

Abstract
Virtually inert sulfur hexafluoride becomes a precious pentafluorosulfanylation agent, if properly activated by photoredox catalysis, to access α‐fluoro and α‐alkoxy SF5‐compounds. This advanced protocol converts SF6 in the presence of alkynols as bifunctional C−C‐ and C−O‐bond forming reagents directly into pentafluorosulfanylated oxygen‐containing heterocycles in a single step from α‐substituted alkenes. The proposed mechanism is supported by theoretical calculations and gives insights not only in the pentafluorosulfanylation step but also into formation of the carbon‐carbon bond and is in full agreement with Baldwin's cyclization rules. The key step is a radical type 5‐, 6‐ respectively 7‐exo‐dig‐cyclization. The synthesized oxaheterocycles cannot be simply prepared by other synthetic methods, show a high level of structural complexity and significantly expand the scope of pentafluorosulfanylated building blocks valuable for medicinal and material chemistry.
Keywords: electron transfer, pentafluorosulfanylation, phenothiazine, photocatalysis, photochemistry, sulfur hexafluoride
Become complex! The advanced one‐step photoredox catalytic activation of SF6 allows the preparation of pentaflurosulfanylated oxepans, tetrahydropyrans and tetrahydrofurans as products with high structural complexity.

Since the initial report on the preparation of the first organic pentafluorosulfanyl compounds SF5CF3 using excessive fluorination of CS2 by Cady in 1950, [1] the pentafluorosulfanyl (SF5) group has found increasing interest in agro‐ und medicinal chemistry as well as in functional materials due to its unique steric and electronic properties.[ 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 ] However, this research field was hampered by the very limited accessibility of pentafluorosulfanylated compounds for about 60 years. In particular, the highly toxic sulfur fluorides SF5Cl, SF5Br und S2F10 have been the only preparative sources of the SF5 radical. However, these reagents are extremely toxic, and their commercial availability is limited. Dolbier[ 10 , 11 , 12 ] and Umemoto [13] reported independently methods to access SF5‐arenes from disulfides by oxidation with AgF2 or chlorine in the presence of KF and ZnF2. Recently, Togni further turned this method into a gas‐free approach by in‐situ chlorine formation from trichloroisocyanuric acid. [14] In contrast, sulfur hexafluoride (SF6) so far is only rarely considered as a reagent in organic synthesis[ 15 , 16 , 17 , 18 , 19 , 20 , 21 ] due to its intrinsic inertness; its potential usefulness as pentafluorosulfanylation agent has even been negatively evaluated. [16] In contrast, SF6 was applied for three deoxyfluorination methods developed independently by Jamison, Rueping and Dielmann.[ 17 , 18 , 19 ] The pentafluorosulfanylation activity of SF6 in photoredox catalysis could only recently be unlocked in our lab (Figure 1).[ 20 , 21 ] We showed that the photoredox catalytic activation of SF6 allows the preparation of pentaflurosulfanylated styrenes and their ethers. [21] Herein, we establish an advanced one‐step pentafluorosulfanylative C−C bond forming protocol based on previous work using a radical cascade sequence. Alkynols are used as bifunctional C−C‐ and C−O‐bond forming reagents to prepare oxaheterocyclic compounds, namely oxepans, tetrahydropyrans and tetrahydrofurans. Following up our previous work on the activation of sulfur hexafluoride to access new chemical space,[ 20 , 21 ] this new method expands the scope of accessible pentafluorosulfanylated products with high complexity that is characteristic for natural products or advanced pharmaceuticals (Figure 1).
Figure 1.

The recent advancement of photoredox catalytic methods to convert SF6 into pentafluorosulfanylated organic compounds as valuable synthesis building blocks.
In particular, oxaheterocycles play a key role in medicinal chemistry. Tetrahydropyrans and tetrahydrofurans are the 6th and 11th, respectively, most frequently used ring substructures listed in the FDA Orange Book in 2014. [22] Moreover, tetrahydropyrans are highly important structural motifs in marine toxins and other natural products with often cytotoxic activity.[ 23 , 24 , 25 ] Oxepanes are not yet quite important in medicinal chemistry although they often show cytotoxic activity [26] and they are found in several classes of natural compounds isolated from marine organisms like algae, fungi or corals.[ 27 , 28 ] Our method to activate SF6 uses N‐phenylphenohiazine 3 as organic photoredox catalyst.[ 20 , 29 , 30 , 31 ] The estimated strong reduction potential of −2.5 V (vs. SCE) in the excited state is crucial for the desired fragmentation channel of SF6 into the SF5 radical.[ 29 , 32 , 33 , 34 , 35 , 36 , 37 ]
Such single electron reduction of SF6 was previously accomplished by a thermal reaction with TEMPO lithium in order to pentafluorosulfanylate alkenes. [15] We recently investigated the addition of both SF5 and 5‐pentyn‐1‐ol to 1,1‐diphenylethylene 1. After irradiation (368 nm LED) of 1 (0.20 M) in the presence of 3 equiv. of 4‐pentynol, 10 mol% photoredox catalyst 3 and 10 mol % of triethylborane as radical stabilizer and fluoride trap, we found not only the expected acyclic ether 4 in 26 % yield, but also the new reaction product 8 in 14 % yield. According to 1H‐, 13C and 19F‐NMR spectroscopy as well as XRD structure and mass spectrometry this product is oxepane 8 that carries the SF5‐substituent in the remote vinylic position. It is remarkable with respect to its complex structure that it could be prepared from the α‐substituted alkene 1 in just one step.
The fundamental steps of photoredox catalytic activation of SF6 by N‐phenylphenothiazine were spectroscopically investigated (Figure 2) and include two consecutive electron transfer steps [38] to activate both SF6 and the substrate 1. The first electron transfer forms radical cation 3 .+ and generates the SF5 radical. After excitation of 3 .+ the back electron transfer closes the photoredox catalytic cycle and generates the key radical cation 1 .+ which can be trapped either by the in‐situ generated fluoride anion [20] or by alcohols as external nucleophiles to yield product 4. The fluoride trapping is suppressed by triethylborane as additive. We propose an intramolecular radical‐alkyne addition for the cyclization reaction to the new product 8. [21] In particular, we postulate a competitive trapping of the spin center of the substrate radical cation 1 .+ after photoredox catalytic activation by the free SF5 radical or the alkynol.
Figure 2.

Overview of reaction pathways after photoredox catalytic activation of SF6 by irradiation at 368 nm and pentafluorosulfanylation of α‐methyl (1) and α‐phenyl styrene (2) with novel intramolecular radical‐alkyne addition to cyclization products 8 and 9. Insert: Structure of the isolated sulfone 24.
To get more insight into the reaction mechanism and to optimize the reaction towards cyclization product 8 we varied the concentration of 4‐pentynol with substrate 1. Based on our mechanistic proposal we expected that an increase of the alcohol concentration causes the faster trapping of the radical cation 1 .+ and preferential formation of the cyclic product 8 and vice versa for the acyclic product 4. This assumption could be experimentally confirmed by increasing the amount of alcohol in the solution from 3 equiv. to 20 equiv. Indeed, the ratio of the two competing reaction products 4 : 8 changed from 1 : 1 to 3 : 1 while forming 32 % of product 8 (Figure 3). The total yields of 40–51 % are in good agreement of the yields previously reported for acyclic SF5 compounds.[ 20 , 21 ] The best total yield of 51 % for both products was achieved with 7–10 equiv. 4‐pentynol. The relatively constant total yield of products 4 and 8 over the whole range of different 4‐pentynol concentrations indicates a high stability of the SF5‐radical in the presence of relatively high alcohol concentrations. Similar results were obtained using α‐methyl styrene 2 as the second substrate. Here the relative yields of products 5 : 9 could be changed from more than 3 : 1 using 3 equiv. of 4‐pentynole to 1 : 1 using 10 equiv. (Figure S91). Due to the quite aggressive reaction conditions paired with the sensitivity of the radicals involved, the yields could not be further increased due to several side reactions and photocatalyst decomposition as reported in previous studies.[ 20 , 21 ] Higher photocatalyst concentrations lower the product yields likely due to overreduction of the transients. This hypothesis is supported by formation of sulfone 24 that we isolated as a side‐product during the synthesis of 8 (see Figure 2, insert).
Figure 3.

Yields of 4 and 8 and the total yields of both products depending on the concentration of 4‐pentynol. The yields were determined by 19F NMR spectroscopy. Reaction conditions: 0.20 M 1, 10 mol % BEt3, 10 mol% 3, 22 h, 20 °C, MeCN, 368 nm LED. 2.8 bar SF6.
At first glance, the observed concentration dependence hints a competitive trapping of radical cation 1 .+ by either 4‐pentynole as nucleophile or the SF5 radical. In this scenario, the relative rate of formation of the methylene‐type radical 6 . and cation 12+ decide on the formation of the cyclic product 8 vs. the acyclic product 4. However, this reaction pathway does not seem to be reasonable due to the initial formation of the oxonium‐type radical cation 16 .+. Therefore, we performed theoretical investigations applying DFT on the level of the M06‐2X functional as well as DLPNO‐CCSD(T) to shine light on the mechanism.[ 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ] All investigated levels of theory are in qualitative alignment on each single reaction step except for the almost isoenergetic isomers of vinyl radical cation 14+. As expected, only trapping of 1 + by the SF5 radical forming 12 + is thermodynamically strongly favored (−37.5 kcal/mol), whereas the direct addition of the alkyne under formation of 16 .+ is strongly endergonic, which has been confirmed by theory (+22.7 kcal/mol). These results strongly support the proposed reaction pathway to the open chain product 4 but cannot explain the concentration dependence of both reaction products. To explain the observed behavior one need to take into consideration the strong basicity of anhydrous fluoride which has been shown previously in several reports; its pKa (HF) in MeCN has been estimated to be 25.2. [48] We therefore propose a protolytic pre‐equilibrium between the in‐situ generated anhydrous fluoride anion (from SF6) and 4‐pentynol that could fully explain the observed concentration dependence of the reaction outcome. Indeed, the Gibbs free energy for the addition of the 4‐pentoxide anion generating the methylene‐type radical 6 . is about −37.0 kcal/mol, clearly exergonic, and the reaction therefore fully irreversible. Furthermore, 6 . is rapidly converted into the intermediate 10 . by 7‐exo‐dig‐cyclisation according to Baldwin's rules.[ 49 , 50 , 51 ] The calculated Gibbs free energy difference between 6 . and 10 . is −13.2 kcal/mol indicates a strongly exergonic cyclisation forming the seven‐membered ring 10 .. This result is in qualitative alignment with similar radical cyclization reactions reported in literature.[ 52 , 53 ] It is therefore unreasonable that a bimolecular trapping by the SF5 radical can compete with the ring closure reaction, but it cannot be fully excluded. We therefore propose the vinyl radical 10 . as key intermediate to the cyclization product. To rule out the direct alkyne addition to radical cation 1 .+ we carried out a control reaction using 1‐octyne as substrate lacking the tethered hydroxyl function for intramolecular cyclization.
This reaction does not show significant amounts of any pentafluorosulfanylation product. It agrees well with our theoretical prediction that this step to 14+ is entropically highly unfavourable by TΔSR=−14.3 kcal/mol. This clearly indicates that the tethering of the hydroxy function of 4‐pentynol onto the radical cation 1 .+ is required prior to the alkyne addition reaction. Furthermore, this result also confirms the effective abstraction of the fluoride anion by triethylborane preventing the formation of the competing addition products of SF6 to 15 and 17, respectively. We assume that this reaction mechanism applies also for substrate 2.
The configuration of the pentafluorosulfanylated double bond was investigated by NOE measurements as well as by XRD analysis of 8. Remarkably, all products showed exclusively (E)‐configuration of the double bond. To investigate the reason for the pronounced stereoselectivity minimum energies of both intermediate radicals have been calculated. Both configurations, (E)‐isomer 10 .‐E and (Z)‐isomer 10 .‐Z, differ only by about 0.1 kcal/mol in energy. Hence, the stereoselectivity cannot be attributed to thermodynamic control by the vinyl radical intermediate 10 .. Regarding the trajectories of the radical‐radical recombination, the selectivity could be attributed to pure kinetic differences caused by the steric bulk of the quartenary carbon center (Figure 4).
Figure 4.

Proposed mechanism controlling the formation of the acylic products 4 and 5 vs. the ring closed products 8 and 9 during photoredox catalytic pentafluorosulfanylation of substrates 1 and 2, respectively, in the presence of 1‐pentynol. Geometry optimizations and frequency calclations: DFT/def2‐TZVP/M06‐2X, 293 K, CPCM (MeCN). Refinement of electronic energies: DLPNO‐CCSD(T)/def2‐TZVP/CPCM (MeCN).
The yields were generally determined by 19F NMR spectroscopy due to the fact that the fluorinated compounds are hard to purify. However, to validate our 19F‐NMR quantification protocol we recently could show a good alignment of the isolated yield of a representative SF5‐product with the yield determined by 19F NMR spectroscopy. [21] The substrate scope of this reaction was elucidated with different 1‐alkynols, and the corresponding products are formed according to Baldwin's cyclization rules. 5‐, 6‐ and 7‐exo‐dig ring closures were obtained (Figure 5). Our method is not restricted to the formation of the achiral oxepane 8, but was also be used to prepare the chiral oxepane 9 as racemic mixture. The variation of the chain length of the 1‐alkynol yielded differently sized heterocycles, including tetrahydropyranes 18 and 21, as well as tetrahydrofurans 19 and 22. A significant decrease in the product yields was observed by introducing α‐substitution to the 1‐alkynol, shutting down the reaction by quaternization of the α‐carbon due to excess steric bulk and rigidity. While the monosubstituted 4‐methylheptyn‐3‐ol reduced the yield of product 20 to below 10 %, the use of 2‐methylbut‐3‐yn‐2‐ol generated only the addition product of SF6 instead of 23. This effect could be attributed to the steric bulk and decreased nucleophilicity. Our attempts to further expand the substrate scope by 5‐hexyn‐1‐ol as reagent towards oxocanes yielded a different and unexpected product. We isolated only the labile double substitution products 25 and 26 that are formed after ring opening of the unfavorably strained ring and consecutive trapping of the cation by a second 5‐hexynole.
Figure 5.
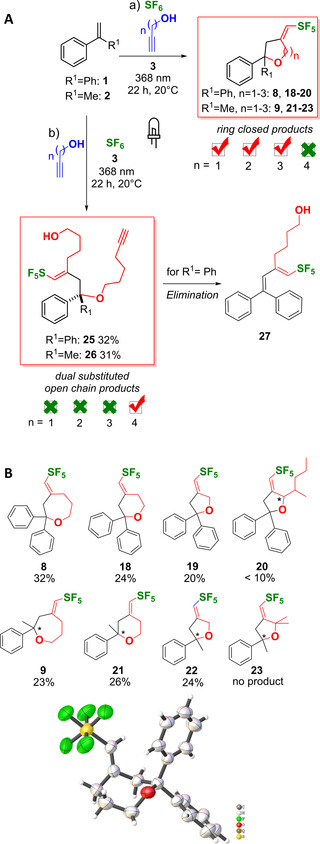
A: Overview of reactions: a) 0.20 mmol 1 or 2 (0.2 M in MeCN), 20.0 equiv. 1‐alkynol, 10 mol% BEt3, 10 mol% 3, MeCN, 20 °C, 368 nm, 2.8 bar SF6. b) 1.00 mmol 1 or 2 (0.2 M in MeCN), 20.0 eq. 1‐hexynol, 10 mol% BEt3, 10 mol% 3, MeCN, 20 °C, 368 nm, 2.8 bar SF6. B: Substrate scope. Image: XRD structure of the isolated product 8. Volume corrections (alcohol volume) neglected except for optimization process of the lead compound to facilitate the protocol. The yields were determined by 19F NMR spectroscopy.
Furthermore, formation of the elimination product 27 was observed after storage for several days or during purification of 25 on silica. Remarkably, during this study we also isolated sulfone 24 as side product that was identified by XRD, NMR spectroscopy and FAB‐MS (Figure S41–S44, S79–S80, Table S2). This is an important result because the formation of 24 hints for the first time at the “overreduction” of the primarily formed SF5 species, however the detailed mechanism of formation is not clear yet.
In conclusion, we present a new protocol to synthesize pentafluorosulfanylated and oxygen‐containing heterocyclic compounds ranging from five‐ to seven‐membered rings in a one‐step protocol by photoredox catalysis. Although the yields are only in the range of 20 % to 32 %, it is important to point out that this advanced method shows proof of concept that even complex transformations could be realized under SF6 activating conditions by precise finetuning of the kinetics. The method therefore expands the scope of accessible SF5 containing chemical space. However, the method suffers from catalyst decomposition under the highly aggressive reaction conditions and further improvements of substrate scope and ‐tolerance need to be carried out in the future. The method utilizes SF6 as non‐toxic pentafluorosulfanylation reagent and could be applied to the preparation of pentafluorosulfanylated achiral and chiral products with remarkable structural complexity, in particular including precious 5‐, 6‐ and 7‐membered heterocycles. The corresponding eight‐membered cyclic product is not formed due to significant ring strain; instead, ring‐opened products have been formed in comparable yields. We support, for the first time, the proposed reaction pathways after photoredox SF6 activation by extensive theoretical calculations. While the method today is still limited to the use of α‐substituted styrenes, the substrate scope is broad with respect to the alcohols as external nucleophiles and can potentially be broadened in the future by tackling the highly reactive reaction conditions. Our photoredox catalytic method combines the disposal of SF6 (after any technical applications) and completely avoids the highly toxic “conventional” SF5‐transfer reagents SF5Cl, SF5Br and S2F10. The combined experimental and computational approach allowed to gain important insights into the operating reaction mechanism, indicating that the addition of the nucleophile precedes the spin trapping step to yield the heterocyclic products. These pentafluorosulfanylated products of high structural complexity cannot be simply synthesized by other methods and shows once more that SF6 can act as a precious pentafluorosulfanylation reagent. We hope that this work will encourage further investigations on the use of SF6 in pentafluorosulfanylation chemistry pushing the frontiers towards the development of less aggressive and more selective protocols in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/16‐2) and KIT is gratefully acknowledged. D. R. thanks the Landesgraduiertenstiftung Baden‐Württemberg for a doctoral fellowship and the GRK 1626 “Chemical photocatalysis” for participation in their qualification program. D. R. acknowledges the Swiss National Science Foundation (SNSF, CRSK‐2 195863) for financial support of this research and thanks Prof. Antonio Togni for fruitful discussions and continuous support. D. R. further thanks Prof. Erick. M. Carreira for his support. Furthermore, we thank Prof. Frank Breher and Prof. Michael A. R. Meier for sharing parts of their infrastructure and ETH Zurich for providing access to the Euler HPC Cluster. Open access funding enabled and organized by Projekt DEAL.
D. Rombach, B. Birenheide, H.-A. Wagenknecht, Chem. Eur. J. 2021, 27, 8088.
Dedicated to Professor Antonio Togni on the occasion of his 65th birthday.
Contributor Information
Dr. David Rombach, Email: drombach@ethz.ch.
Prof. Hans‐Achim Wagenknecht, Email: Wagenknecht@kit.edu.
References
- 1. Silvey G. A., Cady G. H., J. Am. Chem. Soc. 1950, 72, 3624–3626. [Google Scholar]
- 2. Sowaileh M. F., Hazlitt R. A., Colby D. A., ChemMedChem 2017, 12, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 3. Welch J. T., Lim D. S., Bioorg. Med. Chem. 2007, 15, 6659–6666. [DOI] [PubMed] [Google Scholar]
- 4. Altomonte S., Baillie G. L., Ross R. A., Riley J., Zanda M., RSC Adv. 2014, 4, 20164–20176. [Google Scholar]
- 5. Zhang J., Zhou J. Z., Xu Z. P., Li Y., Cao T., Zhao J., Ruan X., Liu Q., Qian G., Environ. Sci. Technol. 2014, 48, 599–606. [DOI] [PubMed] [Google Scholar]
- 6. Kirsch P., Binder J. T., Lork E., Röschenthaler G.-V., J. Fluorine Chem. 2006, 127, 610–619. [Google Scholar]
- 7. Kirsch P., J. Fluorine Chem. 2015, 177, 29–36. [Google Scholar]
- 8. Gautam P., Yu C. P., Zhang G., Hillier V. E., Chan J. M. W., J. Org. Chem. 2017, 82, 11008–11020. [DOI] [PubMed] [Google Scholar]
- 9. Savoie P. R., Welch J. T., Chem. Rev. 2015, 115, 1130–1190. [DOI] [PubMed] [Google Scholar]
- 10. Sergeeva T. A., W. R. Dolbier Jr. , Org. Lett. 2004, 2417–2419. [DOI] [PubMed] [Google Scholar]
- 11. Aït-Mohand S., W. R. Dolbier Jr , Org. Lett. 2002, 4, 3013–3015. [DOI] [PubMed] [Google Scholar]
- 12. W. R. Dolbier Jr. , Aït-Mohand S., Schertz T. D., Sergeeva T. A., Cradlebaugh J. A., Mitani A., Gard G. L., Winter R. W., Thrasher J. S., J. Fluorine Chem. 2006, 127, 1302–1310. [Google Scholar]
- 13. Umemoto T., Garrick L. M., Saito N., Beilstein J. Org. Chem. 2012, 8, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pitts C. R., Bornemann D., Liebing P., Santschi N., Togni A., Angew. Chem. Int. Ed. 2019, 58, 1950–1954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1970–1974. [Google Scholar]
- 15. Iakobson G., Pošta M., Beier P., J. Fluorine Chem. 2018, 213, 51–55. [Google Scholar]
- 16. Altomonte S., Zanda M., J. Fluorine Chem. 2012, 143, 57–93. [Google Scholar]
- 17. Rueping M., Nikolaienko P., Lebedev Y., Adams A., Green Chem. 2017, 19, 2571–2575. [Google Scholar]
- 18. McTeague T. A., Jamison T. F., Angew. Chem. Int. Ed. 2016, 55, 15072–15075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15296–15299; [Google Scholar]; Angew. Chem. 2016, 128, 15296–15299; [Google Scholar]; Angew. Chem. Int. Ed. 2016, 55, 15072–15075. [DOI] [PubMed] [Google Scholar]
- 19. Buß F., Mück-Lichtenfeld C., Mehlmann P., Dielmann F., Angew. Chem. Int. Ed. 2018, 57, 4951–4955; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5045–5049; [Google Scholar]; Angew. Chem. 2018, 130, 5045–5049; [Google Scholar]; Angew. Chem. Int. Ed. 2018, 57, 4951–4955. [DOI] [PubMed] [Google Scholar]
- 20. Rombach D., Wagenknecht H.-A., ChemCatChem 2018, 10, 2955–2961. [Google Scholar]
- 21. Rombach D., Wagenknecht H.-A., Angew. Chem. Int. Ed. 2020, 59, 300–303; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 306–310. [Google Scholar]
- 22. Taylor R. D., MacCoss M., Lawson A. D. G., J. Med. Chem. 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- 23. Lee K.-S., Li G., Kim S. H., Lee C.-S., Woo M.-H., Lee S.-H., Jhang Y.-D., Son J.-K., J. Nat. Prod. 2002, 65, 1707–1708. [DOI] [PubMed] [Google Scholar]
- 24. Mayer A. M. S., Gustafson K. R., Eur. J. Cancer 2008, 44, 2357–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alvarez E., Candenas M.-L., Perez R., Ravelo J. L., Delgado M., Chem. Rev. 1995, 95, 1953–1980. [Google Scholar]
- 26. Barbero H., Díez-Poza C., Barbero A., Barbero H., Díez-Poza C., Barbero A., Mar. Drugs 2017, 15, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shimizu Y., Chou H. N., Bando H., Van Duyne G., Clardy J., J. Am. Chem. Soc. 1986, 108, 514–515. [DOI] [PubMed] [Google Scholar]
- 28. Basu S., Ellinger B., Rizzo S., Deraeve C., Schürmann M., Preut H., Arndt H.-D., Waldmann H., Proc. Natl. Acad. Sci. USA 2011, 108, 6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Speck F., Rombach D., Wagenknecht H.-A., Beilstein J. Org. Chem. 2019, 15, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poelma S. O., Burnett G. L., Discekici E. H., Mattson K.-M., Treat N. J., Luo Y., Hudson Z. M., Shankel S. L., Clark P. G., Kramer J. W., Hawker C. J., Read de Alaniz J., J. Org. Chem. 2016, 81, 7155–7160. [DOI] [PubMed] [Google Scholar]
- 31. Wagner C., Wagenknecht H.-A., Chem. Eur. J. 2005, 11, 1871–1876. [DOI] [PubMed] [Google Scholar]
- 32. Akhgarnusch A., Höckendorf R. F., Beyer M. K., J. Phys. Chem. A 2015, 119, 9978–9985. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Sauers I., Harman G. A., J. Phys. D. Appl. Phys. 1992, 25, 761–773; [Google Scholar]
- 33b. Sauers I., Harman G. A., J. Phys. D. Appl. Phys. 1992, 25, 774–782. [Google Scholar]
- 34. Chen C. L., Chantry P. J., J. Chem. Phys. 1979, 71, 3897–3907. [Google Scholar]
- 35. Streit G. E., J. Chem. Phys. 1982, 77, 826–833. [Google Scholar]
- 36. Christophorou L. G., Olthoff J. K., Int. J. Mass Spectrom. 2001, 205, 27–41. [Google Scholar]
- 37. Troe J., Miller T. M., Viggiano A. A., J. Chem. Phys. 2007, 127, 244304. [DOI] [PubMed] [Google Scholar]
- 38. Ghosh I., Ghosh T., Bardagi J. I., König B., Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]
- 39. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 40. Zhao Y., Truhlar D. G., J. Phys. Chem. A 2008, 112, 1095–1099. [DOI] [PubMed] [Google Scholar]
- 41. Bartlett R. J., Musail M., Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar]
- 42. Neese F., Hansen A., Liakos D. G., J. Chem. Phys. 2009, 131, 064103. [DOI] [PubMed] [Google Scholar]
- 43. Riplinger C., Sandhoefer B., Hansen A., Neese F., J. Chem. Phys. 2013, 139. 134101. [DOI] [PubMed] [Google Scholar]
- 44. Liakos D. G., Sparta M., Kesharwani M. K., Martin J. M. L., Neese F., J. Chem. Theory Comput. 2015, 11, 1525–1539. [DOI] [PubMed] [Google Scholar]
- 45. Liakos D. G., Neese F., J. Chem. Theory Comput. 2015, 11, 4054–4063. [DOI] [PubMed] [Google Scholar]
- 46. Neese F., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- 47. Neese F., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar]
- 48. Nicoleti C. R., Marini V. G., Zimmermann L. M., Machado V. G., J. Braz. Chem. Soc. 2012, 23, 1488–1500. [Google Scholar]
- 49. Baldwin J. E., J. Chem. Soc. Chem. Commun. 1976, 18, 734–736. [Google Scholar]
- 50. Alabugin I. V., Gilmore K., Manoharan M., J. Am. Chem. Soc. 2011, 133, 12608–12623. [DOI] [PubMed] [Google Scholar]
- 51. Gilmore K., Mohamed R. K., Alabugin I. V., WIREs Comput. Mol. Sci. 2016, 6, 487–514. [Google Scholar]
- 52. Marco-Contelles J., Opazo E., Tetrahedron Lett. 2000, 41, 5341–5345. [Google Scholar]
- 53. Horning B. D., MacMillan D. W. C., J. Am. Chem. Soc. 2013, 135, 6442–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary