

Abstract
Targeting protein‐protein interactions (PPIs) with small‐molecule inhibitors has become a hotbed of modern drug development. In this review, we describe a new class of PPI inhibitors that block menin from binding to MLL proteins. Menin is encoded by the MEN1 tumor suppressor, but acts as an essential cofactor for MLL/KMT2A‐rearranged leukemias. The most promising menin‐MLL inhibitors belong to the thienopyrimidine class and have recently entered phase I/II clinical trials for treating acute leukemias characterized by MLL/KMT2A translocations or NPM1 mutations. As single agents, thienopyrimidine compounds eradicate leukemia in a xenograft models of primary leukemic cells belonging to the MLL‐rearranged or NPM1‐mutant subtypes. These compounds are well tolerated with few or no side effects, which is remarkable given the tumor‐suppressor function of menin. The menin‐MLL inhibitors highlight how leukemia patients could benefit from a targeted epigenetic therapy with novel PPI inhibitors obtained by directed chemical evolution.
Keywords: acute leukemias, chromatin regulation, menin, MLL, thienopyrimidines
Menin inhibition: This review gives a background on the MEN1 gene and its context‐dependent dual role as a tumor suppressor for endocrine tissues and as a promoter for certain acute leukemias. We discuss the functions of wild‐type MLL and oncogenic MLL fusion proteins. Finally, we detail the discovery and development of menin‐MLL interaction inhibitors in acute leukemias.

1. Introduction to Menin‐MLL PPI Inhibitors
Protein‐protein interactions (PPIs) are abundant in human proteomes and they play crucial roles in the regulation of gene transcription and chromatin function. These regulatory pathways are directly involved in many human pathologies including several types of cancers and immunological or neurodegenerative diseases, which raised the prospects of targeting transcription and chromatin PPIs by tailored chemical probes. However, due to their large and flat interaction surface areas, PPIs were believed for many years to be unsuitable for the development of small molecule inhibitors. This paradigm was challenged by several successes including natural compounds like rapamycin and cyclosporin. This revealed that, in principle, PPI surfaces are “druggable”. These surfaces can center around so‐called “hotspots” conferring most of the binding energy. [1] Interaction hotspots typically cover a surface area compatible with small molecule binding, tend to be hydrophobic and display conformational flexibility. These molecular features have motivated the many discovery and development programs for small‐molecule PPI inhibitors of today.
Computational, biophysical and chemical approaches such as fragment‐based lead discovery facilitated the development of potent PPI inhibitors. Several inhibitors have now reached clinical trials or have already been approved for clinical use. To exemplify this, anti‐apoptotic BCL family members are targeted by small molecule inhibitors called BH3 mimetics (e. g., ABT‐263, ABT‐199, WEHI‐539 and UMI‐77). ABT‐199 (Venetoclax) is now used in the clinic to treat various hematological malignancies. [2] An epigenetic class of PPI inhibitors targets the binding of bromodomains (BrDs) to acetylated lysines on histones. This post‐translational modification is directly involved in the regulation of transcription and chromatin function. BrD inhibitors (e. g., I‐BET762, CPI‐0610, OTX15, ABBV‐075 and ZEN‐2906) can show a remarkable specificity and in vivo efficiency. Several of the BrD inhibitors have entered clinical trials in cancer patients. [3] A novel class of PPI inhibitors, which is the subject of this review, is represented by small molecules targeting the interaction of the menin tumor suppressor with the MLL (KMT2A, also known as ALL1, CXXC7, HRX or HTRX) protein, which is essential for MLL‐rearranged (MLLr) leukemias. These aggressive leukemias represent subsets of acute myeloid leukemias (AML) and acute lymphoid leukemias (ALL), which harbor reciprocal translations of the MLL gene on chromosome 11q23 with a variety of other translocation partner genes (TPGs; e. g., AF4, AF9, ENL, AF10) on other chromosomes (Figure 1). The resultant MLL fusion proteins (MLL FP) act as dominant oncogenes for AML and ALL. Treatment outcomes remain poor with event‐free survival rates of ∼50 % for pediatric and adult MLLr patients. [4]
Figure 1.
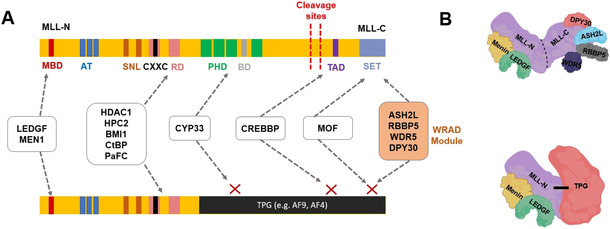
Schematic structures of A) wild‐type MLL protein and B) MLL FPs. The functions of the MLL‐N fragment are described in the text. MLL‐C includes a SET domain, which mediates methylation of histone H3K4 at target gene promoters and binds the WRAD module (ASH2 L, WDR5, RBBP5 and DPY30). MLL FP adopts novel functions from the TPG, but also preserves some MLL functions. MBD: Menin‐binding domain, AT: AT hooks, SNL: speckled nuclear localization domains, RD: repression domains, PHD: PHD fingers, BD: bromodomain, TAD: transactivation domain. Note that the bars are not drawn to scale. Adapted from ref. [58].
MLL is the catalytic subunit of the MLL1 histone methyltransferase complex. The enzymatic activity is specific for Lys4 of histone H3 (H3K4) and located in the C‐terminal SET domain, which is not retained in MLL FPs. The molecular mechanism for the oncogenic activity of MLL FPs involves their interaction with chromatin‐associated proteins, which include menin, LEDGF (official name PSIP1), DOT1L and members of the transcription elongation complexes. The interaction of MLL FPs with menin and indirectly with the LEDGF/PSIP1 protein has been shown to be critical for the oncogenic activity of MLL FPs. [5] Recent work with a novel class of PPI inhibitors targeting the menin‐MLL interaction bears great promise for improved clinical treatment of MLLr leukemias but also of NPM1‐mutated leukemias characterized by aberrant cytoplasmic retention of the encoded nucleophosmin.[ 6 , 7 , 8 ] In this review, we shall discuss the development and findings with this class of menin‐MLL PPI inhibitors. We shall provide a solid background of menin as a tumor suppressor in familial and sporadic forms of endocrine cancer and how this can be hijacked by MLL FPs. Discovery and developments of menin inhibitors will be described and we shall end with an outlook on the future challenges in basic and clinical research to provide a better molecular understanding of targeting menin in various forms of cancer.
2. The MEN1 Gene: A Tumor Suppressor for Endocrine Tissue but a Tumor Promotor for MLLr
The transforming ability of MLL FPs in acute leukemias relies on their interaction with the menin protein. [9] Menin is an integral subunit of MLL1, MLL2 and MLL FP complexes, which interact through two short N‐terminal sequences (Figure 1): menin binding motif 1 (MBM1) and 2 (MBM2), which encompass the RWRFP sequence. [9] MBM1 possesses higher affinity to menin in comparison to MBM2 (K D values of 56 nM and 1 μM, respectively). Both motifs bind in a competitive manner indicating that they use the same binding pocket of menin. Both MBM1 and MBM2 are crucial for the oncogenic activities of MLL FPs and target gene regulation. [10]
The menin protein is encoded by the MEN1 gene, which was identified in 1997 as the tumor suppressor gene of the familial multiple endocrine neoplasia type 1 (MEN1) syndrome. [11] MEN1 patients are predisposed to develop tumors with a high frequency in the parathyroid glands, which are concomitant but at lower frequencies with neoplasias in the pancreas, and pituitary gland. [12] Female MEN1 patients have an elevated risk of breast cancer development. [13] In addition, somatic MEN1 gene mutations have been observed in a large proportion of sporadic non‐functioning, pancreatic neuroendocrine tumors. [14]
MEN1 is a prototypical tumor suppressor gene as MEN1 patients display loss of heterozygosity (LOH) at the MEN1 locus. [15] Other than by LOH, inactivation of second allele can also be achieved by somatic mutations [16] or post‐transcriptional decrease in menin levels via microRNAs. [17] Germline MEN1 mutations are invariably loss‐of‐function mutations scattered over its ten exons or in pre‐mRNA splice sites. [18] Frameshift and nonsense mutations constitute the majority of mutations, which mostly result in premature stop codons leading to truncated proteins susceptible to degradation and to truncated mRNAs suppressed by nonsense‐mediated mRNA decay.[ 18 , 19 ] Missense mutations (∼20 % of the total) are often located in hydrophobic core of menin resulting in unstable proteins subjected to ubiquitin‐dependent proteasomal degradation. [20] The small number of mis‐sense mutations in the surface‐exposed residues of menin are most informative. In analogy with the other mutants, this group of mutations might result in the loss of menin protein function(s).
Menin is ubiquitously expressed in both endocrine and non‐endocrine tissues, [21] but its tumor‐suppressor function is exclusive for endocrine organs. [22] The 610‐residue menin protein resides predominantly in the nucleus and it has been involved in various DNA‐mediated processes such as gene transcription, DNA repair and DNA replication. For gene transcription menin can act both as an activator and a repressor. And as it lacks a DNA binding domain, menin relies on bona fide transcription factors to achieve gene specificity. Indeed, protein interactions with many DNA sequence‐specific transcription have been reported, [23] including the transcription factors JUND, MYC, NF‐κB, SMAD1, SMAD3, SMAD5, TCF3 and the Forkhead box proteins, FOXN1, FOXO1 and FOXA1. Ligand‐dependent interactions with receptors for estrogens, vitamin D3 and glucocorticoids and with PPARγ have also been found.[ 23 , 24 ] Interactions with these activated nuclear receptors depend on a conserved LXXLL motif of menin. [24] Interestingly, menin can both act as an adapter for estrogen receptor 1 (ESR1)‐regulated transcription and as a direct activator of ESR1 transcription.[ 24 , 25 ] Many of the transcription factor interactions with menin have been found in targeted assays. Unbiased quantitative proteomics in HeLa cervical carcinoma cells showed, that only JUND‐containing AP1 and ATF transcription factors form stable menin complexes.[ 26 , 27 ] Interestingly, menin only interacts with the long JUND isoform to mediate transcriptional repression of JUND target genes like hTERT.[ 27 , 28 ]
A breakthrough in menin research came from the Meyerson and Cleary laboratories, which reported that menin interacts with the MLL2 (official name KMT2B) protein in 293 human embryonic kidney cells [29] and with MLL (KMT2A) in K652 erythroleukemia cells. [30] These results were confirmed and extended by quantitative proteomics showing that menin is an integral but sub‐stoichiometric subunit of the MLL1 and MLL2 complexes and not of the other SET1/MLL complexes. [26] LEDGF was also confirmed as a menin interactor, but other chromatin regulators like SIN3A, SIRT1, PRMT5, HDACs or SUV39H1 were not stable interactors in this proteomic screen.[ 5 , 26 ] Structural studies detailed how the hydrophobic pocket of menin interacts with MBM1 of MLL (Figure 2A). [31] The MBM1 motif (RWRFPARP) adopts an almost cyclical form to display hydrophobic interactions with Tyr 319, Tyr323 and M278 and electrostatic interactions of acidic residues (D285/E288/E290 and E366/D370) with the several arginines of MBM1.[ 32 , 33 , 34 ] In addition, mutation of H139, A182 and C241 indicated their involvement in MLL‐MBM1 interaction.
Figure 2.
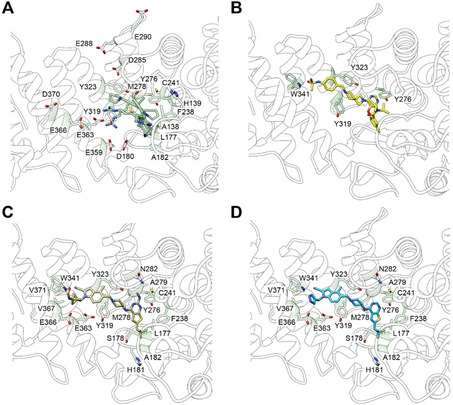
Structural models for menin bound A) to the MBM1 peptide of MLL or to analogs of menin‐MLL PPI inhibitors now in clinical trials: B) VTP‐50469, C) MI‐503, and D) MI‐3454. The menin residues of the central cavity contacting the MLL peptide or PPI inhibitors are indicated by numbers. The structures are based on PDB IDs: 3 U85, 6PKC, 6O5I and 4X5Y.
The prevalent model is that menin acts as an epigenetic activator of transcription by recruiting the MLL1 and MLL2 complexes to deposit the activating H3K4me3 mark.[ 24 , 29 ] Menin‐MLL target genes are within the HOX clusters,[ 30 , 35 ] MEIS1, PBX1, [36] the cyclin‐dependent kinase (CDK) inhibitors CDKN1B and CDKN2D, [37] and estrogen‐regulated genes. [23] Furthermore, menin‐MLL1/2 complexes also activate the Wnt‐signaling pathway by changing chromatin accessibility of several WNT genes [38] and the expression of the GATA3 [39] and MNX1/HLXB9 [40] transcription factors. Menin links the MLL1 and MLL2 complexes to LEDGF, which interacts with methylated H3K36 nucleosomes through its PWWP domain. [41] Thereby, LEDGF is crucial for stable interaction of MLL1, MLL2 complexes as well as MLL FPs with target chromatin to activate transcription of HOX and CDKN genes. [5] HOXA9 transcription is reduced by menin mutations affecting the LEDGF interaction. [5] Intact LEDGF binding is indispensable for leukemic transformation driven by MLLr.[ 5 , 42 ] An artificial construct tethering the PWWP domain of LEDGF to MLL‐ENL can transform myeloid progenitors regardless of menin. This would imply that the PWWP domain of LEDGF and CXXC domain [43] of MLL proteins are the minimum targeting module for transcription activation by MLL. [5] In this scenario, chromatin with unmethylated CpGs and H3K36me2/3 mark would be the major targets for MLL FP complexes.
3. The Normal Functions of the Mixed‐Lineage Leukemia Gene
MLL is ubiquitously expressed in diverse tissue types including, colon, lungs, spleen, liver, brain, thyroid, kidneys, heart, testes. The MLL protein is cleaved into the MLL‐N and MLL‐C polypeptides, [44] which associate with each other (Figure 1). MLL‐N harbors the binding motifs for menin protein and LEDGF [5] and also contains a CXXC domain mediating binding to nonmethylated CpG DNA sites. [45] Sequences on both side of CXXC are crucial for binding to polymerase‐associated factor complex (PAFc), which facilitates recruitment of MLL to target gene loci. [46] Other regions of MLL‐N, namely a bromodomain (BrD) and plant homology domains (PHDs) are responsible for interaction with acetylated lysines or H3K4me2/3 modified histones, respectively. [47] MLL‐N also possess AT‐hook motifs participating in DNA binding [48] as well as the homodimerization‐facilitating FYRN domain. [49] MLL‐C consists of transactivator domain (TAD) and functional SET [Su(Var)3–9, enhancer‐of‐zeste, trithorax] domain. This domain carries the H3K4 methylation function, [50] which requires integration of the WRAD module (Figure 1).
Similar to MLL FPs, wild‐type MLL regulates expression of genes important for embryonic development [51] and differentiation like the genes in the HOXA cluster (e. g., Hoxa7, Hoxa9 and Hoxa10)[ 30 , 35 ] as well as HOX cofactor genes, MEIS1 and PBX1. [36] HOX gene regulation by MLL and MLL2 requires the menin and LEDGF binding motifs. [42] The HOXA and MEIS1 genes are highly transcribed in hematopoietic stem cells to maintain undifferentiated and proliferative state, whereas their expression decline during differentiation. [52] While MLL expression is crucial for stem cell self‐renewal in adult bone marrow, the gene is dispensable for the mature adult hematopoietic lineages. [53] Mis‐regulation of HOXA and MEIS1 in hematopoietic progenitor cells trigger leukemogenesis [54] and HOXA9 and MEIS1 overexpression can drive the myeloid phenotype in mice. [55]
4. MLL Rearrangements and Oncogenic Fusion Proteins
MLL chromosomal rearrangements are observed frequently in patients with de novo AML, ALL and myelodysplastic syndrome (MDS). [56] AML is characterized by defective differentiation, maturation and accelerated proliferation of cells in the bone marrow and blood, which interferes with normal hematopoiesis. [57] ALL, on the other hand, arises from transformed lymphoid cells. In adult patients MLLr leukemia constitutes 5 % of ALL cases and 5–10 % of AML cases. [58] MLLr is much more prevalent in pediatric leukemias with 70 % of ALL infants [59] and 50–66 % of AML infants. [60] MLLr leukemia patients have a very poor prognosis for recovery. The five‐year event free survival of MLLr infant ALL patients is 34–39 % depending on the treatment regimen and is much worse than for non‐MLLr ALL infants. [61] Similarly, adult MLLr leukemia patients also have a poor prognosis with current treatments.[ 62 , 63 ]
MLL rearrangements are mostly triggered by DNA damage. [64] MLLr can involve exposure to mutagens including ionizing radiation, cytotoxic agents, DNA alkylating and intercalating agents, DNA topoisomerase II inhibitors and/or anti‐tubulin agents used as chemotherapeutic agents for non‐hematological cancers. [65] The non‐homologous end joining DNA repair system predominantly causes reciprocal chromosome translocations, which result in MLL recombination with other genes. Partial tandem duplications (PTD) of MLL are also encountered in AML patients. [66] Although identified rarely in AML, MLL amplification is another mechanism of oncogenic transformation. [67]
MLL FPs resulting from MLLr received broad interest as they are capable of initiating tumorigenesis with none to very few accompanying mutations. [68] The translocation partner bestows novel functions to MLL FPs, which also preserve (some of the) MLL functions in target recognition. In general, MLL FPs constitutively activate transcriptional programs of MLL target genes involved in the hematopoietic program (e. g., HOXA and MEIS1 genes as well as TERT in case of AF4 fusions).[ 69 , 70 , 71 ] The elevated levels of MLL target genes enhance proliferation and block hematopoietic differentiation, which consequently drives leukemia.[ 72 , 73 ] At present, 94 TPGs for MLLr have been identified. [74] These genes regulate diverse cellular processes including DNA binding, RNA decay, chromatin, transcription elongation, cell adhesion, metabolism, apoptosis and the cell cycle. [56] Despite the large number of MLLr TPGs only a subset represents >80 % of all clinical cases. Among those, most frequently encountered TPGs are AF4/AFF1 (36 %), AF9/MLLT3 (19 %), ENL/MLLT1 (13 %), AF6 (4 %), ELL (4 %), and AF10/MLLT10 (8 %; Figure 3). [74] This set of most frequent MLL FPs acts within same protein complex. The leukemogenic activity of the MLL‐AF4, MLL‐AF9 and MLL‐ENL fusions has been recapitulated in various mouse models,[ 75 , 76 ] which also represent valid models for drug testing.
Figure 3.
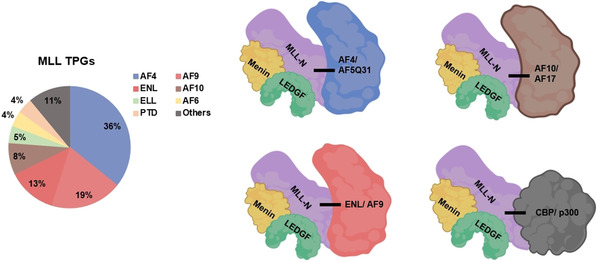
Pie chart of the prevalence of each MLL TPG in MLLr leukemia cases. Abundant fusion partners of MLL explanatory for clinical leukemia cases are AF4/AFF1 (36 %), AF9/MLLT3 (19 %), ENL/MLLT1 (13 %), AF6 (4 %), ELL (4 %), and AF10/MLLT10 (8 %) genes. PTD, partial tandem duplication. The right part of the figure depicts the protein interactions maintained and formed by the most frequent MLL FPs. Adapted from ref. [58].
5. Formation and Function of MLL/AEP, MLL/SEC and MLL/DOT1L Hybrid Complexes
The most prevalent MLLr fusion partners belong to the AF4 family (AF4/AF5q31 and AFF3/LAF4) and ENL family (MLLT1/ENL and MLLT3/AF9). MLL FP members of these families form the AEP (AF4 family/ENL family/P‐TEFb) complex together with the P‐TEFb transcription elongation factor (Figure 3). [77] P‐TEFb can release paused RNA polymerase II (pol II) by phosphorylating its CTD at Ser2, DSIF and NELF. [78] The pol II elongation factor ELL interacts with AF4 family members to form the super‐elongation complex (SEC). [79] This SEC is crucial for MLL FP‐mediated HOX gene expression in leukemic cells. [79]
MLL‐ENL and MLL‐AF9 fusions also interact with disruptor of telomeric silencing 1‐like (DOT1L; Figure 4). [80] DOT1L is a histone H3K79 methyltransferase capable of mono‐, di‐, and tri‐methylation, which may counteract repressive histone deacetylases like SIRT1 to maintain transcriptional activity. [81] The DOT1L link is supported by modulated H3K79me2 levels at MLL target genes after MLL‐ENL expression or mutation.[ 80 , 82 ]
Figure 4.
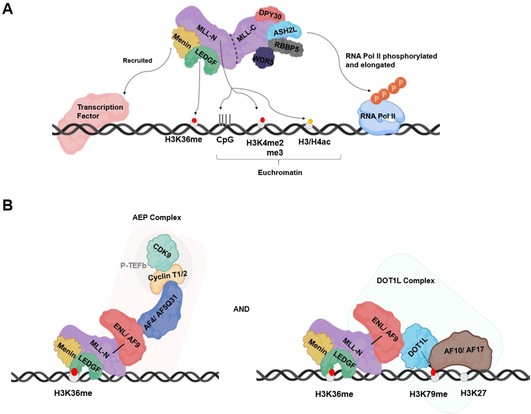
A model for the roles of MLL complexes in transcription regulation and chromatin binding. Recruitment of A) wild‐type MLL and B) AEP and DOT1L complexes by MLL‐AF9 FP. Adapted from ref. [52].
Other MLL FPs like MLL‐AF10 and MLL‐AF17 also nucleate formation of a MLL/DOT1L hybrid complex.[ 83 , 84 ] AF10 contains a “reader” domain PZP that senses and binds unmodified H3K27 to regulate DOT1L‐mediated methylation of the H3K79. [85] The MLL/DOT1L complex also recruits AEP to activate transcription through interactions with AEP component ENL. [52] The interactions of multiple MLL FPs with DOT1L underscore the importance of this histone methyltransferase during hematopoietic transformation, which is supported by observations that genetic perturbation of DOT1L results in loss of the transforming activity of MLL‐AF9, MLL‐AF10 and MLL‐AF4.[ 86 , 87 , 88 ] Not surprisingly, DOT1L inhibitors (e. g., Pinometostat) are promising candidates for therapeutic interventions in MLLr leukemias. [89]
The low‐frequency MLLr partners EP300 [90] and CREBBP [91] encode the p300 and CBP histone acetyltransferases capable of modifying H3K18, H3K27 and H3K36. The YEATS domain of ENL can bind these activating marks, [92] which further recruits AEP and DOT1L to link histone acetylation to oncogenic gene expression in AML. [93] ELL interacts with p300, [94] which represents a similar pathway for MLL‐ELL mediated leukemogenesis.
6. Therapeutic Targeting of the Menin‐MLL Interaction in Acute Leukemias
Targeting menin‐MLL interaction would be general approach to inhibit the leukemogenic activity of MLL FPs regardless of the fusion partner. Indeed, pharmacological inhibition of this interaction abrogates progression of AML in vivo,[ 9 , 10 , 35 ] and now represents a promising strategy for the treatment of MLLr leukemias. Various small molecule inhibitors against menin‐MLL have been developed, which were shown to modulate expression of MLL FP target genes involved in proliferation, differentiation and transformation process. Below, we describe and discuss the discovery and development of several chemical classes of menin‐MLL inhibitors. All available structures of these PPI inhibitors are depicted in Figure 5.
Figure 5.
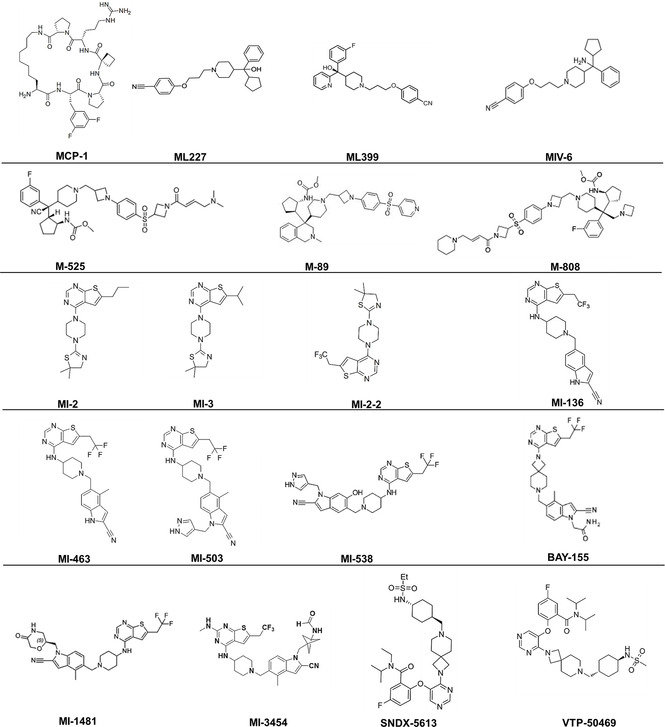
Chemical structures of menin‐MLL PPI inhibitors, as taken from ref. [6], [7], [95]–[108].
6.1. Peptidomimetics
Peptidomimetics are small molecule menin‐MLL interaction inhibitors, which probe the MLL1 binding pocket of menin. MCP‐1 was initially discovered in 2012 through synthesis and screening of optimized macrocyclic peptidomimetics mimicking MBM1 region of MLL‐N, which displays high‐affinity binding to menin. [95] MCP‐1 possessed low‐nanomolar inhibitory potential (IC50=18.6 nM), low molecular weight (815 Da) and high cell‐permeability. Cellular activity of the peptidomimetic inhibitors have not been reported up to date and their further development has been stalled.
6.2. Hydroxy‐/aminomethylpiperidine scaffold
The second class of menin inhibitors are compounds with hydroxy or amino methylpiperidine scaffold. ML227, which is the first with sub‐micromolar inhibitory potential (IC50=390 nM), was discovered via high‐throughput screening of small molecules. [96] However, ML227 displays poor metabolic stability and off target activities. Through optimization the more potent ML399 (IC50=90 nM) with a hydroxymethylpiperidine scaffold was discovered and shown to inhibit cell growth in MLL‐AF9 leukemia cells and to have increased half‐life in vivo. [97] Introduction of an amino moiety to hydroxymethylpiperidine scaffold resulted in the discovery of the MIV‐6 amino methylpiperidine compound, which showed increased inhibitory potential (IC50=67 nM). [98] MIV‐6 downregulated expression of HOXA9 and MEIS1, selectively blocked proliferation and induced differentiation in MLL‐AF9 leukemic cells. [98]
Modification of the MIV‐6 resulted in the covalent menin‐MLL inhibitor M‐525. [99] This compound inhibited menin much more potently (IC50=3 nM), reduced expression of MEIS1, HOXA9 and HOXA11 in tumor tissues and prohibited cell growth in leukemic cell lines carrying distinct MLL FPs. M‐525 demonstrated high cellular selectivity over non‐MLL leukemia cells. M‐89 was developed through structure‐based optimization of MIV‐6. [100] M‐89 was shown to be more potent than MIV‐6 (IC50=25 and 54 nM, respectively, in the MV‐4‐11 and MOLM‐13 cells) and 100 times more selective on MLLr leukemia cells over non‐MLL leukemia cells. Its high cellular selectivity and potency encouraged testing menin‐MLL inhibitors on cell line‐derived xenograft tumor models of mice. Xenograft tumor models rely on the subcutaneous or orthotopic implantation of human tumor cells into immunocompromised mice to examine in vivo potency of drug candidates. M‐89 efficiently downregulated HOXA9 and MEIS1 expression in xenograft tumors and induced apoptosis and differentiation in leukemic cells. By optimization of M‐525, Xu et al. described another covalent inhibitor, M‐808, in 2020. [101] M‐808 had elevated inhibitory potential (IC50=1 and 4 nM in the MV‐4‐11 and MOLM‐13 cell lines, respectively) and blocked proliferation of MLLr leukemic cells. This inhibitor also resulted in partial tumor regression in an AML xenograft mouse model.
6.3. Thienopyrimidines and their derivatives
Thienopyrimidine inhibitors were the first menin‐MLL inhibitors discovered in 2012 and their cellular activity was illustrated in various MLLr leukemia models.[ 102 , 103 ] These compounds were initially identified through a high‐throughput screening of 49 000 small molecules using a fluorescence polarization (FP) assay with a fluorescein‐labeled MBM1 peptide and recombinant menin. Chemical optimization of screen‐derived compounds led to discovery of rigid MI‐2 (IC50=446 nM) and MI‐3 (IC50=648 nM), which bind reversibly to the central cavity of menin and mimic interaction with MBM1 of MLL. MI‐2 and MI‐3 reversed the oncogenic potential of MLL FPs through reducing promoter occupancy and downmodulating HOXA9 and MEIS1, inhibiting proliferation, inducing hematopoietic differentiation and boosting apoptosis of MLLr leukemia cells in vitro. [102] These findings further encouraged development of analogs of the thienopyrimidine class of inhibitors. The more potent analog MI‐2‐2 was obtained through substitution of n‐propyl with trifluoroethyl moiety, which resulted in affinity enhancement (IC50=46 nM) and a four‐fold improvement in cellular activity. [103] MI‐2‐2 strongly reduced Hoxa9 and Meis1 expression resulting in a pronounced hematopoietic differentiation reflected by increased CD11b. Taken together, the initial thienopyrimidine inhibitors were promising with their low molecular weight and high affinity, but the poor pharmacokinetics of these compounds prevented in vivo evaluation and stressed the need for more stable derivatives.[ 103 , 104 ]
Such molecules, which bear an amino‐piperidine linker, were described by Borkin et al. [104] MI‐136 was developed by introduction of cyano‐indole ring and demonstrated increased inhibitory potential on menin‐MLL interaction by a higher affinity for menin (K D=24 nM, IC50=31 nM). MI‐136 was further optimized through substitutions on the indole ring leading to MI‐463 (IC50=15.3 nM) and MI‐503 (IC50=14.7 nM). [104] MI‐503 interacts with the menin‐MLL pocket via hydrogen bonding with Y276, W341 and E366 (Figure 2C). The MI‐436 and MI‐503 compounds are very efficient in promoting differentiation of leukemic blasts in various AML models. Both have anti‐leukemic activities in vivo and they prolong the survival of MLLr leukemic mice. MI‐463 and MI‐503 were also effective against patient xenografts. In addition, these compounds have higher metabolic stability and they do not impair normal murine hematopoiesis, which reduces toxicity concerns and increases the therapeutic window. MI‐503 had profound antileukemic activity in human and murine models of NPM1mut AML, which has mutation‐driven elevated expression of HOX and MEIS1 genes. [105] However, high concentrations (>2 μM) were required to modulate disease‐related gene expression and only modest in vivo effects were observed, which underscored the need for further development.
By substitutions introduced to the indole ring of MI‐136, the hydroxylated derivative MI‐538 was identified. [106] MI‐538 is a stronger inhibitor of menin‐MLL interactions with higher binding affinity (IC50=21 nM, K D=6.5 nM) and demonstrated a more pronounced inhibition of MLL‐AF9‐dependent cell proliferation. The compound has over 100‐fold selectivity of MLL leukemic cells over control cells, increased cell membrane permeability, high oral bioavailability and consequently pronounced in vivo efficacy in a mice xenograft model for AML. Substitution of the amino‐piperidine linker moiety of MI‐503 with suitable spirocyclic amines resulted in the menin inhibitor BAY‐155. This compound displays a higher potency (IC50=8 nM) and three‐ to six‐fold improved inhibition effect on the proliferation of MLLr AML cell lines. BAY‐155 led to more pronounced downregulation of menin‐MLL target genes and reduced tumor volume in xenograft models. This orally bioavailable compound combines a high permeability with a high metabolic stability, which improved in vitro and in vivo bioavailability. All leukemic cell lines tested were sensitive to BAY‐155 and this inhibitor does not have anti‐proliferative effects on breast, prostate or bone cancer cell lines as opposed to MI‐503. [107]
Another thienopyrimidine derivative, namely MI‐1481, has been identified by structure‐based optimization through introduction of saturated six‐membered rings at the indole nitrogen of MI‐463 and MI‐503 compounds. [108] MI‐1481 demonstrated low nanomolar activity (IC50=3.6 nM) and more than ten‐fold improved inhibitory activity over MI‐503 due to polar and hydrophobic interactions with menin. In comparison to MI‐463 and MI‐503, MI‐1481 has profoundly increased activity on MLLr leukemia cells (∼10‐fold) with high selectivity. The compound resulted in downmodulation of HOXA9 and MEIS1 mRNA expression and elevated CD11b expression in AML cell models and in xenograft models of MLLr leukemias.
A novel orally bioavailable, low‐toxicity menin inhibitor, the thienopyrimidine derivative KO‐539 (structure has not been disclosed yet) was developed through high‐throughput screening and chemical optimizations by Kura Oncology in 2018. KO‐539 is very potent growth inhibitor of MLLr leukemic cells (IC50=22 nM). The compound induced differentiation of leukemic cells and provided survival benefit both in AML cell line and in patient‐derived xenografts. KO‐539 earned the FDA Orphan Drug Designation and entered in phase I/II clinical trials for relapsed or refractory AML (NCT04067336). The structurally related analog, MI‐3454 displays sub‐nanomolar inhibitory properties (IC50=0.51 nM) and binds to the menin‐MLL pocket via hydrogen bonds with Y276, W341, and E366 on menin as well as hydrophobic interactions with M278, C241, Y276, and A279 (Figure 2D). Furthermore, the methyl‐bicyclo[1.1.1]pentane group introduced at the indole nitrogen of MI‐3454 is engaged in hydrophobic interactions including V367, V371 and E366, while the terminal amide is involved in hydrogen bonding with E366. [7] MI‐3454 diminished clonogenic ability and induced differentiation of AML cell lines as well as MLLr or NPM1 leukemic cells from AML patients. Through downregulating key regulatory genes (e. g., MEIS1), MI‐3454 blocked leukemia progression in PDX models for MLLr leukemia. This compound was not toxic to mice and did not interfere with normal hematopoiesis, which adds another inhibitor for clinical exploration.
In 2019, another thienopyrimidine derivative, orally bioavailable and selective menin inhibitor, VTP‐50469 (Syndax Pharmaceuticals’ analog SNDX‐5613) was described by Krivtsov et al. [6] VTP‐50469 has nanomolar inhibitory potential (IC50<40 nM) on menin‐MLL interaction and its binding to the menin‐MLL pocket involves H‐bonding with Y276 and W341 and pi‐cation interactions with the Y319 and W323 side chains (Figure 2B). VTP‐50469 showed potent antiproliferative activity on MLLr leukemic cell lines by modulating MEIS1 expression and by inducing differentiation and apoptosis. Interestingly, this inhibitor did not repress expression of the HOXA locus. VTP‐50469 strongly reduced leukemic burden in PDX models of MLLr leukemias (AML or ALL) [6] as well as NPM1c AML. [8] After a three‐week treatment regime, engrafted mice remained disease‐free for longer than 1 year. Encouraged by PDX studies, SNDX‐5613 phase I clinical trials were initiated for treatment of relapsed or refractory acute leukemias, whereas phase II trials are recruiting patients with MLLr AML, NPM1c AML or MLLr ALL (NCT04065399).
7. Summary and Outlook
The continued development of menin‐MLL PPI inhibitors has resulted now in the testing of the most promising candidates in clinical trials for treatment of ALL and AML subtypes. These subtypes are characterized by 11q23 translocations involving the MLL gene or by specific NPM1 mutations. Similar to MLLr leukemic cells, NPM1‐mutant cells display dysregulated HOXA and MEIS1 gene transcription, which can be reverted by effective menin‐MLL inhibitors. While for MLLr, the molecular action of the inhibitors relates to removing the menin cofactor from the dominant MLL FP oncoprotein, their effects in NPM1‐mutant leukemias seems to be linked to inhibiting menin to interact with the wild‐type MLL protein. The latter effects could also indicate that besides blocking menin‐MLL interaction to regulate gene transcription, these inhibitors could affect other aspects of menin biology, like the interactions of menin with (ligand‐dependent) gene‐specific transcription factors or with ubiquitin ligases. A recent study showed that menin‐MLL inhibitors could reduce menin levels by Hsp70/CHIP‐mediated proteasomal degradation. [109] In this respect, it is interesting to note that menin‐MLL inhibitors have been tested in other cancer modalities. For example, it was published that menin is a critical cofactor of androgen receptor (AR) signaling in prostate cancer through direct interactions and MLL recruitment. [110] On the other hand, treatment of mice with menin‐MLL inhibitors did not affect normal hematopoiesis, weight gain and did not reveal any toxicity,[ 6 , 7 ] which is remarkable for a tumor suppressor inhibitor.
Decades of cancer research have shown that a single drug is seldom efficient and that drug resistance develops easily. Despite a remarkable single‐agent activity, [6] there is little reason to assume that treatment with menin‐MLL PPI inhibitors will remain the exception. But when resistance to menin‐MLL inhibitors is evolving in patients, the molecular nature of resistance mechanisms will provide directions for the development of effective drug combination approaches. Candidates to increase clinical effectiveness include inhibitors of MLLr‐relevant pathways (e. g., HDAC, p300/CBP, DOT1L and/or kinase inhibitors) or future inhibitors of menin‐LEDGF interactions. A recent study combined HDAC inhibitor chidamide with MI‐3 to show showed increased inhibition of human MLLr AML cell lines in vitro and in the xenograft model. [111] In addition, synergistic effects of the DOT1L inhibitor EPZ004777 and MI‐2‐2 have been observed in MLLr xenografts. [112]
From our discussions, it is clear that questions on the exact molecular pathway involved in the growth‐suppressing, differentiation‐ and apoptosis‐inducing effects of menin‐MLL inhibitors still remain. With the entrance of the most promising of these PPI inhibitors into clinical trials, research spanning from fundamental mechanism and medicinal chemistry to pharmaceutical development and clinical applications is entering an exciting time. Will the current promises of menin inhibitors bear fruit for the companies involved and, most importantly, improve the life expectancy and quality of life of patients diagnosed with MLLr and NPM1‐mutated leukemias? The MEN1 gene was cloned as a tumor suppressor gene in endocrine tissues in 1997. Who could have thought back then that inhibiting the molecular function of this tumor suppressor would have potential for the clinical treatment of aggressive forms of leukemias? In analogy to Tom Hanks in Forrest Gump: “Biomedical research is like a box of chocolates. You never know what you are gonna get.”
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Ezgi Özyerli Göknar graduated in molecular biology and genetics from Bilkent University and obtained her PhD in cellular and molecular medicine from Koç University in 2019. In her PhD thesis she mainly focused on identification of epigenetic vulnerabilities in glioblastoma multiforme through chemical screens as well as CRISPR‐Cas9 mediated genomic screens. Since 2020, she has been receiving post‐doctoral training from Prof. Marc Timmers at the University of Freiburg and conducting research on Menin‐MLL inhibitors’ mechanism of action to inhibit acute leukemias.

Biographical Information
Sheikh Nizamuddin received his PhD in life sciences from Jawaharlal Nehru University in 2016. As a graduate student at CSIR‐CCMB, he explored the genetic diversity of genes coding for drug‐metabolizing enzymes and identified putative functional novel haplotypes in CYP2C9, which metabolizes range of drugs including phenytoin and warfarin. He joined laboratory of Prof. Marc Timmers in Freiburg as a post‐doctoral fellow in 2018. He is currently working to understand the biology of proteins complexes involved in transcription regulation and chromatin function relevant to cancer.

Biographical Information
Marc Timmers graduated in Chemistry and obtained his PhD from the University of Leiden. He received post‐doctoral training from Prof. Phillip Sharp at MIT in the biochemistry of basal transcription. As an independent researcher at the Utrecht University, he studied the dynamic regulation of transcription, which included discoveries of menin as a coactivator for nuclear receptor‐activated transcription and of the increased breast cancer risk for women from MEN1 families. Since 2017, Marc has been the DKTK Professor of Medical Epigenetics at the University of Freiburg.

Acknowledgements
We apologize to the colleagues, whose primary research data could not be cited due to the lack of space. Clearly, their data made much of the described progress possible. The research of E.O.G. and H.T.M.T. is financially supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through projects 192904750‐SFB 992 and TI688/1‐1. We thank our colleagues Koen Dreijerink (Amsterdam UMC), Laura Pulido Cortes, Simona Capponi and Timothy En Haw Chan for critical reading of the manuscript and all other members of the Timmers lab for discussions. Open access funding enabled and organized by Projekt DEAL.
E. Ozyerli-Goknar, S. Nizamuddin, H. T. M. Timmers, ChemMedChem 2021, 16, 1391.
References
- 1. Arkin M. R., Tang Y., Wells J. A., Chem. Biol. 2014, 21, 1102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gangat N., Tefferi A., Blood Cancer J. 2020, 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alqahtani A., Choucair K., Ashraf M., Hammouda D. M., Alloghbi A., Khan T., Senzer N., Nemunaitis J., Future Sci. OA 2019, 5, FS0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsai C. T., So C. W. E., Oncogene. 2017, 36, 1753–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yokoyama A., Cleary M. L., Cancer Cell 2008, 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krivtsov A. V., Evans K., Gadrey J. Y., et al., Cancer Cell 2019, 36, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klossowski S., Miao H., Kempinska K., et al., J. Clin. Invest. 2020, 130, 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uckelmann H. J., Kim S. M., Wong E. M., Hatton C., Giovinazzo H., Gadrey J. Y., Krivtsov A. V., Rücker F. G., Döhner K., McGeehan G. M., Levine R. L., Bullinger L., Vassiliou G. S., Armstrong S. A., Science 2020, 367, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yokoyama A., Somervaille T. C. P., Smith K. S., Rozenblatt-Rosen O., Meyerson M., Cleary M. L., Cell 2005, 123, 207–18. [DOI] [PubMed] [Google Scholar]
- 10. Caslini C., Yang Z., El-Osta M., Milne T. A., Slany R. K., Hess J. L., Cancer Res. 2007, 67, 7275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandrasekharappa S. C., Guru S. C., Manickam P., et al., Science 1997, 276, 404–407. [DOI] [PubMed] [Google Scholar]
- 12. Thakker R. V., Newey P. J., Walls G. V., Bilezikian J., Dralle H., Ebeling P. R., Melmed S., Sakurai A., Tonelli F., Brandi M. L., J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [DOI] [PubMed] [Google Scholar]
- 13. Dreijerink K. M. A., Goudet P., Burgess J. R., Valk G. D., N. Engl. J. Med. 2014, 371, 583–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiao Y., Shi C., Edil B. H., de Wilde R. F., Klimstra D. S., Maitra A., Schulick R. D., Tang L. H., Wolfgang C. L., Choti M. A., Velculescu V. E., L. A. Diaz Jr , Vogelstein B., Kinzler K. W., Hruban R. H., Papadopoulos N., Science 2011, 331,1199-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Debelenko L. V., Zhuang Z., Emmert-Buck M. R., Chandrasekharappa S. C., Manickam P., Guru S. C., Marx S. J., Skarulis M. C., Spiegel A. M., Collins F. S., Jensen R. T., Liotta L. A., Lubensky I. A., Cancer Res. 1997, 57, 2238–43. [PubMed] [Google Scholar]
- 16. Pannett A. A. J., Thakker R. V., J. Clin. Endocrinol. Metab. 2001, 86, 4371–4. [DOI] [PubMed] [Google Scholar]
- 17. Luzi E., Marini F., Giusti F., Galli G., Cavalli L., Brandi M. L., PLoS One 2012, 7, e39767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemos M. C., Thakker R. V., Hum. Mutat. 2008, 29, 22–32. [DOI] [PubMed] [Google Scholar]
- 19. Zetoune A. B., Fontanière S., Magnin D., Anczuków O., Buisson M., Zhang C. X., Mazoyer S., BMC Genet. 2008, 9, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Canaff L., Vanbellinghen J. F., Kanazawa I., Kwak H., Garfield N., Vautour L., Hendy G. N., J. Clin. Endocrinol. Metab. 2012, 97, E282–E291. [DOI] [PubMed] [Google Scholar]
- 21. Stewart C., Parente F., Piehl F., et al., Oncogene 1998, 17, 2485–93. [DOI] [PubMed] [Google Scholar]
- 22. Gracanin A., Dreijerink K. M. A., Van Der Luijt R. B., Lips C. J. M., Höppener J. W. M., Cancer Res. 2009, 69, 6371–6374. [DOI] [PubMed] [Google Scholar]
- 23. Dreijerink K. M. A., Timmers H. T.M, Brown M., Endocr.-Relat. Cancer 2017, 24, T135–T145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dreijerink K. M. A., Mulder K. W., Winkler G. S., Höppener J. W. M., Lips C. J. M., Timmers H. T. M., Cancer Res. 2006, 66, 9. [DOI] [PubMed] [Google Scholar]
- 25. Dreijerink K. M. A., Groner A. C., Vos E. S. M., Font-Tello A., Gu L., Chi D., Reyes J., Cook J., Lim E., Lin C. Y., de Laat W., Rao P. K., Long H. W., Brown M., Cell Rep. 2017, 18, 2359–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Nuland R., Smits A. H., Pallaki P., Jansen P. W. T. C., Vermeulen M., Timmers H. T. M., Mol. Cell. Biol. 2013, 33, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agarwal S. K., Guru S. C., Heppner C., Erdos M. R., Collins R. M., Park S. Y., Saggar S., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J., Burns A. L., Cell. 1999, 96, 143–152. [DOI] [PubMed] [Google Scholar]
- 28. Lin S. Y., Elledge S. J., Cell 2003, 113, P881–P889. [DOI] [PubMed] [Google Scholar]
- 29. Hughes C. M., Rozenblatt-Rosen O., Milne T. A., Copeland T. D., Levine S. S., Lee J. C., Hayes D. N., Shanmugam K. S., Bhattacharjee A., Biondi C. A., Kay G. F., Hayward N. K., Hess J. L., Meyerson M., Mol. Cell 2004, 13, 587–97. [DOI] [PubMed] [Google Scholar]
- 30. Yokoyama A., Wang Z., Wysocka J., Sanyal M., Aufiero D. J., Kitabayashi I., Herr W., Cleary M. L., Mol. Cell. Biol. 2004, 24, 5639–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang J., Gurung B., Wan B., Matkar S., Veniaminova N. A., Wan K., Merchant J. L., Hua X., Lei M., Nature 2012, 482, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thiel A. T., Huang J., Lei M., Hua X., BioEssays 2012, 34, 771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matkar S., Thiel A., Hua X., Trends Biochem. Sci. 2013, 38, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ehrlich L., Hall C., Meng F., Lairmore T., Alpini G., Glaser S., Gene Expression 2017, 17, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y. X., Yan J., Keeshan K., Tubbs A. T., Wang H., Silva A., Brown E. J., Hess J. L., Pear W. S., Hua X., Proc. Natl. Acad. Sci. USA 2006, 103, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rozovskaia T., Feinstein E., Mor O., Foa R., Blechman J., Nakamura T., Croce C. M., Cimino G., Canaani E., Oncogene 2001, 20, 874–8. [DOI] [PubMed] [Google Scholar]
- 37. Milne T. A., Hughes C. M., Lloyd R., Yang Z., Rozenblatt-Rosen O., Dou Y., Schnepp R. W., Krankel C., LiVolsi V. A., Gibbs D., Hua X., Roeder R. G., Meyerson M., Hess J. L., Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L. S., Kang X., Lu J., et al., EBioMedicine 2019, 39, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Onodera A., Yamashita M., Endo Y., Kuwahara M., Tofukuji S., Hosokawa H., Kanai A., Suzuki Y., Nakayama T., J. Exp. Med. 2010, 207, 2493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shi K., Parekh V. I., Roy S., Desai S. S., Agarwal S. K., Endocr.-Relat. Cancer 2013, 20, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Nuland R., van Schaik F. M., Simonis M., van Heesch S., Cuppen E., Boelens R., Timmers H. M., van Ingen H., Epigenetics Chromatin 2013, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. El Ashkar S., Schwaller J., Pieters T., Goossens S., Demeulemeester J., Christ F., Van Belle S., Juge S., Boeckx N., Engelman A., Van Vlierberghe P., Debyser Z., De Rijck J., Blood 2018, 131, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cierpicki T., Risner L. E., Grembecka J., Lukasik S. M., Popovic R., Omonkowska M., Shultis D. D., Zeleznik-Le N. J., Bushweller J. H., Nat. Struct. Mol. Biol. 2010, 17, 62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yokoyama A., Kitabayashi A. I., Ayton P. M., Cleary M. L., Ohki M., Blood 2002, 100, 3710–8. [DOI] [PubMed] [Google Scholar]
- 45. Ayton P. M., Chen E. H., Cleary M. L., Mol. Cell. Biol. 2004, 24, 10470–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Muntean A. G., Tan J., Sitwala K., Huang Y., Bronstein J., Connelly J. A., Basrur V., Elenitoba-Johnson K. S., Hess J. L., Cancer Cell 2010, 17, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fair K., Anderson M., Bulanova E., Mi H., Tropschug M., Diaz M. O., Mol. Cell. Biol. 2001, 21, 3589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zeleznik-Le N. J., Harden A. M., Rowley J. D., Proc. Natl. Acad. Sci. USA 1994, 91, 10610–10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. García-Alai M. M., Allen M. D., Joerger A. C., Bycroft M., Protein Sci. 2010, 19, 1432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dou Y., Milne T. A., Ruthenburg A. J., Lee S., Lee J. W., Verdine G. L., Allis C. D., Roeder R. G., Nat. Struct. Mol. Biol. 2006, 13, 713–719. [DOI] [PubMed] [Google Scholar]
- 51. Yu B. D., Hess J. L., Horning S. E., Brown G. A. J., Korsmeyer S. J., Nature 1995, 378, 505–8. [DOI] [PubMed] [Google Scholar]
- 52. Takahashi S., Yokoyama A., Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 7. [DOI] [PubMed] [Google Scholar]
- 53. McMahon K. A., Hiew S. Y., Hadjur S., Veiga-Fernandes H., Menzel U., Price A. J., Kioussis D., Williams O., Brady H. J., Cell Stem Cell 2007, 1, 338–45. [DOI] [PubMed] [Google Scholar]
- 54. Kroon E., Krosl J., Thorsteinsdottir U., Baban S., Buchberg A. M., Sauvageau G., EMBO J. 1998, 17, 3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faber J., Krivtsov A. V., Stubbs M. C., Wright R., Davis T. N., van den Heuvel-Eibrink M., Zwaan C. M., Kung A. L., Armstrong S. A., Blood 2009, 113, 2375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meyer C., Kowarz E., Hofmann J., et al., Leukemia 2009, 23, 1490–9. [DOI] [PubMed] [Google Scholar]
- 57. Kawamoto H., Minato N., Int. J. Biochem. Cell Biol. 2004, 36, 1374–9. [DOI] [PubMed] [Google Scholar]
- 58. Winters A. C., Bernt K. M., Front. Pediatr. 2017, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heerema N. A., Sather H. N., Ge J., Arthur D. C., Hilden J. M., Trigg M. E., Reaman G. H., Leukemia 1999, 13, 679–686. [DOI] [PubMed] [Google Scholar]
- 60. Satake N., Maseki N., Nishiyama M., Kobayashi H., Sakurai M., Inaba H., Katano N., Horikoshi Y., Eguchi H., Miyake M., Seto M., Kaneko Y., Leukemia 1999, 13, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 61. Hilden J. M., Dinndorf P. A., Meerbaum S. O., Sather H., Villaluna D., Heerema N. A., McGlennen R., Smith F. O., Woods W. G., Salzer W. L., Johnstone H. S., Dreyer Z., Reaman G. H., Blood 2006, 108, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cox M. C., Panetta P., Lo-Coco F., Del Poeta G., Venditti A., Maurillo L., Del Principe M. I., Mauriello A., Anemona L., Bruno A., Mazzone C., Palombo P., Amadori S., Am. J. Clin. Pathol. 2004, 122, 298–306. [DOI] [PubMed] [Google Scholar]
- 63. Slany R. K., Hematol. Oncol. 2005, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- 64. Reichel M., Gillert E., Nilson I., Siegler G., Greil J., Fey G. H., Marschalek R., Oncogene 1998, 17, 3035–3044. [DOI] [PubMed] [Google Scholar]
- 65. Kayser S., Döhner K., Krauter J., et al., Blood 2011, 117, 2137–45. [DOI] [PubMed] [Google Scholar]
- 66. Schnittger S., Kinkelin U., Schoch C., Heinecke A., Haase D., Haferlach T., Büchner T., Wörmann B., Hiddemann W., Griesinger F., Leukemia 2000, 14, 796–804. [DOI] [PubMed] [Google Scholar]
- 67. Tang G., DiNardo C., Zhang L., Ravandi F., Khoury J. D., Huh Y. O., Muzzafar T., Medeiros L. J., Wang S. A., Bueso-Ramos C. E., Hum. Pathol. 2015, 46, 65–73. [DOI] [PubMed] [Google Scholar]
- 68. Greaves M., Cancer Cell 2015, 27, 433–434. [DOI] [PubMed] [Google Scholar]
- 69. Armstrong S. A., Staunton J. E., Silverman L. B., Pieters R., den Boer M. L., Minden M. D., Sallan S. E., Lander E. S., Golub T. R., Korsmeyer S. J., Nat. Genet. 2002, 30, 41–47. [DOI] [PubMed] [Google Scholar]
- 70. Ferrando A.A., Armstrong S. A., Neuberg D. S., Sallan S. E., Silverman L. B., Korsmeyer S. J., Look A. T., Blood 2003, 102, 262–8. [DOI] [PubMed] [Google Scholar]
- 71. Gessner A., Thomas M., Castro P. G., Büchler L., Scholz A., Brümmendorf T. H., Soria N. M., Vormoor J., Greil J., Heidenreich O., Leukemia 2010, 24, 1751–9. [DOI] [PubMed] [Google Scholar]
- 72. Ayton P. M., Cleary M. L., Genes Dev. 2003, 17, 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong P., Iwasaki M., Somervaille T. C. P., So C. W. E., Cleary M. L., Genes Dev. 2007, 21, 2762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Meyer C., Burmeister T., Gröger D., et al., Leukemia 2018, 32, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Milne T. A., Blood 2017, 129, 2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barabé F., Kennedy J. A., Hope K. J., Dick J. E., Science 2007, 316, 600–604. [DOI] [PubMed] [Google Scholar]
- 77. Yokoyama A., Lin M., Naresh A., Kitabayashi I., Cleary M. L., Cancer Cell 2010, 17, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Peterlin B. M., Price D. H., Mol. Cell 2006, 23, 297–305. [DOI] [PubMed] [Google Scholar]
- 79. Lin C., Smith E. R., Takahashi H., Lai K. C., Martin-Brown S., Florens L., Washburn M. P., Conaway J. W., Conaway R. C., Shilatifard A., Mol. Cell 2010, 37, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kuntimaddi A., Achille N. J., Thorpe J., Lokken A. A., Singh R., Hemenway C. S., Adli M., Zeleznik-Le N. J., Bushweller J. H., Cell Rep. 2015, 11, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen C. W., Koche R. P., Sinha A. U., Deshpande J. A., Zhu N., Eng R., Doench J. G., Xu H., Chu S. H., Qi J., Wang X., Delaney C., Bernt K. M., Root D. E., Hahn W. C., Bradner J. E., Armstrong S. A., Nat. Med. 2015, 21, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Milne T. A., Martin M. E., Brock H. W., Slany R. K., Hess J. L., Cancer Res. 2005, 65, 11367–11374. [DOI] [PubMed] [Google Scholar]
- 83. Okada Y., Feng Q., Lin Y., Jiang Q., Li Y., Coffield V. M., Su L., Xu G., Zhang Y., Cell 2005, 121, 167–78. [DOI] [PubMed] [Google Scholar]
- 84. Zhang H., Zhou B., Qin S., Xu J., Harding R., Tempel W., Nayak V., Li Y., Loppnau P., Dou Y., Min J., Genes Dev. 2018, 32, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen S., Yang Z., Wilkinson A. W., Deshpande A. J., Sidoli S., Krajewski K., Strahl B. D., Garcia B. A., Armstrong S. A., Patel D. J., Gozani O., Mol. Cell 2015, 60, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bernt K. M., Zhu N., Sinha A. U., Vempati S., Faber J., Krivtsov A. V., Feng Z., Punt N., Daigle A., Bullinger L., Pollock R. M., Richon V. M., Kung A. L., Armstrong S. A., Cancer Cell 2011, 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Deshpande A. J., Deshpande A., Sinha A. U., Chen L., Chang J., Cihan A., Fazio M., Chen C. W., Zhu N., Koche R., Dzhekieva L., Ibáñez G., Dias S., Banka D., Krivtsov A., Luo M., Roeder R. G., Bradner J. E., Bernt K. M., Armstrong S. A., Cancer Cell 2014, 26, 896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Krivtsov A. V., Feng Z., Lemieux M. E., Faber J., Vempati S., Sinha A. U., Xia X., Jesneck J., Bracken A. P., Silverman L. B., Kutok J. L., Kung A. L., Armstrong S. A., Cancer Cell 2008, 14, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Daigle S. R., Olhava E. J., Therkelsen C. A., Basavapathruni A., Jin L., Boriack-Sjodin P. A., Allain C. J., Klaus C. R., Raimondi A., Scott M. P., Waters N. J., Chesworth R., Moyer M. P., Copeland R. A., Richon V. M., Pollock R. M., Blood 2013, 122, 1017–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ida K., Kitabayashi I., Taki T., Taniwaki M., Noro K., Yamamoto M., Ohki M., Hayashi Y., Blood. 1997, 90, 4699–4704. [PubMed] [Google Scholar]
- 91. Taki T., Sako M., Tsuchida M., Hayashi Y., Blood 1997, 89, 3945–3950. [PubMed] [Google Scholar]
- 92. Erb M. A., Scott T. G., Li B. E., Xie H., Paulk J., Seo H. S., Souza A., Roberts J. M., Dastjerdi S., Buckley D. L., Sanjana N. E., Shalem O., Nabet B., Zeid R., Offei-Addo N. K., Dhe-Paganon S., Zhang F., Orkin S. H., Winter G. E., Bradner J. E., Nature 2017, 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wan L., Wen H., Li Y., Lyu J., Xi Y., Hoshii T., Joseph J. K., Wang X., Loh Y. H. E., Erb M. A., Souza A. L., Bradner J. E., Shen L., Li W., Li H., Allis C. D., Armstrong S. A., Shi X., Nature 2017, 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Byun J. S., Fufa T. D., Wakano C., Fernandez A., Haggerty C. M., Sung M. H., Gardner K., Nat. Commun. 2012, 3, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou H., Liu L., Huang J., Bernard D., Karatas H., Navarro A., Lei M., Wang S., J. Med. Chem. 2013, 56, 1113–23. [DOI] [PubMed] [Google Scholar]
- 96.J. Manka, R. N. Daniels, E. Dawson, J. S. Daniels, N. Southall, A. Jadhav, W. Zheng, C. Austin, J. Grembecka, T. Cierpicki, C. W. Lindsley, S. R. Stauffer, Bethesda (MD): Probe Reports from the NIH Molecular Libraries Program, 2010.
- 97. Senter T., Gogliotti R. D., Han C., Locuson C. W., Morrison R., Daniels J. S., Cierpicki T., Grembecka J., Lindsley C. W., Stauffer S. R., Bioorg. Med. Chem. Lett. 2015, 25, 2720–2725. [DOI] [PubMed] [Google Scholar]
- 98. He S., Senter T. J., Pollock J., Han C., Upadhyay S. K., Purohit T., Gogliotti R. D., Lindsley C. W., Cierpicki T., Stauffer S. R., Grembecka J., J. Med. Chem. 2014, 57, 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xu S., Aguilar A., Xu T., Zheng K., Huang L., Stuckey J., Chinnaswamy K., Bernard D., Fernández-Salas E., Liu L., Wang M., McEachern D., Przybranowski S., Foster C., Wang S., Angew. Chem. Int. Ed. 2018, 57, 1601–1605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1617–1621. [Google Scholar]
- 100. Aguilar A., Zheng K., Xu T., Xu S., Huang L., Fernandez-Salas E., Liu L., Bernard D., Harvey K. P., Foster C., McEachern D., Stuckey J., Chinnaswamy K., Delproposto J., Kampf J. W., Wang S., J. Med. Chem. 2019, 62, 6015–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xu S., Aguilar A., Huang L., Xu T., Zheng K., McEachern D., Przybranowski S., Foster C., Zawacki K., Liu Z., Chinnaswamy K., Stuckey J., Wang S., J. Med. Chem. 2020, 63, 4997–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Grembecka J., He S., Shi A., Purohit T., Muntean A. G., Sorenson R. J., Showalter H. D., Murai M. J., Belcher A. M., Hartley T., Hess J. L., Cierpicki T., Nat. Chem. Biol. 2012, 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shi A., Murai M. J., He S., Lund G., Hartley T., Purohit T., Reddy G., Chruszcz M., Grembecka J., Cierpicki T., Blood 2012, 120, 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Borkin D., He S., Miao H., et al., Cancer Cell 2015, 27, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kühn M. W., Song E., Feng Z., Sinha A., Chen C. W., Deshpande A. J., Cusan M., Farnoud N., Mupo A., Grove C., Koche R., Bradner J. E., de Stanchina E., Vassiliou G. S., Hoshii T., Armstrong S. A., Cancer Dis. 2016, 6, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Borkin D., Pollock J., Kempinska K., Purohit T., Li X., Wen B., Zhao T., Miao H., Shukla S., He M., Sun D., Cierpicki T., Grembecka J., J. Med. Chem. 2016, 59, 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brzezinka K., Nevedomskaya E., Lesche R., Haegebarth A., Ter Laak A., Fernández-Montalván A. E., Eberspaecher U., Werbeck N. D., Moenning U., Siegel S., Haendler B., Eheim A. L., Stresemann C., Cancers (Basel). 2020, 12, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Borkin D., Klossowski S., Pollock J., Miao H., Linhares B. M., Kempinska K., Jin Z., Purohit T., Wen B., He M., Sun D., Cierpicki T., Grembecka J., J. Med. Chem. 2018, 61, 4832–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wu Y., Doepner M., Hojnacki T., Feng Z., Katona B. W., He X., Ma J., Cao Y., Busino L., Zhou F., Hua X., Am. J. Cancer Res. 2019, 9, 1682–1694. [PMC free article] [PubMed] [Google Scholar]
- 110. Malik R., Khan A. P., Asangani I. A., et al., Nat. Med. 2015, 21, 344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ye J., Zha J., Shi Y., Li Y., Yuan D., Chen Q., Lin F., Fang Z., Yu Y., Dai Y., Xu B., Clin. Epigenet. 2019, 11, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dafflon C., Craig V. J., Méreau H., Gräsel J., Engstler B. S., Hoffman G., Nigsch F., Gaulis S., Barys L., Ito M., Aguadé-Gorgorió J., Bornhauser B., Bourquin J. P., Proske A., Stork-Fux C., Murakami M., Sellers W. R., Hofmann F., Schwaller J., Tiedt R., Leukemia 2017, 31, 1269–1277. [DOI] [PubMed] [Google Scholar]