

Abstract
Objective
Blood tests to monitor disease activity, attack severity, or treatment impact in neuromyelitis optica spectrum disorder (NMOSD) have not been developed. This study investigated the relationship between serum glial fibrillary acidic protein (sGFAP) concentration and NMOSD activity and assessed the impact of inebilizumab treatment.
Methods
N‐MOmentum was a prospective, multicenter, double‐blind, placebo‐controlled, randomized clinical trial in adults with NMOSD. sGFAP levels were measured by single‐molecule arrays (SIMOA) in 1,260 serial and attack‐related samples from 215 N‐MOmentum participants (92% aquaporin 4‐immunoglobulin G‐seropositive) and in control samples (from healthy donors and patients with relapsing–remitting multiple sclerosis).
Results
At baseline, 62 participants (29%) exhibited high sGFAP concentrations (≥170 pg/ml; ≥2 standard deviations above healthy donor mean concentration) and were more likely to experience an adjudicated attack than participants with lower baseline concentrations (hazard ratio [95% confidence interval], 3.09 [1.6–6.1], p = 0.001). Median (interquartile range [IQR]) concentrations increased within 1 week of an attack (baseline: 168.4, IQR = 128.9–449.7 pg/ml; attack: 2,160.1, IQR = 302.7–9,455.0 pg/ml, p = 0.0015) and correlated with attack severity (median fold change from baseline [FC], minor attacks: 1.06, IQR = 0.9–7.4; major attacks: 34.32, IQR = 8.7–107.5, p = 0.023). This attack‐related increase in sGFAP occurred primarily in placebo‐treated participants (FC: 20.2, IQR = 4.4–98.3, p = 0.001) and was not observed in inebilizumab‐treated participants (FC: 1.1, IQR = 0.8–24.6, p > 0.05). Five participants (28%) with elevated baseline sGFAP reported neurological symptoms leading to nonadjudicated attack assessments.
Interpretation
Serum GFAP may serve as a biomarker of NMOSD activity, attack risk, and treatment effects. ANN NEUROL 2021;89:895–910
Neuromyelitis optica spectrum disorder (NMOSD) is a rare, chronic, autoimmune, inflammatory disorder of the central nervous system (CNS) characterized by recurrent attacks of optic neuritis and longitudinally extensive transverse myelitis, whereas brain and brainstem inflammation are less frequently observed. 1 Attacks can be severe, with incomplete recovery leading to cumulative disability.
Traditionally, immunosuppressants, such as corticosteroids, azathioprine, and mycophenolate mofetil, 2 and rituximab, 3 , 4 , 5 are used to prevent attacks, although clinical evidence for their effectiveness is limited and based on uncontrolled, retrospective, or small studies. Several new therapies, including eculizumab, satralizumab, and inebilizumab, were proven to be effective. 6 , 7 , 8 , 9 Inebilizumab is a humanized, affinity‐optimized, afucosylated immunoglobulin G (IgG) 1κ monoclonal antibody that binds to the B‐cell‐specific surface antigen CD19 and depletes a wide range of B cells, as demonstrated in preclinical animal models 10 and in systemic sclerosis, 11 relapsing forms of multiple sclerosis (MS), 12 and NMOSD. 8 The efficacy and safety of inebilizumab treatment were evaluated in participants with NMOSD in the randomized, double‐masked, placebo‐controlled N‐MOmentum study. 8 Compared with placebo, inebilizumab reduced the risk of an attack by 73% (hazard ratio [HR] = 0.272, p < 0.0001) and reduced the risk of disability worsening by 63% (odds ratio = 0.370, p = 0.0049).
The presence of serum autoantibodies against aquaporin 4 (AQP4) is a distinct feature of NMOSD and distinguishes it from MS. 13 , 14 , 15 AQP4 is a water channel protein expressed predominantly on astrocytes and concentrated on the perivascular foot processes. Autoantibodies to AQP4 are pathogenic in NMOSD, 16 , 17 resulting in targeted astrocyte dysfunction and destruction. Astrocyte injury results in the release of astrocyte contents in cerebrospinal fluid (CSF) and serum, 18 , 19 , 20 including glial fibrillary acidic protein (GFAP), an intermediate filament protein predominantly expressed by astrocytes that forms the astrocyte cytoskeleton. 21 Therefore, serum GFAP (sGFAP) could be a biomarker of disease activity in NMOSD.
The aims of the current study were to investigate the relationship between prospectively sampled sGFAP concentration and disease activity in participants from the N‐MOmentum clinical trial and to assess the impact of inebilizumab on sGFAP levels compared with placebo, a predefined, exploratory study outcome.
Methods
Study Design and Participants
The sGFAP concentrations were assessed in participants from the N‐MOmentum study and in reference cohorts of healthy donors and patients with relapsing–remitting MS (RRMS).
Full details of the N‐MOmentum study, including a trial profile, were published. 8 In brief, the N‐MOmentum study was an international, multicenter, randomized, double‐blind, placebo‐controlled, phase II/III trial with an open‐label extension phase (ClinicalTrials.gov, NCT02200770). Among the clinically relevant inclusion criteria that helped define the study population, criteria important for interpreting this manuscript were the requirement for subjects with a recent attack to have at least 4 weeks during which their attack symptoms were stable or improving prior to randomization, and that trial participants were not eligible for the study if they had received intravenous immunoglobulin (IVIG) treatment within 1 month prior to randomization, or had received doses of methotrexate or a range of other immunosuppressive medications (cyclosporin, cyclophosphamide, eculizumab, mitoxantrone, natalizumab, or tocilizumab) in the 3 months prior to randomization. In addition, the study was designed to investigate inebilizumab as monotherapy; no on‐study immunotherapy was allowed beyond the tapering dose of steroids given to all participants after infusion of inebilizumab or placebo. Baseline serum sampling for sGFAP occurred prior to any of these study‐related medications. The primary end point was the time to an adjudicated NMOSD attack during the randomized controlled period (RCP). An attack was defined by protocol‐defined criteria 1 upon neurological evaluation that was adjudicated by an independent committee within 17 days. Attack severity was graded according to the Opticospinal Impairment Scale (OSIS), 22 , 23 which characterizes attacks as “minor” or “major” on the basis of changes in domain‐specific neurological scores relative to the previous assessment. Investigators had the option of providing rescue therapy following an attack, at their discretion and according to clinical need. Investigators were advised to give rescue therapy after the attack assessment (including serum follow‐up sampling), as not to influence the data obtained.
Two reference cohorts of individuals without NMOSD, one comprising age‐ and sex‐matched healthy donors (n = 85) and another comprising patients with moderate‐to‐severe RRMS (baseline Expanded Disability Status Scale [EDSS] score >3.5; n = 23) from the United States and Europe described previously, 12 were used as controls in the assessment of the significance of sGFAP concentration in NMOSD attack prediction and attack severity.
The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the principles of the Declaration of Helsinki. An institutional review board or ethics committee at each study site approved the protocol. Written informed consent was obtained from all participants.
Sample Collection and Analysis
Blood was collected from N‐MOmentum participants during RCP study visits at baseline (day 1) and on days 15, 29, 57, 85, 113, 155, and 197, and during any assessment visit for new or worsening NMOSD symptoms. For the reference cohorts, 12 participants were untreated and blood was collected at the single baseline visit. Ten healthy donors underwent repeated longitudinal sampling for serial sGFAP measurements. As validated in serum samples from patients with MS (aged 18–65 years with confirmed relapsing forms of MS defined by the McDonald 2010 criteria), with at least one relapse in the last 3 years, EDSS score ≤6.5, normal baseline CD19 B‐cell count (>80 cells/μl) and no more than 20 gadolinium (Gd)‐enhancing lesions on magnetic resonance imaging [MRI]) or traumatic brain injury, 24 , 25 the sGFAP concentration was determined using the single‐molecule arrays (SIMOA) GFAP assay by Quanterix (Lexington, MA, USA). Elevated sGFAP concentrations were defined as being ≥2 standard deviations (SDs) above the healthy donor mean concentration (≥170 pg/ml) according to established laboratory procedures. 26
Statistical Analysis
NMOSD attacks were analyzed using Cox proportional hazards regression. Statistical significance of regression coefficients was assessed using Wald's test. Differences in sGFAP concentration between groups were evaluated using the Mann–Whitney U test for continuous measures, and either the Cochran–Armitage test or Fisher's exact test to test differences in proportions. Paired changes in sGFAP concentration from baseline were assessed using the Wilcoxon signed‐rank test. The Bonferroni correction was applied as a multiple testing correction when appropriate and is described in the corresponding figure legends. All statistical analysis was performed in R version 3.6.2.
Results
Study Participants
In total, 215 participants (198 of whom were AQP4‐IgG seropositive) provided 1,260 serial and NMOSD attack‐related samples for sGFAP concentration analysis. Most study participants were women (194/215 [90%]) and approximately half were White (110/215 [51%]). Demographics were largely similar between the inebilizumab (n = 164) and placebo (n = 51) groups (Table 1). In addition, samples for sGFAP concentration analysis were obtained from the reference cohorts of age‐ and sex‐matched healthy donors (n = 85) and untreated individuals with RRMS (n = 23; see Table 1).
TABLE 1.
Participant Demographics and Characteristics at Baseline (ITT sGFAP Analysis Set)
| Demographic/characteristic | NMOSD placebo (n = 51) | NMOSD inebilizumab (n = 164) | RRMS (n = 23) | HD (n = 85) |
|---|---|---|---|---|
| Age, yr | ||||
| Mean (SD) | 43.4 (14.0) | 43.0 (11.2) | 45.3 (12.3) | 43.4 (12.9) |
| Median (range) | 43 (20–74) | 43 (18–73) | 44 (21–63) | 43 (20–72) |
| Sex | ||||
| F | 45 (88) | 149 (91) | 14 (61) | 88 (91) |
| Race/ethnicity | ||||
| Asian | 7 (14) | 38 (23) | 0 (0) | 0 (0) |
| Black or African American | 5 (10) | 14 (9) | 3 (13) | 5 (6) |
| Hispanic or Latino | 14 (27) | 27 (17) | 1 (4) | 13 (15) |
| White | 23 (51) | 82 (51) | 17 (74) | 63 (74) |
| Other | 2 (4) | 3 (8) | 2 (4) | 1 (1) |
| Disease duration, yr | ||||
| Mean (SD) | 2.8 (3.5) | 2.4 (3.3) | 8.9 (9.9) | N/A |
| Median (IQR) | 1.3 (0.2–16.9) | 1.1 (0.1–22.2) | 5.3 (0.2–36.0) | N/A |
| Baseline EDSS score | ||||
| Mean (SD) | 4.2 (1.7) | 3.8 (1.8) | 3.9 (1.7) | N/A |
| Median (IQR) | 4.0 (1.0–8.0) | 3.5 (0.0–8.0) | 4.0 (0.0–6.5) | N/A |
| Serostatus | ||||
| AQP4‐IgG seropositive | 47 (92) | 151 (92) | N/A | N/A |
| MOG‐IgG seropositive | 1 (2) | 6 (4) | N/A | N/A |
| Double seronegative | 3 (6) | 7 (4) | N/A | N/A |
| sGFAP >170 pg/ml (%) | 16 (31) | 46 (28) | 2 (9) | 3 (4) |
Data are n (%) unless stated otherwise. Race/ethnicity was self‐reported by participants.
AQP4‐IgG = aquaporin 4‐immunoglobulin G; EDSS = Expanded Disability Status Scale; HD = healthy donors; IQR = interquartile range; ITT = intention‐to‐treat; MOG‐IgG = myelin oligodendrocyte glycoprotein‐immunoglobulin G; N/A = not applicable; NMOSD = neuromyelitis optica spectrum disorder; RRMS = relapsing–remitting multiple sclerosis; SD = standard deviation; sGFAP = serum glial fibrillary acidic protein.
sGFAP Concentration at Baseline in Participants with NMOSD or RRMS and Healthy Donors
The sGFAP levels were defined as elevated when they reached or surpassed 170 pg/ml, a threshold calculated as 2 SDs from the mean sGFAP concentration in healthy donors. At study baseline, elevated sGFAP levels were observed in significantly more participants with NMOSD (62/215 [29%]) than individuals with RRMS (2/23 [9%]) or healthy donors (2/85 [2.4%]); p < 0.05 and p < 0.001, respectively (Fig 1A). Median (interquartile range [IQR]) sGFAP concentration was 128.3 pg/ml (IQR = 92.0–181.2 pg/ml) for participants with NMOSD, compared with 71.3 pg/ml (IQR = 55.6–102.2 pg/ml) for age‐ and sex‐matched healthy donors and 97.5 pg/ml (76.5–131.4 pg/ml) for individuals with RRMS. Two AQP4‐IgG‐seronegative participants (one of whom was myelin oligodendrocyte glycoprotein (MOG)‐IgG seropositive) had elevated sGFAP levels at baseline (see Fig 1A). For both patients with NMOSD and healthy donors, we observed a modest but clear correlation of sGFAP levels with age (Fig 1B, C) and EDSS score (Table 2), whereas sex or race/ethnicity had no effect.
FIGURE 1.
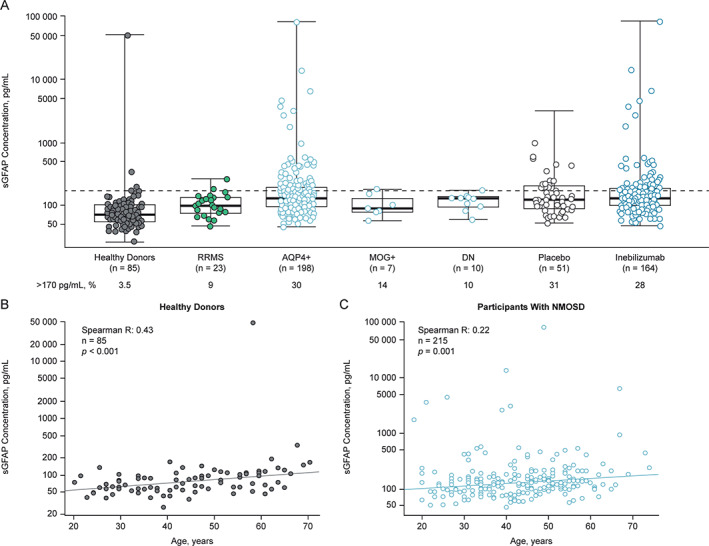
Baseline sGFAP concentration in the reference and NMOSD cohorts, and correlation of sGFAP with age. *p < 0.05; **p < 0.01; **p < 0.001. (A) Graph shows baseline sGFAP concentrations by cohort and antibody serostatus. Dashed line represents 2 SDs from the healthy donor mean (170 pg/ml). Box and whiskers represent sample quartiles. Statistical significance of differences in sGFAP concentration between groups was assessed using the Mann–Whitney U test. (B and C) Graphs show the correlation between age and baseline sGFAP concentration in both heathy donors and NMOSD participants. AQP4+ = aquaporin 4‐immunoglobulin G seropositive; DN = double negative serostatus; MOG+ = myelin oligodendrocyte glycoprotein‐immunoglobulin G seropositive; NMOSD = neuromyelitis optica spectrum disorder; RRMS = relapsing–remitting multiple sclerosis; SD = standard deviation; sGFAP = serum glial fibrillary acidic protein.
TABLE 2.
Hazard Regressions of sGFAP Versus RCP Attack Frequency, Adjusted for Covariates
| Variable name | HR (95% CI) | p value | Association between day 1 RCP sGFAP and continuous variables | Association between day 1 RCP sGFAP and categorical variables | ||
|---|---|---|---|---|---|---|
| Spearman R (95% CI) | Spearman test: p value | AUC (95% CI) | Mann–Whitney U test: p value | |||
| Baseline sGFAP >170 pg/ml | 3.20 (1.63, 6.26) | <0.001 | ‐ | ‐ | ‐ | ‐ |
| Inebilizumab treatment | 0.28 (0.15, 0.53) | <0.001 | ‐ | ‐ | 0.51 (0.42, 0.61) | 0.81 |
| Attack <90 days prior to RCP | 0.45 (0.11, 2.07) | 0.32 | ‐ | ‐ | 0.52 (0.34, 0.70) | 0.80 |
| Age | 0.98 (0.96, 1.01) | 0.16 | 0.22 (0.09, 0.34) | 0.001 | ‐ | ‐ |
| Baseline EDSS | 1.08 (0.89, 1.31) | 0.43 | 0.32 (0.20, 0.44) | <0.001 | ‐ | ‐ |
AUC = area under the curve; CI = confidence interval; EDSS = expanded disability status scale; HR = hazard ratio; RCP = randomized controlled period; sGFAP = serum glial fibrillary acidic protein.
Participants with Elevated sGFAP Concentrations Are at Increased Risk of Attacks
Participants with NMOSD and with an elevated baseline sGFAP concentration were at increased risk of experiencing an adjudicated NMOSD attack. Analysis of all study participants showed that 19 of 62 patients (31%) with an elevated sGFAP concentration at baseline experienced an adjudicated NMOSD attack during the 28‐week RCP versus 19 of 153 patients (12%) without elevated sGFAP, equating to 3 times the risk of an attack during the RCP (HR = 3.09, 95% confidence interval [CI] = 1.57–6.10, p = 0.001). A similar pattern was observed for both the placebo and the inebilizumab groups (for placebo: HR = 2.35, 95% CI = 0.94–5.87, p = 0.06; for inebilizumab: HR = 4.15, 95% CI = 1.67–10.32, p = 0.002; Fig 2A, B).
FIGURE 2.
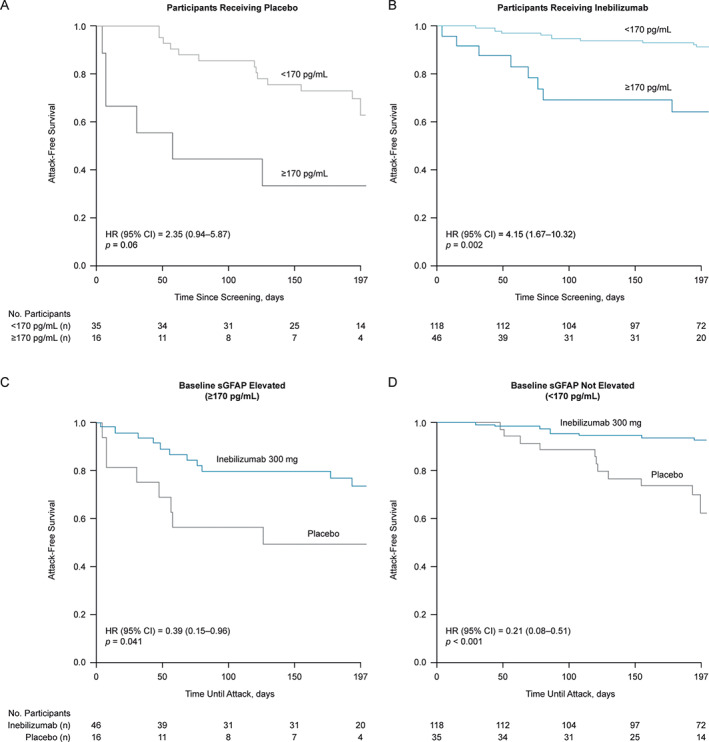
Attack‐free survival in participants with NMOSD according to treatment and baseline sGFAP status. Graphs show Kaplan–Meier plots of time until first NMOSD attack during the RCP in (A) placebo and (B) inebilizumab participants with baseline sGFAP concentration ≥170 pg/ml versus those with baseline sGFAP concentration <170 pg/ml. Statistical significance of difference in time until first adjudicated attack between groups was assessed using Wald's test. (C and D) Graphs show Kaplan–Meier plots of time until first attack between participants receiving inebilizumab or placebo with baseline sGFAP concentration <170 pg/ml or ≥170 pg/ml. Statistical significance of difference in time until first adjudicated attack between groups was assessed using Wald's test. CI = confidence interval; EDSS = Expanded Disability Status Scale; HR = hazard ratio; NMOSD = neuromyelitis optica spectrum disorder; RCP = randomized controlled period; sGFAP = serum glial fibrillary acidic protein.
In participants with elevated baseline sGFAP concentrations, inebilizumab reduced the risk of an adjudicated attack by 61% compared with placebo (HR = 0.39, 95% CI = 0.15–0.96, p = 0.041; Fig 2C). In participants without elevated baseline sGFAP levels, the risk of an adjudicated attack was reduced by 79% with inebilizumab compared with placebo (HR = 0.21, 95% CI = 0.08–0.51, p < 0.001; Fig 2D). The association between sGFAP and upcoming attacks was significant when controlling for recent attacks, age, and baseline EDSS (see Table 2).
sGFAP Concentration Increases during NMOSD Attacks and Correlates with Attack Severity
Significant increases in sGFAP concentrations from baseline were observed for participants within 1 week (either before or after) of adjudicated NMOSD attacks (Fig 3A, median, pre‐attack (baseline) samples: 168.4, IQR = 128.9–449.7 pg/ml; attack samples: 2,160.1, IQR = 302.7–9,455.0 pg/ml, p = 0.0015). Indeed, sGFAP appears to be linearly correlated with upcoming attack risk. Notably, serum sampling was performed prior to initiation of attack therapy, and increases in sGFAP levels were not observed in the majority of samples drawn greater than 1 week prior to attack. An elevated sGFAP concentration (≥170 pg/ml) was observed in 29 of 37 attack samples (78%) compared with 19 of 38 pre‐attack samples (50%). Eight of 37 samples (21.6%) obtained from participants during adjudicated attacks did not have elevated sGFAP levels; 7 were from inebilizumab‐treated participants and 1 was from a placebo‐treated participant receiving placebo (7/20 [35%] vs 1/17 [6%]; Fisher's exact test, p = 0.048).
FIGURE 3.
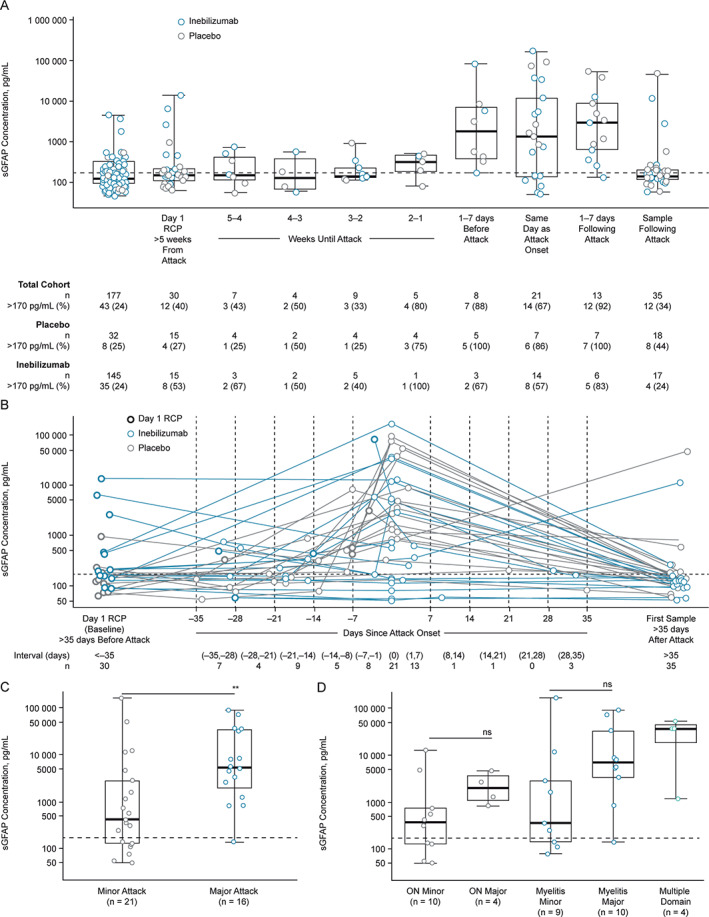
The sGFAP concentration at baseline at visits leading to an adjudicated NMOSD attack, and during attack. (A) Graph shows a boxplot of sGFAP measurements leading up to attack in 38 participants who experienced an adjudicated attack and provided sGFAP measurements. Baseline samples >5 weeks from attack were available in 30 of 38 participants. Eight participants experienced attacks within 5 weeks of the baseline sample collection. (B) Graph shows a profile plot of sGFAP measurements in the days leading up to the attack. Graphs show sGFAP concentration of samples drawn most proximal to attack and within 1 week of adjudicated NMOSD attack by attack severity (C) and organ domain involvement (D), measured by the OSIS. Of the attacks across multiple domains, 4 were minor myelitis attacks, and one sample from myelitis major group displayed sGFAP within the healthy donor range. Box and whiskers represent sample quartiles. Statistical significance between groups was assessed using Mann–Whitney U test. Dashed lines represent 2 SDs from the healthy donor mean (170 pg/ml). NMOSD = neuromyelitis optica spectrum disorder; ON = optic neuritis; OSIS = Opticospinal Impairment Scale; RCP = randomized controlled period; SD = standard deviation; sGFAP = serum glial fibrillary acidic protein.
No significant trend was observed between sGFAP concentration and time since attack within the 1‐week window. Seven samples (5 placebo‐treated and 2 inebilizumab‐treated) of the 8 drawn up to 1 week prior to the attack displayed sGFAP above healthy donor range and were increased relative to each participant's value at baseline (ie, day 1 of the RCP). Similarly, 12 of 13 samples (7 placebo‐ and 6 inebilizumab‐treated participants) drawn in the days after the attack displayed sGFAP above healthy donor range (see Fig 3A). One inebilizumab‐treated subject displayed sGFAP within healthy donor range 2 days after attack onset. Seven out of the 9 samples with sGFAP concentrations below healthy donor range were drawn during attack assessments performed on the same day as attack onset, 6 of which were drawn from inebilizumab‐treated participants (see Fig 3B).
Following the attack assessments, serum samples were drawn again per study protocol at day 15 of the open‐label period (OLP) at the earliest – more than 5 weeks after attack onset for the majority of participants. By this time point, sGFAP largely returned to concentrations comparable to those observed for the given participants at baseline, with 12 of 35 (34%) samples drawn following attack above 170 pg/ml. Eight of these samples were drawn from placebo‐treated participants, and 4 from inebilizumab‐treated subjects. Moreover, 5 samples were drawn earlier than 5 weeks after the attack. Of these 5 samples, 2 (40%) displayed sGFAP concentrations higher than 170 pg/ml, thus similar to the distribution of sGFAP levels in NMOSD samples drawn at baseline (see Fig 3B).
Elevated sGFAP concentration was associated with adjudicated‐attack severity as determined by the OSIS. The sGFAP concentration during an attack was significantly higher in participants who had major attacks than in those who had minor attacks (median fold change [IQR], major attacks: 34.32, IQR = 8.72–107.53; minor attacks: 1.06, IQR = 0.85–7.43, p = 0.023; see Fig 3C). The sGFAP levels were increased during major attacks of both myelitis and optic neuritis (see Fig 3D).
At the time of adjudicated NMOSD attacks, sGFAP concentration increased significantly from baseline in placebo‐treated participants (median fold change, 20.2, IQR = 4.4–98.3, p = 0.001; Fig 4A). This was not the case in the majority of inebilizumab‐treated participants (median fold change, 1.1, IQR = 0.8–24.6, p = 0.31; Fig 4B), although a subset of 7 of 18 (39%) inebilizumab‐treated participants with paired baseline and attack samples displayed significant increases from baseline (median fold change, 75.7, IQR = 24.5–171.4; time course of fold changes from baseline displayed in Fig 4C). Absolute sGFAP levels were also significantly lower in inebilizumab‐treated participants than in the placebo group (median: inebilizumab [n = 20], 653.0, IQR = 139.0–7,227.8 pg/ml; placebo [n = 17], 3,056.1, IQR = 1,091.5–15,858.5 pg/ml, p = 0.048; see Fig 4A, B). None of the 3 inebilizumab‐treated AQP4‐IgG‐seronegative patients who experienced attacks exhibited increases in sGFAP (Fig 4D).
FIGURE 4.
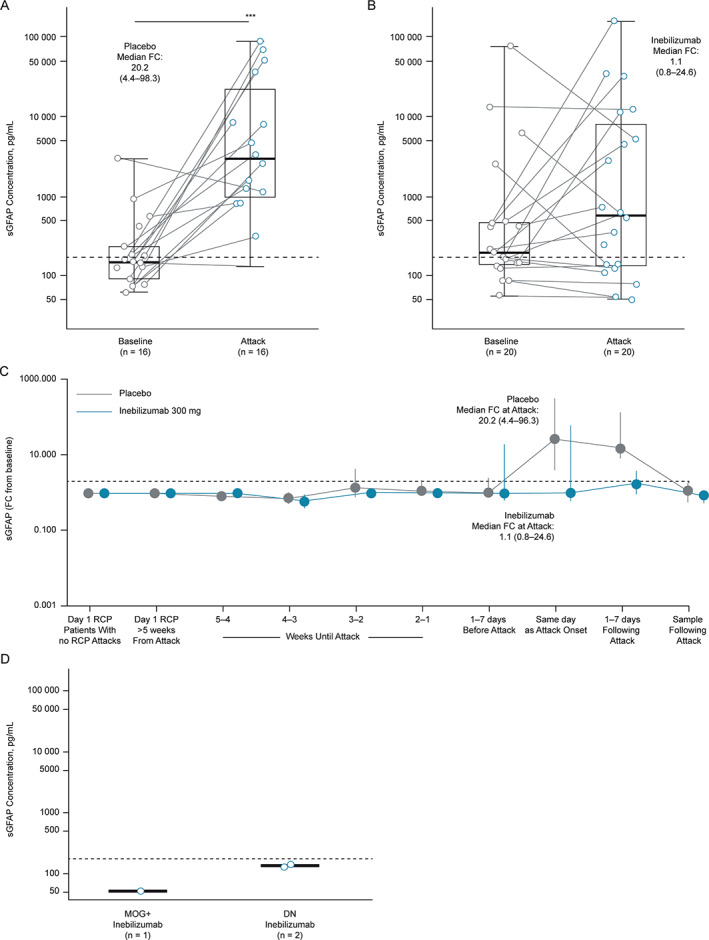
The sGFAP concentration during adjudicated NMOSD attacks. (A and B) Graphs show sGFAP concentration of most proximal samples drawn within 1 week of attack according to treatment in participants with attacks. Statistical significance of increases from baseline was assessed using the Wilcoxon signed‐rank test. Dashed line represents sGFAP concentration 2 SDs from the healthy donor mean (170 pg/ml). (C) Graph displays time course of FC in sGFAP from baseline in the weeks leading up to and proceeding adjudicated attacks. Points represent medians, error bars represent IQR. (D) The sGFAP concentration during adjudicated NMOSD attacks according to treatment group and antibody serostatus. Box and whiskers represent sample quartiles. DN = double negative serostatus; FC = fold change; IQR = interquartile range; MOG+ = myelin oligodendrocyte glycoprotein‐immunoglobulin G seropositive; RCP = randomized controlled period; SD = standard deviation; sGFAP = serum glial fibrillary acidic protein.
sGFAP Concentrations in Participants Who Did Not Experience an Adjudicated NMOSD Attack
For participants who did not experience an adjudicated NMOSD attack during the RCP, sGFAP concentration decreased with inebilizumab treatment after week 4. The percentage reduction from baseline was statistically significant at week 16 (median reduction of 12.9%, IQR = −25.6 to −1.6) and on to the end of the RCP (p < 0.05; Fig 5A). Conversely, there was no significant change in sGFAP levels in participants receiving placebo without attacks (p > 0.05; see Fig 4D). By the end of the RCP, 9 of 26 participants (35%) within the placebo group had elevated sGFAP concentrations, compared with 19 of 117 participants (16%) within the inebilizumab group (Fig 5B).
FIGURE 5.
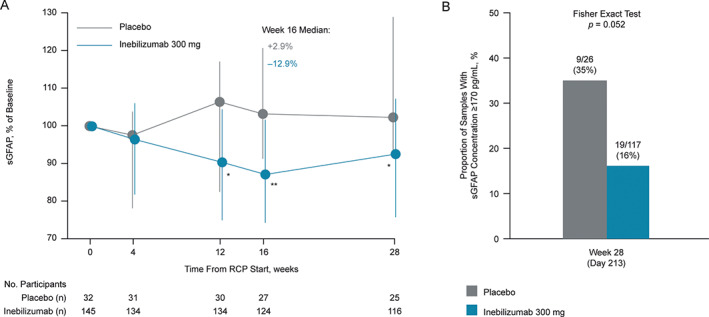
Change in sGFAP concentration from baseline in participants who did not experience an adjudicated NMOSD attack. (A) Graph shows change in sGFAP concentration over time in participants without attacks. Error bars represent interquartile range. Statistical significance of percentage change from baseline, comparing inebilizumab to placebo, was assessed using the Mann–Whitney U test; *p < 0.05; **p < 0.01; ***p < 0.001; week 16 changes from baseline remained significant (adjusted p value < 0.05) between dose groups after the Bonferroni correction was applied to p values. Nineteen of 117 (16%) inebilizumab samples and 9/26 (35%) placebo samples were outside healthy donor range (Fisher's exact test, p = 0.052). (B) Graph shows proportion of participants with elevated sGFAP concentrations (≥170 pg/ml) at week 28 of the RCP in participants receiving placebo or inebilizumab. FC = fold change; NMOSD = neuromyelitis optica spectrum disorder; RCP = randomized controlled period; sGFAP = serum glial fibrillary acidic protein.
Among the 177 participants without an attack, 18 (10.2%) displayed an increase greater than twofold in sGFAP concentration in at least one sample draw. These increases from baseline were comparable to the range observed in participants with attacks (Fig 6A) and well outside the variation observed in longitudinal draws from healthy donors, although the time frames of sample acquisition from healthy donors and N‐MOmentum participants were different (Fig 6B). Five of these 18 participants (28%) with an elevation in sGFAP reported neurological symptoms that were rated as attacks by the treating investigators but not confirmed by the adjudication committee. Of note, for the total cohort of these 18 participants, an increased rate of adverse events in temporal vicinity to sampling was observed (Tables 3 and 4). Moreover, of the subgroup of 16 attack‐free participants who had increases greater than twofold in sGFAP concentration and had spinal cord MRI scans, 9 (56%) presented with new or enlarging T2 lesions versus 9 (6%) of the 143 participants who neither experienced attacks nor displayed longitudinal sGFAP level changes (Fig 6C). A similar pattern was seen for Gd‐enhancing T1 spine lesions (Fig 6D). The proportion of participants with twofold increases in sGFAP levels was reduced by treatment with inebilizumab from week 12 onward (Fig 6E).
FIGURE 6.
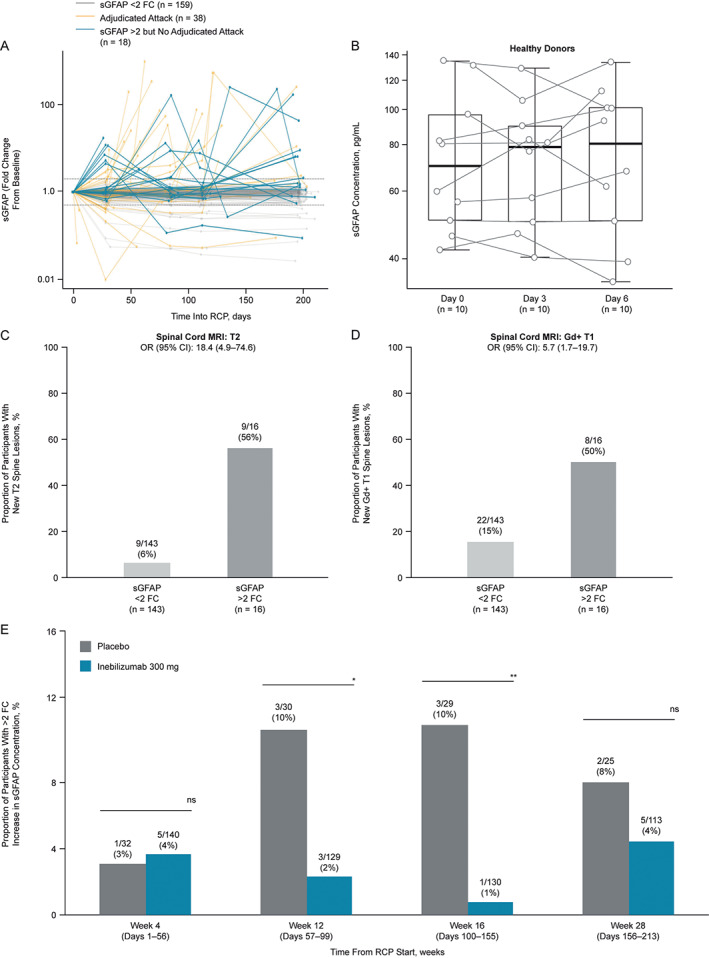
The sGFAP increases in participants without adjudicated attacks are correlated with disease activity. (A) Profile plot of longitudinal fold change from baseline in sGFAP concentration in participants with NMOSD who experienced adjudicated attacks (gold), those who did not experience adjudicated attacks but displayed an increase greater than twofold from baseline (blue), and in those who neither experienced attacks nor displayed increases greater than twofold from baseline (gray) in sGFAP during the RCP. (B) Boxplots displaying sGFAP concentrations observed in 10 healthy controls across 3 blood draws. Proportion of (C) new spinal cord T2 or (D) Gd‐enhancing T1 lesions observed in participants who did not experience committee‐adjudicated attacks and had MRI scans, but either did or did not display a greater than twofold increase in sGFAP during the RCP. (E) Bar chart shows proportion of participants with an increase greater than twofold in sGFAP from baseline. The total number of participants at each time point represent those remaining in the RCP with available sGFAP data. Statistical significance in the between‐group difference was assessed using the Cochran–Armitage test, *p < 0.05; **p < 0.01; ***p < 0.001, week 16 changes from baseline remained significant (adjusted p value < 0.05) after the Bonferroni correction was applied to p values. AC = adjudication committee; CI = confidence interval; FC = fold change; Gd = gadolinium; MRI = magnetic resonance imaging; NMOSD = neuromyelitis optica spectrum disorder; OR = odds ratio; RCP = randomized controlled period; sGFAP = serum glial fibrillary acidic protein.
TABLE 3.
Overview of AEs and SAEs in Attack‐Free Participants with sGFAP Increases
| Field | ||
|---|---|---|
| Any AE in RCP | Any SAE in RCP | |
| Participants with AE and no sGFAP increase, No. (%) | 116/158 (73%) | 7/158 (4%) |
| Participants with AE and sGFAP increase, No. (%) | 15/18 (83%) | 1/18 (6%) |
| No sGFAP increase, median AE count (Q1, Q3) | 2 (0, 4) | 0 (0, 0) |
| sGFAP increase, median AE count (Q1, Q3) | 3 (1, 4) | 0 (0, 0) |
| No sGFAP increase, mean AE count (SEM) | 2.99 (2.66–3.33) | 0.06 (0.03–0.08) |
| sGFAP increase, mean AE count (SEM) | 4.67 (3.24–6.09) | 0.17 (0.00–0.33) |
| RR (95% CI) | 1.56 (2.81–0.87) | 2.93 (26.9–0.32) |
| NB regression p value | 0.139 | 0.343 |
Negative binomial regression of the number of AEs in participants without attacks and with or without sGFAP increases.
AE = adverse event; CI = confidence interval; NB = negative binomial; OR = odds ratio; Q = quartile; RCP = randomized controlled period; RR = risk ratio; SAE = severe AE; SEM = standard error of the mean; sGFAP = serum glial fibrillary acidic protein.
TABLE 4.
Overview of AEs and SAEs in Relation to Time of sGFAP Sample Collection in Attack‐Free Participants with sGFAP Increases
| Field | Samples with AE and no sGFAP increase, No. (%) | Samples with AE and sGFAP increase, No. (%) | OR (95% CI) | Fisher's exact test p value |
|---|---|---|---|---|
| AEs | ||||
| Any AE within 7 days of sample | 220/1,015 (21.7%) | 9/23 (39%) | 2.32 (0.87–5.85) | 0.07 |
| Any AE within 14 days of sample | 257/1,015 (25.3%) | 12/23 (52%) | 3.21 (1.28–8.15) | 0.007 |
| Any AE within 30 days of sample | 328/1,015 (32.3%) | 12/23 (52%) | 2.29 (0.91–5.78) | 0.07 |
| Any AE within 45 days of sample | 386/1,015 (38.0%) | 13/23 (57%) | 2.12 (0.85–5.45) | 0.08 |
| SAEs | ||||
| Any SAE within 7 days of sample | 5/1,015 (0.5%) | 2/23 (9%) | 19.01 (1.72–124) | 0.009 |
| Any SAE within 14 days of sample | 9/1,015 (0.9%) | 2/23 (9%) | 10.57 (1.05–55.8) | 0.02 |
| Any SAE within 30 days of sample | 13/1,015 (1.3%) | 2/23 (9%) | 7.30 (0.75–35.6) | 0.04 |
| Any SAE within 45 days of sample | 16/1,015 (1.6%) | 2/23 (9%) | 5.92 (0.62–27.9) | 0.06 |
AE = adverse event; CI = confidence interval; OR = odds ratio; Q = quartile; SAE = severe adverse event; sGFAP = serum glial fibrillary acidic protein.
Discussion
In NMOSD, astroglial injury results in release of GFAP into the parenchymal interstitial fluid, subsequently detectable in the CSF and serum. 27 , 28 Given that sGFAP is derived predominantly from the CNS, 29 detection of GFAP may be particularly important in NMOSD because it is directly linked to the astrocyte damage underlying lesion formation. 16 , 17
The concentration of GFAP in the CSF is raised in NMOSD and MS compared with healthy conditions. 30 In NMOSD, CSF GFAP concentration correlates with EDSS score and with the length of spinal cord involvement. CSF GFAP concentration increases during attacks and falls following successful corticosteroid treatment. 31 , 32 , 33 However, measurement of CSF GFAP concentrations is unsuitable for frequent sampling. We chose to measure sGFAP concentrations using the SIMOA assay, which allows reliable detection of picomolar concentrations of CNS‐derived proteins in blood. 34 , 35 CSF and serum GFAP concentrations correlate strongly in patients with MS and NMOSD. 36 , 37
Our analyses confirmed that sGFAP concentration is often raised in untreated participants with NMOSD compared with both healthy donors and patients with RRMS. Of note, regarding the definition of physiological ranges, we observed a clear age‐dependent increase of sGFAP in all cohorts, including age‐ and sex‐matched healthy donors, and we were able to confirm the previously reported correlation of sGFAP with EDSS. 37 For NMOSD, neuropathology shows that astrocyte damage is more severe than in MS; consequently, the concentration of GFAP can be far greater than that detected in patients with MS, further supporting the use of sGFAP as a potential biomarker in NMOSD. 37
The risk of an adjudicated attack in participants who had elevated levels of baseline sGFAP was 3 times higher than in those with lower sGFAP concentration. This finding suggests that subclinical astrocyte injury or enhanced blood–brain barrier permeability may occur as a long‐range prelude to an acute attack. Clinical attacks might occur when a certain threshold of astrocytic injury is surpassed or if compensatory mechanisms that would otherwise suppress inflammation or restore neurologic function are overcome. Indeed, sGFAP concentration was raised in some participants without an adjudicated attack. Subclinical tissue injury in patients with NMOSD is not well‐recognized, although the occurrence of clinically silent brain lesions, 38 cervical spinal cord atrophy, 39 retinal thinning, 40 , 41 , 42 and changes in visual evoked potentials 43 , 44 were reported. Indeed, we observed that elevated sGFAP levels detected in participants without adjudicated attacks were associated with new or enhanced MRI activity as well as with adverse events, including patient‐reported complaints. Alternatively, the increased frequency of enhanced MRI activity may also indicate that enhanced blood–brain barrier permeability is facilitating the transit of GFAP to the peripheral circulation.
In clinical practice, the current goal of disease‐modifying therapy is to induce a state of clinical remission. Our data suggest that normalizing sGFAP levels might also guide treatment, in the context of clinical parameters.
Our analysis showed that CD19‐mediated B‐cell depletion by inebilizumab decreased the risk of attack versus placebo regardless of baseline sGFAP concentration, although risk reduction was less prominent in participants with elevated baseline sGFAP than in those with lower baseline levels. This suggests that patients with higher baseline sGFAP concentrations may be undergoing subclinical astrocytic damage prior to onset of clinical symptoms.
We also observed that sGFAP concentration increased during adjudicated attacks compared with baseline. The increase in attack risk in participants was based on a cutoff relative to healthy donors. However, because a linear relationship appears to exist between sGFAP concentration and attack risk, in the future, one could potentially define different strata of attack risk. Furthermore, greater attack severity was associated with higher sGFAP concentrations, even if the attack affected only the optic nerve(s). Our findings are also consistent with the results of a recent study in which sGFAP concentration was shown to increase during an NMOSD attack. 37
In participants receiving inebilizumab who experienced an attack, sGFAP concentration was nevertheless lower than in participants receiving placebo, and in the subset of inebilizumab‐treated participants who experienced an attack, sGFAP did not significantly increase compared to the baseline samples. Inebilizumab decreased sGFAP concentration in participants without an adjudicated attack, and fewer inebilizumab‐treated participants had raised sGFAP levels compared with placebo‐treated participants, indicating a potential protective effect of B‐cell depletion regardless of attack status. B‐cell depletion by inebilizumab could interfere with both the activity of B cells and the detrimental effects of circulating AQP4‐IgG on astrocytic endfeet that are a component of the functional blood–brain barrier. In turn, astrocyte injury (reduced soluble GFAP production) may be lessened, and blood–brain barrier stability (reduced access of soluble GFAP to serum) enhanced. In combination, the result is decreased concentration of GFAP in the circulation. As increased levels of sGFAP were correlated with attack severity, participants who experienced less severe attacks would have reduced levels of sGFAP, which in tandem with the hypothesized beneficial effects of inebilizumab on astrocyte damage and blood–brain barrier permeability, may lead to a stabilization of sGFAP concentrations. Finally, it remains to be clarified whether access of soluble GFAP to the circulation during quiescence and NMOSD activity is mediated exclusively through a leaky blood–brain barrier or is facilitated by CNS glymphatics. 45 , 46
Several questions remain unanswered by the present study. Although an expert adjudication committee confirmed all attacks, a new enhancing MRI lesion was not required for determination of all attacks. Further study is needed to determine whether radiographically silent, clinically adjudicated attacks are associated with sGFAP elevation. Pseudo‐attacks can occur in NMOSD and may be difficult to diagnose clinically. Presumably, sGFAP concentration would not be elevated in pseudo‐attacks and sGFAP measurement could be a less costly and more accessible option for accurate attack diagnosis. Moreover, regarding our data on timing of sGFAP changes during attack onset, it is possible that unspecific but attack‐driven clinical symptoms may have started before formal onset of an attack, depending on an individual patient's awareness and willingness to report such changes.
There are limitations in interpreting our results. NMOSD is a rare disease and recruiting large numbers of participants was challenging. Although the N‐MOmentum study was adequately powered to detect treatment effects for major end points, the study was not designed to show statistically significant effects for all subgroups. For example, sample size limitations are apparent for the AQP4‐IgG‐seronegative participants (N = 17) and the even smaller MOG‐IgG‐seropositive subset (N = 7). Although we presented data on the intention‐to‐treat population, the sGFAP results are driven almost entirely by observations from the AQP4‐IgG‐seropositive subgroup. Furthermore, because of the 3:1 randomization in the study, the size of the placebo group was small and decreased further as the study progressed because of the crossover to open‐label treatment following adjudicated attacks and the subsequent exit from the RCP. Therefore, the limited placebo group data at later time points of the RCP reduced the power to detect treatment differences. N‐MOmentum was also a single trial, and replication in other prospectively gathered data sets with inebilizumab and other treatments is needed. For the time being, sGFAP cannot be assessed by point‐of‐care testing as one might wish for acute attack assessment. Until technological advances enable rapid testing, sGFAP can only be assessed by specialized laboratories. Nonetheless, there is potential for sGFAP's application to stratify risk and assess therapeutic benefit, at least in patients undergoing B‐cell depletion. Despite these important limitations, the trends reported here were notably consistent, and the predefined analyses strengthen the validity of the results.
In conclusion, data from the N‐MOmentum study show that sGFAP may be a clinically informative biomarker for both attack risk and severity. The sGFAP elevation in some participants without clinical attacks but with new MRI lesions reveals that subclinical disease activity and/or blood–brain barrier breakdown may be present in NMOSD. These results underscore the potential of soluble CNS‐derived biomarkers like GFAP for the assessment of ongoing neural injury and thus, the identification of patients with NMOSD at risk for relapse.
Author Contributions
All authors (O.A., M.A.S., W.R., J.L.B., D.S., E.K., and B.A.C.C.) contributed to the conception and design of the study, were involved in the acquisition and/or analysis of data, and drafting text or preparing figures.
This manuscript was submitted on behalf of the N‐MOmentum scientific group: Kazuo Fujihara, MD, Department of Multiple Sclerosis Therapeutics, Fukushima Medical University and Multiple Sclerosis and Neuromyelitis Optica Center, Southern Tohoku Research Institute for Neuroscience, Koriyama, Japan; Friedemann Paul, MD, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine and Charité – Universitätsmedizin Berlin, Berlin, Germany; Hans‐Peter Hartung, MD, Medical Faculty, Heinrich Heine University, Düsseldorf, Germany; Brain and Mind Centre, University of Sydney, Australia; and Department of Neurology, Medical University of Vienna, Austria; Romain Marignier, MD, PhD, Centre de Référence des Maladies Inflammatoires Rares du Cerveau et de la Moelle (MIRCEM), Service de Neurologie, Sclérose en Plaques, Pathologies de la Myéline et Neuro‐inflammation – Hôpital Neurologique Pierre Wertheimer Hospices Civils de Lyon, Lyon, France; Ho Jin Kim, MD, PhD, Research Institute and Hospital of National Cancer Center, Goyang, South Korea; Brian G. Weinshenker, MD, Mayo Clinic, Rochester, MN, USA; Sean J. Pittock, MD, Mayo Clinic, Rochester, MN, USA; Dean M. Wingerchuk, MD, Mayo Clinic, Scottsdale, AZ, USA; Gary R. Cutter, PhD, University of Alabama at Birmingham, Birmingham, AL, USA; Ari J. Green, MD, MCR, UCSF Weill Institute for Neurosciences, Department of Ophthalmology, University of California San Francisco, San Francisco, CA, USA; and UCSF Weill Institute for Neurosciences, Department of Neurology, University of California San Francisco, San Francisco, CA, USA; Maureen A. Mealy, PhD, Viela Bio, Gaithersburg, MD, USA; Jorn Drappa, MD, PhD, Viela Bio, Gaithersburg, MD, USA; and Liangwei Wang, PhD, Viela Bio, Gaithersburg, MD, USA; who contributed to the conception and design of the study, and were involved in the acquisition and/or analysis of data; and this manuscript was submitted on behalf of the principal investigators of the N‐MOmentum study group, who administered the clinical trial. The N‐MOmentum study investigators are listed in Supplementary Online Table 1.
Potential Conflicts of Interest
Viela Bio and MedImmune funded the N‐MOmentum study. Viela Bio is the owner of inebilizumab. Mitsubishi Tanabe Pharma Corporation and Hansoh Pharmaceutical Group Co. Ltd. have partnerships with Viela Bio to develop and commercialize inebilizumab for NMOSD (and other potential indications) in Asia. O.A. serves on a steering committee for Viela Bio and has received funding for travel and fees from Viela Bio. M.A.S., W.R., D.S., and E.K. are employees of Viela Bio. J.L.B. reports payment for study design/consultation from Viela Bio and personal fees from Mitsubishi Tanabe Pharma Corporation. B.A.C.C. has nothing to report.
Supporting information
Table S1. List of investigators for the N‐MOmentum Study
Acknowledgments
The N‐MOmentum trial was funded by MedImmune and Viela Bio. Viela Bio supported the development of this manuscript, provided data analyses according to the direction of the authors, and paid for editorial assistance and clerical support. Dr Ian M. Williams of Oxford PharmaGenesis Ltd, supported the development of the project by providing administrative and editorial assistance, according to the direction of the authors. The final approval of content, drafting and revising of the manuscript, and decision to submit this manuscript rested with the authors.
ClinicalTrials.gov (NCT02200770).
Contributor Information
Orhan Aktas, Email: orhan.aktas@hhu.de.
Bruce A. C. Cree, Email: bruce.cree@ucsf.edu.
the N‐MOmentum scientific group and the N‐MOmentum study investigators:
Kazuo Fujihara, Friedemann Paul, Hans‐Peter Hartung, Romain Marignier, Ho Jin Kim, Brian G. Weinshenker, Sean J. Pittock, Dean M. Wingerchuk, Gary R. Cutter, Ari J. Green, Maureen A. Mealy, and Jorn Drappa
References
- 1. Cree BA, Bennett JL, Sheehan M, et al. Placebo‐controlled study in neuromyelitis optica‐ethical and design considerations. Mult Scler 2016;22:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014;261:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cree BA, Lamb S, Morgan K, et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005;64:1270–1272. [DOI] [PubMed] [Google Scholar]
- 4. Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta‐analysis. JAMA Neurol 2016;73:1342–1348. [DOI] [PubMed] [Google Scholar]
- 5. Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN‐1 study): a multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2020;19:298–306. [DOI] [PubMed] [Google Scholar]
- 6. Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin‐4‐positive neuromyelitis optica spectrum disorder. N Engl J Med 2019;381:614–625. [DOI] [PubMed] [Google Scholar]
- 7. Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med 2019;381:2114–2124. [DOI] [PubMed] [Google Scholar]
- 8. Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N‐MOmentum): a double‐blind, randomised placebo‐controlled phase 2/3 trial. Lancet 2019;394:1352–1363. [DOI] [PubMed] [Google Scholar]
- 9. Fujihara K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol 2019;32:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen D, Gallagher S, Monson NL, et al. Inebilizumab, a B cell‐depleting anti‐CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med 2016;5:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schiopu E, Chatterjee S, Hsu V, et al. Safety and tolerability of an anti‐CD19 monoclonal antibody, MEDI‐551, in subjects with systemic sclerosis: a phase I, randomized, placebo‐controlled, escalating single‐dose study. Arthritis Res Ther 2016;18:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agius MA, Klodowska‐Duda G, Maciejowski M, et al. Safety and tolerability of inebilizumab (MEDI‐551), an anti‐CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: results from a phase 1 randomised, placebo‐controlled, escalating intravenous and subcutaneous dose study. Mult Scler 2019;25:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 15. Zekeridou A, Lennon VA. Aquaporin‐4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. 2015;2:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 17. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 2009;66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liem RK, Messing A. Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest 2009;119:1814–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papa L, Lewis LM, Falk JL, et al. Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann Emerg Med 2012;59:471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petzold A. Glial fibrillary acidic protein is a body fluid biomarker for glial pathology in human disease. Brain Res 2015;1600:17–31. [DOI] [PubMed] [Google Scholar]
- 21. Brenner M, Johnson AB, Boespflug‐Tanguy O, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001;27:117–120. [DOI] [PubMed] [Google Scholar]
- 22. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 23. Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4‐IgG‐positive relapsing neuromyelitis optica spectrum disorders: an open‐label pilot study. Lancet Neurol 2013;12:554–562. [DOI] [PubMed] [Google Scholar]
- 24. Abdelhak A, Huss A, Kassubek J, et al. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep 2018;8:14798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Czeiter E, Amrein K, Gravesteijn BY, et al. Blood biomarkers on admission in acute traumatic brain injury: relations to severity, CT findings and care path in the CENTER‐TBI study. EBioMedicine 2020;56:102785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marshall WJ, Bangert SK. Clinical biochemistry: metabolic and clinical aspects. 2nd ed. London, UK: Churchill Livingstone, 2008. [Google Scholar]
- 27. Lucchinetti CF, Guo Y, Popescu BF, et al. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol 2014;24:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuhle J, Kropshofer H, Meinert R, et al. Plasma glial fibrillary acidic protein correlates with characteristics of advanced disease and treatment response in secondary progressive multiple sclerosis. Paper presented at: 35th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), 11–13 September 2019, Stockholm, Sweden (Abstract P1630).
- 29. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 2010;119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petzold A, Marignier R, Verbeek MM, Confavreux C. Glial but not axonal protein biomarkers as a new supportive diagnostic criteria for devic neuromyelitis optica? Preliminary results on 188 patients with different neurological diseases. J Neurol Neurosurg Psychiatry 2011;82:467–469. [DOI] [PubMed] [Google Scholar]
- 31. Misu T, Takano R, Fujihara K, et al. Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: an astrocytic damage marker. J Neurol Neurosurg Psychiatry 2009;80:575–577. [DOI] [PubMed] [Google Scholar]
- 32. Takano R, Misu T, Takahashi T, et al. A prominent elevation of glial fibrillary acidic protein in the cerebrospinal fluid during relapse in neuromyelitis optica. Tohoku J Exp Med 2008;215:55–59. [DOI] [PubMed] [Google Scholar]
- 33. Uzawa A, Mori M, Sawai S, et al. Cerebrospinal fluid interleukin‐6 and glial fibrillary acidic protein levels are increased during initial neuromyelitis optica attacks. Clin Chim Acta 2013;421:181–183. [DOI] [PubMed] [Google Scholar]
- 34. Rissin DM, Kan CW, Campbell TG, et al. Single‐molecule enzyme‐linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol 2010;28:595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018;14:577–589. [DOI] [PubMed] [Google Scholar]
- 36. Hogel H, Rissanen E, Barro C, et al. Serum glial fibrillary acidic protein correlates with multiple sclerosis disease severity. Mult Scler 2018;26:210–219. [DOI] [PubMed] [Google Scholar]
- 37. Watanabe M, Nakamura Y, Michalak Z, et al. Serum GFAP and neurofilament light as biomarkers of disease activity and disability in NMOSD. Neurology 2019;93:e1299–e1311. [DOI] [PubMed] [Google Scholar]
- 38. Jeong IH, Choi JY, Kim SH, et al. Normal‐appearing white matter demyelination in neuromyelitis optica spectrum disorder. Eur J Neurol 2017;24:652–658. [DOI] [PubMed] [Google Scholar]
- 39. Ventura RE, Kister I, Chung S, et al. Cervical spinal cord atrophy in NMOSD without a history of myelitis or MRI‐visible lesions. Neurol Neuroimmunol Neuroinflamm. 2016;3:e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jeong IH, Kim HJ, Kim NH, et al. Subclinical primary retinal pathology in neuromyelitis optica spectrum disorder. J Neurol 2016;263:1343–1348. [DOI] [PubMed] [Google Scholar]
- 41. Oertel FC, Havla J, Roca‐Fernandez A, et al. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry 2018;89:1259–1265. [DOI] [PubMed] [Google Scholar]
- 42. Oertel FC, Kuchling J, Zimmermann H, et al. Microstructural visual system changes in AQP4‐antibody‐seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm 2017;4:e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ringelstein M, Kleiter I, Ayzenberg I, et al. Visual evoked potentials in neuromyelitis optica and its spectrum disorders. Mult Scler 2014;20:617–620. [DOI] [PubMed] [Google Scholar]
- 44. Ringelstein M, Harmel J, Zimmermann H, et al. Longitudinal optic neuritis‐unrelated visual evoked potential changes in NMO spectrum disorders. Neurology 2020;94:e407–e418. [DOI] [PubMed] [Google Scholar]
- 45. Mestre H, Mori Y, Nedergaard M. The brain's glymphatic system: current controversies. Trends Neurosci 2020;43:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Da Mesquita S, Fu Z, Kipnis J. The meningeal lymphatic system: a new player in neurophysiology. Neuron 2018;100:375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of investigators for the N‐MOmentum Study