

Abstract
We present the first example of macroscalar helices co‐assembled from temperature‐responsive carbohydrate‐based bolaamphiphiles (CHO‐Bolas) and 1,4‐benzenediboronic acid (BDBA). The CHO‐Bolas contained hydrophilic glucose or mannose moieties and a hydrophobic coumarin dimer. They showed temperature‐responsive reversible micelle‐to‐vesicle transition (MVT) in aqueous solutions. After the binding of carbohydrate moieties with boronic acids of BDBA in their alkaline solutions, right‐handed helices were formed via the temperature‐driven chirality transfer of d‐glucose or d‐mannose from the molecular to supramolecular level. These helices were co‐assembled by unreacted BDBA, boronate esters (B−O−C bonds) between CHO‐Bolas and BDBA, as well as boroxine anhydrides (B−O−B bonds) of self‐condensed BDBA. After heating at 300 °C under nitrogen, the helices displayed excellent morphological stability. Moreover, they emitted bright blue luminescence caused by strong self‐condensation of BDBA and decomposition of coumarin dimers.
Keywords: assembly, bolaamphiphile, chirality transfer, helices, temperature-responsive
Macroscalar, thermally stable helices were formed by co‐assembly of temperature‐responsive carbohydrate‐based bolaamphiphiles and 1,4‐benzenediboronic acids accompanying chirality transfer in alkaline solutions during the temperature decrease from 80 °C to room temperature.
Introduction
In living systems, helical self‐assemblies of biomacromolecules including nucleic acids, proteins, and oligo‐/polysaccharides are sophisticated and vital structures formed via non‐covalent interactions. Inspired by these intriguing phenomena, chemists have devoted great efforts to mimic nano‐/macroscalar helical architectures using designed polymer backbones or supramolecular systems.[ 1 , 2 ] Generally, the non‐covalent forces utilized in preparing helical assemblies are diverse, including van der Waals interactions, hydrogen bonding, electrostatic interactions and π–π stacking.[ 3 , 4 ] Both the specific chemical compositions of the molecules and environmental factors, such as solvents, temperature, and pH, greatly influence the formation of helical aggregates during the assembly process.[ 5 , 6 , 7 , 8 ] Well‐controlled helices in the single/multiple‐stranded form have potential applications in medicine and materials fields, such as drug delivery, gene delivery systems and asymmetric catalysis.[ 9 , 10 , 11 ] However, it is still a great challenge to form macroscalar helices from small organic molecules, for example, by using a chiral template.
Bolaamphiphiles (also known as bipolar amphiphiles) are defined as molecules in which two polar functional headgroups are linked by one or more nonpolar hydrocarbon chains. The identical or different polar headgroups of synthetic bolaamphiphiles can be moieties of various types, such as phosphocholine, nucleotide, amino acid, and sugar.[ 12 , 13 , 14 ] In nature, there are typical bolaamphiphiles which form the stable monolayer lipid membranes of thermophilic archaebacteria.[ 15 , 16 ] Over the past four decades, bolaamphiphiles have drawn a great deal of attention due to the formation of diverse supramolecular self‐assemblies via non‐covalent interactions, such as nanofibers, nanotubes, and vesicles.[ 17 , 18 ] They find potential applications in various fields, for example, pharmaceutics and biomaterials.[ 19 , 20 ] Among the nonionic ones, sugar‐based bolaamphiphiles have outstanding properties, including abundance of chiral centers and recognition ability.[ 21 , 22 ] However, to our knowledge, there is no report about helical co‐assembly of sugar‐based chiral bolaamphiphiles and their achiral binding counterpart phenylboronic acids via chirality transfer.
Herein, we report the first macroscalar helices co‐assembled from temperature‐responsive carbohydrate‐based bolaamphiphiles and BDBA. We first synthesized novel nonionic CHO‐Bolas: CHO‐Bolas with d‐glucose moieties and a coumarin dimer (GCCG‐12) 1 as well as CHO‐Bolas with d‐mannose moieties and a coumarin dimer (MCCM‐12) 2. The CHO‐Bolas had two sugar skeletons as hydrophilic heads for further boronic‐acid‐binding sites and one dimer of 7‐alkoxy‐4‐methylcoumarins as the hydrophobic motif. The temperature‐responsive property of CHO‐Bolas allowed their reversible micelle‐to‐vesicle transition (MVT) in aqueous solutions. Due to the chirality and boronic‐acid‐binding ability of the carbohydrate moieties, the CHO‐bolas induced macroscalar helices by co‐assembling with BDBA in alkaline aqueous solutions with the assistance of temperature at more than 80 °C.
Results and Discussion
The novel bolaamphiphiles 1 and 2 contained carbohydrates (glucose or mannose) as hydrophilic headgroups and coumarin dimers as the hydrophobic core. They were synthesized through the simultaneous thiol–ene reaction of 2‐allylethoxyl monosaccharide (7, 8) and 7‐mercaptohexyloxy‐4‐methylcoumarin (11), as well as a [2+2]‐photodimerization reaction of coumarin moieties in a one‐pot reaction by 300–400 nm UV irradiation (Figure 1 a and Schemes S1–S3). It was reported that direct excitation of 7‐alkoxy‐4‐methylcoumarins in organic solvents readily gave the syn head–tail dimer, and triplet sensitization resulted in the anti head–tail dimer. [23] In our work, the photodimerization reactions worked out only under UV irradiation in dichloromethane via a singlet excited state, in the absence of photocatalysis. The chemical shifts belonging to the cyclobutane protons and cyclobutane methyl protons were visible at 3.41 and 1.41 ppm (see Supporting Information for 1H NMR spectra). 1H NMR spectra further confirmed the formation of primarily the syn head–tail product with minor quantities of the anti head–tail conformation, which is in agreement with previous reports.[ 23 , 24 ]
Figure 1.
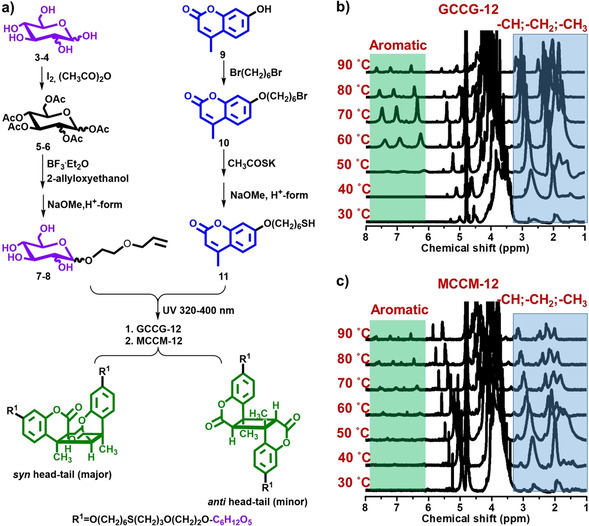
a) Synthesis route to CHO‐Bolas containing glucose and a coumarin dimer (GCCG‐12) 1 or mannose and a coumarin dimer (MCCM‐12) 2. 1H NMR spectra of b) GCCG‐12 and c) MCCM‐12 recorded in D2O at various temperatures between 30 and 90 °C.
1H NMR measurements of water‐soluble CHO‐Bolas in D2O at various temperatures between 30 and 90 °C showed their temperature‐responsive properties at the molecular level. As shown in Figure 1 b,c, the peaks in the aromatic region (δ≈6–7.5 ppm) were absent and the peaks in the alkyl region (δ≈1–3 ppm) were significantly diminished at 30 °C, indicating the formation of aggregates via non‐covalent interactions. [25] When the temperature was raised to 60 °C, the intensities of peaks in both regions were enhanced due to the increased molecular mobility and partial breaking of these interactions. These peaks became gradually weaker with further increasing temperature to more than 80 °C, indicating the formation of another aggregate state. Furthermore, a slight downfield shift of signals of aromatic protons was observed with increasing temperature. This behavior can be ascribed to stronger water exposure and pressure‐induced conformational changes.[ 26 , 27 , 28 ]
The aggregation of the temperature‐responsive CHO‐Bolas at neutral or acidic environments of pH 5 was visually detectable (Figure 2 a and Figure S1). The solutions changed from transparent to turbid by increasing the temperature to 90 °C. The turbid suspensions returned to the transparent solution when cooling down to room temperature. This reversible transparent–turbid change demonstrates a micelle‐to‐vesicle transition (MVT). Dynamic light scattering (DLS) was further used to analyze the temperature‐dependent process and the size development of the aggregates (Figure 2 b). In aqueous solutions of GCCG‐12, the Z‐average diameters of aggregates were 22.2±2.0 nm with a narrow size distribution (PDI=0.152±0.008) at a temperature below 80 °C. By raising the temperature to 85 °C, they increased strongly to 1.5±0.1 μm with a broad size distribution (PDI=0.815±0.163). This huge size enhancement indicates the formation of vesicles from the initial micelles. Compared with GCCG‐12, the MCCM‐12 solution displayed MVT at a lower temperature. The Z‐average diameters of aggregates in MCCM‐12 solution were 54.2±2.9 nm with a PDI of 0.233±0.005 at a temperature below 50 °C. After elevating the temperature to 55 °C, they increased notably to 917.7±49.3 nm with a PDI of 0.735±0.105. The spherical morphologies of nanoparticles dried from micelles formed by CHO‐Bolas were verified with TEM and vesicles recorded with temperature‐controlled PLM measurements, as shown in Figure 2 c,d. The average sizes of the predominantly spherical nanoparticles were 5.4±1.0 nm for dried GCCG‐12 and 7.1±1.1 nm for dried MCCM‐12. By heating solutions of CHO‐Bolas, vesicles were clearly observed under PLM (Figure 2 e,f, Movies S1 and S2). The average sizes of vesicles increased to 6.2±1.9 μm for GCCG‐12 at 95 °C and 9.4±2.2 μm for MCCM‐12 at 70 °C. Therefore, the CHO‐Bolas exhibited temperature‐responsive, reversible transition between micelles and vesicles.
Figure 2.
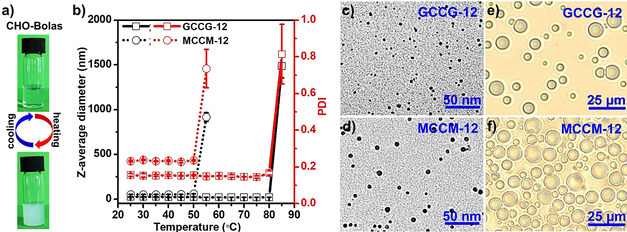
a) Photos of reversible clear–turbid cycles during heating and cooling of CHO‐Bolas in deionized water. b) Z‐average diameters and polydispersity index (PDI) of aggregates formed by GCCG‐12 and MCCM‐12 in aqueous solutions in correlation with temperature. TEM images of nanoparticles formed by dried c) GCCG‐12 and d) MCCM‐12. Polarized light microscopy (PLM) images of vesicles formed by e) GCCG‐12 at 98 °C and f) MCCM‐12 at 70 °C in aqueous solutions.
Apart from the temperature‐responsive behaviors of the reversible transition between micelles and vesicles, CHO‐Bolas had other characteristic properties, such as chirality and recognition via bonding with lectins or boronic acids through the carbohydrate moieties.[ 21 , 22 ] It is known that boronic acids have the potential to interact with diverse molecular motifs in aqueous media, such as reversibly bonding with diols to form boronic esters, self‐condensation to form boroxines, and hydrogen bonding with other active molecules.[ 29 , 30 ]
As shown in Figure 3 a, CHO‐Bolas formed specific helices after the co‐assembly with BDBA. By dissolving them in alkaline aqueous solutions (pH 9) at 80 °C and cooling the solutions slowly down to room temperature, helical aggregates of several hundred microns in length were obtained (Figure 3 b,f). The assembled solid‐state helical structures emitted blue luminescence, as observed under the fluorescence microscope with a UV lamp (λ=365 nm) (Figure 3 c,g). Moreover, the SEM images of the air‐dried samples showed right‐handed helices containing packed thin layers (Figure 3 d,e,h,i). The helices of co‐assembled BDBA/GCCG‐12 had helical pitches of 5.4±1.1 μm and an average lamellae thickness of 0.3±0.1 μm (Figure 3 e). In comparison, co‐assembled helices of BDBA/MCCM‐12 had larger helical pitches of 6.3±1.0 μm and thinner lamellae with an average thickness of 0.2±0.1 μm (Figure 3 i). In contrast to the helices co‐assembled by CHO‐Bolas and BDBA, BDBA alone only self‐assembled into brick‐shaped structures under equal conditions. They were revealed by optical light and fluorescence microscopy as well as SEM images (Figure S2). It should be noted that the mixture of CHO‐Bolas and BDBA in neutral aqueous solutions produced sporadic macroscalar helices and a large amount of brick‐shaped structures by decreasing the temperature from 80 °C to room temperature. Therefore, well‐ordered helices should be formed by the chirality transfer from the CHO‐Bolas to co‐assembled structures via diverse interactions among cis‐diols of carbohydrates and boronic acids of BDBA during cooling down to room temperature. Without the CHO‐Bolas, brick‐shaped structures containing only BDBA did not form helical structures.
Figure 3.
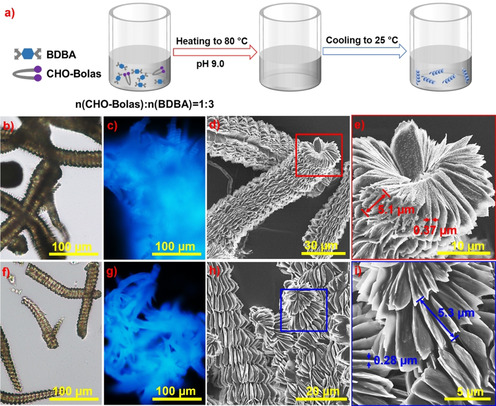
a) Schematic illustration of the formation of helical structures in alkaline aqueous solution of CHO‐Bolas and 1,4‐benzeneboronic acid (BDBA). Optical light and fluorescence microscopy as well as SEM images of the helices with diverse magnifications: b–e) formed by GCCG‐12 and BDBA; f–i) formed by MCCM‐12 and BDBA.
Co‐assembled helices were further analyzed with FTIR and solid‐state 11B/13C NMR spectroscopy in order to reveal their chemical compositions. According to the FTIR spectra of the helices and brick‐shaped aggregates (Figure 4 a), the peaks ascribed to unreacted B−OH groups of BDBA were predominant and strong, for example, the peaks assigned to the BO−H stretching vibration at 3280 and 3400 cm−1, the peaks attributed to B−O at 1335 and 1395 cm−1, the peak of B−C(aryl) at 1030 cm−1, and the peaks corresponding to B−OH stretching vibrations at 636 and 1005 cm−1.[ 31 , 32 ] Within the FTIR spectra of helices, the new emerging peaks at 1621 and 1755 cm−1 were assigned to aromatic rings and C=O groups of coumarin dimers, respectively. Moreover, the greatly increased peak at 1120 cm−1 was attributed to the symmetric C‐O‐B‐O‐C stretching vibration. [33] These results showed the formation of boronate esters between BDBA and CHO‐Bolas within the helices. Moreover, brick‐shaped aggregates contained much more boroxine anhydrides according to the peak at 580 cm−1 attributed to boroxine anhydrides, [34] while the amount of boroxine anhydrides should be marginal within helices based on their very weak peaks.
Figure 4.
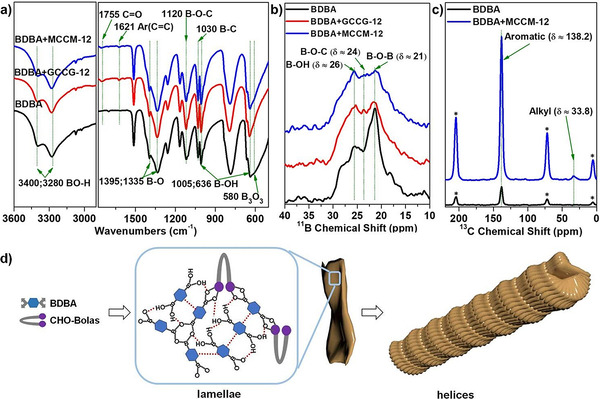
a) FTIR spectra and b) solid‐state 11B NMR spectra of the helical structures formed by the CHO‐Bolas and BDBA, as well as brick‐shaped structure formed by BDBA alone. c) Solid‐state 13C NMR spectra of the helical structures formed by MCCM‐12 and BDBA, as well as brick‐shaped structure formed by BDBA alone. Asterisks indicate spinning side‐band peaks. d) Plausible packing model of the co‐assembled helical structures.
The solid‐state 11B NMR spectrum of the self‐assembled helices exhibited overlapping multiplets due to the boron atoms in different structural environments (Figure 4 b). Compared to the brick‐shaped aggregates formed by BDBA alone, the helical structures formed by BDBA and CHO‐Bolas showed a much broader peak. They consisted of multiple peaks with roughly the same intensity. This can be explained with the overlapping peaks ascribed to boroxine anhydrides at around 21 ppm, newly emerging boronate ester at around 24 ppm, and boronic acid at around 26 ppm, in agreement with the FTIR results shown above. The solid‐state 13C NMR spectrum of brick‐shaped aggregates exhibited one single and weak resonance for aromatic rings of BDBA at 138 ppm (Figure 4 c). In comparison, the solid‐state 13C NMR spectrum of helices formed by MCCM‐12 and BDBA displayed one stronger and broader resonance at 138 ppm and one weak resonance for alkyl chains of MCCM‐12 at approx. 34 ppm. Therefore, the helical structures contained high amounts of bulk BDBA, reversible boronate esters formed between BDBA and CHO‐Bolas, and boroxine anhydrides by self‐condensation of BDBA. In addition, adding an excess of glucose or mannose to the suspensions of these helices led to their gradual disassembly and finally complete dissolution in aqueous solutions. This phenomenon also indicated the pivotal role of reversible boronate esters between BDBA and CHO‐Bolas in chirally guiding the formation of right‐handed helical structures.
Taking the above results into account, we consequently propose a plausible model for the co‐assembly of helical structures (Figure 4 d). In addition to high amounts of unreacted BDBA, the cis‐diols of CHO‐Bolas reversibly and covalently interacted with BDBA, while BDBA self‐condensed into a small amount of boroxine anhydrides. During the cooling process, non‐covalent interactions including hydrogen bonding and π–π stacking further stabilized the structures by forming lamellae. In parallel, the chirality transferred from CHO‐Bolas guided the lamellae into helices.
The helices were further treated at high temperature to analyze their thermal properties and compositions. It is well known that the six‐membered boroxine anhydrides can be easily formed by dehydrative self‐condensation of boronic acid when the temperature exceeds 100 °C. [35] According to the FTIR and solid‐state 11B NMR spectra of the assembled helical structures (Figure 4), there were excessive active groups of B−OH in comparison to hydroxy groups of carbohydrate moieties. As shown in Figure 5 a, the DSC curves of the self‐assembled helices showed endothermic processes. They started at around 123 °C (initial temperature, T i) and ended at around 282 °C (end temperature, T e). The helices formed by both CHO‐Bolas and BDBA exhibited only one single endothermic peak at 225–228 °C. In comparison, the brick‐shaped structures formed by BDBA alone showed two partially overlapping endothermic peaks at 222 °C and 255 °C. Since all DSC measurements of assembled aggregates were carried out in sealed crucibles, no peaks corresponding to water evaporation were observed. The endothermic processes of helices involved multiple reactions and physical changes, such as self‐condensation of B−OH groups into boroxine anhydrides, decomposition of coumarin dimers into coumarin monomers, [36] esterification of B−OH groups with hydroxy groups of mannose moieties, and melting.
Figure 5.
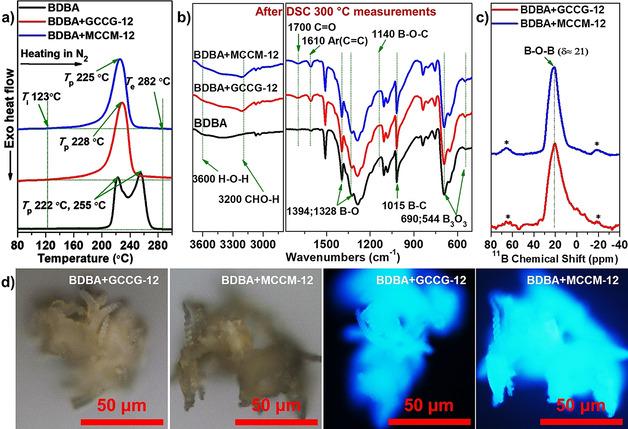
a) DSC curves of assembled structures heated from 20 °C to 300 °C at a heating rate of 5 K min−1 under nitrogen atmosphere. b) FTIR spectra of assembled structures after treatment at 300 °C in nitrogen gas. c) Solid‐state 11B NMR spectra of a helical structure formed by CHO‐Bolas and BDBA after the treatment at 300 °C in nitrogen gas. Asterisks indicate spinning side‐band peaks. d) Optical and fluorescence microscopy images of helices formed by CHO‐Bolas and BDBA after treatment at 300 °C in nitrogen gas.
Moreover, the strong bands within the FTIR spectra at 3000–3580, 1005, and 636 cm−1 ascribed to BO−H and B−OH vibrations sharply decreased (Figure 5 b). In comparison, the bands at 690 and 544 cm−1 attributed to B−O−B of boroxine anhydrides greatly increased.[ 31 , 32 , 37 ] Therefore, boroxine anhydrides were formed via the self‐condensation of BDBA during the heat treatment. Due to the self‐condensation and esterification of B−OH groups with hydroxy groups of glucose or mannose moieties, the signal at 3600 cm−1 ascribed to free hydroxy groups of resulting water was observed. Interestingly, the band at 1755 cm−1 attributed to the saturated ketones and the band at 1621 cm−1 due to aromatic rings of coumarin dimers disappeared. New stronger bands at 1700 and 1610 cm−1 for the unsaturated ketones and aromatic rings of coumarins appeared. They indicated the decomposition of coumarin dimers into coumarin monomers after heat treatment. [36] Besides, signals emerged at 1140 and 1015 cm−1 corresponding to B−O−C of boronate esters and B−C bonds, respectively.
As shown in Figure 5 c and Figure S3, the solid‐state 11B NMR spectra of assembled helices after the treatment at 300 °C under nitrogen atmosphere exhibited only one single broad peak at around 21 ppm. This peak is attributed to resulting B−O−B bonds in high quantities during the heat treatment, which was in good agreement with the FTIR results. Moreover, helices showed great morphological stability after the heat treatment at 300 °C (Figure 5 d). Their luminescence also changed to a bright blue from blue due to the formation of more π–π stacking in the boroxine network upon heating and decomposition of coumarin dimers in helices. This enhanced luminescence was similar to the heat‐treated brick‐shaped structure of bulk BDBA (Figures 5 d and S2 d).
Conclusion
In summary, we found a new approach to prepare macroscalar right‐handed helices via chirality transfer from novel carbohydrate‐based bolaamphiphiles (CHO‐Bolas) after their co‐assembly with 1,4‐benzenediboronic acid (BDBA). The CHO‐Bolas containing chiral glucose/mannose headgroups were synthesized via simultaneous thiol–ene click reaction and [2+2]‐photodimerization of coumarins in a one‐pot synthesis after UV irradiation. The CHO‐Bolas displayed temperature‐responsive behavior by showing reversible micelle‐to‐vesicle transition. Furthermore, the CHO‐Bolas had other characteristic properties including chirality and recognition ability. When mixing with BDBA in alkaline solution at 80 °C, the carbohydrate moieties of CHO‐Bolas transferred chirality to macroscalar right‐handed helices in a co‐assembly process. The resulting helical structures constructed by BDBA were induced by CHO‐Bolas, as demonstrated by FTIR spectra and solid‐state NMR measurements. After treatment at 300 °C in nitrogen gas, the fluorescence of the helices was enhanced due to the enhanced self‐condensation of BDBA and decomposition of coumarin dimers. This work paves the way for the realization of chirality transfer from small organic molecules to assembled supramolecular structures using carbohydrate as chirality templates.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
K.Z. thanks the Fonds der Chemischen Industrie (FCI) for financial support and Georg August University of Goettingen for the Department Start‐up funding. S.W. and B.P. gratefully acknowledge their PhD scholarship from the Chinese Scholarship Council (CSC). Open access funding enabled and organized by Projekt DEAL.
S. Wang, M. C. Forster, K. Xue, F. Ehlers, B. Pang, L. B. Andreas, P. Vana, K. Zhang, Angew. Chem. Int. Ed. 2021, 60, 9712.
References
- 1. Rowan A. E., Nolte R. J., Angew. Chem. Int. Ed. 1998, 37, 63–68; [Google Scholar]; Angew. Chem. 1998, 110, 65–71. [Google Scholar]
- 2. Schmuck C., Angew. Chem. Int. Ed. 2003, 42, 2448–2452; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2552–2556. [Google Scholar]
- 3. Hoeben F. J., Jonkheijm P., Meijer E., Schenning A. P., Chem. Rev. 2005, 105, 1491–1546. [DOI] [PubMed] [Google Scholar]
- 4. Yang Y., Zhang Y., Wei Z., Adv. Mater. 2013, 25, 6039–6049. [DOI] [PubMed] [Google Scholar]
- 5. Murata K., Aoki M., Suzuki T., Harada T., Kawabata H., Komori T., Ohseto F., Ueda K., Shinkai S., J. Am. Chem. Soc. 1994, 116, 6664–6676. [Google Scholar]
- 6. Brizard A., Aimé C., Labrot T., Huc I., Berthier D., Artzner F., Desbat B., Oda R., J. Am. Chem. Soc. 2007, 129, 3754–3762. [DOI] [PubMed] [Google Scholar]
- 7. Zhang X., Zou J., Tamhane K., Kobzeff F. F., Fang J., Small 2010, 6, 217–220. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen T. D., Glotzer S. C., Small 2009, 5, 2092–2098. [DOI] [PubMed] [Google Scholar]
- 9. Gillies E. R., Deiss F., Staedel C., Schmitter J. M., Huc I., Angew. Chem. Int. Ed. 2007, 46, 4081–4084; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4159–4162. [Google Scholar]
- 10. Nakano T., Okamoto Y., Chem. Rev. 2001, 101, 4013–4038. [DOI] [PubMed] [Google Scholar]
- 11. Kimmerlin T., Namoto K., Seebach D., Helv. Chim. Acta 2003, 86, 2104–2109. [Google Scholar]
- 12. Ambrosi M., Fratini E., Alfredsson V., Ninham B. W., Giorgi R., Lo Nostro P., Baglioni P., J. Am. Chem. Soc. 2006, 128, 7209–7214. [DOI] [PubMed] [Google Scholar]
- 13. Köhler K., Förster G., Hauser A., Dobner B., Heiser U. F., Ziethe F., Richter W., Steiniger F., Drechsler M., Stettin H., J. Am. Chem. Soc. 2004, 126, 16804–16813. [DOI] [PubMed] [Google Scholar]
- 14. Iwaura R., Yoshida K., Masuda M., Yase K., Shimizu T., Chem. Mater. 2002, 14, 3047–3053. [Google Scholar]
- 15. Escamilla G. H., Newkome G. R., Angew. Chem. Int. Ed. Engl. 1994, 33, 1937–1940; [Google Scholar]; Angew. Chem. 1994, 106, 2013–2016. [Google Scholar]
- 16. Fuhrhop J. H., Fritsch D., Acc. Chem. Res. 1986, 19, 130–137. [Google Scholar]
- 17. Iwaura R., Yoshida K., Masuda M., Ohnishi-Kameyama M., Yoshida M., Shimizu T., Angew. Chem. Int. Ed. 2003, 42, 1009–1012; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1039–1042. [Google Scholar]
- 18. Jiang J., Meng Y., Zhang L., Liu M., J. Am. Chem. Soc. 2016, 138, 15629–15635. [DOI] [PubMed] [Google Scholar]
- 19. Nuraje N., Bai H., Su K., Prog. Polym. Sci. 2013, 38, 302–343. [Google Scholar]
- 20. Fuhrhop J.-H., Wang T., Chem. Rev. 2004, 104, 2901–2938. [DOI] [PubMed] [Google Scholar]
- 21. Nasir M. N., Crowet J.-M., Lins L., Akong F. O., Haudrechy A., Bouquillon S., Deleu M., Biochimie 2016, 130, 23–32. [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi H., Koumoto K., Jung J. H., Shinkai S., J. Chem. Soc. Perkin Trans. 2 2002, 1930–1936. [Google Scholar]
- 23. Muthuramu K., Ramnath N., Ramamurthy V., J. Org. Chem. 1983, 48, 1872–1876. [Google Scholar]
- 24. Kiskan B., Yagci Y., J. Polym. Sci. Part A 2007, 45, 1670–1676. [Google Scholar]
- 25. Yuan W., Zou H., Guo W., Wang A., Ren J., J. Mater. Chem. 2012, 22, 24783–24791. [Google Scholar]
- 26. McAlpine S. R., Garcia-Garibay M. A., J. Am. Chem. Soc. 1996, 118, 2750–2751. [Google Scholar]
- 27. Morales J. C., Penadés S., Angew. Chem. Int. Ed. 1998, 37, 654–657; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 673–676. [Google Scholar]
- 28. Kalbitzer H. R., Görler A., Li H., Dubovskii P. V., Hengstenberg W., Kowolik C., Yamada H., Akasaka K., Protein Sci. 2000, 9, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pizer R., Tihal C., Inorg. Chem. 1992, 31, 3243–3247. [Google Scholar]
- 30. Korich A. L., Iovine P. M., Dalton Trans. 2010, 39, 1423–1431. [DOI] [PubMed] [Google Scholar]
- 31. Santucci L., Gilman H., J. Am. Chem. Soc. 1958, 80, 193–196. [Google Scholar]
- 32. Snyder H., Konecky M. S., Lennarz W., J. Am. Chem. Soc. 1958, 80, 3611–3615. [Google Scholar]
- 33. Smith M. K., Northrop B. H., Chem. Mater. 2014, 26, 3781–3795. [Google Scholar]
- 34. Rambo B. M., Lavigne J. J., Chem. Mater. 2007, 19, 3732–3739. [Google Scholar]
- 35. Snyder H., Kuck J., Johnson J. R., J. Am. Chem. Soc. 1938, 60, 105–111. [Google Scholar]
- 36. Yonezawa N., Ikebe Y., Yoshida T., Hirai T., Saigo K., Hasegawa M., Bull. Chem. Soc. Jpn. 1984, 57, 1608–1611. [Google Scholar]
- 37. Bao C., Jiang Y. J., Zhang H., Lu X., Sun J., Adv. Funct. Mater. 2018, 28, 1800560. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary