

Abstract
Macromolecular drugs inefficiently cross membranes to reach their cytosolic targets. They require drug delivery vectors to facilitate their translocation across the plasma membrane or escape from endosomes. Optimization of these vectors has however been hindered by the difficulty to accurately measure cytosolic arrival. We have developed an exceptionally sensitive and robust assay for the relative or absolute quantification of this step. The assay is based on benzylguanine and biotin modifications on a drug delivery vector of interest, which allow, respectively, for selective covalent capture in the cytosol with a SNAP‐tag fusion protein and for quantification at picomolar sensitivity. The assay was validated by determining the absolute numbers of cytosolic molecules for two drug delivery vectors: the B‐subunit of Shiga toxin and the cell‐penetrating peptide TAT. We expect this assay to favor delivery vector optimization and the understanding of the enigmatic translocation process.
Keywords: cell-penetrating peptides, drug delivery, endosomal escape, membranes, Shiga toxin B-subunit
Macromolecular drug delivery vectors must translocate at the plasma membrane or escape from endosomes to reach the cytosol. This very inefficient process requires optimization, which is hindered by the difficulty to accurately measure cytosolic arrival. An exceptionally sensitive and robust assay to quantify this step was developed. The assay was validated by determining the absolute numbers of two macromolecular vectors reaching the cytosol.
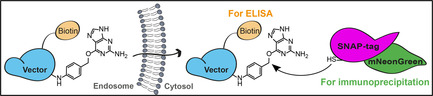
Biological macromolecules such as peptides, proteins or oligonucleotides hold great therapeutic potential. They enable indeed to target “undruggable” proteins which lack cavities for small molecule binding, or to stimulate the immune system by antigen cross‐presentation. However, macromolecules do not readily cross membranes. Most remain trapped at the plasma membrane or in intracellular compartments and do not reach their targets in the cytosol. Cytosolic arrival, via direct translocation across the plasma membrane or endocytosis followed by endosomal escape, is currently one of the main bottlenecks for the development of new macromolecular therapeutics.[ 1 , 2 , 3 ]
For the optimization of cytosolic delivery, the process must be quantified. [4] Existing assays for this have a number of limitations, including the failure to distinguish between cytosolic versus intraluminal localizations and a lack of sensitivity and robustness (Table S1). Of note, two recently developed assays, the Chloroalkane Penetration Assay [5] and the NanoClick assay [6] rely on the HaloTag protein, [7] which covalently reacts to chloroalkanes. The cytosolic localization of the HaloTag reporter protein ensures the cytosolic specificity of the assay. Both assays measure the non‐reacted reporter protein fraction, which is inversely proportional to the extent of translocated molecules. This limits the assay sensitivity. Furthermore, these assays do not provide absolute quantification of translocated molecules. Therefore, the quantification of small amounts of molecules reaching the cytosol remains challenging.
The example of siRNAs for the downregulation of disease‐related proteins might be chosen to illustrate this point. For an efficient therapeutical effect, it is estimated that 2000–4000 siRNA molecules need to reach the cytosol per cell.[ 8 , 9 ] The sensitivity of a cytosolic arrival assay is thus essential to detect such low numbers of molecules per cell and to optimize existing or develop new delivery tools.
To address this challenge, we have designed the Cyto‐SNAP assay. This assay is based on a cytosolic capture protein assembled from SNAP‐tag and mNeonGreen. The SNAP‐tag reacts covalently with the small molecule benzylguanine (BG). [10] A vector of interest (e.g., protein‐ or peptide‐based drug delivery tools) modified with BG will react with the SNAP‐tag only if the vector reaches the cytosol (Figure 1 a).
Figure 1.
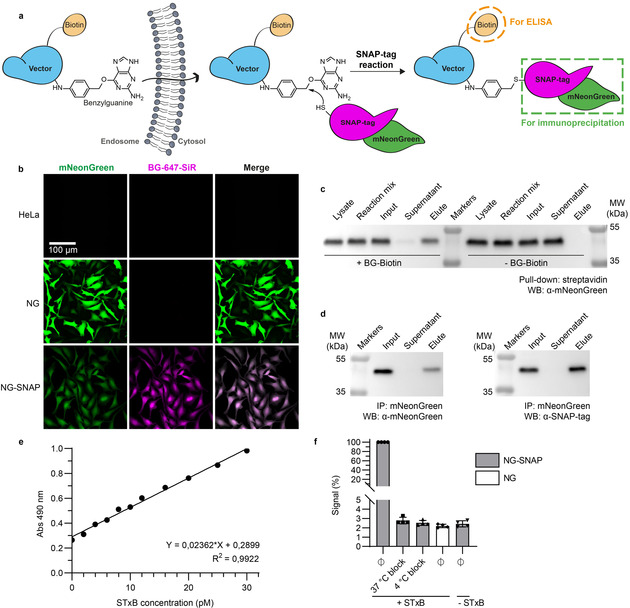
A robust, sensitive, and quantitative cytosolic arrival assay. a) Schematic representation of the Cyto‐SNAP assay. Upon membrane translocation, the BG‐modified vector encounters the cytosolic mNeonGreen‐SNAP‐tag protein with which it reacts covalently. mNeonGreen is exploited for immunoprecipitation on beads coated with anti‐mNeonGreen nanobodies, and the biotin moiety for ELISA. b) Parental, polyclonal mNeonGreen expressing (NG) or monoclonal mNeonGreen‐SNAP‐tag expressing (NG‐SNAP) HeLa cell lines were treated with fluorescent SNAP‐tag ligand BG‐647‐SiR. SiR fluorescence was only observed on NG‐SNAP cells, in which it was homogeneously distributed in the cytosolic space. c) Demonstration of the high efficiency of the SNAP‐tag reaction. Lysate from NG‐SNAP cells was incubated with excess benzylguanine–biotin (BG–biotin) ligand, followed by streptavidin pull‐down. Western blotting analysis showed that the cell lysate was depleted of mNeonGreen‐SNAP‐tag protein, which was indeed recovered on beads. d) Demonstration that the mNeonGreen immunoprecipitation (IP) is complete. The mNeonGreen‐SNAP‐tag protein was totally recovered on mNeonGreen‐Trap beads, which confirmed the efficacy of the immunoprecipitation. e) Sensitivity and linearity of the assay. Known amounts of STxB‐BG‐biotin conjugate C were added into NG‐SNAP cell lysate, followed by incubation at 37 °C, mNeonGreen‐SNAP‐tag immunoprecipitation and ELISA development. The obtained standard curve was linear over a wide range of concentrations. Even low picomolar STxB‐BG‐biotin concentrations were robustly detected. f) Demonstration that non‐reacted SNAP‐tag protein is efficiently quenched before cell lysis. Intact NG‐SNAP and NG cells were incubated for 30 min at 4 °C or 37 °C in presence or absence (Ø) of SNAP‐Cell® Block reagent. The cells were then washed, lysed, and lysates were incubated at 4 °C with or without STxB‐BG‐biotin conjugate A. Both 37 °C and 4 °C block conditions gave ELISA signals comparable to background signal without STxB‐BG‐biotin incubation.
mNeonGreen, [11] the second part of the cytosolic capture protein, is used for high affinity immunoprecipitation on beads coated with anti‐mNeonGreen nanobodies (low K d of 2 nm). In this way, BG‐tagged vector that reacts with the SNAP‐tag of the cytosolic capture protein can be isolated. Finally, a biotin conjugated to the vector serves for quantification by ELISA (Figure S1).
We chose to validate the assay using two different vectors: i) Residues 47–57 from the HIV TAT protein, [12] which is one of the best‐studied cell‐penetrating peptides, and ii) the B‐subunit of Shiga toxin (STxB), a vector for cancer cell targeting and immunotherapy. [13] After binding to its receptor, the glycosphingolipid Gb3, STxB is internalized and follows the retrograde transport route to the endoplasmic reticulum. STxB was shown to be able to translocate to the cytosol [14] and to efficiently deliver antigens for antigen cross‐presentation, [13] making it an interesting model for a cytosolic arrival assay.
A monoclonal HeLa cell line, termed NG‐SNAP, stably expressing the cytosolic mNeonGreen‐SNAP‐tag capture protein was generated for the assay. Diffuse mNeonGreen fluorescence and BG‐fluorophore labeling were observed, which documented the expression of properly folded protein in the cytosol (Figure 1 b). To test whether the cytosolically localized mNeonGreen‐SNAP‐tag was fully functional, immunoprecipitation and pull‐down experiments were performed against each of its two subunits: i) Lysates from NG‐SNAP cells were incubated with an excess of BG–biotin, followed by streptavidin pull‐down. This led to the complete depletion of mNeonGreen‐SNAP‐tag from the lysates, thereby proving that the SNAP‐tag subunit was fully functional (Figure 1 c). ii) The same approach using anti‐mNeonGreen nanobody beads also led to the depletion of mNeonGreen‐SNAP‐tag from the lysates, which further validated the functionality of the fusion protein (Figure 1 d). The combination of highly efficient SNAP‐tag reaction and complete mNeonGreen immunoprecipitation laid the foundation for a fully quantitative assay.
Linearity and sensitivity are key parameters of a quantitative assay. To this end, low picomolar concentrations of BG‐ and biotin‐tagged STxB were added to mNeonGreen‐SNAP‐tag‐containing cell lysate, and incubated at 37 °C to allow for BG‐SNAP‐tag reaction. Subsequent mNeonGreen immunoprecipitation and ELISA development resulted in a linear standard curve, which documented the exquisite sensitivity of the assay (Figure 1 e).
Importantly, on cells, using a membrane‐permeable BG‐derivative, it was possible to quench non‐reacted SNAP‐tag prior to lysis (Figure 1 f). In the real assay format, this step is required to avoid post‐lysis reaction between non‐cytosolic BG‐tagged vector and non‐conjugated SNAP‐tag in the lysate. Finally, STxB showed normal intracellular trafficking in NG‐SNAP cells and in cells stably expressing mNeonGreen in the absence of SNAP‐tag (termed NG cells), which were used as controls (Figure S2). All these findings qualified the engineered NG‐SNAP and NG cell lines for the Cyto‐SNAP assay.
The Cyto‐SNAP assay (Figure S3) was first used to provide relative quantifications of cytosolic arrival between different conditions (incubation times, vector concentrations or inhibitor treatments). STxB (Figure 2 a,b) and TAT (Figure 2 g,h) translocation to the cytosol increased in a time‐ and concentration‐dependent manner. For STxB, the translocation signal started to plateau at concentrations above 100 nm (Figure 2 b and S4a), while TAT translocation continued to increase exponentially beyond at least 20 μm (Figure 2 h and S4b), suggesting that the process was receptor‐independent for TAT, and receptor‐dependent for STxB. Depletion of the STxB receptor, glycosphingolipid Gb3, by incubation of NG‐SNAP cells with a glucosylceramide synthase inhibitor indeed reduced the cytosolic arrival of STxB to background level (Figure 2 c). Background was defined throughout all experiments as ELISA signal observed from NG cells. STxB translocation to the cytosol was also greatly impacted when incubations were performed at low temperatures, i.e., 4 °C or 19.5 °C (Figure 2 d). This observation was likely caused by changes in membrane organization leading to the disappearance of domain boundaries at which membranes are more permeable.[ 15 , 16 ] In ATP depleted cells, STxB arrival in the cytosol was also strongly decreased (Figure 2 e). As for the 4 °C condition, endocytosis of STxB is inhibited in ATP‐depleted cells. [17] Thus, it can be concluded that cellular entry is required for efficient translocation to the cytosol. The arrival of STxB in the cytosol was also found to be partially reduced when endosomal acidification was inhibited (Figure 2 f). Finally, TAT was compared to TAT‐PEG6‐GFWFG, previously reported to have an enhanced capacity to translocate to the cytosol. [18] We found no difference in cytosolic arrival at concentrations below 10 μm (Figure 2 i). In contrast, TAT‐PEG6‐GFWFG translocated much more efficiently than TAT at concentrations above 15 μm (Figure 2 i). TAT undergoes endocytosis at low concentrations, whereas at concentrations above 10 μm direct translocation across the plasma membrane becomes the dominant mechanism (ref. [19]; for a review, see ref. [20]). It therefore appears likely that the strongly enhanced cytosolic arrival of TAT‐PEG6‐GFWFG at concentrations above 15 μm resulted from direct translocation across the plasma membrane. These results qualified the Cyto‐SNAP assay for membrane translocation measurements with different types of vectors.
Figure 2.
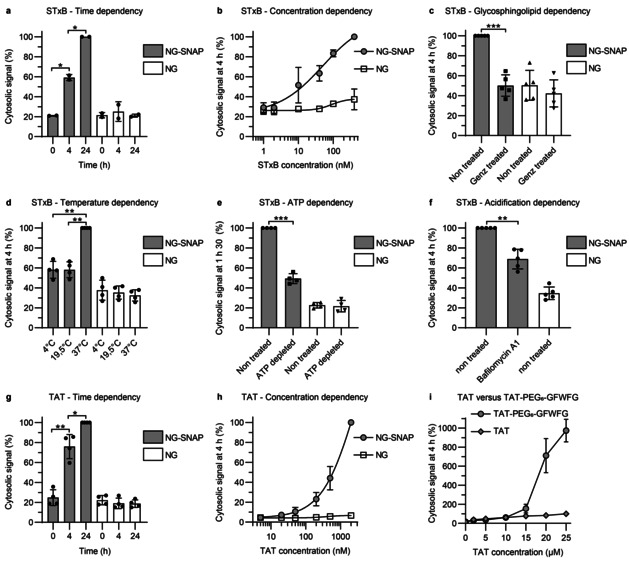
Relative quantifications of STxB and TAT translocation to the cytosol, using the Cyto‐SNAP assay on NG‐SNAP cells. In all experiments, NG cells served as background controls. The cytosolic signal is expressed as percentage of the maximum signal obtained in each individual replicate experiment. a) Time dependency. Cells were incubated with 40 nm STxB‐BG‐biotin conjugate A for 0, 4, or 24 h (continuous incubation). The cytosolic signal increased with time. b) Concentration dependency. Cells were incubated for 4 h with STxB‐BG‐biotin conjugate A at the indicated concentrations. The cytosolic signal increased with concentration. c) Glycosphingolipid dependency. Cells were pre‐treated or not for 5 days with the glycosylceramide synthase inhibitor Genz‐123346, and then incubated for 4 h with 40 nm STxB‐BG‐biotin conjugate A. The translocation process to the cytosol was glycosphingolipid dependent. d) Temperature dependency. Cells were incubated for 4 h with 40 nm STxB‐BG‐biotin conjugate A at the indicated temperatures. Translocation to the cytosol was blocked at 4 °C and 19.5 °C. e) ATP dependency. Cells were incubated for 90 min with 40 nm STxB‐BG‐biotin conjugate A under ATP depletion conditions. Translocation to the cytosol was ATP dependent. f) Acidification dependency. Cells were incubated for 4 h with 40 nm STxB‐BG‐biotin conjugate A in the presence or absence of 100 nm of the V‐ATPase inhibitor bafilomycin A1. Translocation to the cytosol was partly inhibited by bafilomycin A1 treatment. g) Time dependency of TAT translocation to the cytosol. Cells were incubated with 200 nm BG‐biotin‐TAT conjugate C for 0, 4, or 24 h (continuous incubation). The cytosolic signal increased with time. h) Concentration dependency of TAT translocation to the cytosol. Cells were incubated for 4 h with BG‐biotin‐TAT conjugate C at the indicated concentrations. The cytosolic signal increased with concentration. i) Comparison of TAT versus TAT‐PEG6‐GFWFG. NG‐SNAP cells were incubated for 4 h at 37 °C with different concentrations of TAT or TAT‐PEG6‐GFWFG. Cytosolic signal is normalized to signal from 25 μm TAT. TAT‐PEG6‐GFWFG has an increased cytosolic translocation capacity compared to TAT at concentrations above 15 μm. Statistical analysis: two‐tailed paired t‐test; * p<0.05; ** p<0.01 and *** p<0.001.
For absolute quantification of cytosolic arrival, the conjugation of BG and biotin reporter moieties was optimized in order to achieve 1:1 molar ratios of both BG and biotin per vector (or per monomer in the case of homopentameric STxB). We synthesized several scaffolds, comprising each BG, biotin, and maleimide for conjugation to the vectors (Scheme 1 and Figure S5–7). All STxB‐based conjugates showed intracellular retrograde trafficking to the Golgi, as for non‐modified STxB (Figure S8). With increasing PEG linker sizes, STxB conjugates C and D showed higher ELISA signal intensity when added directly in cell lysate (Figure 3 a). This was likely due to reduced steric hindrance for streptavidin binding to biotin on STxB pentamer whose BG had already reacted with the SNAP‐tag. The same effect was not observed on the monomeric TAT (Figure 3 b). In contrast, cytosolic arrival was reduced for both STxB and TAT with the increased PEG linker size of scaffold D (Figure 3 c,d). An effect of long PEG linkers on cytosolic arrival was previously reported for the cell‐penetrating peptide TAT‐PEG‐GWWG: PEG12 or PEG18 diminished the translocation efficiency when compared to PEG6. [18] In order to balance sensitivity and translocation efficiency, we chose to work with the maleimide‐BG‐biotin scaffold C. The resulting STxB conjugate C had a membrane translocation capacity similar to conjugate A, for which BG and biotin were separately coupled to STxB.
Scheme 1.
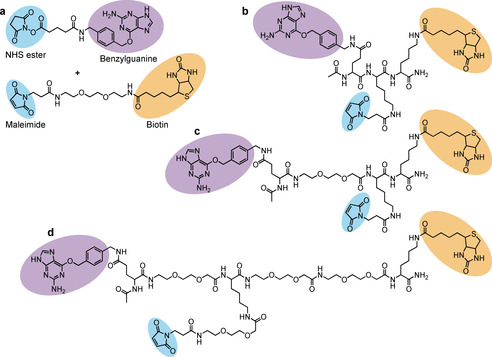
Different strategies used for biotin and BG conjugation to vectors; a) Biconjugation method: Use of commercial NHS ester—BG for conjugation to amines of the vector, and maleimide‐biotin for conjugation to thiol. b–d) Monoconjugation method: Synthesized maleimide‐benzylguanine‐biotin molecules for conjugation to thiol of the vector. Different PEG linker sizes were tested in order to avoid steric hindrance between vector, the BG reaction with the SNAP‐tag, and streptavidin binding to the biotin moiety.
Figure 3.
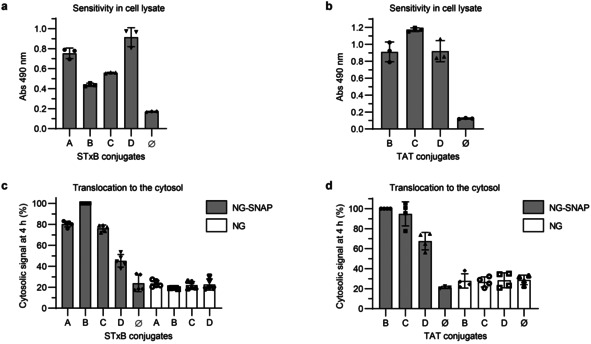
Evaluation of the different STxB and TAT conjugates for the Cyto‐SNAP assay. a,b) Comparison of the ELISA signal obtained with the different STxB and TAT conjugates directly added into cell lysate. 40 pm of each STxB conjugate or 100 pm of each TAT conjugate were added to NG‐SNAP cell lysate and incubated for 1 h at 37 °C for the SNAP reaction to occur before mNeonGreen immunoprecipitation and ELISA development on beads. c,d) Comparison of cytosolic arrival signal from the different STxB conjugates (40 nm) or TAT conjugates (200 nm) after 4 h incubation at 37 °C with NG or NG‐SNAP HeLa cells.
The choice of lysis conditions was also critical for quantification to ensure that vectors are extracted from membranes with which they may remain associated even after translocation to the cytosolic compartment (see ref. [14] for STxB). We used high salt concentrations and sonication as recommended for TAT extraction, [21] and optimized the choice of detergent to achieve complete STxB extraction (Figure S9a). These conditions remained fully compatible with the requirement for SNAP‐tag reaction and immunoprecipitation (Figure S9b,c).
For absolute quantification, a standard curve is essential. For this, we have devised a simple approach (Figure S10): Known amounts of BG and biotin‐tagged vector were mixed into NG‐SNAP cell lysate, leading to complete reaction with the SNAP‐tag protein of the lysate. As shown in Figure 1 e, low picomolar sensitivity was reached with this approach. Using the standard curve, the amount of vector translocated to the cytosol and the total amount of vector associated with NG‐SNAP cells were then determined (Figure S10). For the total cell‐associated amount, the SNAP‐tag quenching step was omitted, such that BG and biotin‐tagged vector molecules fully reacted with unquenched SNAP‐tag in the cell lysate. Of note, immunoprecipitation and subsequent ELISA steps were performed for all samples in parallel under identical conditions such that absorbance readings could be compared directly.
Using this approach, we have for the first time determined the absolute amount of STxB molecules that reach the cytosol of a mammalian cell line—here, NG‐SNAP HeLa cells that were continuously incubated for 4 h at 37 °C with 40 nm STxB. We found that 23 000 STxB molecules were translocated to the cytosol, which corresponded to 0.5 % of total cell‐associated STxB (Table 1). For TAT, 0.1 % of total cell‐associated molecules reached the cytosol upon 4 h incubation at 200 nm (Table 1). The translocation efficiency is concentration‐dependent: an increase in translocation efficiency at higher concentrations was observed for TAT (Table 1). The percentages however remain low, thereby confirming the notion that endosomal escape is in general very inefficient. First‐generation cell‐penetrating peptides such as TAT have indeed been described to mostly remain entrapped in endolysosomal compartments. [3] The second generation TAT‐PEG6‐GFWFG has a translocation efficiency similar to TAT at concentrations up to 10 μm (Table 1). In contrast, its translocation efficiency increased 9‐fold compared to TAT at 20 μm (Table 1). This effect is in line with the previously reported numbers [18] (i.e., 2.3‐fold change at 15 μm and 10.3‐fold change at 30 μm), further validating the Cyto‐SNAP assay.
Table 1.
Absolute quantification of cytosolic arrival after 4 h incubation of STxB‐BG‐biotin conjugate C, BG‐biotin‐TAT conjugate C, or BG‐Biotin‐TAT‐PEG6‐GFWFG conjugate C on NG‐SNAP cells.[a]
|
Vector |
Incubation concentration [μm] |
Total number of molecules/cell |
Number of cytosolic molecules/cell |
% of cytosolic molecules |
|---|---|---|---|---|
|
STxB |
0.04 |
4.9×106 (σ=1.4×106) |
2.3×104 (σ=9.2×103) |
0.47 |
|
|
|
|
|
|
|
TAT |
0.2 |
3.2×107 (σ=9.6×106) |
2.7×104 (σ=7.1×103) |
0.08 |
|
10 |
1.1×108 (σ=1.1×107) |
4.2×105 (σ=1.7×104) |
0.38 |
|
|
20 |
1.1×108 (σ=2.4×107) |
7.3×105 (σ=9.1×104) |
0.66 |
|
|
|
|
|
|
|
|
TAT‐PEG6‐GFWFG |
0.2 |
3.2×107 (σ=5.6×106) |
1.8×104 (σ=3.1×103) |
0.06 |
|
10 |
7.4×107 (σ=2.9×106) |
4.0×105 (σ=1.1×105) |
0.54 |
|
|
20 |
1.2×108 (σ=1.6×107) |
7.4×106 (σ=4.3×106) |
6.2 |
[a] As STxB is a homopentamer, it carries up to 5 substitutions per STxB molecule. A 5‐times higher TAT concentration (0.2 μm) compared to STxB (0.04 μm) was chosen to compare their drug delivery potential from the same drug concentration. Three independent experiments were performed at each concentration.
The Cyto‐SNAP assay is optimized to allow for the absolute quantification of minute amounts of vector molecules that have translocated to the cytosol. The reporter moieties, BG and biotin, are small, which limits their impact on the membrane translocation process. The assay is robust and performed with standard laboratory equipment. The current proof‐of‐concept study was performed with 2 vectors, TAT and STxB, that have very different ways to interact with membranes of target cells. We expect that the Cyto‐SNAP assay will be of use to study the cytosolic delivery of therapeutic macromolecules in general, such as peptides, [22] proteins [23] or oligonucleotides, [24] either free or encapsulated into liposomes, [25] synthetic nanocarriers[ 26 , 27 , 28 ] or even the cellular entry of viruses. [29] When transposed to a high‐throughput format, the assay may also be used to screen for modifiers of the membrane translocation process, which would not only hold the promise for groundbreaking discoveries in fundamental research on the enigmatic membrane translocation process, but would also open new avenues for therapeutic macromolecule delivery.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge Alison Forrester for revision of the manuscript, Marine Ghazarian, Pascal Kessler, Gilles Mourier, and Denis Servent from SIMoS, CEA, for Cys‐TAT synthesis, Raphaël Rodriguez's team from Institut Curie for the use of various instruments, and U830 INSERM (Institut Curie) for the use of the Bioruptor® Pico sonicator. We acknowledge the Cell and Tissue Imaging (PICT‐IBiSA) and Nikon Imaging Centre, Institut Curie, member of the French National Research Infrastructure France‐BioImaging (ANR10‐INBS‐04) for microscopy use, and the Antibody (Table‐IP) and Cytometry platforms from Institut Curie for their assistance. This work received grant support from the Institut National du Cancer (n° 2019‐1‐PLBIO‐05‐1), Agence National de la Recherche (ANR‐16‐CE23‐0005‐02, ANR‐19‐CE13‐0001‐01), Fondation pour la Recherche Médicale (EQU202103012926), Mizutani Foundation for Glycosciences (reference n° 200014), Institut Carnot (Synthetic STxB program). A.B. was funded by the French Ministry of Higher Education and Research (AMX funding) and La Ligue Nationale Contre le Cancer, and received support from “Frontières de l'Innovation en Recherche et Éducation” (FIRE) Doctoral School—Bettencourt Program. M.L. was funded by the ITN ProteinConjugates European Union program H2020‐MSCA‐ITN‐2015 and by La Ligue Nationale Contre le Cancer. J.H. was funded by the Fondation pour la Recherche Médicale (FRM). The Johannes team is member of Labex Cell(n)Scale (ANR‐11‐LABX‐0038) and Idex Paris Sciences et Lettres (ANR‐10‐IDEX‐0001‐02).
M. Lucchino, A. Billet, S.-K. Bai, E. Dransart, J. Hadjerci, F. Schmidt, C. Wunder, L. Johannes, Angew. Chem. Int. Ed. 2021, 60, 14824.
Contributor Information
Christian Wunder, Email: Christian.Wunder@curie.fr.
Ludger Johannes, Email: Ludger.Johannes@curie.fr.
References
- 1. Dowdy S. F., Nat. Biotechnol. 2017, 35, 222–229. [DOI] [PubMed] [Google Scholar]
- 2. Smith S. A., Selby L. I., Johnston A. P. R., Such G. K., Bioconjugate Chem. 2019, 30, 263–272. [DOI] [PubMed] [Google Scholar]
- 3. Erazo-Oliveras A., Muthukrishnan N., Baker R., Wang T.-Y., Pellois J.-P., Pharmaceuticals 2012, 5, 1177–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deprey K., Becker L., Kritzer J., Plückthun A., Bioconjugate Chem. 2019, 30, 1006–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peraro L., Deprey K. L., Moser M. K., Zou Z., Ball H. L., Levine B., Kritzer J. A., J. Am. Chem. Soc. 2018, 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peier A., Ge L., Boyer N., Frost J., Duggal R., Biswas K., Edmondson S., Hermes J. D., Yan L., Zimprich C., Sadruddin A., Kristal Kaan H. Y., Chandramohan A., Brown C. J., Thean D., Lee X. E., Yuen T. Y., Ferrer-Gago F. J., Johannes C. W., Lane D. P., Sherborne B., Corona C., Robers M. B., Sawyer T. K., Partridge A. W., ACS Chem. Biol. 2021, 16, 293–309. [DOI] [PubMed] [Google Scholar]
- 7. Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., Zhu J., Darzins A., Klaubert D. H., Bulleit R. F., Wood K. V., ACS Chem. Biol. 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- 8. Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stöter M., Epstein-Barash H., Zhang L., Koteliansky V., Fitzgerald K., Fava E., Bickle M., Kalaidzidis Y., Akinc A., Maier M., Zerial M., Nat. Biotechnol. 2013, 31, 638–646. [DOI] [PubMed] [Google Scholar]
- 9. Wittrup A., Ai A., Liu X., Hamar P., Trifonova R., Charisse K., Manoharan M., Kirchhausen T., Lieberman J., Nat. Biotechnol. 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K., Nat. Biotechnol. 2003, 21, 86–89. [DOI] [PubMed] [Google Scholar]
- 11. Shaner N. C., Lambert G. G., Chammas A., Ni Y., Cranfill P. J., Baird M. A., Sell B. R., Allen J. R., Day R. N., Israelsson M., Davidson M. W., Wang J., Nat. Methods 2013, 10, 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zou L., Peng Q., Wang P., Zhou B., J. Membr. Biol. 2017, 250, 115–122. [DOI] [PubMed] [Google Scholar]
- 13. Johannes L., Römer W., Nat. Rev. Microbiol. 2010, 8, 105–116. [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Castillo M. D., Tran T., Bobard A., Renard H.-F., Rathjen S. J., Dransart E., Stechmann B., Lamaze C., Lord M., Cintrat J.-C., Enninga J., Tartour E., Johannes L., J. Cell Sci. 2015, 128, 2373–2387. [DOI] [PubMed] [Google Scholar]
- 15. Yang L., Kindt J. T., J. Phys. Chem. B 2016, 120, 11740–11750. [DOI] [PubMed] [Google Scholar]
- 16. Cordeiro R. M., J. Phys. Chem. B 2018, 122, 6954–6965. [DOI] [PubMed] [Google Scholar]
- 17. Römer W., Berland L., Chambon V., Gaus K., Windschiegl B., Tenza D., Aly M. R. E., Fraisier V., Florent J.-C., Perrais D., Lamaze C., Raposo G., Steinem C., Sens P., Bassereau P., Johannes L., Nature 2007, 450, 670–675. [DOI] [PubMed] [Google Scholar]
- 18. Lönn P., Kacsinta A. D., Cui X.-S., Hamil A. S., Kaulich M., Gogoi K., Dowdy S. F., Sci. Rep. 2016, 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R., Brock R., Traffic Cph. Den. 2007, 8, 848–866. [DOI] [PubMed] [Google Scholar]
- 20. Ruseska I., Zimmer A., Beilstein J. Nanotechnol. 2020, 11, 101–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Illien F., Rodriguez N., Amoura M., Joliot A., Pallerla M., Cribier S., Burlina F., Sagan S., Sci. Rep. 2016, 6, 36938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qian Z., Rhodes C. A., McCroskey L. C., Wen J., Appiah-Kubi G., Wang D. J., Guttridge D. C., Pei D., Angew. Chem. Int. Ed. 2017, 56, 1525–1529; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1547–1551. [Google Scholar]
- 23. Herce H. D., Schumacher D., Schneider A. F. L., Ludwig A. K., Mann F. A., Fillies M., Kasper M.-A., Reinke S., Krause E., Leonhardt H., Cardoso M. C., Hackenberger C. P. R., Nat. Chem. 2017, 9, 762–771. [DOI] [PubMed] [Google Scholar]
- 24. Setten R. L., Rossi J. J., Han S.-P., Nat. Rev. Drug Discovery 2019, 18, 421–446. [DOI] [PubMed] [Google Scholar]
- 25. Filipczak N., Pan J., Yalamarty S. S. K., Torchilin V. P., Adv. Drug Delivery Rev. 2020, 156, 4–22. [DOI] [PubMed] [Google Scholar]
- 26. Dahlman J. E., Barnes C., Khan O. F., Thiriot A., Jhunjunwala S., Shaw T. E., Xing Y., Sager H. B., Sahay G., Speciner L., Bader A., Bogorad R. L., Yin H., Racie T., Dong Y., Jiang S., Seedorf D., Dave A., Singh Sandhu K., Webber M. J., Novobrantseva T., Ruda V. M., Lytton-Jean A. K. R., Levins C. G., Kalish B., Mudge D. K., Perez M., Abezgauz L., Dutta P., Smith L., Charisse K., Kieran M. W., Fitzgerald K., Nahrendorf M., Danino D., Tuder R. M., von Andrian U. H., Akinc A., Panigrahy D., Schroeder A., Koteliansky V., Langer R., Anderson D. G., Nat. Nanotechnol. 2014, 9, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitchell M. J., Billingsley M. M., Haley R. M., Wechsler M. E., Peppas N. A., Langer R., Nat. Rev. Drug Discovery 2020, 19, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng Q., Wei T., Farbiak L., Johnson L. T., Dilliard S. A., Siegwart D. J., Nat. Nanotechnol. 2020, 15, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Staring J., Raaben M., Brummelkamp T. R., J. Cell Sci. 2018, 131, jcs216259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary