iodo species to heterobimetallic Au–Ag species A. Red: concerted pathway; blue: stepwise pathway.
Geometry optimizations of the postulated active complex A show a distance of of 3.17 Å between gold and silver. Given the van der Waals radii of both metals (r(Au)=1.66 Å and r(Ag)=1.72 Å), this indicates a metallophilic interaction between the two metal centers. This and the ligation of the Ag(Phen) fragment lead to the increased reactivity of complex A. [17]
On the basis of these observations and DFT calculations, a plausible mechanism [18] is outlined in Scheme 5. First the active heterobimetallic species Ph3PAuIClAg(Phen) (C) is formed. This species then undergoes cis‐oxidative addition with 2 to afford alkynyl gold(III) compound D. At this stage, attack of the phenol at the alkynyl AuIII species D, with liberation of PhenAgCl and H+, delivers the quinone‐type intermediate E. Reductive elimination then affords the alkynyl phenol F after tautomerization. In the second cycle the alkyne in F is activated by AuIII‐acetylide species D, which triggers the intramolecular oxyauration to afford the gold(III) complex H. Upon reductive elimination of H, the desired product I is formed. [19] The reaction of the released catalyst G then regenerates C with liberation of C6H4IC(CF3)2OH (which leads to a low proton activity in the reaction mixture and thus avoids proto‐deauration processes).
In summary, by taking advantage of the unique redox property and carbophilic π acidity of gold, we have developed a novel direct synthesis of 3‐alkynyl benzofurans involving a Au–Ag bimetallic‐catalyzed tandem reaction of commercially available phenols with alkynylbenziodoxole reagents. The available mechanistic data are consistent with the proposed oxidative addition of alkynyl hypervalent iodine reagent onto the Au–Ag bimetallic active catalyst. This study also enhances our understanding of the gold catalytic reaction and draws attention to the actual role of AgCl in organic transformations. [17]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
L. Hu and C. Han are grateful to the CSC (China Scholarship Council) for a Ph.D. fellowship. The authors acknowledge support by the state of Baden–Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467‐1 FUGG (JUSTUS cluster). Open access funding enabled and organized by Projekt DEAL.
L. Hu, M. C. Dietl, C. Han, M. Rudolph, F. Rominger, A. S. K. Hashmi, Angew. Chem. Int. Ed. 2021, 60, 10637.
Contributor Information
Long Hu, http://www.hashmi.de.
Prof. Dr. A. Stephen K. Hashmi, Email: hashmi@hashmi.de.
References
- 1.
- 1a. Cagniant P., Cagniant D., Adv. Heterocycl. Chem. 1975, 18, 337–482; [Google Scholar]
- 1b. Inoue M., Carson M. W., Frontier A. J., Danishefsky S. J., J. Am. Chem. Soc. 2001, 123, 1878–1889. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Rizzo S., Rivière C., Piazzi L., Bisi A., Gobbi S., Bartolini M., Andrisano V., Morroni F., Tarozzi A., Monti J.-P., Rampa A., J. Med. Chem. 2008, 51, 2883–2886; [DOI] [PubMed] [Google Scholar]
- 2b. Bharate S. B., Sawant S. D., Singh P. P., Vishwakarma R. A., Chem. Rev. 2013, 113, 6761–6815; [DOI] [PubMed] [Google Scholar]
- 2c. Kirilmis C., Ahmedzade M., Servi S., Koca M., Kizirgil A., Kazaz C., Eur. J. Med. Chem. 2008, 43, 300–308; [DOI] [PubMed] [Google Scholar]
- 2d. Aslam S. N., Stevenson P. C., Phythian S. J., Veitch N. C., Hall D. R., Tetrahedron 2006, 62, 4214–4226. [Google Scholar]
- 3.
- 3a. Walker B., Tamayo A. B., Dang X.-D., Zalar P., Seo J. H., Garcia A., Tantiwiwat M., Nguyen T.-Q., Adv. Funct. Mater. 2009, 19, 3063–3069; [Google Scholar]
- 3b. Tsuji H., Mitsui C., Ilies L., Sato Y., Nakamura E., J. Am. Chem. Soc. 2007, 129, 11902–11903. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Anderson K. W., Ikawa T., Tundel R. E., Buchwald S. L., J. Am. Chem. Soc. 2006, 128, 10694–10695; [DOI] [PubMed] [Google Scholar]
- 4b. Katritzky A. R., Rachwal S., Chem. Rev. 2011, 111, 7063–7120; [DOI] [PubMed] [Google Scholar]
- 4c. Wu X.-F., Neumann H., Beller M., Chem. Rev. 2013, 113, 1–35; [DOI] [PubMed] [Google Scholar]
- 4d. Khan I., Ibrar A., Shehzadi S. A., Coord. Chem. Rev. 2019, 380, 440–470. [Google Scholar]
- 5.
- 5a. Guo X., Yu R., Li H., Li Z., J. Am. Chem. Soc. 2009, 131, 17387–17393; [DOI] [PubMed] [Google Scholar]
- 5b. Maimone T. J., Buchwald S. L., J. Am. Chem. Soc. 2010, 132, 9990–9991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Kobatake T., Fujino D., Yoshida S., Yorimitsu H., Oshima K., J. Am. Chem. Soc. 2010, 132, 11838–11840; [DOI] [PubMed] [Google Scholar]
- 5d. Lee D.-H., Kwon K.-H., Yi C. S., J. Am. Chem. Soc. 2012, 134, 7325–7328; [DOI] [PubMed] [Google Scholar]
- 5e. Zhu R., Wei J., Shi Z., Chem. Sci. 2013, 4, 3706–3711; [Google Scholar]
- 5f. Okamoto K., Hori M., Yanagi T., Murakami K., Nogi K., Yorimitsu H., Angew. Chem. Int. Ed. 2018, 57, 14230–14234; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14426–14430; [Google Scholar]
- 5g. Kuram M. R., Bhanuchandra M., Sahoo A. K., Angew. Chem. Int. Ed. 2013, 52, 4607–4612; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4705–4710; [Google Scholar]
- 5h. Sharma U., Naveen T., Maji A., Manna S., Maiti D., Angew. Chem. Int. Ed. 2013, 52, 12669–12673; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12901–12905. [Google Scholar]
- 6.
- 6a. Fürstner A., Davies P. W., J. Am. Chem. Soc. 2005, 127, 15024–15025; [DOI] [PubMed] [Google Scholar]
- 6b. Hirner J. J., Faizi D. J., Blum S. A., J. Am. Chem. Soc. 2014, 136, 4740–4745; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Han Z., Zhang L., Li Z., Fan R., Angew. Chem. Int. Ed. 2014, 53, 6805–6809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6923–6927; [Google Scholar]
- 6d. Nakamura M., Ilies L., Otsubo S., Nakamura E., Angew. Chem. Int. Ed. 2006, 45, 944–947; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 958–961; [Google Scholar]
- 6e. Sakiyama N., Noguchi K., Tanaka K., Angew. Chem. Int. Ed. 2012, 51, 5976–5980; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6078–6082; [Google Scholar]
- 6f. Iqbal N., Iqbal N., Maiti D., Cho E. J., Angew. Chem. Int. Ed. 2019, 58, 15808–15812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15955–15959; [Google Scholar]
- 6g. Nakamura I., Mizushima Y., Yamamoto Y., J. Am. Chem. Soc. 2005, 127, 15022–15023; [DOI] [PubMed] [Google Scholar]
- 6h. Zhang H., Ferreira E. M., Stoltz B. M., Angew. Chem. Int. Ed. 2004, 43, 6144–6148; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6270–6274; [Google Scholar]
- 6i. Xia Z., Corcé V., Zhao F., Przybylski C., Espagne A., Jullien L., Le Saux T., Gimbert Y., Dossmann H., Mouriès-Mansuy V., Ollivier C., Fensterbank L., Nat. Chem. 2019, 11, 797–805. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Hashmi A. S. K., Hutchings G. J., Angew. Chem. Int. Ed. 2006, 45, 7896–7936; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8064–8105; [Google Scholar]
- 7b. Hashmi A. S. K., Chem. Rev. 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]
- 7c. Gorin D. J., Toste F. D., Nature 2007, 446, 395–403; [DOI] [PubMed] [Google Scholar]
- 7d. Li Z., Brouwer C., He C., Chem. Rev. 2008, 108, 3239–3265; [DOI] [PubMed] [Google Scholar]
- 7e. Corma A., Leyva-Pérez A., Sabater M. J., Chem. Rev. 2011, 111, 1657–1712; [DOI] [PubMed] [Google Scholar]
- 7f. Hashmi A. S. K., Toste F. D., Modern gold-catalyzed synthesis, Wiley, Hoboken, 2012; [Google Scholar]
- 7g. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7h. Zheng Z., Wang Z., Wang Y., Zhang L., Chem. Soc. Rev. 2016, 45, 4448–4458; for early results, see: [DOI] [PubMed] [Google Scholar]
- 7i. Hashmi A. S. K., Schwarz L., Choi J.-H., Frost T. M., Angew. Chem. Int. Ed. Engl. 2000, 39, 2285–2288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2382–2385; [Google Scholar]
- 7j. Hashmi A. S. K., Frost T. M., Bats J. W., J. Am. Chem. Soc. 2000, 122, 11553–11554. [Google Scholar]
- 8.
- 8a. Hashmi A. S. K., Bührle M., Aldrichim. Acta 2010, 43, 27–33; [Google Scholar]
- 8b. Rudolph M., Hashmi A. S. K., Chem. Commun. 2011, 47, 6536–6544; [DOI] [PubMed] [Google Scholar]
- 8c. Fürstner A., Angew. Chem. Int. Ed. 2018, 57, 4215–4233; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4289–4308; [Google Scholar]
- 8d. Tian X., Song L., Hashmi A. S. K., Chem. Eur. J. 2020, 26, 3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Hashmi A. S. K., Rudolph M., Chem. Soc. Rev. 2008, 37, 1766–1775; [DOI] [PubMed] [Google Scholar]
- 9b. Rudolph M., Hashmi A. S. K., Chem. Soc. Rev. 2012, 41, 2448–2462; [DOI] [PubMed] [Google Scholar]
- 9c. Pflästerer D., Hashmi A. S. K., Chem. Soc. Rev. 2016, 45, 1331–1367. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Brand J. P., Charpentier J., Waser J., Angew. Chem. Int. Ed. 2009, 48, 9346–9349; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9510–9513; [Google Scholar]
- 10b. Brand J. P., Waser J., Angew. Chem. Int. Ed. 2010, 49, 7304–7307; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7462–7465; [Google Scholar]
- 10c. Li Y., Brand J. P., Waser J., Angew. Chem. Int. Ed. 2013, 52, 6743–6747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6875–6879; [Google Scholar]
- 10d. Ma Y., Zhang S., Yang S., Song F., You J., Angew. Chem. Int. Ed. 2014, 53, 7870–7874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8004–8008; [Google Scholar]
- 10e. Li X., Xie X., Sun N., Liu Y., Angew. Chem. Int. Ed. 2017, 56, 6994–6998; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7098–7102; [Google Scholar]
- 10f. de Haro T., Nevado C., J. Am. Chem. Soc. 2010, 132, 1512–1513; [DOI] [PubMed] [Google Scholar]
- 10g. Peng H., Xi Y., Ronaghi N., Dong B., Akhmedov N. G., Shi X., J. Am. Chem. Soc. 2014, 136, 13174–13177. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Yang Y., Antoni P., Zimmer M., Sekine K., Mulks F. F., Hu L., Zhang L., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2019, 58, 5129–5133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5183–5187; [Google Scholar]
- 11b. Yang Y., Eberle L., Mulks F. F., Wunsch J. F., Zimmer M., Rominger F., Rudolph M., Hashmi A. S. K., J. Am. Chem. Soc. 2019, 141, 17414–17420; [DOI] [PubMed] [Google Scholar]
- 11c. Huang L., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 4808–4813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4888–4893; [Google Scholar]
- 11d. Huang L., Rominger F., Rudolph M., Hashmi A. S. K., Chem. Commun. 2016, 52, 6435–6438; [DOI] [PubMed] [Google Scholar]
- 11e. Yu Y., Yang W., Pflästerer D., Hashmi A. S. K., Angew. Chem. Int. Ed. 2014, 53, 1144–1147; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1162–1165. [Google Scholar]
- 12.
- 12a. Xiao B., Gong T.-J., Liu Z.-J., Liu J.-H., Luo D.-F., Xu J., Liu L., J. Am. Chem. Soc. 2011, 133, 9250–9253; [DOI] [PubMed] [Google Scholar]
- 12b. Li X., Hewgley J. B., Mulrooney C. A., Yang J., Kozlowski M. C., J. Org. Chem. 2003, 68, 5500–5511; [DOI] [PubMed] [Google Scholar]
- 12c. Smrcina M., Polakova J., Vyskocil S., Kocovsky P., J. Org. Chem. 1993, 58, 4534–4538; [Google Scholar]
- 12d. Hay A. S., Blanchard H. S., Endres G. F., Eustance J. W., J. Am. Chem. Soc. 1959, 81, 6335–6336. [Google Scholar]
- 13. Wang D., Cai R., Sharma S., Jirak J., Thummanapelli S. K., Akhmedov N. G., Zhang H., Liu X., Petersen J. L., Shi X., J. Am. Chem. Soc. 2012, 134, 9012–9019. [DOI] [PubMed] [Google Scholar]
- 14. Deposition Number 2067515 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 15.We also tested 3-alkynyl benzothiophenes and thiophenols, but no product formation was observed, with thiophenols immediately a precipitate was formed. Silyl-protected hypervalent iodine reagents also did not react.
- 16.
- 16a. Melcher A. C., J. Am. Chem. Soc. 1910, 32, 50–66; [Google Scholar]
- 16b. Jonte J. H., Martin D. S., J. Am. Chem. Soc. 1952, 74, 2052–2054; [Google Scholar]
- 16c. Alexander R., Ko E. C. F., Mac Y. C., Parker A. J., J. Am. Chem. Soc. 1967, 89, 3703–3712. [Google Scholar]
- 17.A more detailed computational analysis and the further exploitation of this concept are currently under investigation.
- 18.
- 18a. Hashmi A. S. K., Angew. Chem. Int. Ed. 2010, 49, 5232–5241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5360–5369; [Google Scholar]
- 18b. Lauterbach T., Asiri A. M., Hashmi A. S. K., Adv. Organomet. Chem. 2014, 62, 261–297. [Google Scholar]
- 19.A competing hydrophenoxylation of the product is not observed, it is known to typically proceed at 100 °C, while our conversion already occurs at 45 °C, a selectivity which is based on the significant energy difference of the barriers for both reactions. For hydrophenoxylations, see: Kuram M. R., Bhanuchandra M., Sahoo A. K., J. Org. Chem. 2010, 75, 2247–2258; For other alternative pathways, see:20205371 [Google Scholar]; Adak T., Schulmeister J., Dietl M. C., Rudolph M., Rominger F., Hashmi A. S. K., Eur. J. Org. Chem. 2019, 3867–3876. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary