

Abstract
Objective
Many multiple sclerosis (MS) genetic susceptibility variants have been identified, but understanding disease heterogeneity remains a key challenge. Relapses are a core feature of MS and a common primary outcome of clinical trials, with prevention of relapses benefiting patients immediately and potentially limiting long‐term disability accrual. We aim to identify genetic variation associated with relapse hazard in MS by analyzing the largest study population to date.
Methods
We performed a genomewide association study (GWAS) in a discovery cohort and investigated the genomewide significant variants in a replication cohort. Combining both cohorts, we captured a total of 2,231 relapses occurring before the start of any immunomodulatory treatment in 991 patients. For assessing time to relapse, we applied a survival analysis utilizing Cox proportional hazards models. We also investigated the association between MS genetic risk scores and relapse hazard and performed a gene ontology pathway analysis.
Results
The low‐frequency genetic variant rs11871306 within WNT9B reached genomewide significance in predicting relapse hazard and replicated (meta‐analysis hazard ratio (HR) = 2.15, 95% confidence interval (CI) = 1.70–2.78, p = 2.07 × 10−10). A pathway analysis identified an association of the pathway “response to vitamin D” with relapse hazard (p = 4.33 × 10−6). The MS genetic risk scores, however, were not associated with relapse hazard.
Interpretation
Genetic factors underlying disease heterogeneity differ from variants associated with MS susceptibility. Our findings imply that genetic variation within the Wnt signaling and vitamin D pathways contributes to differences in relapse occurrence. The present study highlights these cross‐talking pathways as potential modulators of MS disease activity. ANN NEUROL 2021;89:884–894
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS). Despite progress in identifying genetic variants associated with disease susceptibility, 1 , 2 , 3 , 4 the genetic drivers of disease severity and heterogeneity in MS remain largely unknown. Relapses are a key feature of relapsing remitting MS and patients with a higher relapse rate have, at the group level, a more severe disease course than patients with a lower relapse rate. 5 Preventing relapses benefits patients immediately and is a common primary outcome measure in clinical trials testing the efficacy of MS disease‐modifying drugs. 5 To our knowledge, only one genomewide association study (GWAS) for relapse risk has been performed previously, reporting an association of genetic variants in the gene LRP2 with relapse risk. 6 This finding has been replicated in a larger, independent cohort. 7 Similar to the early days of disease susceptibility GWASs, the identification of genetic factors controlling disease heterogeneity is still in the initial discovery phase, with novel and strong associations emerging from these studies. 6 , 8 We performed a within‐cases GWAS for relapse hazard in a Belgian cohort, the largest study population to date, with extensive genomewide coverage. For the first time, both common (>5%) and less frequent (2–5%) variants were analyzed. Replication was conducted in an independent, large German cohort of the Technical University of Munich (TUM).
Materials and Methods
Discovery Phase
Study Population
Patients with MS fulfilled McDonald diagnostic criteria 2017 9 and were recruited at the Department of Neurology of the University Hospitals Leuven upon providing written informed consent. The study was approved by the Ethics Committee of the University Hospitals Leuven (S60222). Clinical data, including relapse and treatment history, were collected during clinical follow‐up by the same treating clinician (author B.D.). Relapse was defined as the patient reported symptoms or objectively observed signs typical of an acute inflammatory demyelinating event in the CNS with a duration of at least 24 hours, in the absence of fever or infection, according to the 2017 McDonald criteria. 9 Onset was defined as the occurrence of first symptoms. A total of 506 patients with bout‐onset MS with a median duration of 4 years between disease onset and either the start of any immunomodulatory treatment or the end of follow‐up in untreated patients were included. A description of the study cohort is provided in Table 1.
TABLE 1.
Clinical Description of Study Cohorts
| Characteristics | Discovery cohort | Replication cohort |
|---|---|---|
| (N = 506) | (N = 485) | |
| Gender: % F:M | 70:30 | 67:33 |
| Age at onset, yr: median (IQR) | 31 (24–39) | 33 (27–41) |
| Total relapses before treatment | 1412 | 819 |
| Baseline duration, yr: median (IQR) | 4 (1.35–12.07) | 0.50 (0.25–2.57) |
Baseline = follow‐up time from disease onset prior to any treatment; IQR = interquartile range; N = number of individuals.
Genotyping
Participants of this study were genotyped for 700,078 variants using the Infinium HTS assay on Illumina Global Screening Array BeadChips (version 1.0) in 2 batches as part of genotyping of a larger cohort of individuals, including participants of a companion study published previously 10 (batch 1 = 216 cases, 503 controls; and batch 2 = 477 cases, 96 controls). Genotype calling was conducted in GenomeStudio version 2011.1.
Genotype Quality Control
Genotype quality control (QC) was performed jointly for all individuals. Sample and variant QC were performed with PLINK version 1.09b5.4, 11 as described previously, 10 and following guidelines from Anderson CA et al. 12 For the individual‐level QC, a subset of common, high‐quality variants with a minor allele frequency (MAF) ≥5%, a genotyping success rate ≥98% and a Hardy–Weinberg equilibrium (HWE) test p > 10−6 was used. In total, 14 individuals who showed either a genotype call rate <98% or excess heterozygosity (>3 standard deviations from the sample mean) were removed. The mean homozygosity rate across X chromosome markers was calculated to exclude individuals with discordant sex information (n = 9). The biological relationship of all individuals was verified based on a pairwise identity by descent (IBD) estimate, and duplicate (n = 11) or related (PI‐HAT >0.1875) individuals (n = 12) were removed. For each pair showing cryptic relatedness, the individual with the lower genotyping call rate was removed from further analysis or, for MS cases, the individuals with the shortest follow‐up or the least characterized. Genetic ancestry multidimensional scaling (MDS) components were calculated and plotted combined with the 1,000 Genomes project phase I reference panel 13 to identify individuals with ancestry different from the European cluster (n = 12). For both IBD and MDS analyses, regions of extended linkage disequilibrium (LD) 12 , 14 , 15 were removed, and the dataset was pruned so that no pair of single nucleotide polymorphisms (SNPs) within a given window of 50 kb was correlated (ie, LD r 2 > 0.2). A total of 660 MS cases and 574 controls remained after sample QC.
Variant‐level QC was performed in the cleaned dataset. Alleles were aligned to the National Center for Biotechnology Information (NCBI) build 37 forward strand alleles to match the 1,000 Genomes phase III version 5 imputation haplotype reference panel. Variants with an MAF <1%, a call rate <98% for common (MAF >5%) or <99% for less frequent variants (1% ≤ MAF ≤5%), or a significant deviation from the HWE (HWE test p < 10‐ 6) were excluded (n = 197,497). In addition, the following variants were excluded: A/T and G/C SNPs with an MAF >0.4 (n = 407), SNPs missing in the 1,000 Genomes reference panel (n = 229), sex‐chromosome and mitochondrial SNPs (n = 876), indels (n = 108), SNPs with non‐matching alleles in 1,000 Genomes (n = 495), duplicate SNPs (n = 256), and SNPs showing an allele frequency difference >0.2 with the reference panel (n = 859). Finally, variants with batch effects were removed (n = 61). For this purpose, MS cases and controls were extracted from the cleaned sample set, with the batch number in which they were genotyped as the phenotype. For both MS cases and control individuals, a logistic regression analysis for the genotyping batch was performed. Outlier genetic variants were defined as reaching p < 0.05 (Bonferroni adjusted p value for the number of SNPs) in cases and/or controls. A total of 4 individuals were genotyped in both batch 1 and batch 2. Both batches were merged with PLINK with a report of mismatching non‐missing calls. Outliers were defined as variants appearing different between 2 batches in more than one sample. A total of 487,395 SNPs remained after QC.
Genotype Imputation
Genotypes were pre‐phased with SHAPEIT2 (version 2.12) 16 using the 1,000 Genomes phase III EUR reference panel (October 2014). Imputation was performed using IMPUTE version 2.0 17 , 18 and all 1,000 Genomes populations in approximately 5 Mb chunks (http://dougspeed.com/imputation-regions/). The INFO metric reflects the SNP imputation accuracy. Any missing genotypes for directly genotyped variants (INFO = 1) were filled in with the ‐pgs_miss parameter. The HWE test p value was obtained using QCTOOL version 2. Variants with an imputation INFO metric ≥0.9, an MAF ≥2% (corresponding to an expected minimum of 20 effect alleles), and an HWE test p > 1 × 10−6 were retained for GWAS. The final dataset contained 7,344,935 autosomal variants, including 6,118,705 common (MAF >5%) and 1,226,230 less common variants (MAF 2–5%). Genotype probabilities were converted into minor allele dosages. When <6 individuals showed a dosage >1.5 for a specific variant, all dosages >1 were set to 1. Thereby, we used dominant inheritance models for low frequency variants, comparing carriers with noncarriers. All remaining variants were analyzed using additive models.
Top‐associated SNPs that were directly genotyped were validated by inspection of cluster plots. For imputed SNPs, the discordance rate of the nonreference allele was assessed via Sanger sequencing and TaqMan genotyping. For the low frequency SNP rs11871306, genotyping of imputed calls in the entire discovery cohort was validated using a TaqMan assay (C_1139290_10; Life Technologies) and Sanger sequencing (Forward primer: 5′‐GGGTTATGCACACTCACCCA‐3′, Reverse primer: 5′‐GCCAGGGCAAAAGCCTTAAC‐3′).
GWAS Analysis
The association analysis of genetic variants coded as minor allele dosages and the time to relapses were assessed using a Cox proportional hazards model calculated with the coxph function in the “survival” package in R version 3.4.2. Only relapses at baseline (before treatment initiation) were included. The disease onset was defined as the first relapse, and any subsequent relapse was considered as an event. Each relapse resets the time to zero, as described previously. 6 As censor, either the start of immunomodulatory treatment or – for untreated patients – the end of follow‐up was considered. The different time periods were analyzed separately and adjustment of standard errors (SEs) for clusters of correlated observations (relapses within the same individual) was performed by applying the cluster function in the Cox proportional hazards model and by calculating robust SEs. The hazard ratio (HR) was estimated by considering each time at which an event occurred; the overall HR across the baseline duration was averaged over the event times. 19 Model significance was assessed using the Wald test, not assuming independence of observations within clusters. Likewise, time to treatment initiation, based on clinical assessment by the same treating neurologist, was analyzed with the Cox proportional hazards model, using the end of follow‐up as the censor. We confirmed the proportional hazards assumption with the Schoenfeld residuals test. For plots of the predicted survival curve (time to relapse), the Cox proportional hazards model was fitted using genotypes determined via TaqMan and Sanger sequencing. To estimate the median time to relapse, corresponding to the time at which 50% of the individuals (carriers vs noncarriers of the rs11871306*C allele) remain relapse‐free after the previous event, we passed the fitted Cox proportional hazards model to the survfit function in R.
Genetic MDS ancestry components were calculated in PLINK version 1.09b6.9, based on the set of variants with an MAF ≥5%, genotyping success rate ≥98% and HWE test p > 10−6 and after removing regions of extended LD 12 , 14 , 15 and dataset pruning so that no pair of SNPs within a given window of 50 kb was correlated (ie, LD r 2 > 0.2). As eigenvalues were <2.0, the primary analysis was performed without MDS components as covariates. 20 Sensitivity analyses were performed for the replicated genetic variants, including age at onset, sex, and the first 8 ancestry MDS components as covariates.
The Manhattan plot was created using the “CMplot” package in R version 3.6.1. Regional association plots were generated using the LocusZoom 21 webtool. Independent genomewide significant (p < 5 × 10−8) SNPs were defined as having an r 2 < 0.1 in the 1,000 Genomes phase III EUR super population in LDmatrix (https://ldlink.nci.nih.gov/?tab=ldmatrix). Functional annotation was done using RegulomeDB version 1.1 22 and HaploReg version 4.1. 23
MS Genetic Risk Score
The latest MS genomic map includes 200 independent autosomal susceptibility variants outside the major histocompatibility complex (MHC), of which 138 are primary, independent effects, as well as 31 statistically independent variants within the MHC region. 2 In a primary analysis, the non‐MHC MS genetic risk score was calculated starting from the 138, primary, independent non‐MHC effects. Strand‐ambiguous SNPs with an effect allele frequency between 40% and 60% were replaced by non‐strand‐ambiguous proxy SNPs showing r 2 > 0.9 with the published lead variants. A total of 135 non‐MHC SNPs (including 5 proxy SNPs showing r 2 > 0.9 with the published lead variants) were present in the imputed genetic data and hence included in the MS genetic risk score (Table S1). In addition, 22 MHC risk variants were present in the imputed genetic data and included in the MHC MS genetic risk score (Table S2). In a secondary analysis, we included both primary and secondary independent effects in the non‐MHC region. The secondary effects emerged from conditional modeling in the original susceptibility analysis. 2 The genetic risk score was weighted using the marginal odds ratios. A total of 195 non‐MHC SNPs were included in our secondary analysis (including 6 proxy SNPs showing r 2 > 0.9 with the published lead variants; Table S3).
Classical human leukocyte antigen (HLA) alleles, amino acid polymorphisms, and SNPs in the MHC region were imputed with SNP2HLA version 1.0.3 and the T1DGC reference panel (build 37). 24 To investigate a potential association between the burden of MS susceptibility variants and relapse hazard, weighted MS genetic risk scores for the non‐MHC SNPs and MHC risk variants were calculated using PRSice version 2.3.1.e, 25 assuming an additive genetic model. The association between the weighted MS genetic risk scores and time to relapses was assessed using a Cox proportional hazards model, as described above.
Gene Ontology Pathway Analysis
Genetic variants were assigned to their corresponding genes based on their position according to the NCBI 37.3 (hg19) build with MAGMA version 1.07. For each gene, a p value was calculated using a SNP‐wise multi model (ie, by integrating the p values for the mean and top SNP association within a gene into an aggregated p value for that gene). Subsequently, a competitive gene‐set analysis was performed to test whether the mean association with relapse hazard of genes in a specific gene set is greater than that of genes not in the gene set while conditioning on gene size, gene density, inverse minor allele count, log (gene size), log (gene density), and log (inverse minor allele count) according to standard settings. This analysis was repeated for each of the 10,181 Gene Ontology gene sets from the Molecular Signatures Database website (MSigDB, release 7.1, March 2020). We applied a Bonferroni correction for the number of gene sets tested (α = 0.05/10,181 = 4.91 × 10−6).
Replication Phase
Study Population
For replication, 485 patients with relapsing–remitting MS were analyzed. This cohort included all patients with available genotype and longitudinal relapse data diagnosed after 2010 at the Klinikum rechts der Isar of the Technical University of Munich. Diagnoses and the definition of relapses were based on the 2017 McDonald criteria. 9 Among these 485 patients, 819 relapses were captured over a median baseline duration of 0.5 years before receiving any treatment (see Table 1). Written informed consent was obtained from all patients according to the Declaration of Helsinki and samples were collected with ethical approval of the Klinikum rechts der Isar.
Genotyping, QC, and Imputation
SNPs were genotyped using Illumina OmniExpress BeadChips. QC was conducted using PLINK version 1.90b7, as described previously. 4 QC and imputation were carried out within a larger dataset of patients with MS. In brief, individuals were removed if fulfilling any of the following criteria: sex mismatches; genotyping rate <98%; cryptic relatedness >1/8; genetic outliers (distance in the first 8 MDS components of the identity‐by‐state matrix >4 standard deviations); a significant deviation in autosomal heterozygosity from the mean (>4.4 standard deviations); and X‐chromosomal heterozygosity ≤0.2. Variants were excluded according to the following criteria: call rate <98%; MAF <1%; HWE test p < 1 × 10‐ 6; A/T and G/C variants; variants not present in the 1,000 Genomes phase III reference panel. Genotype data were imputed to the 1,000 Genomes phase III reference panel using SHAPEIT2 16 and IMPUTE2. 17 , 18 The resulting dataset contained 8,964,526 high‐quality variants with an MAF ≥0.01 and INFO ≥0.8.
Association Analysis
A total of 8 SNPs reaching genomewide significance in the discovery cohort were present in the replication cohort. For these SNPs, association analysis between the minor allele dosages and time to relapses was assessed using the Cox proportional hazards model, including the first 8 MDS components as covariates.
Meta‐Analysis
A variant was considered as replicated if the two‐sided p in the replication sample <0.006 (= 0.05/8 SNPs in replication phase) with an effect in the same direction, and a meta‐analysis p < 5 × 10−8 using the inverse variance‐weighted method and the fixed‐effects model in META version 1.7.
Results
MS Genetic Risk Scores are not Associated With Relapse Hazard
Among 506 Belgian patients with MS, 1,412 relapses were captured over a median baseline duration of 4 years before receiving any treatment (see Table 1). Neither the MS genetic risk score consisting of 135 primary, independent SNPs outside the MHC region (HR = 0.95, 95% confidence interval [CI] = 0.87–1.05, p = 0.33) nor the MHC genetic risk score (HR = 1.01, 95% CI = 0.94–1.10, p = 0.76) was associated with relapse hazard at baseline. Including both primary and secondary independent SNPs outside the MHC region in the MS genetic risk score, resulted in very similar findings (HR = 0.95, 95% CI = 0.87–1.03, p = 0.24). Thus, genetic variants conveying MS disease risk did not appear to be major drivers of relapse hazard in our cohort, warranting genomewide approaches.
Genomewide Association Study Identifies rs11871306 as a Novel SNP Affecting Relapse Hazard
We analyzed 7,344,935 SNPs with an MAF ≥2%, HWE test p > 1 × 10−6, and INFO ≥0.9 after imputation and QC, with the Manhattan plot shown in Figure 1. The genomic inflation factor (λ) of the GWAS was 1.02, indicating no inflation of test statistics. In this discovery stage GWAS, 10 independent loci passed the threshold for genomewide significance (p < 5 × 10−8; Table 2). For imputed SNPs, nonreference allele discordance rates (4–18%), calculated based on direct genotyping and Sanger sequencing, were in line with expectations, especially for low‐frequency variants. 26
FIGURE 1.
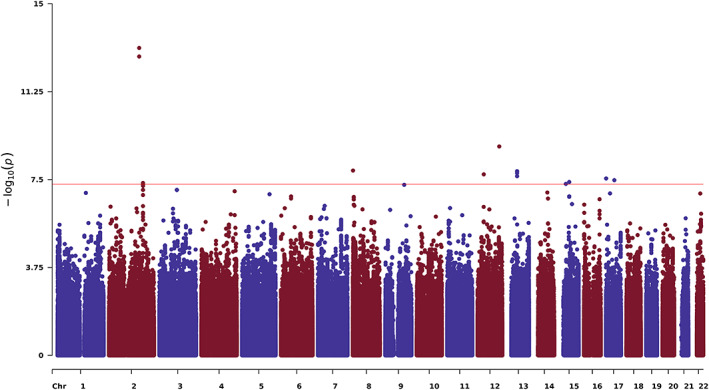
Manhattan plot showing genomewide association results of the survival analysis for time to relapse in the discovery phase. Each dot represents a genetic variant, with the x‐axis depicting chromosomal position and the y‐axis showing the negative logarithm of the p value for an association with relapse hazard. The red line indicates the genomewide significance threshold (p = 5 × 10−8).
TABLE 2.
Independent (r 2 < 0.1) Genomewide Significant Associations With Relapse Hazard in the Discovery Cohort
| SNP | CHR:POS | A1 | A2 | Discovery cohort | Replication cohort | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR | SE | P | MAF | INFO | DISC | HR | SE | P | MAF | INFO | ||||
| rs139075191 | 2:183736241 | G | A | 1.85 | 0.11 | 4.44 × 10−8 | 0.03 | 0.98 | 14 | 1.06 | 0.24 | 0.80 | 0.03 | 0.93 |
| rs36084110 | 2:163160798 | T | C | 2.58 | 0.13 | 7.72 × 10−14 | 0.02 | 0.91 | 5 | 1.22 | 0.24 | 0.41 | 0.03 | 0.89 |
| 8:1197491 | 8:1197491 | C | G | 2.36 | 0.15 | 1.31 × 10−8 | 0.02 | 0.95 | NA | 0.57 | 0.33 | 0.09 | 0.02 | 0.92 |
| rs79434188 | 12:33587295 | A | T | 0.49 | 0.13 | 1.90 × 10−8 | 0.03 | 0.95 | 4 | 1.02 | 0.37 | 0.96 | 0.02 | 0.84 |
| rs117185095 | 12:117089904 | G | T | 2.58 | 0.16 | 1.23 × 10−9 | 0.02 | 1 | NA | NA | NA | NA | NA | NA |
| rs77836326 | 13:50601566 | G | A | 0.52 | 0.12 | 1.42 × 10−8 | 0.03 | 1 | NA | 0.99 | 0.36 | 0.98 | 0.02 | 0.91 |
| rs17817182 | 15:33716899 | C | A | 2.00 | 0.13 | 4.85 × 10−8 | 0.03 | 1 | NA | NA | NA | NA | NA | NA |
| rs79719335 | 15:51835376 | A | G | 2.13 | 0.14 | 4.04 × 10−8 | 0.02 | 0.94 | 18 | 1.43 | 0.24 | 0.14 | 0.02 | 0.94 |
| rs76915261 | 17:774611 | T | C | 2.00 | 0.13 | 2.85 × 10−8 | 0.03 | 1 | NA | 1.70 | 0.29 | 0.06 | 0.03 | 0.87 |
| rs11871306 | 17:44954984 | C | T | 2.08 | 0.17 | 3.37 × 10−8 | 0.02 | 0.97 | 10 | 2.53 | 0.24 | 1.01 × 10−4 | 0.02 | 1.00 |
All variants with an INFO value of 1 were genotyped.
A1 = minor allele (effect allele); A2 = major allele; CHR = chromosome; DISC = percentage of discordant non‐reference alleles between direct genotyping and imputation; HR = hazard ratio; INFO = imputation INFO metric; MAF = minor allele frequency; NA = not applicable/not available; P = p‐value; POS = position (hg19); SE = robust standard error.
Eight of the genomewide significant loci in the discovery cohort were available and passed postimputation QC in the replication cohort, capturing 819 relapses over a median baseline duration of 0.5 years in 485 treatment‐naïve patients (see Tables 1 and 2). For the directly genotyped variant rs11871306 on chromosome 17, the association with relapse hazard replicated after Bonferroni correction for multiple testing (p = 1.01 × 10−4). Sensitivity analyses indicated that including covariates did not substantially alter the results (Table S4) and that the Cox proportional hazards assumption was fulfilled (Schoenfeld residual test p value for discovery cohort = 0.27). In an inverse variance‐weighted fixed‐effects meta‐analysis using direct genotypes for both the discovery and replication cohorts, variant rs11871306 was associated with relapse hazard with HR = 2.15, 95% CI = 1.70–2.78, and p = 2.07 × 10−10 (Fig 2).
FIGURE 2.
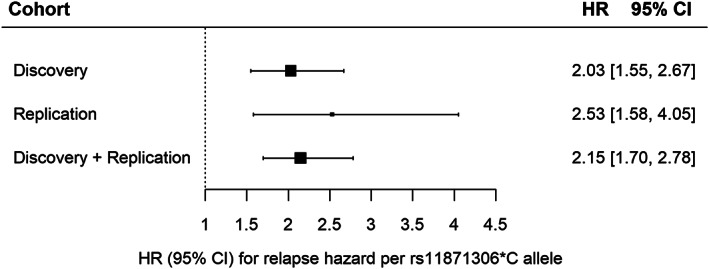
Forest plot of meta‐analysis of rs11871306*C association with relapse hazard. The Cox proportional hazards model was fitted with genotypes obtained by direct genotyping: genotypes obtained through TaqMan genotyping and Sanger sequencing for the discovery cohort, and through genotyping array for the replication cohort. CI, confidence interval; HR, hazard ratio.
WNT9B rs11871306 Minor Allele Carriers Show More Disease Activity
Depending on the isoform, rs11871306 is either located in an intron or the 3′ untranslated region (UTR) of the gene Wnt family member 9B (WNT9B; Fig 3). The variant maps to a DNaseI hypersensitivity cluster identified in several cell types, including both immune and brain cells from the ENCODE and Roadmap Epigenomics projects. The minor allele C, with a frequency of 2% in European but up to 29% in African populations, disrupts a binding motif for the transcription factor c‐Ets‐1. The associated region is located toward the telomeric end of a well‐known large 900 kb inversion (H1/H2), but is not in linkage disequilibrium (LD) with SNPs (rs1800547 and rs9468) tagging this inversion (r 2 = 0.0003). 27 Of note, in both the discovery and replication phase of our study, rs11871306 was associated with relapse hazard but this variant is not associated with MS susceptibility (OR = 1.01, p = 0.90) in the most recent published MS risk GWAS. 2
FIGURE 3.
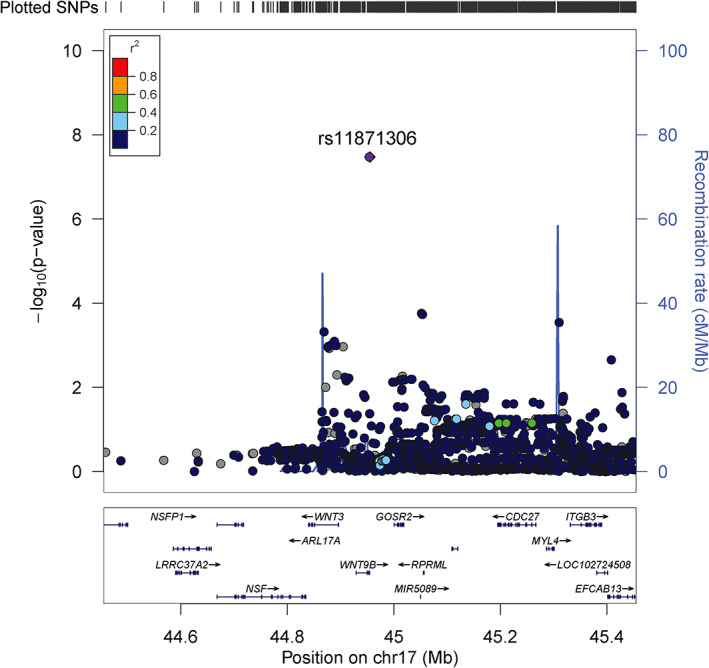
Regional association plot of chromosome 17q21.32 with lead single nucleotide polymorphism (SNP) rs11871306 demonstrating an association with relapse hazard. Association results (primary y‐axis) are shown for genetic variants with a minor allele frequency (MAF) ≥2% and imputation INFO metric ≥0.9, along with recombination rates (secondary y‐axis), for a 500 kb region (250 kb region flanking the lead SNP rs11871306 (chr17:44954984 (hg19)). Each dot represents the ‐log10 p value from the survival analysis using the Cox proportional hazards model including baseline relapses before any immunomodulatory treatment. Genetic variants are colored according their linkage disequilibrium (LD; r 2) with the lead SNP using the 1,000 Genomes Phase III EUR superpopulation.
In both the discovery and replication cohorts, individuals carrying the minor rs11871306*C allele had a shorter time to relapse compared with noncarriers (Fig 4). The median time that patients remained relapse‐free in the discovery cohort was 0.95 years for carriers versus 2.22 years for noncarriers. In the replication cohort, individuals carrying the rs11871306*C allele similarly showed a shorter time to relapse compared with noncarriers, with a median time to relapse of 0.59 versus 2.00 years. A secondary analysis in the discovery cohort showed that rs11871306*C carriers had received treatment earlier than noncarriers (HR = 1.90, 95% CI = 1.16–3.10, p = 0.01), based on standard clinical evaluation by the same treating neurologist.
FIGURE 4.
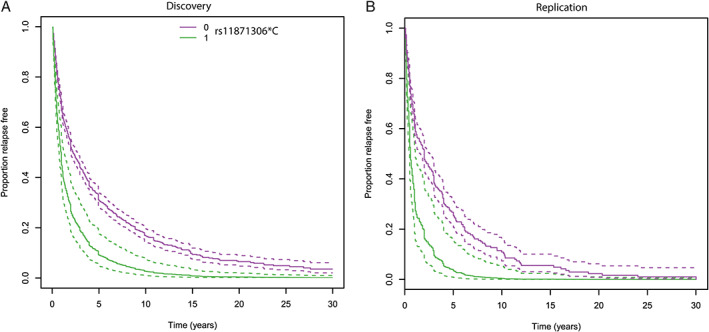
Survival curves for time to relapse by rs11871306 genotype. (A) In the discovery cohort, individuals carrying the rs11871306*C allele have a shorter time to relapse compared with noncarriers, with a median relapse‐free interval of 0.95 versus 2.22 years. (B) In the replication cohort, individuals carrying the rs11871306*C allele have a shorter time to relapse compared with noncarriers with a median relapse‐free interval of 0.59 versus 2.00 years. Dotted lines represent 95% confidence intervals.
Pathway Analysis Implicates Vitamin D Response in Susceptibility to Relapses
Using the discovery‐stage GWAS summary statistics, a gene‐set analysis based on Gene Ontology pathways was performed to further explore the underlying biology of relapses. The only significant pathway after correction for multiple testing was “Response to vitamin D” (GO: 0033280; uncorrected p = 4.33 × 10−6; Table 3). This pathway is defined in Gene Ontology as “reflecting processes that result in a change in state or activity of a cell or an organism as a result of a vitamin D stimulus.” Other pathways ranking among the top 10 results but not meeting the Bonferroni correction threshold include response to viruses and interferon‐α secretion (see Table 3). Of note, the biological process “Cell‐cell signaling by WNT” (GO: 0198738), including WNT9B, ranked at 210 of 10,181 (2.06%; Table S5).
TABLE 3.
Top‐10 Gene Ontology Terms Enriched for Associations with Relapse Hazard
| GO terms | GO ID | N GENES | BETA | SE | p |
|---|---|---|---|---|---|
| Response to vitamin D | GO:0033280 | 32 | 0.76 | 0.17 | 4.33 × 10 −6 |
| Detection of virus | GO:0009597 | 6 | 1.53 | 0.37 | 1.55 × 10−5 |
| Positive regulation of heterotypic cell–cell adhesion | GO:0034116 | 16 | 0.93 | 0.23 | 2.79 × 10−5 |
| αβ T‐cell receptor complex | GO:0042105 | 5 | 1.59 | 0.40 | 3.64 × 10−5 |
| Origin recognition complex | GO:0000808 | 9 | 0.86 | 0.22 | 3.89 × 10−5 |
| Interferon‐α secretion | GO:0072642 | 9 | 1.17 | 0.30 | 4.59 × 10−5 |
| Negative regulation of fibroblast migration | GO:0010764 | 6 | 1.25 | 0.32 | 5.60 × 10−5 |
| Presynaptic active zone cytoplasmic component | GO:0098831 | 16 | 0.94 | 0.24 | 5.82 × 10−5 |
| Regulation of cell chemotaxis to fibroblast growth factor | GO:1904847 | 6 | 1.29 | 0.33 | 6.13 × 10−5 |
| Positive regulation of mRNA processing | GO:0050685 | 29 | 0.53 | 0.14 | 8.82 × 10−5 |
P values that passed Bonferroni correction (alpha = 0.05/10,181) are highlighted in bold font.
BETA = the regression coefficient of association with relapse hazard for gene sets in the GO pathway; GO = Gene Ontology; ID = identifier, N GENES = number of genes in the data that are part of the GO pathway; P = p‐value; SE = standard error.
Discussion
In the present study, we identified low‐frequency variation in the gene WNT9B to increase relapse hazard in MS cases. Combining 2 longitudinal cohorts, capturing a total of 2,231 relapses in 991 treatment‐naïve individuals, rs11871306, mapping to the WNT9B gene, reached genomewide significance. Individuals carrying the minor allele showed a more than doubled relapse hazard.
The protein Wnt‐9b functions in the canonical Wnt/β‐catenin signaling pathway as a component of the Wnt‐Fzd‐LRP5‐LRP6 signaling complex. Wnt/β‐catenin signaling plays a critical role in nervous system development, axonal guidance, and synapse formation. 28 It has been implicated in neuropsychiatric disorders, such as autism and schizophrenia, 28 , 29 , 30 and in neurodegenerative disorders, such as Parkinson's disease 31 and Alzheimer's disease. 32
In the mouse model Experimental Autoimmune Encephalitis (EAE), the effects of the Wnt/β‐catenin pathway depend on the specific cell types in which the pathway is active. Endothelial Wnt/β‐catenin signaling protects cerebrovascular integrity, 33 and reduces immune cell infiltration by decreasing vascular cell adhesion molecule (VCAM)‐1 and Caveolin1 expression. 34 Downregulation of Wnt signaling in oligodendrocytes after white matter injury may, on the other hand, support regenerative myelination. 35 , 36 Upregulation of the Wnt pathway in oligodendrocyte precursor cells induces a negative feedback loop involving production of the Wnt inhibitory factor (Wif1), which may add to a disruption of the blood‐brain barrier. 37 Overall, the Wnt/β‐catenin pathway appears to have a beneficial effect on EAE: deletion of the Wnt co‐receptors LRP5 and LRP6 or of β‐catenin in dendritic cells exacerbates EAE disease severity, 38 whereas β‐catenin agonist treatment of healthy mice before disease induction results in a delayed EAE onset and reduced EAE severity. Likewise, therapeutic activation of β‐catenin after disease induction reduces clinical severity of EAE and CNS pathology. 38
Data on the Wnt pathway in the context of human MS are limited, but whole‐blood RNA gene expression analysis in cases and controls revealed an upregulation of Wnt signaling in MS cases versus controls. 39 Similarly, upregulation of the Wnt pathway in MS lesions 37 and brain blood vessels 34 has been observed. Our data now add that genetic variation in the Wnt pathway contributes to differences in relapse hazard between patients.
Following the GWAS identifying an association of a variant in the WNT9B gene with relapse hazard, we conducted a pathway‐based association analysis. Pathways ranking highly within the top 10 association results included “detection of virus” and “Interferon‐α secretion.” As a role of viruses in triggering relapses and of type I interferon in preventing relapses has been well described, 40 , 41 this finding supports the validity of our analysis. Our pathway analysis identified vitamin D response as significantly associated after the highly conservative Bonferroni correction for multiple testing, given that pathways were partially overlapping. Vitamin D is a known immunomodulator, skewing cells of the adaptive immune system toward a more tolerogenic status in vitro. 42 Increased levels of 25‐hydroxyvitamin D (25(OH)D) are associated with a decrease in relapses in MS observational studies. 43 , 44 In addition, the low‐density lipoprotein receptor‐related protein 2 (LRP2), previously associated with relapse hazard 6 , 7 and in the discovery cohort reaching HR = 1.17, 95% CI = 1.02–1.32, and p = 0.02, is a transmembrane receptor that mediates uptake and activation of 25(OH)D through endocytosis in renal 45 and mesenchymal stem cells. 46 Crosstalk between vitamin D and the Wnt pathway is suggested, with the direction of effect depending on the biological system. In cancer cells, vitamin D can act as an antagonist of Wnt/β‐catenin, but it may act as a co‐activator in other cell types, such as osteoblasts and keratinocytes. 47 In mice, maternal vitamin D deficiency led to lower levels of Wnt3a, β‐catenin, and the tight junction protein claudin‐1 and to a higher permeability in the offspring's intestinal epithelial barrier. 48 This barrier shows similarities with the blood–brain‐barrier. 49
Neither the WNT9B variant we report here nor the previously identified LRP2 variant are associated with MS susceptibility. 2 The genetic risk burden is a more powerful instrument than the analysis of single variants for distinguishing differences in susceptibility, and has previously been associated with the presence of oligoclonal bands in the cerebrospinal fluid. 50 Our findings do not confirm the previously reported trend for an association of the non‐MHC genetic risk burden with higher baseline relapse rate, 50 in line with other studies investigating the association between weighted genetic MS risk scores and relapses. 51 Similarly, analyses for other diseases, such as Crohn's disease, emphasize the difference between susceptibility and disease course heterogeneity after onset. 52 Our data suggest that the genetic basis for relapse hazard is distinct from genetic factors driving disease susceptibility. However, larger studies are required to exclude a more modest effect of MS‐associated variants on relapse.
Our main association results point to the involvement of less common variants (MAF 2–5%) in relapse hazard with relatively large effect sizes. The previously published GWAS investigating the association between genetic variants and relapse hazard was limited to common SNPs (MAF >5%). 6 A second strength of the present study is that we only included relapses before treatment to prevent the confounding effect of treatment on relapse hazard, as effective disease‐modifying treatments reduce relapse rates. In the pooled analysis, 991 individuals were included, rendering this the largest study population to date for relapse hazard in MS. However, to fully identify the genetic architecture of relapse in MS, larger study cohorts with even longer follow‐up periods are required. Limitations of our study include the higher complexity of disease heterogeneity phenotypes compared with susceptibility. 53 However, all relapses in our discovery cohort were registered by one expert clinician and our findings were replicated in an independent replication cohort.
The HR of 2 observed in both cohorts in this study indicates clinically meaningful differences, comparable to primary outcome effect sizes expected in clinical trials. 54 In the discovery cohort, this translates in carriers of the minor allele remaining relapse‐free for a median duration of 1 year versus more than 2 years in noncarriers. The converging impact of the Wnt and vitamin D pathways on blood–brain barrier integrity and immune cell infiltration provides an immediate parallel with existing efficacious treatments. Blockade of VCAM‐1 interactions with its cognate lymphocyte ligand integrin α4 decreases immune cell infiltration into the CNS and reduces the clinical severity of EAE. 55 This constitutes the basis for the MS disease modifying treatment (DMT) natalizumab, which reduces the rate of relapse by 68% 56 and is regarded as a high efficacy DMT, albeit with safety considerations. 57
In conclusion, we have identified an association between genetic variation in WNT9B and in the vitamin D pathway with relapse hazard. Further functional assessment of the Wnt/β‐catenin and vitamin D pathway in MS disease activity is necessary to provide an improved mechanistic understanding of the association underlying patient‐to‐patient variation in relapse hazard, potentially leading to improved opportunities for therapeutic intervention.
Author Contributions
M.V., T.A., Y.Z., B.T., B.H., B.D., and A.G. contributed to the conception and design of the study. M.V., T.A., K.M., F.H., L.A., B.H., and B.D. contributed to the acquisition and analysis of data. M.V., T.A., and A.G. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
The authors declared no conflict of interest.
Supporting information
Supplementary Table S1. Primary, independent, non‐major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the non‐MHC MS genetic risk score in the primary analysis (N = 135)
Supplementary Table S2. Major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the MHC‐based MS genetic risk score (N = 22)
Supplementary Table S3. Primary and secondary independent, non‐major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the non‐MHC MS genetic risk score in the secondary analysis (N = 195)
Supplementary Table S4. Association of rs11871306 with relapse hazard, including age at onset, sex, and the first 8 ancestry components as covariates
Supplementary Table S5. Competitive gene‐set analysis using Gene Ontology terms
Acknowledgments
B.D. is a Clinical Investigator and M.V. a Research Fellow (11ZZZ21N) of the Research Foundation Flanders (FWO‐Vlaanderen). M.V. received travel grants from the Research Foundation Flanders (FWO‐Vlaanderen) (V414719N) and from YouReCa Junior Mobility Programme of KU Leuven (JUMO/18/040). The Belgian part of this study was supported by the Research Fund KU Leuven (C24/16/045), the Research Foundation Flanders (FWO G.07334.15), the Belgian Charcot Foundation and MS Liga Vlaanderen. We thank the High‐Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z) for the generation of the genotyping data and the McCarthy group at the Wellcome Trust Centre for Human Genetics for providing the tools for imputation preparation (https://www.well.ox.ac.uk/~wrayner/tools/index.html). The computational resources and services used in this work were provided by the Flemish Supercomputer Center (VSC), funded by the Research Foundation Flanders (FWO‐Vlaanderen) and the Flemish Government (department EWI). The German part of this study was supported by a grant from the German Federal Ministry of Education and Research (BMBF; grant 01GI1601D for the German Competence Network Multiple Sclerosis, KKNMS) and is associated with DIFUTURE (Data Integration for Future Medicine, BMBF grant 01ZZ1804[A‐I]). A.G., B.D., and B.H. have received funding for the present study from the Horizon2020 "MultipleMS" consortium (grant EU RIA 733161). B.H. has received funding for the present study from the German Research Foundation (DFG) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198). Y.Z. was supported by an MS Research Australia postdoctoral fellowship as well as NHMRC investigator grant L1 (GNT1173155). B.V.T. was supported by a MS Research Australia‐Macquarie Foundation Senior clinical Fellowship, the Australian Medical Research Future Fund, the National Health and Medical Research Council Australia and MS Research Australia.
References
- 1. International Multiple Sclerosis Genetics Consortium . Low‐frequency and rare‐coding variation contributes to multiple sclerosis risk. Cell 2018;175:1679–1687.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. International Multiple Sclerosis Genetics Consortium . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019;365:eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moutsianas L, Jostins L, Beecham AH, et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet 2015;47:1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andlauer TF, Buck D, Antony G, et al. Novel multiple sclerosis susceptibility loci implicated in epigenetic regulation. Sci Adv 2016;2:e1501678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lavery AM, Verhey LH, Waldman AT. Outcome measures in relapsing‐remitting multiple sclerosis: capturing disability and disease progression in clinical trials. Mult Scler Int 2014;2014:262350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou Y, Graves JS, Simpson S Jr, et al. Genetic variation in the gene LRP2 increases relapse risk in multiple sclerosis. J Neurol Neurosurg Psychiatry 2017;88:864–868. [DOI] [PubMed] [Google Scholar]
- 7. Hilven K, Vandebergh M, Smets I, et al. Genetic basis for relapse rate in multiple sclerosis: association with LRP2 genetic variation. Mult Scler 2018;24:1773–1775. [DOI] [PubMed] [Google Scholar]
- 8. Goris A, Pauwels I, Gustavsen MW, et al. Genetic variants are major determinants of CSF antibody levels in multiple sclerosis. Brain 2015;138:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 10. Lagou V, Garcia‐Perez JE, Smets I, et al. Genetic architecture of adaptive immune system identifies key immune regulators. Cell Rep 2018;25:798–810 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang CC, Chow CC, Tellier LC, et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson CA, Pettersson FH, Clarke GM, et al. Data quality control in genetic case‐control association studies. Nat Protoc 2010;5:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. The 1000 Genomes Project Consortium , An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Price AL, Weale ME, Patterson N, et al. Long‐range LD can confound genome scans in admixed populations. Am J Hum Genet 2008;83:132–135; author reply 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weale ME. Quality control for genome‐wide association studies. Methods Mol Biol 2010;628:341–372. [DOI] [PubMed] [Google Scholar]
- 16. Delaneau O, Zagury JF, Marchini J. Improved whole‐chromosome phasing for disease and population genetic studies. Nat Methods 2013;10:5–6. [DOI] [PubMed] [Google Scholar]
- 17. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howie B, Fuchsberger C, Stephens M, et al. Fast and accurate genotype imputation in genome‐wide association studies through pre‐phasing. Nat Genet 2012;44:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bellera CA, MacGrogan G, Debled M, et al. Variables with time‐varying effects and the Cox model: some statistical concepts illustrated with a prognostic factor study in breast cancer. BMC Med Res Methodol 2010;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amare AT, Schubert KO, Hou L, et al. Association of polygenic score for major depression with response to lithium in patients with bipolar disorder. Mol Psychiatry 2020. Online ahead of print. 10.1038/s41380-020-0689-5. [DOI] [PubMed] [Google Scholar]
- 21. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40:D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jia X, Han B, Onengut‐Gumuscu S, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 2013;8:e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi SW, O'Reilly PF. PRSice‐2: polygenic risk score software for biobank‐scale data. Gigascience. 2019;8(7):giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitt M, Kals M, Parn K, et al. Improved imputation accuracy of rare and low‐frequency variants using population‐specific high‐coverage WGS‐based imputation reference panel. Eur J Hum Genet 2017;25:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet 2005;37:129–137. [DOI] [PubMed] [Google Scholar]
- 28. Okerlund ND, Cheyette BN. Synaptic Wnt signaling‐a contributor to major psychiatric disorders? J Neurodev Disord 2011;3:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwan V, Unda BK, Singh KK. Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord. 2016;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoseth EZ, Krull F, Dieset I, et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl Psychiatry 2018;8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berwick DC, Javaheri B, Wetzel A, et al. Pathogenic LRRK2 variants are gain‐of‐function mutations that enhance LRRK2‐mediated repression of beta‐catenin signaling. Mol Neurodegener 2017;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu CC, Tsai CW, Deak F, et al. Deficiency in LRP6‐mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron 2014;84:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tran KA, Zhang X, Predescu D, et al. Endothelial beta‐catenin signaling is required for maintaining adult blood‐brain barrier integrity and central nervous system homeostasis. Circulation 2016;133:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lengfeld JE, Lutz SE, Smith JR, et al. Endothelial Wnt/beta‐catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci U S A 2017;114:E1168–E1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee HK, Laug D, Zhu W, et al. Apcdd1 stimulates oligodendrocyte differentiation after white matter injury. Glia 2015;63:1840–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fancy SP, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 2009;23:1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niu J, Tsai HH, Hoi KK, et al. Aberrant oligodendroglial‐vascular interactions disrupt the blood‐brain barrier, triggering CNS inflammation. Nat Neurosci 2019;22:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suryawanshi A, Manoharan I, Hong Y, et al. Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol 2015;194:3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nickles D, Chen HP, Li MM, et al. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum Mol Genet 2013;22:4194–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steelman AJ. Infection as an environmental trigger of multiple sclerosis disease exacerbation. Front Immunol 2015;6:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rudick RA, Goelz SE. Beta‐interferon for multiple sclerosis. Exp Cell Res 2011;317:1301–1311. [DOI] [PubMed] [Google Scholar]
- 42. Peelen E, Knippenberg S, Muris AH, et al. Effects of vitamin D on the peripheral adaptive immune system: a review. Autoimmun Rev 2011;10:733–743. [DOI] [PubMed] [Google Scholar]
- 43. Mowry EM, Krupp LB, Milazzo M, et al. Vitamin D status is associated with relapse rate in pediatric‐onset multiple sclerosis. Ann Neurol 2010;67:618–624. [DOI] [PubMed] [Google Scholar]
- 44. Simpson S Jr, Taylor B, Blizzard L, et al. Higher 25‐hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol 2010;68:193–203. [DOI] [PubMed] [Google Scholar]
- 45. Nykjaer A, Dragun D, Walther D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25‐(OH) vitamin D3. Cell 1999;96:507–515. [DOI] [PubMed] [Google Scholar]
- 46. Gao Y, Zhou S, Luu S, Glowacki J. Megalin mediates 25‐hydroxyvitamin D3 actions in human mesenchymal stem cells. FASEB J 2019;33:7684–7693. [DOI] [PubMed] [Google Scholar]
- 47. Gomez‐Oliva R, Geribaldi‐Doldan N, Dominguez‐Garcia S, et al. Vitamin D deficiency as a potential risk factor for accelerated aging, impaired hippocampal neurogenesis and cognitive decline: a role for Wnt/beta‐catenin signaling. Aging (Albany NY) 2020;12(13):13824–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang K, Zhu J, Wu J, et al. Maternal vitamin D deficiency increases intestinal permeability and programs Wnt/beta‐catenin pathway in BALB/C mice. JPEN J Parenter Enteral Nutr 2020;45(1):102–114. [DOI] [PubMed] [Google Scholar]
- 49. Daneman R, Rescigno M. The gut immune barrier and the blood‐brain barrier: are they so different? Immunity 2009;31:722–735. [DOI] [PubMed] [Google Scholar]
- 50. Hilven K, Patsopoulos NA, Dubois B, Goris A. Burden of risk variants correlates with phenotype of multiple sclerosis. Mult Scler 2015;21:1670–1680. [DOI] [PubMed] [Google Scholar]
- 51. Sondergaard HB, Petersen ER, Magyari M, et al. Genetic burden of MS risk variants distinguish patients from healthy individuals but are not associated with disease activity. Mult Scler Relat Disord 2017;13:25–27. [DOI] [PubMed] [Google Scholar]
- 52. Lee JC, Biasci D, Roberts R, et al. Genome‐wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn's disease. Nat Genet 2017;49:262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Axisa PP, Hafler DA. Multiple sclerosis: genetics, biomarkers, treatments. Curr Opin Neurol 2016;29:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sormani MP, Signori A, Siri P, De Stefano N. Time to first relapse as an endpoint in multiple sclerosis clinical trials. Mult Scler 2013;19:466–474. [DOI] [PubMed] [Google Scholar]
- 55. Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992;356:63–66. [DOI] [PubMed] [Google Scholar]
- 56. Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 57. Brandstadter R, Katz Sand I. The use of natalizumab for multiple sclerosis. Neuropsychiatr Dis Treat 2017;13:1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Primary, independent, non‐major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the non‐MHC MS genetic risk score in the primary analysis (N = 135)
Supplementary Table S2. Major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the MHC‐based MS genetic risk score (N = 22)
Supplementary Table S3. Primary and secondary independent, non‐major histocompatibility complex (MHC) multiple sclerosis (MS) risk variants used for the non‐MHC MS genetic risk score in the secondary analysis (N = 195)
Supplementary Table S4. Association of rs11871306 with relapse hazard, including age at onset, sex, and the first 8 ancestry components as covariates
Supplementary Table S5. Competitive gene‐set analysis using Gene Ontology terms